

ABSTRACT
Mammalian cells are thought to protect themselves and their host organisms from DNA double strand breaks (DSBs) through universal mechanisms that restrain cellular proliferation until DNA is repaired. The Cyclin D3 protein drives G1-to-S cell cycle progression and is required for proliferation of immature T and B cells and of mature B cells during a T cell-dependent immune response. We demonstrate that mouse thymocytes and pre-B cells, but not mature B cells, repress Cyclin D3 protein levels in response to DSBs. This response requires the ATM protein kinase that is activated by DSBs. Cyclin D3 protein loss in thymocytes coincides with decreased association of Cyclin D3 mRNA with the HuR RNA binding protein that ATM regulates. HuR inactivation reduces basal Cyclin D3 protein levels without affecting Cyclin D3 mRNA levels, indicating that thymocytes repress Cyclin D3 expression via ATM-dependent inhibition of Cyclin D3 mRNA translation. In contrast, ATM-dependent transcriptional repression of the Cyclin D3 gene represses Cyclin D3 protein levels in pre-B cells. Retrovirus-driven Cyclin D3 expression is resistant to transcriptional repression by DSBs; this prevents pre-B cells from suppressing Cyclin D3 protein levels and from inhibiting DNA synthesis to the normal extent following DSBs. Our data indicate that immature B and T cells use lymphocyte lineage- and developmental stage-specific mechanisms to inhibit Cyclin D3 protein levels and thereby help prevent cellular proliferation in response to DSBs. We discuss the relevance of these cellular context-dependent DSB response mechanisms in restraining proliferation, maintaining genomic integrity, and suppressing malignant transformation of lymphocytes.
KEYWORDS: ATM, Cyclin D3, DNA damage response, HuR, lymphocytes
Introduction
External and internal stimuli regulate cellular proliferation. These stimuli control the activities of cyclin and cyclin-dependent kinase (CDK) proteins that function as heterodimeric enzymes to move cells through the cell cycle.1-3 Growth promoting stimuli activate G1 phase cyclin:CDK complexes to drive G1 progression and S phase entry. After DNA replication in S phase, other types of cyclin:CDK complexes transition cells through the G2 and M cell cycle phases where chromosomes are condensed and segregated into daughter cells, respectively. In response to DSBs or other types of damage, cells activate mechanisms to inhibit cyclin:CDK complexes and thereby block cell cycle progression at G1/S or G2/M checkpoints.4 The repair of damage inactivates these checkpoints, enabling resumption of cellular proliferation. The ability of cells to repress activities of cyclin:CDK complexes following stress is critical to maintain cellular viability and function and suppress malignant cellular transformation.4
Mitogenic stimuli cause proliferation of mammalian cells by inducing expression of one or more D-type cyclins, which bind and activate CDK4 and/or CDK6 to drive G1 phase progression and commit cells to enter the cell cycle.1-3 Cyclin D:CDK4/6 complexes phosphorylate the Retinoblastoma (Rb) family proteins that sequester and inhibit E2F transcription factors. Rb phosphorylation triggers E2F release, leading to E2F-mediated transcription of the Cyclin E gene. Resultant CyclinE:CDK2 complexes further phosphorylate Rb proteins to initiate a positive feedback loop that ultimately allows E2F proteins to transcribe S phase genes and/or phosphorylate DNA replication proteins. When Cyclin D:CDK4/6-dependent Rb phosphorylation passes a threshold, cells become irreversibly committed to exit G1 and transit through S, G2, and M phases. After this “restriction point,” D-type cyclins are no longer needed for cells to progress through the cell cycle and back into G1 phase. However, cells only commit to another cell cycle if D-type cyclin protein levels are re-induced in G1 phase by growth promoting stimuli. Observations that increased expression of D-type cyclins shortens G1 phase, occurs in diverse human cancers, and promotes malignant transformation of mouse cells highlight the major role of these proteins in driving cellular proliferation.1-3
The three mammalian D-type cyclin proteins (D1, D2, and D3) are expressed in overlapping tissue-specific and developmental stage-specific patterns and exhibit context-dependent redundant or unique functions in driving cell cycle progression.5 Analyses of mice with global inactivation of Cyclin D2 (Ccnd2−/− mice) or Cyclin D3 (Ccnd3−/− mice) established the paradigm for context-dependent roles of D-type cyclins in stimulating cellular proliferation. The most obvious phenotypes of Ccnd3−/− mice are reduced numbers of developing and mature T and B lineage lymphocytes6,7 and impaired ability of mature B cells to participate in a T cell-dependent immune response.8,9 Consistent with Cyclin D3 being the only D-type cyclin expressed in pro-T cells that have assembled Tcrβ genes, Ccnd3−/− mice exhibit reduced TCRβ-mediated cycling and expansion of thymocytes.7 Mice expressing a Cyclin D2 cDNA from the Ccnd3 locus have equivalently defective proliferation and expansion of pro-T cells as Ccnd3−/− mice,10 indicating that Cyclin D3 has unique function in directing cell cycle progression of Tcrβ-selected thymocytes. Although both Cyclin D2 and Cyclin D3 are expressed in pro-B cells that have assembled Igh genes and in IgH-selected large cycling pre-B cells, only Ccnd3−/− mice display impaired cycling and expansion of these types of immature B cells.6 Similarly, while both Cyclin D2 and Cyclin D3 are expressed in mature B cells, only B cells from Ccnd3−/− mice exhibit impaired ability to promote IgH class switch recombination and participate in a T cell-dependent immune response.8,9 The impaired proliferation of immature and mature B lymphocytes of Ccnd3−/− mice occurs despite compensatory increased Cyclin D2 protein levels in these cells,6 indicating that Cyclin D3 also has unique function in driving proliferation of B lineage lymphocytes at specific developmental stages. Notably, these above-mentioned studies revealed that Cyclin D3 is critical for expansion of lymphocytes during rapid bursts of proliferation associated with genetically programmed DSBs induced in G1 phase cells during antigen receptor gene rearrangements.
Mammalian cells protect themselves and their host organisms from DSBs through universal mechanisms that restrain cell cycle progression until DNA is repaired. Mammalian cells experiencing DSBs in G1 activate the ATM kinase to restrict S phase entry until DSBs are repaired or apoptosis is induced.4 In all non-malignant mammalian cell types analyzed, ATM activates complementary pathways that inhibit phosphorylation of CDK2 substrates and thereby block cell cycle progression in late G1 at the G1/S checkpoint. ATM inactivates the Cdc25a phosphatase that removes inhibitory phosphates from CDK2 proteins.4 ATM activates the p53 transcription factor, which transcriptionally induces expression of the p21 CDK inhibitor (CKI) that binds and inhibits Cyclin E:CDK2 complexes.4 The p53-independent arm of the G1/S checkpoint is activated more rapidly than the p53-dependent arm, which requires transcription and is more important for G1/S checkpoint maintenance.4 Despite complementary mechanisms to arrest cells with DSBs in G1, a significant fraction of G1 cells bearing DSBs enters S phase and repairs their DSBs in S phase or arrests at the G2/M checkpoint until DNA is repaired or apoptosis is induced.4 In non-lymphoid mammalian cells, ATM also helps prevent S phase entry in response to DSBs by stimulating Cyclin D1 proteolysis11-15 and possibly repressing transcription of the Cyclin D1 gene.16 In contrast to the canonical function of CKIs in inhibiting Cyclin E:CDK2 complexes, p21 and the related CKI, p27, promote the assembly and activation of Cyclin D:CDK4/6 complexes.17 Accordingly, increased Cyclin D1 proteolysis following DSBs may help block S phase entry by freeing CKIs to inhibit Cyclin E:CDK2 complexes and thereby rapidly trigger G1 arrest.11 Despite repressing Cyclin D1 expression and predominantly arresting in G1 phase, non-lymphoid cells that experience DSBs in G1 maintain Cyclin D2 and Cyclin D3 protein levels because these 2 cyclin proteins lack the amino acid motif that targets Cyclin D1 for proteolysis.11 This observation indicates that downregulation of Cyclin D2 and Cyclin D3 expression is not necessary for non-lymphoid cells to prevent S phase entry following DSBs, possibly reflecting tissue-specific functions of the D-type cyclins in directing cell cycle progression.
Since Cyclin D3 has unique roles in driving proliferation of developing B and T lymphocytes and of activated mature B cells that induce programmed DSBs in G1 phase, we investigated whether DSBs induced in these cells signal through ATM to repress Cyclin D3 expression and thereby help prevent S phase entry. Analogous to experiments on Cyclin D1 in non-lymphoid cells,11-15 we used ionizing radiation (IR) to induce DSBs. We found that DSBs signal through ATM to suppress Cyclin D3 protein levels in thymocytes and pre-B cells, but not mature B cells. We show that loss of Cyclin D3 protein in thymocytes coincides with decreased interaction between Cyclin D3 mRNA and the HuR RNA binding protein that is a downstream ATM effector. Thymocytes lacking HuR express lower than normal levels of Cyclin D3 protein but normal levels of Cyclin D3 mRNA, suggesting that thymocytes experiencing DSBs downregulate Cyclin D3 expression via ATM-dependent inhibition of Cyclin D3 mRNA translation. In contrast, pre-B cells experiencing DSBs downregulate Cyclin D3 expression through ATM-dependent transcriptional repression. Retrovirus-driven Cyclin D3 expression, which is refractory to transcriptional repression by DSBs, prevents pre-B cells from suppressing Cyclin D3 protein levels and from inhibiting DNA synthesis to the normal extent following DSBs. Our data indicate that developing B and T cells use lymphocyte lineage-specific and developmental stage-specific mechanisms to suppress Cyclin D3 expression and thereby help inhibit cell cycle progression in response to DSBs. We discuss the relevance of these cellular context-dependent DSB response mechanisms in restraining proliferation, maintaining genomic integrity, and suppressing malignant transformation of lymphocytes.
Results
Lymphocyte developmental stage-specific responses to IR suppress cyclin D3 expression
We sought to determine whether DSBs induced in proliferating immature B and T lymphocytes or mature B cells downregulate Cyclin D3 expression. We first studied primary thymocytes immediately isolated from mice, reasoning sufficient levels of Cyclin D3 protein is expressed in the subset of proliferating thymocytes to detect changes of Cyclin D3 expression in total thymocytes. As a means to induce DSBs, we exposed thymocytes to IR since this type of radiation induces DSBs and IR was used to demonstrate DSBs signal downregulation of Cyclin D1 protein levels in non-lymphoid cells.11-15 To circumvent potential loss of Cyclin D3 expression from DSB-induced apoptosis or DSB-induced feedback inhibition of Tcrβ recombination,18-20 we used mice expressing the pro-survival EμBCL2 transgene and the Vβ14NT pre-assembled functional Tcrβ gene. Although we could detect Cyclin D3 protein in total thymocytes, we found removal of these immature T cells from the thymus caused loss of Cyclin D3 protein even without IR exposure. Therefore, we instead exposed live mice to IR and waited up to 4 hours before harvesting thymocytes. We then performed Western blot analysis to measure Cyclin D3 protein levels in thymocytes isolated from unirradiated or irradiated EμBCL2:Vβ14NT/NT mice. We observed substantial reduction of Cyclin D3 protein 4 hours after IR (Fig. 1A). We confirmed that this response also occurs in thymocytes from wild-type mice (Fig. 1B). Our data suggest DSBs induced by IR in primary mouse thymocytes signal downregulation of Cyclin D3 expression.
Figure 1.

Lymphocyte developmental stage-specific downregulation of Cyclin D3 expression in response to DSBs. (A) Representative Western blot analyses of Cyclin D3 and actin protein and graphical quantification of Cyclin D3 protein expression in thymocytes of unirradiated or irradiated EμBCL2:Vβ14NT/NT mice at 4 hours after exposure of mice to 9 Gy of IR. Each data point is from a single mouse. This experiment was conducted 3 times with more than one unirradiated mouse and irradiated mouse each time. Error bars are standard error. **p < 0.01. (B) Representative Western blot analyses of Cyclin D3 and actin protein in thymocytes of unirradiated or irradiated wild-type mice at 4 hours after exposure of mice to 9 Gy of IR. (C) Representative Western blot analyses of Cyclin D3, Cyclin D2, and actin protein and graphical quantification of Cyclin D3 protein expression in unirradiated or irradiated IL7-cultured EμBCL2 pre-B cells at indicated times after exposure to 10 Gy of IR. Cyclin D3 expression levels are set to 1.0 for the unirradiated control in each experiment and other timepoints are normalized to control. (D) Graphical quantification of Cyclin D3 protein expression in unirradiated or irradiated IL7-cultured wild-type pre-B cells at indicated times after exposure to 10 Gy of IR. Cyclin D3 expression levels are set to 1.0 for the unirradiated control in each experiment and other timepoints are normalized to the control. (E) Representative Western blot analyses of Cyclin D3, Cyclin D2, and actin protein in unirradiated or irradiated LPS/IL4-stimulated EμBCL2 mature B cells at indicated times following their exposure to 9 Gy of IR. This experiment was conducted 3 times. (F) Representative Western blot analyses of Cyclin D3 and actin protein in IL7-cultured EμBCL2 pre-B cells untreated or 4 hours following addition of etoposide to media at a final concentration of 10 μg/mL. This experiment was conducted 3 times.
We next studied primary pre-B cells cultured from mouse bone marrow in media containing the IL7 cytokine, which promotes proliferation and expansion of IgH-selected large cycling pre-B cells.21 Although primary pre-B cells stimulated in vitro by IL7 express both Cyclin D3 and Cyclin D2 (Fig. 1C), Cyclin D3 has unique function in driving proliferation of these immature B cells.10 In EμBCL2 pre-B cells, we detected substantial reduction of mean Cyclin D3 protein levels at 4 and 6 hours after IR (Fig. 1C). We confirmed this response in wild-type pre-B cells (Fig. 1D). In contrast, we observed no decrease, but a modest increase, of Cyclin D2 protein (Fig. 1C). This increase likely is caused by loss of Cyclin D3 protein releasing CKI proteins to assemble CyclinD2:CDK4/6 complexes and thereby stabilize Cyclin D2 protein. Regardless, our data reveal IR exposure of cycling pre-B cells signals loss of Cyclin D3 but not Cyclin D2 protein, suggesting pro-T and pre-B cells have evolved mechanisms to downregulate expression of the D-type cyclin that has unique functions in driving their expansion.
Finally, we analyzed splenic B cells stimulated in vitro with LPS and IL4 since these mitogens drive proliferation and induce IgH class switch recombination. In stimulated EμBCL2 B cells, we found no change in mean Cyclin D3 protein levels at any time assayed following IR (Fig. 1E). These data suggest that DSBs induced in proliferating mature B cells during in vitro stimulation with LPS and IL4 do not repress Cyclin D3 protein levels. Collectively, our data indicate that immature T and B lymphocytes downregulate Cyclin D3 expression upon exposure to IR and, at least in B lineage cells, signal this response through developmental stage-specific mechanisms.
Although IR induces DSBs, this type of radiation also generates additional types of DNA and cellular damage. Thus, to demonstrate that DSBs downregulate Cyclin D3 expression, we assayed Cyclin D3 protein levels in pre-B cells exposed to the genotoxic drug etoposide, which causes only DSBs. We observed substantially decreased levels of Cyclin D3 protein at 4 hours after exposure to etoposide (Fig. 1F), indicating that DSBs suppress Cyclin D3 protein levels in primary pre-B cells. Therefore, we conclude that IR downregulates Cyclin D3 expression in immature T and B cells by inducing DSBs.
DSBs signal ATM-dependent repression of cyclin D3 protein levels through lineage-specific mechanisms in immature B and T cells
In all mammalian cells studied, DSBs generated by IR or other factors activate the ATM protein kinase, which controls gene expression via transcriptional, post-transcriptional, and post-translational mechanisms.22 Thus, we next evaluated whether DSBs induced by IR in immature T and B cells downregulate Cyclin D3 expression through ATM-dependent mechanisms. For this purpose, we generated and analyzed Atm−/− mice expressing the EμBCL2 transgene alone (for pre-B cells) or with a pre-assembled functional TCRβ gene (for thymocytes). We observed no difference in mean Cyclin D3 protein levels between thymocytes from irradiated or unirradiated EμBCL2:Vβ14NT/NT:Atm−/− mice (Fig. 2A) and between irradiated or unirradiated EμBCL2:Atm−/− pre-B cells (Fig. 2B). These data indicate that DSBs induced by IR in immature T and B cells signal via ATM to downregulate Cyclin D3 expression. We also performed qRT-PCR to quantify Cyclin D3 mRNA levels in irradiated and unirradiated thymocytes and pre-B cells. We detected no change of Cyclin D3 mRNA levels in thymocytes following IR of EμBCL2:Vβ14NT/NT or EμBCL2:Vβ14NT/NT:Atm−/− mice (Fig. 2C, D). In contrast, we observed lower Cyclin D3 mRNA levels after IR of EμBCL2 pre-B cells (Fig. 2E). Notably, this DSB-induced downregulation of Cyclin D3 mRNA was obvious at 30 minutes following IR and therefore preceded loss of Cyclin D3 protein. In addition, this response is ATM-dependent as evidenced by no change in Cyclin D3 mRNA levels after IR of EμBCL2:Atm−/− pre-B cells (Fig. 2F). We also observe loss of Cyclin D3 mRNA in total bone marrow cells following irradiation of wild-type mice (Fig. S1). These data indicate that thymocytes and pre-B cells utilize lymphocyte lineage-specific ATM-dependent mechanisms to downregulate Cyclin D3 expression in response to DSBs .
Figure 2.
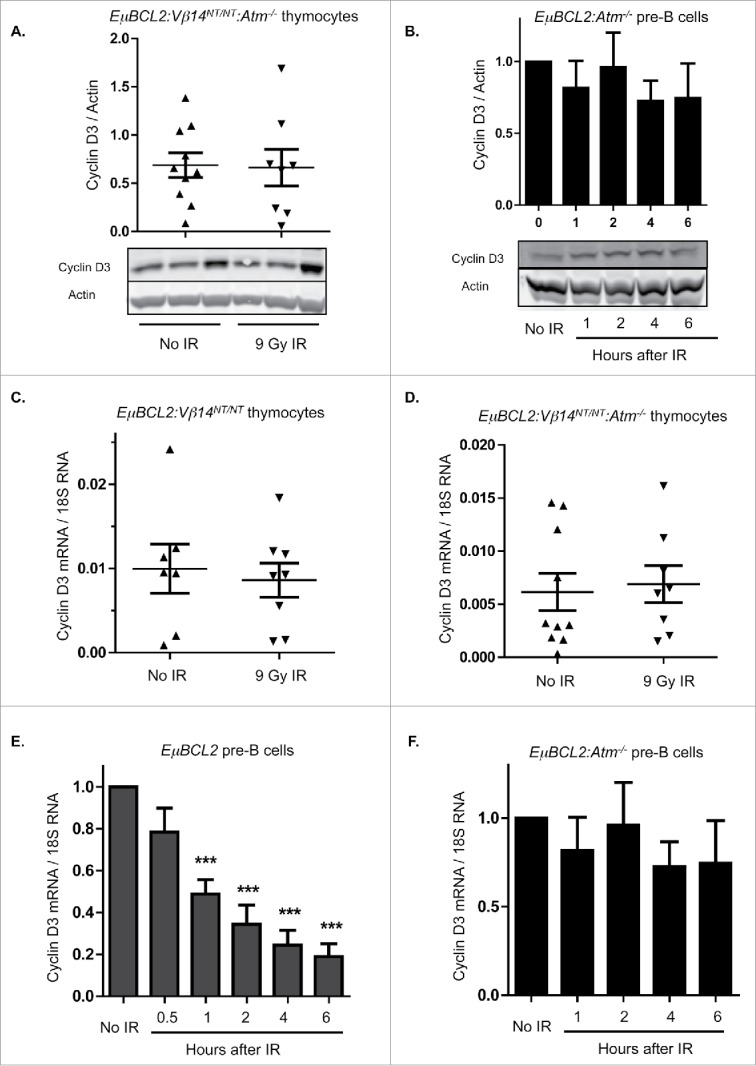
DSBs suppress Cyclin D3 expression via ATM-dependent lymphocyte lineage-specific mechanisms. (A) Representative Western blot analyses of Cyclin D3 and actin protein and graphical quantification of Cyclin D3 protein expression in thymocytes of unirradiated or irradiated EμBCL2:Vβ14NT/NT:Atm−/− mice at 4 hours following exposure of mice to 9 Gy of IR. Each lane contains thymocyte protein from a single mouse. This experiment was conducted 4 times with a total of 10 unirradiated and 8 irradiated mice. Error bars are standard error. (B) Representative Western blot analyses of Cyclin D3 and actin protein and graphical quantification of Cyclin D3 protein expression in unirradiated or irradiated IL7-cultured EμBCL2:Atm−/− pre-B cells at indicated times after exposure to 9 Gy of IR. This experiment was conducted 4 times with more than one unirradiated mouse and more than one irradiated mouse each time. Error bars are standard error. ((C)- D) Graphical quantification of Cyclin D3 mRNA levels relative to 18S RNA levels in thymocytes from unirradiated or irradiated EμBCL2:Vβ14NT/NT mice (C) or EμBCL2:Vβ14NT/NT:Atm−/− (D) mice at 4 hours after exposure to 9 Gy IR. Each data point is from a single mouse. Each of these experiments was conducted 4 times with a total of 10 unirradiated and 8 irradiated mice. Error bars are standard error. (E- F) Graphical quantification of Cyclin D3 mRNA levels relative to 18S RNA levels in unirradiated or irradiated IL7-cultured EμBCL2 (E) or EμBCL2:Atm−/− (F) pre-B cells at indicated times after exposure to 10 Gy of IR. Cyclin D3 expression levels are set to 1.0 for the unirradiated control in each experiment and other time points are normalized to this control. These experiments were performed 4 times. Error bars are standard error. ***p < 0.001 in comparison to the unirradiated control.
DSBs induced in thymocytes downregulate cyclin D3 protein levels and cellular proliferation by disrupting association of the HuR ATM substrate with cyclin D3 mRNAs
Our data indicate DSBs induced in thymocytes downregulate Cyclin D3 expression by signaling through ATM to block translation of Cyclin D3 mRNAs and/or increase degradation of Cyclin D3 protein. The need to irradiate live mice to study DSB-induced repression of Cyclin D3 expression in thymocytes is an obstacle to evaluate the effects of DSBs on translation of Cyclin D3 mRNA and stability of Cyclin D3 protein. To overcome this obstacle, we attempted to identify immature T lymphocyte cell lines that express Cyclin D3 and suppress Cyclin D3 protein levels in response to DSBs. Although we identified cell lines that express Cyclin D3 protein, we discovered they all maintained Cyclin D3 protein following exposure to IR (unpublished observations, A.D. and C.H.B.). Therefore, we employed a candidate approach to test roles of known ATM effectors in blocking translation of Cyclin D3 mRNAs following induction of DSBs. In response to DSBs induced by IR, ATM promotes phosphorylation of the HuR RNA binding protein to regulate association of HuR with its target mRNAs.23 HuR functions in context-dependent manners to positively or negatively regulate stability and/or translation of its target mRNAs.24 In primary human T cells, HuR binds and stabilizes Cyclin D3 mRNAs to enhance Cyclin D3 protein expression.25 To measure HuR association with Cyclin D3 mRNAs in primary mouse immature T cells, we conducted RNA immunoprecipitation (RIP) using an anti-HuR antibody or a control antibody and then performed qRT-PCR to quantify Cyclin D3 mRNA precipitated from thymcytes by each antibody. We detected greater association between HuR and Cyclin D3 mRNAs in unirradiated thymocytes as compared to irradiated thymocytes (Fig. 3A). These data are consistent with a model that HuR binds and stimulates translation of Cyclin D3 mRNAs in unirradiated thymocytes and reduced association of HuR with Cyclin D3 mRNAs after IR-induced DSBs leads to downregulation of Cyclin D3 protein levels.
Figure 3.

DSBs induced in thymocytes inhibit Cyclin D3 protein levels and cellular proliferation by disrupting association of HuR with Cyclin D3 mRNAs. (A) Graph quantifying association of HuR with Cyclin D3 mRNAs in thymocytes of unirradiated or irradiated EμBCL2:Vβ14NT/NT mice at 4 hours after exposure to 10 Gy of IR. Fold enrichment is the amount of Cyclin D3 mRNA relative to the amount of Gapdh mRNA precipitated by the anti-HuR antibody normalized to the amount of Cyclin D3 mRNA relative to the amount of Gapdh mRNA precipitated by normal IgG antibody. Each data point is from a single mouse. This experiment was conducted 3 times. Error bars are standard error. *p < 0.05. (B-C) Graphs depicting the average numbers of thymocytes (B) and splenic αβ T cells (C) in HuRf/f and HuRΔ/Δ mice. This experiment was conducted 5 times. Error bars are standard error. ***p < 0.001. (D) Representative flow cytometry analysis and graphical quantification of thymocytes at the DN3 and DN4 stages in HuRf/f and HuRΔ/Δ mice. This experiment was conducted 5 times. Error bars are standard error. *p < 0.05. (E) Representative flow cytometry analysis and graphical quantification of thymocytes at the DN, DP, and CD4+ or CD8+ SP stages in HuRf/f or HuRΔ/Δ mice. This experiment was conducted 5 times. Error bars are standard error. *p < 0.05 and ***p < 0.001. (F) Representative Western blot analyses of HuR, Cyclin D3, and actin protein and graphical quantification of Cyclin D3 protein expression in thymocytes of HuRf/f or HuRΔ/Δ mice. This experiment was conducted 4 times on a total of 8 HuRf/f and 10 HuRΔ/Δ mice. Error bars are standard error. *p < 0.05. (G) Graphical quantification of Cyclin D3 mRNA levels relative to 18S RNA levels in thymocytes from HuRf/f and HuRΔ/Δ mice. Each data point is from a single mouse. This experiment was conducted twice with 3 mice of each genotype each time.
As a means to test whether HuR binding to Cyclin D3 mRNAs regulates Cyclin D3 protein levels in immature T cells, we generated and analyzed mice with LckCre-mediated conditional inactivation of HuR initiating in CD4−CD8− (“double-negative” or DN) thymocytes concomitant with Tcrβ recombination. We used this approach since inherited HuR inactivation in mice is embryonic lethal.26 The assembly and expression of functional Tcrβ genes in CD25−CD117+CD4−CD8− (DN3) thymocytes activates signals that promote differentiation of these cells into CD25−CD117−CD4−CD8− (DN4) thymocytes and then into CD4+CD8+ (DP) thymocytes.27 Tcrβ expression in DN3 cells also activates Cyclin D3 expression to drive proliferation and expansion as these cells develop into DP thymocytes.7 Following arrest in G1 phase, DP thymocytes can further differentiate without proliferation into CD4+ or CD8+ (SP) thymocytes, which exit the thymus as quiescent mature αβ T cells.27 We performed cell counting and flow cytometry analyses for T cell markers on thymocytes and splenocytes of 4 to 6 weeks old Lck-Cre+HuRflox/flox (HuRΔ/Δ) and littermate HuRflox/flox (HuRf/f) mice. Compared to HuRf/f mice, HuRΔ/Δ mice have ∼3-fold and ∼4.5-fold lower numbers of total thymocytes and splenic αβ T cells, respectively (Fig. 3B, C). In addition, HuRΔ/Δ mice contain ∼2-4-fold decreased numbers of thymocytes at the DN4 stage (Fig. 3D) and the DP and SP stages (Fig. 3E). Western blot analysis confirmed loss of HuR protein in HuRΔ/Δ thymocytes (Fig. 3F). These data demonstrate that inactivation of HuR leads to reduced numbers of T lineage cells at all developmental stages beyond the DN3 thymocyte stage, similar to inactivation of Cyclin D3.7 We conducted Western blot and qRT-PCR analyses to quantify levels of Cyclin D3 protein and mRNA, respectively, in HuRΔ/Δ and HuRf/f thymocytes. We detected a substantially lower level of Cyclin D3 protein (Fig. 3F) but an equivalent amount of Cyclin D3 mRNA in HuRΔ/Δ thymocytes relative to HuRf/f thymocytes (Fig. 3G). These data indicate that HuR is required to maintain normal levels of Cyclin D3 protein but not Cyclin D3 mRNA in developing T cells. Notably, the levels of Cyclin D3 protein expressed in unirradiated HuRΔ/Δ thymocytes and irradiated wild-type thymocytes are comparable (Compare Fig. 3F with Fig. 1A). Accordingly, our data are consistent with a model where HuR binds Cyclin D3 mRNAs in unirradiated thymocytes to increase Cyclin D3 protein levels, helping drive expansion of immature T cells; while DSBs in thymocytes signal ATM-dependent loss of HuR binding to Cyclin D3 mRNAs, decreasing Cyclin D3 protein levels to help prevent S phase entry and DNA synthesis until DSBs are repaired.
DSBs induced in immature B Cells downregulate cyclin D3 protein levels via ATM-dependent repression of Ccnd3 transcription
While our data indicate DSBs induced in pre-B cells repress Cyclin D3 expression by signaling through ATM to block transcription of the Ccnd3 gene and/or stimulate degradation of Cyclin D3 mRNA, our results do not rule out a contribution of increased Cyclin D3 proteolysis. Therefore, to elucidate mechanisms by which DSBs signal ATM-dependent downregulation of Cyclin D3 expression in pre-B cells, we first evaluated the effects of IR-induced DSBs on Cyclin D3 protein turnover in EμBCL2 pre-B cells. For this purpose, we added the protein synthesis inhibitor cycloheximide to primary pre-B cell cultures immediately following IR. We then conducted Western blot analysis to measure the amount of Cyclin D3 protein remaining over time in unirradiated and irradiated cells. At all time points assayed, we detected equivalent levels of Cyclin D3 protein in irradiated and unirradiated pre-B cells (Fig. 4A), indicating that pre-B cells do not increase Cyclin D3 proteolysis in response to IR-induced DSBs. We next utilized Click-iT® mRNA capture to measure turnover of Cyclin D3 mRNA in irradiated and unirradiated pre-B cells. Click-iT® mRNA capture incorporates ethylene uridine (EU) into RNAs during transcription; then EU-labeled RNAs are conjugated with biotin, isolated by streptavidin beads, and quantified by qRT-PCR. We cultured EμBCL2 pre-B cells in media with EU for 16 hours to incorporate EU into existing mRNAs. We then washed out EU immediately before IR so that we could measure the amount of Cyclin D3 mRNA remaining over time in irradiated and unirradiated pre-B cells. At all times assayed, we detected no significant difference in the levels of labeled Cyclin D3 mRNAs between irradiated and unirradiated cells (Fig. 4B), indicating pre-B cells do not increase turnover of Cyclin D3 mRNA in response to DSBs generated by IR. Finally, we utilized Click-iT® mRNA capture to measure the effect of IR-induced DSBs on transcription of Ccnd3. We added EU to IL7-cultured EμBCL2 pre-B cells immediately after IR. Since EU-labeled RNA is only generated following IR in this experiment, we were able to quantify the effect of IR on Ccnd3 transcription. As a positive control, we quantified the amount of p21 mRNA synthesized since DSBs stimulate transcription of the p21 gene and this response is diminished in Atm-deficient lymphocytes.28 At all times assayed, we detected reduced levels of new Cyclin D3 mRNAs in irradiated cells as compared to unirradiated cells (Fig. 4C), demonstrating pre-B cells repress Ccnd3 transcription in response to DSBs arising from IR. This DSB response is ATM-dependent as evidenced by no difference in the levels of new Cyclin D3 mRNAs at all times assayed between irradiated and unirradiated EμBCL2:Atm−/− pre-B cells (Fig. 4C). Notably, DSB-induced repression of Cyclin D3 mRNA synthesis was obvious 30 minutes following IR and therefore precedes loss of Cyclin D3 mRNA levels (Compare Fig. 4C and Fig. 2D), which in turn occurs before loss of Cyclin D3 protein levels (Compare Fig. 4C and Fig. 1B). Consequently, our data suggest that repression of Ccnd3 transcription is the mechanism by which pre-B cells signal via ATM to downregulate Cyclin D3 expression in response to DSBs.
Figure 4.

DSBs induced in pre-B Cells suppress Cyclin D3 protein levels through ATM-dependent inhibition of Ccnd3 transcription. (A) Western blot quantification of Cyclin D3 protein levels relative to actin protein levels in unirradiated or irradiated IL7-cultured EμBCL2 pre-B cells at indicated times after addition of cycloheximide and/or exposure to 10 Gy of IR. This experiment was performed 6 times. Values are normalized to 1.0 for unirradiated cells immediately after cycloheximide addition within each experiment. (B) qRT-PCR quantification of EU-labeled Cyclin D3 mRNA levels relative to EU-labeled 18S RNA levels in unirradiated or irradiated IL7-cultured EμBCL2 pre-B cells at indicated times after EU washout and/or exposure to 10 Gy of IR. Values are normalized to 1.0 for cells immediately before IR within each experiment. This experiment was performed 4 times. (C) qRT-PCR quantification of EU-labeled Cyclin D3 and p21 mRNA levels relative to total/EU-labeled Hprt levels in unirradiated and irradiated IL7-cultured EμBCL2 and EμBCL2:Atm−/− pre-B cells at indicated times after addition of EU and exposure to 10 Gy of IR. Data are presented as the ratio of relative levels of each mRNA in irradiated cells compared to unirradiated cells. The dotted line represents a value of 1, which would indicate that IR had no effect on the transcription rate of an assayed gene(s). This experiment was performed 3 times. Error bars are standard error. Statistical analysis using the 2-way ANOVA method indicates all differences are significant (p < 0.001).
Retroviral expression of cyclin D3 in Pre-B cells is resistant to transcriptional repression and prevents normal inhibition of DNA synthesis in response to DSBs
To validate that repression of Ccnd3 transcription is the mechanism by which IR-induced DSBs signal downregulation of Cyclin D3 expression, we investigated the effect of DSBs on Cyclin D3 protein levels in pre-B cells expressing the Cyclin D3 cDNA from a heterologous promoter that is not subject to transcriptional repression upon DSBs. For this purpose, we transduced IL7-cultured EμBCL2:Ccnd3−/− pre-B cells with a MIGR1 retrovirus carrying a full-length Ccnd3 cDNA expressed by the retroviral promoter (MIGR-D3) or with an empty MIGR1 retrovirus. Transcripts from each of these viruses contain an IRES-GFP so transduced cells can be monitored by GFP expression. We conducted Western blot analysis to monitor Cyclin D3 protein in unirradiated and irradiated EμBCL2:Ccnd3−/− pre-B cells transduced with MIGR1-D3. We observed equivalent levels of Cyclin D3 protein between unirradiated and irradiated cells (Fig. 5A), indicating that DSBs generated by IR in pre-B cells cannot repress Cyclin D3 expression when the MIGR1 retroviral promoter drives transcription of the Ccnd3 cDNA. These data demonstrate that suppression of Ccnd3 transcription is the mechanism by which pre-B cells downregulate Cyclin D3 expression in response to genotoxic DSBs.
Figure 5.

Retroviral expression of Cyclin D3 in pre-B cells prevents repression of Cyclin D3 Protein and impairs G1 arrest in response to DSBs. (A) Representative Western blot analyses of Cyclin D3 and actin protein in unirradiated or irradiated IL7-cultured EμBCL2:Ccnd3−/− pre-B cells transduced with MIGR1 or MIGR1-D3 at indicated times following exposure to 10 Gy of IR. (B) Representative flow cytometry analysis of the cell cycle of unirradiated or irradiated IL7-cultured EμBCL2 pre-B cells transduced with MIGR1 or MIGR1-D3 at 6 hours after exposure to 10 Gy of IR. (C) Graph depicting quantification of the fraction of unirradiated or irradiated IL7-cultured EμBCL2 pre-B cells transduced with MIGR1 or MIGR1-D3 that are BrdU+ at 6 hours following IR. This experiment was conducted 3 times. Error bars are standard error. *p < 0.05. (D) Graph depicting the ratios of the fraction of each cell type in B and C that synthesizes DNA after IR vs. before IR. Error bars are standard error. *p < 0.05.
Our findings are consistent with a model wherein DSB-induced downregulation of Cyclin D3 protein expression helps prevent immature T and B cells from entering S phase. To test this model, we evaluated the effect of DSBs on DNA synthesis in pre-B cells incapable of inhibiting Cyclin D3 protein levels following DSB. Notably, this approach was used to demonstrate that DSB-induced repression of Cyclin D1 protein expression helps prevent non-lymphoid cells with DSBs from entering S phase.11 For our experiment, we transduced IL7-cultured EμBCL2 pre-B cells with MIGR1-D3 or MIGR1 before IR exposure. We then conducted cell cycle analysis, assessing BrdU incorporation by flow cytometry in conjunction with DNA content staining to identify cells in G1, S, or G2/M phases. We observed that transduction of MIGR-D3 caused a 1.5-fold higher fraction of unirradiated pre-B cells to synthesize DNA in a 6 hour time period following IR (Fig. 5B, C). However, we also observed that MIGR-D3 transduction enabled ∼2-fold greater fraction of irradiated cells to synthesize DNA (Fig. 5B, C), and therefore have entered S phase. Since the ratio of the fraction of irradiated versus unirradiated GFP+ pre-B cells that incorporate BrdU is ∼2-fold greater for cells transduced with MIGR-D3 relative to MIGR (Fig. 5D), the greater DNA synthesis in MIGR-D3 pre-B cells following IR is not strictly from a higher basal rate of S phase entry. Therefore, these data are consistent with the prediction of our model that an inability of pre-B cells to downregulate Cyclin D3 protein expression in response to DSBs increases the frequency at which pre-B cells with DSBs enter S phase.
Discussion
The Cyclin D3 protein has a unique function in driving expansion of developing B and T lineage lymphocytes during rapid bursts of proliferation associated with DSBs induced during antigen receptor gene rearrangements in G1 phase cells. We sought to determine whether these cell types repress Cyclin D3 expression in response to DSBs. We have shown that proliferating thymocytes and pre-B cells, but not mature B cells stimulated by LPS and IL4, downregulate Cyclin D3 protein levels in response to DSBs generated by ionizing radiation or etoposide. These data indicate that pre-B cells use a B lineage developmental stage-specific mechanism to inhibit Cyclin D3 expression following the incursion of DSBs. Although cycling pre-B cells and thymocytes each require ATM kinase to suppress Cyclin D3 expression, these cell types use distinct lineage-specific mechanisms to control this shared DSB response. Pre-B cells inhibit Ccnd3 transcription without altering turnover of Cyclin D3 mRNA or protein. In contrast, thymocytes downregulate Cyclin D3 protein levels, at least in part by reducing association of Cyclin D3 mRNAs with the HuR RNA binding protein that is regulated by ATM. We also have shown in pre-B cells that ectopic Cyclin D3 expression from a retroviral promoter is refractory to repression by DSBs, which in turn prevents pre-B cells from suppressing Cyclin D3 protein levels and from normally blocking DNA synthesis in response to ionizing radiation. Our findings indicate that immature B and T cells have evolved lymphocyte lineage-specific, ATM-dependent mechanisms to suppress Cyclin D3 expression and thereby help regulate G1-to-S phase progression in response to DSBs. Considering that Cyclin D3 expression causes translocations in developing B and T cells of Atm−/− mice by driving aberrant repair of DSBs induced in G1 phase with DSBs arising in S phase,19 these ATM-dependent mechanisms are likely important for maintaining genomic stability of immature B and T lymphocytes. However, directly testing this notion requires development of experimental models with specific disruption of lymphocyte lineage-specific mechanisms that downregulate Cyclin D3 protein expression in response to DSBs.
Our findings provide new insights into ATM-dependent mechanisms by which immature B and T lymphocytes arrest in G1 following DSBs and thereby restrict the frequency of cells that enter S phase and replicate broken DNA. Mouse thymocytes lacking the p21 and p27 CKIs express lower than wild type levels of Cyclin D3 protein and do not assemble and activate Cyclin D3:CDK6 complexes,29 indicating that p21 and p27 normally bind Cyclin D3 and CDK4/6 in developing T cells. Accordingly, ATM-dependent downregulation of Cyclin D3 protein in immature B and T cells experiencing DSBs likely helps suppress CDK2-driven S phase entry by promoting re-localization of p21 and p27 from Cyclin D3:CKD2 complexes to Cyclin E:CDK2 complexes. It has been known for years that developing B and T cells and all other cells experiencing DSBs use complementary ATM-dependent mechanisms to inhibit CDK2 activity and thereby prevent G1-to-S progression.4 These mechanisms involve rapid post-translational inactivation of Cdc25a to trigger G1 arrest at the G1/S checkpoint and slower transcriptional activation of p21 to maintain this checkpoint.4 Our data suggest that pre-B cells and thymocytes utilize ATM-dependent downregulation of Cyclin D3 protein levels as another mechanism to rapidly inhibit CDK2 activity and thereby prevent S phase entry following DSBs. This loss of Cyclin D3 protein also would decrease the number of Cyclin D3:CDK4/6 complexes available to phosphorylate Rb and thereby drive G1 phase progression and S phase entry. Thus, we propose that ATM-dependent downregulation of Cyclin D3 expression also helps antagonize S phase entry and replication of broken DNA by providing more time in G1 for additional ATM-dependent pathways to inhibit Cyclin E:CDK2 complexes and thereby activate the G1/S cell cycle checkpoint. This model introduces the concept that developing B and T cells have evolved complementary and non-redundant DNA damage response mechanisms to rapidly inhibit G1-to-S phase progression.
Our discovery that immature B and T lymphocytes employ distinct mechanisms to downregulate Cyclin D3 expression in response to DSBs illustrates another difference between the genetic programs of developing B and T cells. The assembly and expression of functional Igh or Tcrβ genes in pro-lymphocytes activates Cyclin D3 expression to drive these cells into S phase and through additional G1-to-S transitions as they differentiate into pre-lymphocytes.6,7 Igh expression in pro-B cells and pre-B cells inhibits Cyclin D3 proteolysis,6 whereas Tcrβ expression in pro-T cells stimulates Ccnd3 transcription.7 We have provided evidence that DSBs induced in pre-B cells downregulate Ccnd3 transcription, while DSBs induced in thymocytes inhibit translation of Cyclin D3 mRNAs. Assuming no post-translational regulation of Cyclin D3 protein in our experimental conditions, our findings imply that immature T cells upregulate slower and downregulate faster Cyclin D3 activity than immature B cells. It is possible that there is no significance that immature B and T cells evolved distinct mechanisms to control Cyclin D3 protein levels and thereby help regulate G1 progression and S phase commitment. Alternatively, factors unique to developing T cells might have selected for mechanisms that more slowly increase and more rapidly downregulate Cyclin D3 protein levels. For example, transcriptional activation of Cyclin D3 might be important to provide Tcrβ-selected pro-T cells more time in G1 phase to repair DSBs induced during Tcrγ and Tcrδ gene assembly, while post-transcriptional inactivation of Cyclin D3 could be necessary to antagonize G1 progression of Tcrβ-selected pro-T cells that initiate Tcrγ and Tcrδ recombination before cessation of V(D)J recombination concomitant with S phase entry.
Our demonstration that expression of Cyclin D3 from a heterologous promoter prevents pre-B cells from suppressing Cyclin D3 protein levels and normally halting S phase entry in response to DSBs has novel implications for how CCND3 alterations might promote human lymphoid malignancies. Translocations that fuse CCND3 to IgH or other genes are observed in human B lineage lymphomas including Mantle Cell Lymphoma, Multiple Myeloma, and Diffuse Large B Cell Lymphoma.30-32 These translocations have been proposed to drive cancer by accelerating proliferation through transcriptional upregulation of CYCLIN D3 protein. Considering that B lineage cancers with CCND3 translocations harbor additional genomic lesions arising from aberrant repair of DSBs, our data suggest that formation of CCND3 translocations in immature B cells may lead to mature B cell malignancies by disrupting regulation of G1-to-S progression in developing B cells. CCND3 mutations that generate highly stable, C-terminal truncated CYCLIN D3 proteins and drive cell cycle progression are common in human Burkitt's Lymphoma.33 The ability of immature B cells to rapidly trigger the G1/S checkpoint by repressing Ccnd3 transcription likely depends on the relative short half-life of Cyclin D3 protein in these cells. If carboxy-truncating CCND3 mutations arise in immature B lymphocytes, they also might cause mature B cell malignancies by disrupting the control of S phase entry during B cell development. Although we have demonstrated that immature B and T lymphocytes suppress Cyclin D3 expression in response to DSBs, it is conceivable that these cells also inactivate Cyclin D3 or Cyclin D3:CDK4/6 complexes prior to downregulation of Cyclin D3 protein levels. In support of this possibility, the carboxy-terminus of Cyclin D3 contains 2 conserved motifs that are consensus target sites for the ATM kinase. Future experiments will be needed to determine whether immature and even mature B and T lymphocytes also regulate G1-to-S progression via ATM-mediated post-translational modification of Cyclin D3 protein.
Materials and methods
Mice
All mice were on a 129S1/SvImJ and C57BL6 mixed background, and housed, bred, and used under pathogen-free conditions at the Children's Hospital of Philadelphia (CHOP). We made the genetically modified mouse strains for this study by breeding mice containing the EμBCL2 transgene,34 Vβ14NT pre-assembled functional Tcrβ gene,35 LckCre transgene,36 Atm−/− alleles,37 HuRflox/flox alleles,26 and/or Ccnd3−/− alleles.7 All experiments were performed on 4 to 6 week old mice. Similar numbers of male and female mice were analyzed for each experiment. For some experiments, mice were placed in a pie restrainer (Braintree) and exposed to 9 Gy of ionizing radiation. Mice were returned to their holding cages for indicated times before being euthanized and analyzed. Animal husbandry and experiments were performed in accordance with national guidelines and regulations and approved by the CHOP Institutional Animal Care and Use Committee.
Western blotting
Cells were resuspended in a Tween-20 containing lysis buffer and sonicated at intervals of 30 seconds on and 30 seconds off for 5 minutes at 4°C. Cells were incubated for 5 minutes on ice and then spun to remove insoluble material. 100 μg of lysates prepared under reducing conditions were loaded in each well of a NuPage 10% Bis-Tris gel (Life Technologies). Electrophoresed proteins were transferred to PVDF. These membranes were blocked with Odyssey blocking buffer (Li-Cor) and then incubated with anti-Cyclin D3 (Santa Cruz polyclonal C-16), anti-Cyclin D2 (Santa Cruz, polyclonal M-20), anti-actin (Sigma polyclonal), or anti-HuR (Santa Cruz 3A2) antibodies. After washing, blots were incubated with appropriate IRDye800 secondary antibodies (Li-Cor). Following washing, blots were scanned on an Odyssey infrared scanner (Li-Cor).
B lineage cell culture
In vitro stimulations were performed in media consisting of RPMI-1640 supplemented with 10% heat-inactivated FBS, antibiotics, 50 μM β-mercaptoethanol, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, and non-essential amino acids. For IL-7 pre-B cell cultures, bone marrow was harvested from mice of indicated genotypes and cultured at a density of 5 × 106 per ml in medium containing 5 ng/ml IL-7 for 4-5 d. Cells were re-plated in fresh IL-7 containing medium each day except for day 2 (48h after harvest). For mature B cell assays, splenic B cells were isolated using EasySep negative selection B cell isolation kits (Stem Cell Technologies). Cells were stimulated at a density of 1 × 106 per ml in media containing 25 μg/ml LPS (0111:B4, Sigma) and 80 ng/ul recombinant mouse IL-4 (R&D Systems) for 3 d. To induce DSBs, cells were exposed to 10 μg/ml etoposide (Sigma), 10 Gy IR, or other noted doses of IR.
qRT-PCR
Total cellular RNA was isolated using Trizol reagent (Life technologies) and DNase treated according to manufacturer directions (Promega), primed with random nonamer (New England Biolabs), and reverse transcribed with M-MuLV (NEB). qRT-PCR reactions were performed with SYBR green mastermix (Applied Biosystems) and run on an Applied Biosystems 7500 Fast machine. The primers used for qRT-PCR reactions are: Cyclin D3 forward 5′-AGGAGATCAAGCCGCACATG-3′, Cyclin D3 reverse 5′-GGTAGTTCATAGCCAGAGGGAAGA-3′, p21 forward 5′-GACATTCAGAGCCACAGGCAC-3′, p21 reverse 5′-GTCAAAGTTCCACCGTTCTCG-3′, actin forward 5′- TCATCACTATTGGCAACGAGCGGTTC-3′, and actin reverse 5′- TACCACCAGACAGCACTGTGTTGGCA-3′.
RNA-Immunoprecipitation
RNA-Immunoprecipitation for HuR was performed using published procedures.38 Briefly, 100 μl of protein G dynabeads (Life Technologies) were incubated with 15 μg anti-HuR (3A2, Santa Cruz) or 15 μg normal mouse IgG (Santa Cruz). Thymocytes were lysed in polysome lysis buffer containing 0.5% NP-40 supplemented with protease inhibitor (Roche) and RNase inhibitor (NEB). Half of each sample lysate was added to dynabeads coated with one of the antibodies and incubated with shaking for 2 hours at 4°C. After washing beads, RNA was extracted using Trizol and then analyzed by qRT-PCR.
Flow cytometry
Single cell suspensions were prepared from thymuses or spleens of mice as described.39 Cells were stained with antibodies against surface antigens in PBS with 3% FBS and then washed before flow cytometry. The antibodies used from BD PharMingen were: anti-TCRβ (553172), anti-CD4 (553653), anti-CD8a (553033), anti-CD25 (552880), anti-CD117 (553356). Samples were run on a FACSCalibur or LSR Fortessa cytometer (BD Biosciences) and analyzed with Flowjo software (Treestar). BrdU incorporation assays were performed by incubating cells in medium containing 10 μM BrdU for indicated amounts of time before fixing and staining as instructed (BD Biosciences).
Click-It nascent RNA labeling
Click-it nascent RNA labeling kits were obtained from Life Technologies. For mRNA turnover assays, ethynyl uridine (EU) was added to medium of IL-7 cultures at a final concentration of 0.2 mM for the last 16 hours of culture time. Immediately before irradiation, the cells were washed and placed into media lacking EU. Cells were collected for RNA isolation immediately following EU removal or at the indicated times after EU removal. For transcriptional assays, cells were grown in media lacking EU. Cells were either irradiated or left unirradiated. Immediately following irradiation of half the cells, EU was added to the media of all cells at a final concentration of 0.5 mM. After the indicated time, cells were collected and RNA was isolated using Trizol reagent as described above. Click chemistry and streptavidin pull down of EU-labeled RNA were performed per kit instructions. This RNA was analyzed by qRT-PCR as outlined above.
Cycloheximide chase assays
Cycloheximide chase assays were performed on IL-7 pre-B cell cultures by treating cells with 10 Gy IR or no IR and then immediately adding cycloheximide (Sigma) to culture media at a final concentration of 100 ng/ml. Cells were harvested for Western blots immediately after addition of cycloheximide (t = 0) or at indicated times after cycloheximide addition.
Cloning and retroviral transduction
The full-length Ccnd3 cDNA including its 3′UTR (NM_007632.2) was cloned into the MIGR1 retroviral vector40 by PCR amplification using BAC RP-160K24 as template. EcoRI restriction sites were included on the end of each PCR primer. PCR products and MIGR1 were digested with EcoRI and ligated together. Individual clones were screened for correct orientation and sequenced to ensure integrity of the PCR-amplified Ccnd3 cDNA. Viral supernatants were generated by co-transfection of 293T cells with either MIGR1 or MIGR1-Ccnd3 along with helper plasmids pCGP and pHIT123. IL-7 pre-B cultures were transduced by spinfection at 48 hours and 72 hours after being cultured from bone marrow. Spinfections were performed as described40 with final concentrations of 10 μg/ml polybrene. Experiments were performed on these cells 48 hours following the second spinfection
Statistics
Except where indicated in figure legends, all p-values were generated by 2-tailed unpaired Student's t test using Prism (GraphPad Software).
Supplementary Material
Disclosure of potential conflicts of interest
C.H.B. is a consultant for Regeneron Pharmaceuticals. None of the other authors have any conflicts of interest.
Acknowledgments
The authors thank Marta Rowh, Laura Vaites, Alan Diehl, and Warren Pear for providing advice and reagents.
Funding
This research was supported by the Training Program in Cell and Molecular Biology 5T32GM007229 of the University of Pennsylvania (A.D. and A.R.-R.); NCI F31 Pre-doctoral Fellowship CA177092, and a Patel Family Scholar Award of the Abramson Cancer Center of the Perelman School of Medicine at the University of Pennsylvania (A.D.); the Rheumatology Training Grant T32 AR007442 of the University of Pennsylvania, and F31 Predoctoral Fellowship CA183551 (M.R.F.); and the Department of Pathology and Laboratory Medicine of the Children's Hospital of Philadelphia, a Leukemia and Lymphoma Society Scholar Award, a grant from the W.W. Smith Charitable Trust, a grant from the William Lawrence and Blanche Hughes Foundation, and NIH R01 grants CA125195, CA136470, and AI112621 (C.H.B).
References
- [1].Sherr CJ, Roberts JM. Living with or without cyclins and cyclin-dependent kinases. Genes Dev 2004; 18(22):2699-711; PMID:15545627; http://dx.doi.org/ 10.1101/gad.1256504 [DOI] [PubMed] [Google Scholar]
- [2].Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 2009; 9(3):153-66; PMID:19238148; http://dx.doi.org/ 10.1038/nrc2602 [DOI] [PubMed] [Google Scholar]
- [3].Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 2011; 11(8):558-72; PMID:21734724; http://dx.doi.org/ 10.1038/nrc3090 [DOI] [PubMed] [Google Scholar]
- [4].Deckbar D, Jeggo PA, Lobrich M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit Rev Biochem Mol Biol 2011; 46(4):271-83; PMID:21524151; http://dx.doi.org/ 10.3109/10409238.2011.575764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Diehl JA. The cyclin D3 knockout: a pound of redundancy with a dash of tissue specificity. Cancer Biol Ther 2004; 3(2):162-4; PMID:14752277; http://dx.doi.org/ 10.4161/cbt.3.2.774 [DOI] [PubMed] [Google Scholar]
- [6].Cooper AB, Sawai CM, Sicinska E, Powers SE, Sicinski P, Clark MR, Aifantis I. A unique function for cyclin D3 in early B cell development. Nat Immunol 2006; 7(5):489-97; PMID:16582912; http://dx.doi.org/ 10.1038/ni1324 [DOI] [PubMed] [Google Scholar]
- [7].Sicinska E, Aifantis I, Le Cam L, Swat W, Borowski C, Yu Q, Ferrando AA, Levin SD, Geng Y, von Boehmer H, et al.. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003; 4(6):451-61; PMID:14706337; http://dx.doi.org/ 10.1016/S1535-6108(03)00301-5 [DOI] [PubMed] [Google Scholar]
- [8].Cato MH, Chintalapati SK, Yau IW, Omori SA, Rickert RC. Cyclin D3 is selectively required for proliferative expansion of germinal center B cells. Mol Cell Biol 2011; 31(1):127-37; PMID:20956554; http://dx.doi.org/ 10.1128/MCB.00650-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peled JU, Yu JJ, Venkatesh J, Bi E, Ding BB, Krupski-Downs M, Shaknovich R, Sicinski P, Diamond B, Scharff MD, et al.. Requirement for cyclin D3 in germinal center formation and function. Cell Res 2010; 20(6):631-46; PMID:20404856; http://dx.doi.org/ 10.1038/cr.2010.55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sawai CM, Freund J, Oh P, Ndiaye-Lobry D, Bretz JC, Strikoudis A, Genesca L, Trimarchi T, Kelliher MA, Clark M, et al.. Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 2012; 22(4):452-65; PMID:23079656; http://dx.doi.org/ 10.1016/j.ccr.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Agami R, Bernards R. Distinct initiation and maintenance mechanisms cooperate to induce G1 cell cycle arrest in response to DNA damage. Cell 2000; 102(1):55-66; PMID:10929713; http://dx.doi.org/ 10.1016/S0092-8674(00)00010-6 [DOI] [PubMed] [Google Scholar]
- [12].Santra MK, Wajapeyee N, Green MR. F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 2009; 459(7247):722-5; PMID:19412162; http://dx.doi.org/ 10.1038/nature08011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choo DW, Baek HJ, Motoyama N, Cho KH, Kim HS, Kim SS. ATM is required for rapid degradation of cyclin D1 in response to gamma-irradiation. Biochem Biophys Res Commun 2009; 378(4):847-50; PMID:19071090; http://dx.doi.org/ 10.1016/j.bbrc.2008.11.132 [DOI] [PubMed] [Google Scholar]
- [14].Hitomi M, Yang K, Stacey AW, Stacey DW. Phosphorylation of cyclin D1 regulated by ATM or ATR controls cell cycle progression. Mol Cell Biol 2008; 28(17):5478-93; PMID:18606783; http://dx.doi.org/ 10.1128/MCB.02047-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Pontano LL, Aggarwal P, Barbash O, Brown EJ, Bassing CH, Diehl JA. Genotoxic stress-induced cyclin D1 phosphorylation and proteolysis are required for genomic stability. Mol Cell Biol 2008; 28(23):7245-58; PMID:18809569; http://dx.doi.org/ 10.1128/MCB.01085-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wang X, Arai S, Song X, Reichart D, Du K, Pascual G, Tempst P, Rosenfeld MG, Glass CK, Kurokawa R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nature 2008; 454(7200):126-30; PMID:18509338; http://dx.doi.org/ 10.1038/nature06992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13(12):1501-12; PMID:10385618; http://dx.doi.org/ 10.1101/gad.13.12.1501 [DOI] [PubMed] [Google Scholar]
- [18].Steinel NC, Lee BS, Tubbs AT, Bednarski JJ, Schulte E, Yang-Iott KS, et al.. The ataxia telangiectasia mutated kinase controls Igkappa allelic exclusion by inhibiting secondary Vkappa-to-Jkappa rearrangements. J Exp Med 2013; 210(2):233-9; PMID:23382544; http://dx.doi.org/ 10.1084/jem.20121605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Steinel NC, Fisher MR, Yang-Iott KS, Bassing CH. The ataxia telangiectasia mutated and cyclin D3 proteins cooperate to help enforce TCRbeta and IgH allelic exclusion. J Immunol 2014; 193(6):2881-90; PMID:25127855; http://dx.doi.org/ 10.4049/jimmunol.1302201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yang-Iott KS, Carpenter AC, Rowh MA, Steinel N, Brady BL, Hochedlinger K, Jaenisch R, Bassing CH. TCR beta feedback signals inhibit the coupling of recombinationally accessible V beta 14 segments with DJ beta complexes. J Immunol 2010; 184(3):1369-78; PMID:20042591; http://dx.doi.org/ 10.4049/jimmunol.0900723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Clark MR, Mandal M, Ochiai K, Singh H. Orchestrating B cell lymphopoiesis through interplay of IL-7 receptor and pre-B cell receptor signalling. Nat Rev Immunol 2014; 14(2):69-80; PMID:24378843; http://dx.doi.org/ 10.1038/nri3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14(4):197-210; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- [23].Mazan-Mamczarz K, Hagner PR, Zhang Y, Dai B, Lehrmann E, Becker KG, Keene JD, Gorospe M, Liu Z, Gartenhaus RB. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011; 117(8):2441-50; PMID:21209379; http://dx.doi.org/ 10.1182/blood-2010-09-310987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Srikantan S, Gorospe M. HuR function in disease. Front Biosci (Landmark Ed) 2012; 17:189-205; PMID:22201738; http://dx.doi.org/ 10.2741/3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rodriguez PC, Hernandez CP, Morrow K, Sierra R, Zabaleta J, Wyczechowska DD, Ochoa AC. L-arginine deprivation regulates cyclin D3 mRNA stability in human T cells by controlling HuR expression. J Immunol 2010; 185(9):5198-204; PMID:20889542; http://dx.doi.org/ 10.4049/jimmunol.1001224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ghosh M, Aguila HL, Michaud J, Ai Y, Wu MT, Hemmes A, Ristimaki A, Guo C, Furneaux H, Hla T. Essential role of the RNA-binding protein HuR in progenitor cell survival in mice. J Clin Invest 2009; 119(12):3530-43; PMID:19884656; http://dx.doi.org/ 10.1172/JCI38263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhandoola A, von Boehmer H, Petrie HT, Zuniga-Pflucker JC. Commitment and developmental potential of extrathymic and intrathymic T cell precursors: plenty to choose from. Immunity 2007; 26(6):678-89; PMID:17582341; http://dx.doi.org/ 10.1016/j.immuni.2007.05.009 [DOI] [PubMed] [Google Scholar]
- [28].Xu Y, Yang EM, Brugarolas J, Jacks T, Baltimore D. Involvement of p53 and p21 in cellular defects and tumorigenesis in Atm-/- mice. Mol Cell Biol 1998; 18(7):4385-90; PMID:9632822; http://dx.doi.org/ 10.1128/MCB.18.7.4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 1999; 18(6):1571-83; PMID:10075928; http://dx.doi.org/ 10.1093/emboj/18.6.1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shaughnessy J Jr., Gabrea A, Qi Y, Brents L, Zhan F, Tian E, Sawyer J, Barlogie B, Bergsagel PL, Kuehl M. Cyclin D3 at 6p21 is dysregulated by recurrent chromosomal translocations to immunoglobulin loci in multiple myeloma. Blood 2001; 98(1):217-23; PMID:11418483; http://dx.doi.org/ 10.1182/blood.V98.1.217 [DOI] [PubMed] [Google Scholar]
- [31].Wlodarska I, Dierickx D, Vanhentenrijk V, Van Roosbroeck K, Pospisilova H, Minnei F, Verhoef G, Thomas J, Vandenberghe P, De Wolf-Peeters C. Translocations targeting CCND2, CCND3, and MYCN do occur in t(11;14)-negative mantle cell lymphomas. Blood 2008; 111(12):5683-90; PMID:18391076; http://dx.doi.org/ 10.1182/blood-2007-10-118794 [DOI] [PubMed] [Google Scholar]
- [32].Sonoki T, Harder L, Horsman DE, Karran L, Taniguchi I, Willis TG, et al.. Cyclin D3 is a target gene of t(6;14)(p21.1;q32.3) of mature B-cell malignancies. Blood 2001; 98(9):2837-44; PMID:11675358; http://dx.doi.org/ 10.1182/blood.V98.9.2837 [DOI] [PubMed] [Google Scholar]
- [33].Schmitz R, Young RM, Ceribelli M, Jhavar S, Xiao W, Zhang M, Wright G, Shaffer AL, Hodson DJ, Buras E, et al.. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012; 490(7418):116-20; PMID:22885699; http://dx.doi.org/ 10.1038/nature11378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Strasser A, Harris AW, Cory S. bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 1991; 67(5):889-99; PMID:1959134; http://dx.doi.org/ 10.1016/0092-8674(91)90362-3 [DOI] [PubMed] [Google Scholar]
- [35].Steinel NC, Brady BL, Carpenter AC, Yang-Iott KS, Bassing CH. Posttranscriptional silencing of VbetaDJbetaCbeta genes contributes to TCRbeta allelic exclusion in mammalian lymphocytes. J Immunol 2010; 185(2):1055-62; PMID:20562258; http://dx.doi.org/ 10.4049/jimmunol.0903099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pérez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al.. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001; 15(5):763-74; PMID:11728338; http://dx.doi.org/ 10.1016/S1074-7613(01)00227-8 [DOI] [PubMed] [Google Scholar]
- [37].Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, et al.. Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell 1996; 86(1):159-71; PMID:8689683; http://dx.doi.org/ 10.1016/S0092-8674(00)80086-0 [DOI] [PubMed] [Google Scholar]
- [38].Yoon JH, Abdelmohsen K, Srikantan S, Yang X, Martindale JL, De S, Huarte M, Zhan M, Becker KG, Gorospe M. LincRNA-p21 suppresses target mRNA translation. Mol Cell 2012; 47(4):648-55; PMID:22841487; http://dx.doi.org/ 10.1016/j.molcel.2012.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rowh MA, DeMicco A, Horowitz JE, Yin B, Yang-Iott KS, Fusello AM, Hobeika E, Reth M, Bassing CH. Tp53 deletion in B lineage cells predisposes mice to lymphomas with oncogenic translocations. Oncogene 2011; 30(47):4757-64; PMID:21625223; http://dx.doi.org/ 10.1038/onc.2011.191 [DOI] [PubMed] [Google Scholar]
- [40].Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, Pendergast AM, Bronson R, Aster JC, Scott ML, et al.. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood 1998; 92(10):3780-92; PMID:9808572 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.