

ABSTRACT
Cisplatin is the most potent and widespread used chemotherapy drug for lung cancer treatment. However, the development of resistance to cisplatin is a major obstacle in clinical therapy. The principal mechanism of cisplatin is the induction of DNA damage, thus the capability of DNA damage response (DDR) is a key factor that influences the cisplatin sensitivity of cancer cells. Recent advances have demonstrated that miRNAs (microRNAs) exerted critical roles in DNA damage response; nonetheless, the association between DNA damage responsive miRNAs and cisplatin resistance and its underlying molecular mechanism still require further investigation. The present study has attempted to identify differentially expressed miRNAs in cisplatin induced DNA damage response in lung cancer cells, and probe into the effects of the misexpressed miRNAs on cisplatin sensitivity. Deep sequencing showed that miR-33b-3p was dramatically down-regulated in cisplatin-induced DNA damage response in A549 cells; and ectopic expression of miR-33b-3p endowed the lung cancer cells with enhanced survival and decreased γH2A.X expression level under cisplatin treatment. Consistently, silencing of miR-33b-3p in the cisplatin-resistant A549/DDP cells evidently sensitized the cells to cisplatin. Furthermore, we identified CDKN1A (p21) as a functional target of miR-33b-3p, a critical regulator of G1/S checkpoint, which potentially mediated the protection effects of miR-33b-3p against cisplatin. In aggregate, our results suggested that miR-33b-3p modulated the cisplatin sensitivity of cancer cells might probably through impairing the DNA damage response. And the knowledge of the drug resistance conferred by miR-33b-3p has great clinical implications for improving the efficacy of chemotherapies for treating lung cancers.
KEYWORDS: cisplatin resistance, cell survival, DNA damage response, microRNA, miR-33b-3p, p21
Introduction
DNA damage response (DDR) is an evolutionarily conserved, widespread functional network to maintain the genomic integrity, which is pivotal for the viability of cells and the health of organisms.1 The DDR detects DNA lesions arose from numerous intrinsic and extrinsic genotoxic stresses, signals their presence, and promotes DNA repair, otherwise triggers apoptosis or cellular senescence while the DNA damage is beyond repair.2,3 Genomic instability and specific DNA repair defects are the most pervasive characteristics of tumor cells, which are exploited by DNA damaging chemotherapy drugs for cancer therapy,4 including platinum-containing compounds, alkylating agents, and anthracyclines.5 For instance, homologous recombination (HR)-deficient tumor cells can be effectively targeted by DNA double-stranded breaks (DSBs)-inducing chemotherapy agents,5 and platinum based drug (such as cisplatin) is more applied to treat tumors with nucleotide excision repair (NER) defect.6,7 However, tumor cells often acquire drug resistance during chemotherapy treatment by altering DDR pathways involved in DNA repair, apoptosis and cellular senescence.8 Thus, deepening the understanding of the regulation of DDR pathways in tumor cells will provide novel insights and instructions for drug selection for diverse cancer treatment, to maximize the efficacy of chemotherapy drugs and minimize the occurrence of drug resistance.
The platinum-based anticancer drugs, in particular cisplatin, are the most potent and wide used chemotherapeutic agents for the treatment of various solid malignancies, including lung cancers.9,10 Cisplatin exerts the anticancer effects through multiple mechanisms, its most prominent mode of action is the generation of DNA lesions (platinum-DNA adducts), which followed trigger several cellular processes involved in the signaling of DNA damage, cell cycle checkpoints, DNA repair and cell death.10,11 Though cisplatin has a central role in cancer chemotherapy, the development of chemoresistance has become the major limitations for its clinical application. And the underlying molecular mechanisms of cisplatin resistance still far to be elucidated.
MicroRNAs (miRNAs) are a large class of tiny noncoding RNAs (approximately 22∼25nt) generated from the primary hairpin-shaped transcripts through the Drosha/Dicer RNase III endonuclease process, which negatively regulates gene expression at the posttranscriptional level by imperfect base pairing with mRNA 3′ untranslated regions (UTRs), leading to target mRNA cleavage or translational repression.12,13 One single miRNA potentially regulates hundreds of mRNA targets, thus orchestrating diverse biological processes and physiological pathways.14,15 Additionally, accumulating evidences have unraveled that miRNAs exerted critical roles in modulating the DNA damage response.16-19 Thus, it's reasonable to speculate that DNA damage responsive miRNAs may exert a crucial role in modulating cisplatin sensitivity and drug resistance. In this study, we sought to screen differentially expressed miRNAs against cisplatin treatment, and further investigate into the effects of the identified DNA damage responsive miRNAs on cisplatin sensitivity, elucidating a novel molecular mechanism in the development of cisplatin resistance.
Materials and methods
Cell lines
A549 was a non-small cell lung cancer cell line, A549/DDP was a cisplatin-resistant lung cancer cell line derived from A549, and HEK293T was a SV40-transformed embryonic kidney cell line. All the cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) (GIBCO).
RNA isolation, small RNA library construction and sequencing
Total RNA was extracted from A549 cells treated with DMF or cisplatin using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocol. The quality and quantity of the extracted RNAs were evaluated by A260/280 nm reading using NanoDrop1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA integrity was assessed by electrophoresis on a denaturing agarose gel stained with SYBR Green I.
Small-RNA sequencing for the DMF or cisplatin treated A549 cells was then performed by CapitalBio Corporation, Beijing, China. Two small RNA libraries were constructed utilizing TruSeq Small RNA Sample Prep Kit (Illumina) according to the manufacturer's protocol. Briefly, 4 μg of total RNA was ligated to the the 5′-adaptor and 3′-adaptor, and was reverse-transcribed to synthesize single-stranded cDNA. Fragments arranged from 140 to 160 bp were then selected by gel purification to produce small RNA libraries for cluster generation and sequencing. The primary data analysis and base calling were performed utilizing the Illumina instrument's software.
RNA oligoribonucleotides and cell transfections
The RNA duplex mimiced miR-33b-3p was designated as miR-33b-3p mimimcs (Sense Strand: 5′-CAGUGCCUCGGCAGUGCAGCCC-3′). The control RNA duplex, designated as NC (Sense Strand: 5′-UUCUCCGAACGUGUCACGUTT-3′) was nonhomologous to any human genome sequences and used for miR-33b-3p mimics. The inhibitor of miR-33b-3p was designated as miR-33b-3p inhibitor (Sequence: 5′-GGGCUGCACUGCCGAGGCACUG-3′), and the negative control was named as inhibitor NC (Sequence: 5′-CAGUACUUUUGUGUAGUACAA-3′). The small interference RNAs (siRNA) targeting ERCC1 mRNA (Genbank accession no. NM_202001.2) was designated as siERCC1-1 (Sense Strand: 5′-CAGCAAGGAAGA AAUUUGUTT-3′) and siERCC1-2 (Sense Strand: 5′-CGACGUAAUUCCCGACUA UTT-3′). All the above RNA oligoribonucleotides were purchased from Genepharma (China).
Transfection of RNA oligoribonucleotide(s) was done utilizing Lipofectamine RNAiMAX (Invitrogen, USA) according to the manufacturer's protocol. 50nM of RNA duplex or miRNA inhibitor were used for each transfection, unless otherwise indicated. In the experiment of expressing exogenous p21 or Sirt6 protein, 24h after RNA transfection, cells were transfected with 400 ng plasmids in a 24-well plate, using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's protocol.
Cell viability assays
Cell viability was determined by the Alamar blue assay (AbD Serotec, UK). Briefly, cells were firstly transfected with NC and miR-33b-3p mimics, or inhibitor NC and miR-33b-3p inhibitor, 24h later the transfected cells were seeded in a 96-well plate at 50% confluence, and followed by the Alamar blue assay at indicated times. Fluorescence of the reduced Alamar blue dye was measured using Synergy 2 microplate fluorescence reader (BioTek, USA) at excitation wavelength of 540 nm and emission wavelength of 590 nm.
Cell proliferation assay
Cell proliferation was analyzed by measuring DNA synthesis with the EdU cell proliferation assay (RiboBio, China) according to the manufacturer's instructions. Briefly, cells (5 × 104 cells per well) were cultured in triplicate in 24-well plates and transfected with 50 nM of NC and miR-33b-3p mimics, or 50 nM inhibitor NC and miR-33b-3p inhibitor for 48 h. Then cells were incubated with 40 μM of 5-ethynyl-2′-deoxyuridine (EdU) for an additional 2 h at 37°C. Cells were then fixed with 4% formaldehyde for 30 min and treated with 0.5% Triton X-100 for 10 min at room temperature to permeabilize cells. After washing with PBS 3 times, cells were incubated with 1× Apollo reaction cocktail for 30 min. Finally, Cells were stained with 10 μg/ml of Hoechst 33342 for 30 min as counterstain, and read under a fluorescence microscope (Olympus, Japan).
Analysis of cell cycle distribution
Cells were harvested, washed with 1×PBS and fixed by prechilled 70% ethanol at 4°C overnight. Then the fixed cells were resuspended in the propidium iodide (PI) staining solution (0.05 mg/ml PtdIns, 0.01 mg/ml ribonuclease A, 0.2% Triton X-100), incubated at room temperature for 30 min. Flow cytometry analysis was performed on a BD FACSCanto II flow cytometer (BD Biosciences, USA), and the distribution of cells in the G1, S, and G2/M phases of the cell cycle were analyzed utilizing Wincycle software (Phoenix Flow Systems, USA).
Real-time quantitative PCR
For microRNA detection, 1 μg total RNA was reverse-transcribed utilizing All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia, USA) according to the manufacturer's protocol. Real-time qPCR was performed in a LightCycler 96 (Roche, Switzerland) with All-in-One miRNA qRT-PCR Detection Kit (GeneCopoeia, USA) as well. Primers specific for miR-33b-3p and U6 detection were all purchased from GeneCopoeia.
For mRNA detection, 1 μg total RNA was reverse transcribed using PrimeScript RT Reagent Kit with gDNA Eraser (Takara, Japan) according to the manufacturer's protocol. Real-time qPCR was performed in a LightCycler 96 (Roche, Switzerland) with SYBR Select Master Mix (Invitrogen, USA). PCR primer sequences for p21, ERCC1, ERCC4, SIRT6, SREBP1 and GAPDH were listed in Table 2. GAPDH gene was used as internal control.
Table 2.
Sequences of DNA Oligonucleotides.
| Name | Sense primer sequence (5′-3′) | Antisense primer sequence (5′-3′) |
|---|---|---|
| Primers for Gene or 3′UTR Cloning | ||
| p21△3′UTR | AGTGAATTCGTTCCTTGTGGAGCCGGA | AGTTCTAGATGGGCGGATTAGGGCTT |
| P21 with 3′UTR | AGTAAGCTTGTTCCTTGTGGAGCCGGA | AGTTCTAGATTCAGCATTGTGGGAGGAGC |
| p21 3′UTR | AGTGAATTCGGCACCCTAGTTCTACCTCA | AGTTCTAGATTCAGCATTGTGGGAGGAGC |
| Primers for RT-PCR | ||
| p21 | GCCGAAGTCAGTTCCTTGTG | CCATTAGCGCATCACAGTCG |
| ERCC1 | CTACGCCGAATATGCCATCTC | GTACGGGATTGCCCCTCTG |
| ERCC4 | GGAACTGCTCGACACTGACG | GCGAGGGAGGTGTTCAACTC |
| SREBP1 | TGCATTTTCTGACACGCTTC | GATGTTCCCGGAATAGCTGA |
| SIRT6 | CCCACGGAGTCTGGACCAT | CTCTGCCAGTTTGTCCCTG |
| GAPDH | GAGTCAACGGATTTGGTCGT | TTGATTTTGGAGGGATCTCG |
Western blotting
Cell protein lysates were separated on 10% SDS polyacrylamide gels, electrophoretically transferred to polyvinylidene difluoride membranes (0.2 μm pore size)(Millipore, USA), and then incubated with primary antibodies: γH2A.X (2577s, Cell Signaling Technology, USA), ERCC1 (ab129267, Abcam, USA), p21 (Cell Signaling Technology, 2947s), and further with the respective secondary antibodies conjugated with HRP (Bethyl Laboratories, USA). The proteins were detected with a commercial enhanced chemiluminescence (ECL) kit (Pierce, USA). Protein loading was estimated using mouse anti-tubulin monoclonal antibody (Sigma-Aldrich, USA).
Vector construction
To construct a luciferase reporter vector (designated as p21-3′UTR-WT), a wild-type 3′ UTR fragment of p21 containing the putative binding sites for miR-33b-3p was amplified and then inserted downstream of the stop codon of firefly luciferase in pGL3cm as described previously (Promega, USA). p21-3′UTR -MUT, which carried a mutated sequence in the complementary site for the seed region of miR-33b-3p, was generated using the fusion PCR method.
To construct the p21 expression vector (pcDNA3.0-p21△3′UTR or pcDNA3.0-p21 with 3′UTR), the full-length coding sequence of p21 (GenBank accession number NM_000389.4) without or with 3′UTR region was amplified and then cloned into pcDNA3.0 (Invitrogen, USA).
Dual luciferase reporter assay
Dual luciferase reporter assay was comprised of 2 reporters, one is a firefly luciferase expression construct in pGL3cm containing the p21 3′UTR sequences, and another one is Renilla luciferase expression construct pRL-TK, which provides the constitutive expression of Renilla luciferase. Briefly, 293T cells (4 × 104) were plated in a 48-well plate and then cotransfected with 10 nM either NC or miR-33b-3p mimics, 20 ng of either p21-3′UTR-WT or p21-3′UTR-MUT, and 4 ng of pRL-TK (Promega), using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Cells were collected 48 h after transfection and analyzed using the Dual-Luciferase Reporter Assay System (Promega). Luciferase activity was detected by FB12 Luminometer (Berthold). The pRL-TK vector was cotransfected as an internal control to correct the differences in both transfection and harvest efficiencies. Firefly luciferase activity of each sample was normalized by Renilla luciferase activity.
Bioinformatics
The analysis of microRNA target binding sites was performed utilizing the miRWalk software (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk/).
Statistical analysis
Data are presented as mean±SEM from at least 3 separate experiments. Unless otherwise noted, the differences between groups were analyzed using Student's 2-tailed t test when only 2 groups were compared or assessed by one-way analysis of variance (ANOVA) when more than 2 groups were compared. Differences were considered statistically significant at P < 0.05.
Accession number
Genbank Accession numbers for p21 mRNA sequence (NM_000389.4), SIRT6 mRNA sequence (NM_001193285.1), ERCC1 mRNA sequence (NM_202001.2), ERCC4 mRNA sequence (NM_005236.2) and SREBP1 mRNA sequence (NM_001005291.2) are found at the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). The sequences of miR-33b-3p (MIMAT0004811) described in this paper have been deposited in miRBase (http://www.mirbase.org/).
Results
miR-33b-3p was dramatically downregulated in cisplatin treated A549 cells
To characterize the differentially expressed miRNAs upon DNA damage, the A549 lung cancer cells were treated with 50 μM cisplatin for 8h, which can effectively induce high levels of γH2A.X in A549 cells (SFig. 1 and Fig. 1A). Then, a next-generation miRNA sequence based on Illumina HiSeq 2000 platform was performed, and yielded a total reads of 11.64 × 106 reads from the DMF treated A549 cells (Mock) and 12.58 × 106 reads from 50 μM cisplatin treated A549 cells (Cisplatin). After removal of the low quality reads and reads with size < 18 nt, we obtained a total of 10.42 × 106 and 11.38 × 106 high quality reads for Mock and Cisplatin group respectively. A complete list of miRNAs sequenced in total and normalized read counts, and fold differences between Mock and Cisplatin group was provided in Table S1.
Figure 1.
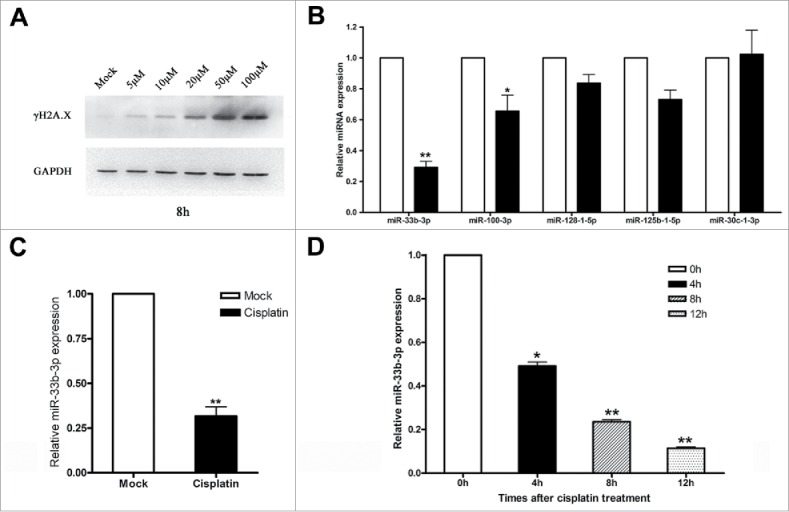
miR-33b-3p was dramatically down-regulated in cisplatin induced DNA damage response. (A) Western blot was performed to monitor the expression levels of γH2A.X in A549 cells treated with cisplatin in indicated concentrations. (B) Real-time qPCR was used to detect the expression levels of differential expressed microRNAs from deep sequence in A549 cells with or without cisplatin treatment. Columns, mean of at least 3 independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01, comparison between 2 groups as indicated. (C) Expression of miR-33b-3p in A549 cells with or without cisplatin treatment. Columns, mean of at least 3 independent experiments; bars, SEM. **, P < 0.01, comparison between 2 groups as indicated. (D) Expression of miR-33b-3p in A549 cells with or without cisplatin treatment in indicated times. Columns, mean of at least 3 independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01, comparison between 2 groups as indicated.
We identified a total of 12 differentially expressed known miRNAs with a fold change ≥2 .0 (Table 1). Of the 12 differentially expressed miRNAs, 2 were up-regulated and 10 were down-regulated in cisplatin treated A549 cells compared to the controls. Real-time qPCR was then preformed to detect the expression of miR-33b-3p, miR-100-3p, miR-128-1-5p, miR-125b-1-3p and miR-30c-1-3p (read counts ≥ 100), and unraveled that miR-33b-3p was the most significantly downregulated in cisplatin treated A549 cells compared with that in DMF treated A549 cells (Fig. 1B and C). Moreover, the down-regulation of miR-33b-3p expression levels was in a time-dependent manner (Fig. 1D), which suggested that miR-33b-3p was a DNA damage responsive miRNA, indicating the potential effect of miR-33b-3p on cisplatin sensitivity.
Table 1.
Differential expressed miRNAs in cisplatin treated A549 cells.
| Reads |
Normalized Counts |
||||||
|---|---|---|---|---|---|---|---|
| miRNA | Total | Cisplatin | Mock | Cisplatin Mock | Fold change | P-Value | |
| hsa-miR-3687 | 19 | 1 | 18 | 0.1881 | 2.8995 | −15.4154136* | 0.004066437 |
| hsa-miR-551b-5p | 56 | 4 | 52 | 0.7524 | 8.3763 | −11.13335426 | 0.000499278 |
| hsa-miR-181b-3p | 27 | 2 | 25 | 0.3762 | 4.0271 | −10.70514833 | 0.002811985 |
| hsa-miR-33b-3p | 329 | 38 | 291 | 7.1474 | 46.8749 | −6.558311924 | 0.000279913 |
| hsa-miR-6724-5p | 24 | 3 | 21 | 0.5643 | 3.3827 | −5.994883065 | 0.018258403 |
| hsa-let-7c-3p | 53 | 7 | 46 | 1.3166 | 7.4098 | −5.627849408 | 0.006732608 |
| hsa-miR-100-3p | 1266 | 183 | 1083 | 34.4204 | 174.452 | −5.068273504 | 0.000932153 |
| hsa-miR-128-1-5p | 942 | 149 | 793 | 28.0253 | 127.7381 | −4.557950403 | 0.001991331 |
| hsa-miR-125b-1-3p | 630 | 124 | 506 | 23.3231 | 81.5076 | −3.494712939 | 0.010495757 |
| hsa-miR-30c-1-3p | 327 | 77 | 250 | 14.4829 | 40.2705 | −2.780558008 | 0.040511526 |
| hsa-miR-4286 | 38 | 28 | 10 | 5.2665 | 1.6108 | 3.269454931 | 0.045980594 |
| hsa-miR-1247-5p | 42 | 31 | 11 | 5.8308 | 1.7719 | 3.290685158 | 0.043422342 |
-means downregulated miRNAs in cisplatin treated A549 cells.
Overexpression of miR-33b-3p induced resistance to cisplatin
To unveil the probable functions of miR-33b-3p, we firstly transfected A549 cells with miR-33b-3p mimics or NC oligoribonucleotides; NC was nonhomologous to human genome sequences (Table 2). Twenty-four hours after transfection, cells were treated with or without cisplatin and evaluated for their viabilities using an Alamar blue assay. In the absence of cisplatin treatment, the RNA mimics did not have evident effect on cell viability; while in the presence of cisplatin, miR-33b-3p gave significantly higher viability than NC group at 48h (Fig. 2B). Fascinatingly, the morphology of A549 cells transfected with miR-33b-3p mimics was greatly altered, and more similar to the morphology of the cisplatin-resistant A549/DDP cells (Fig. 2A). This data showed that miR-33b-3p might exert an important role in the development of cisplatin resistance of lung cancer cells.
Figure 2.
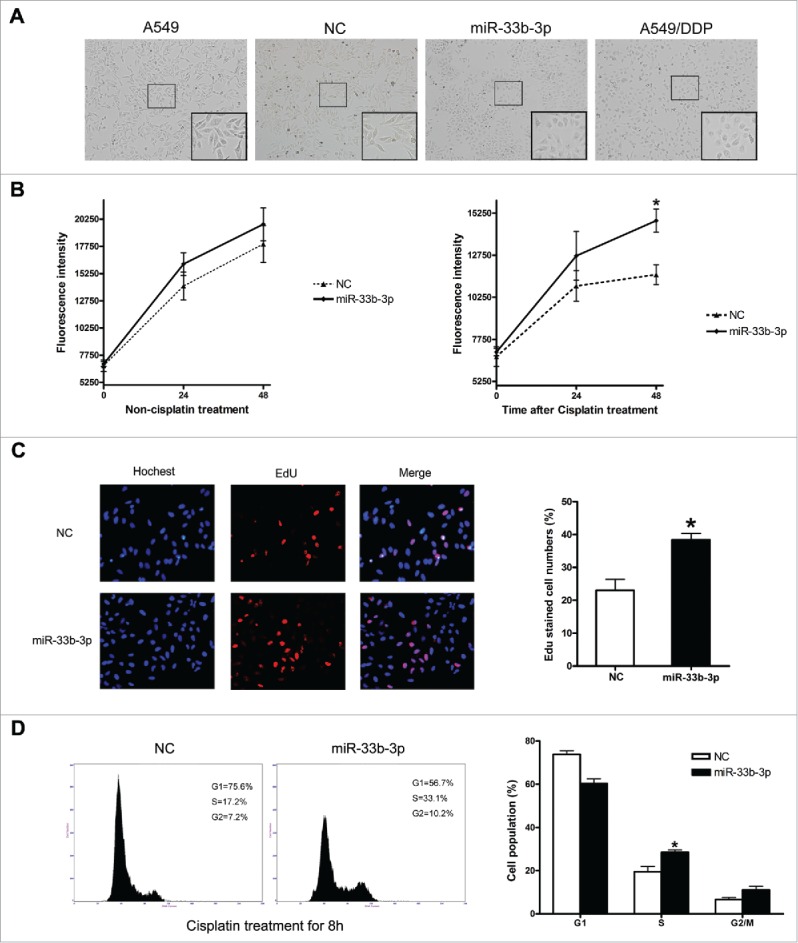
Ectopic expression of miR-33b-3p induced resistance to cisplatin. (A) Representative photographs of the morphology of A549 cells transfected with NC or miR-33b-3p mimics. (B) Effect of miR-33b-3p on A549 cell viability in the absence or under cisplatin treatment. (C) Representative images of cells stained with DAPI (blue fluorescence) and EdU, as a measurement of DNA synthesis (red fluorescence). The average EdU-stained cells were from 3 experiments; bars, SEM. *, P < 0.05, comparison between 2 groups as indicated. (D) Cell cycle analysis was performed at 48 h after transfection with NC or miR-33b-3p mimics under cisplatin treatment. The percentage of G1, S, and G2/M are demonstrated as shown. Columns, mean of at least 3 independent experiments; bars, SEM. *, P < 0.05, comparison between 2 groups as indicated.
The increased cell viability with miR-33b-3p under cisplatin treatment might be the result of elevated cell proliferation and/or reduced apoptosis. The possibility that miR-33b-3p might suppress the apoptosis of A549 cells was excluded by apoptotic morphology examination and PARP1 cleavage detection; no significant difference of apoptotic rates was observed between the A549 cells transfected with miR-33b-3p mimics and the controls (SFig. 2). We then investigated into the effects of miR-33b-3p on A549 cell proliferation by EdU assay and cell cycle analysis. The results of EdU assay showed that A549 cells transfected with miR-33b-3p mimics exhibited much more EdU-positive cells than A549 cells transfected with NC (Fig. 2C). What's more, the cell cycle analysis revealed that ectopic expression of miR-33b-3p significantly promoted G1/S transition, compared with A549 cells transfected with NC (Fig. 2D). These results strongly demonstrated that miR-33b-3p conferred the A549 cells with enhanced cell proliferation, and thus suppressed the cisplatin sensitivity of A549 cells.
miR-33b-3p potentially facilitated DNA damage repair against cisplatin treatment in A549 cells
As the capability of DNA damage response always determined the cell fate following DNA damage, we further probed into whether miR-33b-3p suppressed the cisplatin sensitivity through directly impacting on the cisplatin triggered DNA damage response. The phosphorylation of histone H2A.X at Ser139 (γH2A.X) marked the sites of DNA lesions caused by irradiation, UV and alkylation agents, and provided a nucleation site for recruitment of DNA damage checkpoint and DNA repair proteins.20-22 Thus, we examined the γH2A.X levels in A549 cells transfected with miR-33b-3p mimics or NC. As the result revealed, A549 cells transfected with miR-33b-3p mimics displayed lower phosphorylated levels of γH2A.X compared with that in NC transfected cells after cisplatin treatment (Fig. 3A and B), which indicated that miR-33b-3p might facilitate DNA damage repair.
Figure 3.
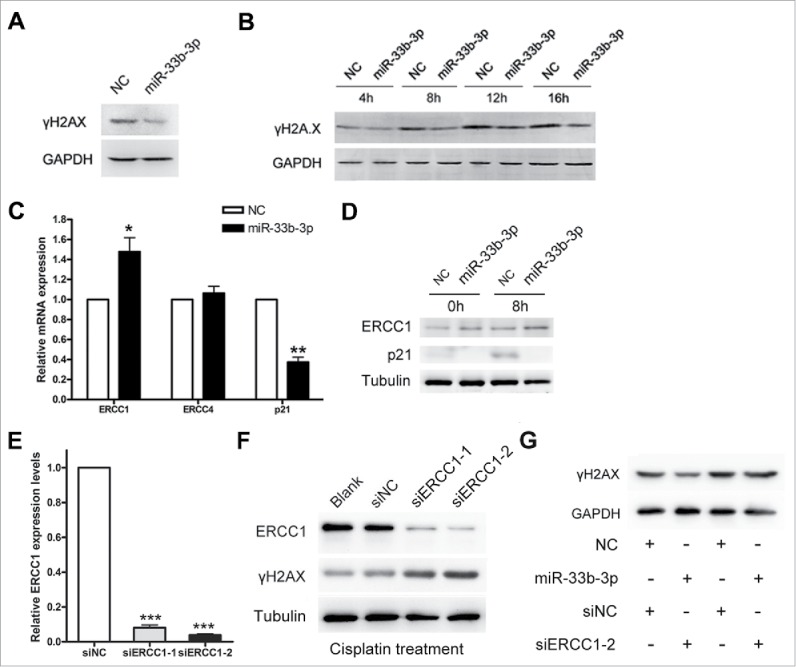
miR-33b-3p potentially facilitated DNA damage repair against cisplatin treatment. (A) Western blot was performed to evaluate the expression of γH2A.X in A549 cells transfected with NC or miR-33b-3p mimics after cisplatin treatment. (B) TheγH2A.X expression levels in A549 cells transfected with NC or miR-33b-3p mimics under cisplatin treatment for indicated times. (C) Real-time qPCR was used to detect the mRNA expression levels of ERCC1, ERCC4 and p21 in NC or miR-33b-3p mimics transfected A549 cells with or without cisplatin treatment. Columns, mean of at least 3 independent experiments; bars, SEM. *, P < 0.05; **, P < 0.01, comparison between 2 groups as indicated. (D) The protein expression levels of ERCC1 and p21 in NC or miR-33b-3p mimics transfected A549 cells with or without cisplatin treatment. (E) siERCC1-1 and siERCC-2 efficiently suppressed the mRNA (E) and protein (F) expressions of ERCC1 in A549 cells. Columns, mean of at least 3 independent experiments; bars, SEM. ***, P < 0.001, compared with A549 cells transfected with siNC. (F) Western blot was performed to examine the expression of γH2A.X in A549 cells transfected with siNC, siERCC1-1 or siERCC1-2 after cisplatin treatment. (G) Western blot was carried out to evaluate the γH2A.X expression levels in A549 cells co-transfected with miR-33b-3p mimics/NC and siERCC1-2/siNC under cisplatin treatment.
Previous studies have established that nucleotide excision repair (NER), was the major pathway responsible for the recognition and removal of cisplatin-DNA adducts.23 We thus further detected the expression of the ERCC1/ERCC4 complex that played a central role in NER pathway, and several checkpoint proteins (such as p21). The results from both the real-time qPCR and western blot showed that, miR-33b-3p modestly increased the ERCC1 expression level (Fig. 3C and D). More interestingly, the p21 expression level was almost abolished in miR-33b-3p transfected A549 cells compared with that in controls (Fig. 3C and D), which provided a probable explanation to the elevated G1/S transition in miR-33b-3p transfected A549 cells.
We then knocked down ERCC1 expression utilizing designed siERCC-1 and siERCC1-2 in A549 cells, in order to validate the effect of miR-33b-3p in NER pathway. The knock-down of endogenous ERCC1 in A549 cells was confirmed (Fig. 3E and F). Silencing of ERCC1 endowed A549 cells with elevated γH2A.X levels under cisplatin treatment, which further validate the pivotal role of ERCC1 in NER (Fig. 3F). Moreover, A549 cells were co-transfected with miR-33b-3p/NC and siERCC1/siNC, and then were treated by cisplatin 24h later. The results showed that knock-down of ERCC1 significantly reversed the effect of miR-33b-3p on promoting the repair of cisplatin-induced DNA damage (Fig. 3G), which further suggested that miR-33b-3p facilitated DNA damage repair against cisplatin treatment potentially through activating the NER pathway.
Inhibition of miR-33b-3p sensitized A549/DDP cells to cisplatin
To further confirm the effect of miR-33b-3p on cisplatin resistance, we then monitor the miR-33b-3p expression levels in the cisplatin-resistant A549/DDP cells with or without cisplatin treatment. Encouragingly, distinct from the downregulation in cisplatin-sensitive A549 cells, miR-33b-3p was significantly upregulated in cisplatin treated A549/DDP cells (Fig. 4A), indicating the potential role of miR-33b-3p in modulating cisplatin resistance. We following transfected the A549/DDP cells with miR-33b-3p inhibitor or inhibitor NC, in order to knock down the miR-33b-3p expression in A549/DDP cells. As expected, silencing of miR-33b-3p endowed A549/DDP cells with diminished cell survival under cisplatin treatment (Fig. 4B), and thus sensitized A549/DDP cells to cisplatin. Additionally, A549/DDP cells transfected with miR-33b-3p inhibitor exhibited a modest G1 arrest (Fig. 4C), which might due to the elevated expression level of p21 (Fig. 4D).
Figure 4.
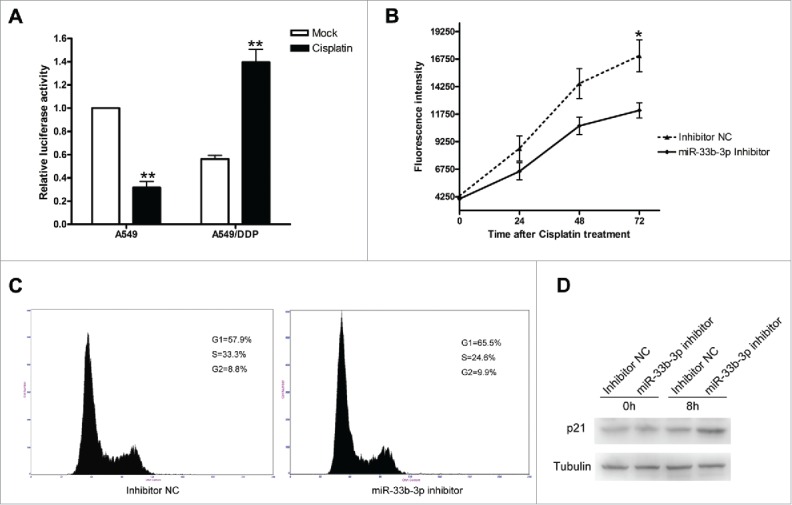
Inhibition of miR-33b-3p sensitized A549/DDP cells to cisplatin. (A) Real-time qPCR was performed to detect the expression levels of miR-33b-3p in the cisplatin-resistant cell line - A549/DDP cells with or without cisplatin treatment. Columns, mean of at least 3 independent experiments; bars, SEM. **, P < 0.01, comparison between 2 groups as indicated. (B) Effect of miR-33b-3p on A549/DDP cell viability in the absence or under cisplatin treatment. (C) Cell cycle analysis of A549/DDP cells was performed at 48 h after transfection with inhibitor NC or miR-33b-3p inhibitor under cisplatin treatment. The percentage of G1, S, and G2/M are demonstrated as shown. (D) The protein expression levels of p21 in inhibitor NC or miR-33b-3p inhibitor transfected A549/DDP cells with or without cisplatin treatment.
p21 was a direct target of miR-33b-3p
To uncover the underlying molecular mechanisms by which miR-33b-3p exerted its effect on cisplatin sensitivity, we then attempted to identify the targets of miR-33b-3p. Firstly, the target prediction analysis was performed using miRWalk software (http://zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk/). Coincidentally, among the predicted targets with high score, p21 had a perfect match with the ‘seed region’ of miR-33b-3p (Fig. 5A), which was negatively correlated with miR-33b-3p expression levels (Figs. 3D and 4D). Thus, p21 stood out as an attractive candidate target of miR-33b-3p.
Figure 5.

p21 was a direct target of miR-33b-3p. (A) Alignments of p21-3′UTR WT, miR-33b-3p and p21-3′UTR MUT, where the complementary site for the seed region of miR-33b-3p is indicated. (B) Analysis of luciferase activity. 293T cells were cotransfected with p21-3′UTR-WT with either miR-33b-3p or NC, and p21-3′UTR-MUT with either miR-33b-3p or NC. Columns, mean of at least 3 independent experiments done in duplicate; bars, SEM. **, P < 0.01, compared with NC-transfected cells. (C) Suppression of endogenous p21 mRNA expression by miR-33b-3p in A549 cells with or without cisplatin treatment. Columns, mean of at least 3 independent experiments done in duplicate; bars, SEM. **, P < 0.01, compared with NC-transfected cells. (D) Suppression of endogenous p21 protein expression by miR-33b-3p in A549 cells with or without cisplatin treatment in indicate times.
p21 as a direct target of miR-33b-3p was explored in the following ways. Initially, A549 cells were transfected with miR-33b-3p mimics or NC; and p21 mRNA and protein expressions were assayed 48h after transfection with or without cisplatin treatment. miR-33b-3p mimics but not NC significantly suppressed the expression of endogenous p21 at both mRNA and protein level (Fig. 5C and D). In addition, a dual-luciferase reporter system was prepared by cloning a wildtype p21-3′UTR-WT or p21-3′UTR-MUT (no complementarity with miR-33b-3p (Fig. 5A)) fragment (∼500 bp with a predicted target site of miR-33b-3p) downstream of the firefly luciferase reporter, and 293T cells were then cotransfected with the dual reporters and miR-33b-3p mimics or NC. miR-33b-3p significantly diminished the relative luciferase activity of the firefly luciferase reporter containing p21-3′UTR-WT (P < 0.001) but not p21-3′UTR-MUT, while NC had no effects on both reporters (Fig. 5B). Taken together, p21 was a direct and authentic target of miR-33b-3p.
miR-33b-3p promoted cisplatin resistance of lung cancer cells via targeting p21
P21 was shown to be an authentic target of miR-33b-3p, but further investigation was required of whether miR-33b-3p impacted on the cisplatin resistance of lung cancer cells through direct down-regulation p21. Therefore, we examined whether constitutive expression of p21 could counteract the effect of miR-33b-3p on cisplatin sensitivity. The p21 expression vector pcDNA3.0-p21△3′UTR was constructed with complete deletion of the 3′UTR, and its ability of overexpression in A549 cells was confirmed (Fig. 6A). A549 cells were first transfected with miR-33b-3p mimics or NC, then 24h later transfected with pcDNA3.0 or pcDNA3.0-p21△3′UTR, and then 24h later treated by cisplatin. The results revealed that ectopic expression of exogenous p21 significantly reversed the effect of miR-33b-3p on promoting cell proliferation under cisplatin treatment (Fig. 6B-D). In all, our data suggested that miR-33b-3p influenced the cisplatin sensitivity of lung cancer cells might probably through targeting p21.
Figure 6.
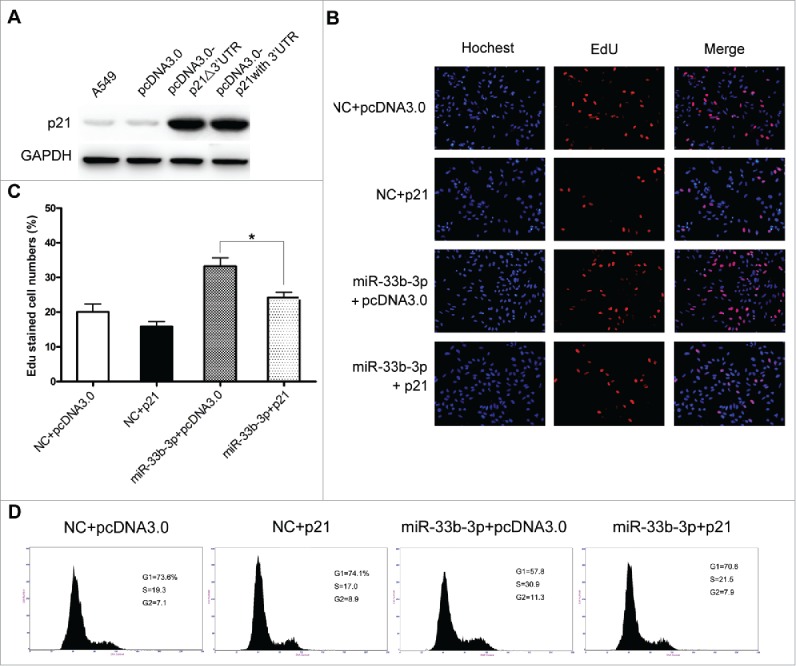
Ectopic expression of exogenous p21 substantially reversed the effect of miR-33b-3p on cisplatin resistance. (A) Over-expression of p21 in A549 cells. Western blot was used to monitor the expression level of p21 in A549 cells 48h after transfection with pcDNA3.0-p21△3′UTR (also designated as p21), pcDNA3.0-p21 with 3′UTR or empty pcDNA3.0 vector, GAPDH was used as an internal control. (B) and (C) Expression of exogenous p21 protein significantly abrogated the enhanced proliferation conferred by miR-33b-3p under cisplatin treatment. Columns, mean of at least 3 independent experiments; bars, SEM. *, P < 0.05; comparison between 2 groups as indicated. (D) Cell cycle analysis of of cells treated as (B). The percentage of G1, S, and G2/M are demonstrated as shown.
Discussion
Increasing number of DNA damage responsive miRNAs have recently been identified. However, the molecular mechanisms by which these miRNAs modulate the DNA damage response and chemoresistance of cancer cells are largely unknown. In this study, we identified that miR-33b-3p was dramatically down-regulated after cisplatin. Our results demonstrated that miR-33-3b-3p exerted a critical role in modulating the cisplatin sensitivity of lung cancer cells, which might probably through suppressing the p21 expression.
Previous studies have established that DNA lesions induced the deregulation of a variety of miRNAs, which played an important role in DNA damage response. For example, one group reported that UV irradiation triggered the up-regulation of miR-16 in HeLa cells, which caused the cell cycle arrest through targeting CDC25A.18 Another group showed that irradiation (IR) lead to the downregulation of miR-335 in an ATM dependent manner, and miR-335 was involved in homologous recombination repair (HRR) via suppressing CtIP expression.24 In addition, miR-15b has been established to be up-regulated following exposure to diverse stress-inducing agents, including ionizing radiation, etoposide, and hydrogen peroxide, which significantly influenced the cell cycle progression by targeting WIP1.25 The demonstration that miR-33b-3p was dramatically downregulated when exposed to cisplatin and impacted on DDR by negative regulation of p21, further enlarged the knowledge and confirmed the effects of miRNAs on DNA damage response.
Multiple miRNAs have been reported to be involved in DNA damage repair by targeting DNA repair genes. miR-192 inhibited NER by suppressing ERCC3 and ERCC4 in HepG2 cells.26 miR-103 and miR-107 directly targeted RAD51, which was an essential protein for catalyzing HR repair of DNA double-strand breaks (DSBs).27 Additionally, another critical component of non-homologous end joining (NHEJ) for DSBs repair-Ku80, was a direct target of miR-526b. Our data unraveled that A549 cells with miR-33b-3p overexpression exhibited lower levels of γH2A.X under cisplatin treatment, indicating that miR-33b-3p promoted DNA damage repair. What's more, ectopic expression of miR-33b-3p enhanced the expression of ERCC1 in A549 cells (Fig. 3C and D), which played a pivotal role in NER for the cisplatin-DNA adducts repair. Nonetheless, the molecular mechanisms of how miR-33b-3p regulates ERCC1 expression still require further investigations.
Accumulating evidences have unveiled the close associations between miRNAs and cisplatin sensitivity of lung cancer cells.28 miR-31 and miR-155 significantly inhibited the cisplatin sensitivity in non-small cell lung cancer cells by suppressing ABCB9 and Apaf-1, respectively,29,30 while miR-451 elevated the cisplatin sensitivity of A549 cells.31 Moreover, miR-98 sensitized cisplatin-resistant A549/DDP cells via upregulation of HMGA2.32 Our results that miR-33b-3p promoted non-small cell lung cancer cells survival when exposed to cisplatin, further confirmed the crucial effects of miRNAs on the chemoresistance of cancer cells, and provided a novel candidate for improving chemotherapy efficacy.
P21 was a member of cyclin-dependent kinase inhibitor, which played an important role in G1/S checkpoint control in response to DNA damage.33 Beyond inducing cell cycle arrest, the relationship of p21 and chemoresistance has been surrounded by controversy and conflicting results. Multiple studies have reported that p21 protected cells from death following anticancer treatments, and thus p21 knockdown could sensitize cancer cells to IR or chemotherapeutic agents.34,35 Nonetheless, there's a growing body of evidence uncovered that functional loss of p21 can mediate a drug-resistance phenotype in cancer therapy.36-38 Enforced expression of p21 inhibited cell growth and enhanced chemosensitivity to cisplatin in lung carcinoma cells.36 Moreover, miR-106b induced cell radioresistance, and miR-520g conferred drug resistance in colorectal cancer both by regulating p21 expression.39,40 And, miR-224 promoted the chemoresistance of A549 cells to cisplatin by targeting p21 as well.41 Our data that miR-33b-3p attenuated the cisplatin sensitivity of A549 cells by direct diminishing the p21 expression, was in consistent with above published literatures.
SIRT6 has been established to suppress the transcription of SREBP1 by H3K56 deacetylation in the promoter, which was the host gene of miR-33b-3p.42 SIRT6, similar as NFκB, was a stress-responsive chromatin modifier, which shaped stress -related transcriptional networks.43 Thus, we speculated that the down-regulation of miR-33b-3p by cisplatin treatment might probably through the SIRT6-mediated deacetylation in the promoter as well. Thus, we examined the expression level of SREBP1 in A549 cells after cisplatin treatment. The results showed that A549 cells treated with cisplatin displayed diminished mRNA level of SREBP1 compared with that in DMF treated cells, which was in consistent to the downregulation of miR-33b-3p (SFig. 3A), though the protein expression level of SIRT6 exhibited no significant difference after cisplatin treatment (SFig. 3B). In addition, the altered expression of SIRT6 by overexpression vector did not evidently influenced miR-33b-3p expression (SFig. 3D). Therefore, we inferred that the reduced miR-33b-3p expression was more derived from the affected miRNA biogenesis than the deacetylation in the promoter by SIRT6. However, further investigations still required to fully elucidate the detailed molecular mechanisms underneath the cisplatin responsive down-regulation of miR-33b-3p.
In aggregate, we herein provided the first evidence that miR-33b-3p was involved in the cisplatin induced DNA damage response, and thus significantly impacted on the cisplatin sensitivity of lung cancer cells by downregulation of p21. The identification that miR-33b-3p promoted the chemoresistance of lung cancer cells has great practical and clinical implications, providing new therapeutic candidates for improving chemotherapy efficacy.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (81170327, 81370456), the Natural Science Foundation of Guangdong Province (2014A030310027, 2016A030313684, 9252402301000002), the Medical Scientific Research Foundation of Guangdong Province (A2015288), the Science & Technology Planning Project for Medical and Health Organizations of Dongguan City (2012108102022), the Science & Technology Innovation Fund of Guangdong Medical University (STIF201102), Scientific Research Foundation of Guangdong Medical University (B2013002), the “Climbing” Program of Guangdong Province (pdjh2015b0239, pdjh2016b0219).
References
- [1].Cline SD, Hanawalt PC. Who's on first in the cellular response to DNA damage? Nat Rev Mol Cell Biol 2003; 4:361-72; PMID:12728270; http://dx.doi.org/ 10.1038/nrm1101 [DOI] [PubMed] [Google Scholar]
- [2].Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361:1475-85; PMID:19812404; http://dx.doi.org/ 10.1056/NEJMra0804615 [DOI] [PubMed] [Google Scholar]
- [4].Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287-94; PMID:22258607; http://dx.doi.org/ 10.1038/nature10760 [DOI] [PubMed] [Google Scholar]
- [5].Bouwman P, Jonkers J. The effects of deregulated DNA damage signalling on cancer chemotherapy response and resistance. Nat Rev Cancer 2012; 12:587-98; PMID:22918414; http://dx.doi.org/ 10.1038/nrc3342 [DOI] [PubMed] [Google Scholar]
- [6].Reed E. Platinum-DNA adduct, nucleotide excision repair and platinum based anti-cancer chemotherapy. Cancer Treat Rev 1998; 24:331-44; PMID:9861196; http://dx.doi.org/ 10.1016/S0305-7372(98)90056-1 [DOI] [PubMed] [Google Scholar]
- [7].Sears CR, Cooney SA, Chin-Sinex H, Mendonca MS, Turchi JJ. DNA damage response (DDR) pathway engagement in cisplatin radiosensitization of non-small cell lung cancer. DNA Repair (Amst) 2016; 40:35-46; PMID:26991853; http://dx.doi.org/ 10.1016/j.dnarep.2016.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].van Jaarsveld MT, Wouters MD, Boersma AW, Smid M, van Ijcken WF, Mathijssen RH, Hoeijmakers JH, Martens JW, van Laere S, Wiemer EA, et al.. DNA damage responsive microRNAs misexpressed in human cancer modulate therapy sensitivity. Mol Oncol 2014; 8:458-68; PMID:24462518; http://dx.doi.org/ 10.1016/j.molonc.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qian W, Wang J, Roginskaya V, McDermott LA, Edwards RP, Stolz DB, Llambi F, Green DR, Van Houten B. Novel combination of mitochondrial division inhibitor 1 (mdivi-1) and platinum agents produces synergistic pro-apoptotic effect in drug resistant tumor cells. Oncotarget 2014; 5:4180-94; PMID:24952704; http://dx.doi.org/ 10.18632/oncotarget.1944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov 2005; 4:307-20; PMID:15789122; http://dx.doi.org/ 10.1038/nrd1691 [DOI] [PubMed] [Google Scholar]
- [11].Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, Castedo M, Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene 2012; 31:1869-83; PMID:21892204; http://dx.doi.org/ 10.1038/onc.2011.384 [DOI] [PubMed] [Google Scholar]
- [12].Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281-97; PMID:14744438; http://dx.doi.org/ 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- [13].Kim VN. MicroRNA biogenesis: coordinated cropping and dicing. Nat Rev Mol Cell Biol 2005; 6:376-85; PMID:15852042; http://dx.doi.org/ 10.1038/nrm1644 [DOI] [PubMed] [Google Scholar]
- [14].Shenoy A, Blelloch RH. Regulation of microRNA function in somatic stem cell proliferation and differentiation. Nat Rev Mol Cell Biol 2014; 15:565-76; PMID:25118717; http://dx.doi.org/ 10.1038/nrm3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Goedeke L, Aranda JF, Fernandez-Hernando C. microRNA regulation of lipoprotein metabolism. Curr Opin Lipidol 2014; 25:282-8; PMID:24978143; http://dx.doi.org/ 10.1097/MOL.0000000000000094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wan G, Mathur R, Hu X, Zhang X, Lu X. miRNA response to DNA damage. Trends Biochem Sci 2011; 36:478-84; PMID:21741842; http://dx.doi.org/ 10.1016/j.tibs.2011.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mueller AC, Sun D, Dutta A. The miR-99 family regulates the DNA damage response through its target SNF2H. Oncogene 2013; 32:1164-72; PMID:22525276; http://dx.doi.org/ 10.1038/onc.2012.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pothof J, Verkaik NS, van IW, Wiemer EA, Ta VT, van der Horst GT, Jaspers NG, van Gent DC, Hoeijmakers JH, Persengiev SP. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J 2009; 28:2090-9; PMID:19536137; http://dx.doi.org/ 10.1038/emboj.2009.156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Liu Y, Li Y, Lu X. Regulators in the DNA damage response. Arch Biochem Biophys 2016; 594:18-25; PMID:26882840; http://dx.doi.org/ 10.1016/j.abb.2016.02.018 [DOI] [PubMed] [Google Scholar]
- [20].Heacock ML, Stefanick DF, Horton JK, Wilson SH. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair (Amst) 2010; 9:929-36; PMID:20573551; http://dx.doi.org/ 10.1016/j.dnarep.2010.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Li S. Implication of posttranslational histone modifications in nucleotide excision repair. Int J Mol Sci 2012; 13:12461-86; PMID:23202908; http://dx.doi.org/ 10.3390/ijms131012461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li A, Yu Y, Lee SC, Ishibashi T, Lees-Miller SP, Ausio J. Phosphorylation of histone H2A.X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. J Biol Chem 2010; 285:17778-88; PMID:20356835; http://dx.doi.org/ 10.1074/jbc.M110.116426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Furuta T, Ueda T, Aune G, Sarasin A, Kraemer KH, Pommier Y. Transcription-coupled nucleotide excision repair as a determinant of cisplatin sensitivity of human cells. Cancer Res 2002; 62:4899-902; PMID:12208738 [PubMed] [Google Scholar]
- [24].Martin NT, Nakamura K, Davies R, Nahas SA, Brown C, Tunuguntla R, Gatti RA, Hu H. ATM-dependent MiR-335 targets CtIP and modulates the DNA damage response. PLoS Genet 2013; 9:e1003505; PMID:23696749; http://dx.doi.org/ 10.1371/journal.pgen.1003505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rahman M, Lovat F, Romano G, Calore F, Acunzo M, Bell EH, Nana-Sinkam P. miR-15b/16-2 regulates factors that promote p53 phosphorylation and augments the DNA damage response following radiation in the lung. J Biol Chem 2014; 289:26406-16; PMID:25092292; http://dx.doi.org/ 10.1074/jbc.M114.573592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Xie QH, He XX, Chang Y, Sun SZ, Jiang X, Li PY, Lin JS. MiR-192 inhibits nucleotide excision repair by targeting ERCC3 and ERCC4 in HepG2.2.15 cells. Biochem Biophys Res Commun 2011; 410:440-5; PMID:21672525; http://dx.doi.org/ 10.1016/j.bbrc.2011.05.153 [DOI] [PubMed] [Google Scholar]
- [27].Huang JW, Wang Y, Dhillon KK, Calses P, Villegas E, Mitchell PS, Tewari M, Kemp CJ, Taniguchi T. Systematic screen identifies miRNAs that target RAD51 and RAD51D to enhance chemosensitivity. Mol Cancer Res 2013; 11:1564-73; PMID:24088786; http://dx.doi.org/ 10.1158/1541-7786.MCR-13-0292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rolfo C, Fanale D, Hong DS, Tsimberidou AM, Piha-Paul SA, Pauwels P, Van Meerbeeck JP, Caruso S, Bazan V, Cicero G, et al.. Impact of microRNAs in resistance to chemotherapy and novel targeted agents in non-small cell lung cancer. Curr Pharm Biotechnol 2014; 15:475-85; PMID:24846062; http://dx.doi.org/ 10.2174/1389201015666140519123219 [DOI] [PubMed] [Google Scholar]
- [29].Dong Z, Zhong Z, Yang L, Wang S, Gong Z. MicroRNA-31 inhibits cisplatin-induced apoptosis in non-small cell lung cancer cells by regulating the drug transporter ABCB9. Cancer Lett 2014; 343:249-57; PMID:24099915; http://dx.doi.org/ 10.1016/j.canlet.2013.09.034 [DOI] [PubMed] [Google Scholar]
- [30].Zang YS, Zhong YF, Fang Z, Li B, An J. MiR-155 inhibits the sensitivity of lung cancer cells to cisplatin via negative regulation of Apaf-1 expression. Cancer Gene Ther 2012; 19:773-8; PMID:22996741; http://dx.doi.org/ 10.1038/cgt.2012.60 [DOI] [PubMed] [Google Scholar]
- [31].Bian HB, Pan X, Yang JS, Wang ZX, De W. Upregulation of microRNA-451 increases cisplatin sensitivity of non-small cell lung cancer cell line (A549). J Exp Clin Cancer Res 2011; 30:20; PMID:21329503; http://dx.doi.org/ 10.1186/1756-9966-30-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xiang Q, Tang H, Yu J, Yin J, Yang X, Lei X. MicroRNA-98 sensitizes cisplatin-resistant human lung adenocarcinoma cells by up-regulation of HMGA2. Pharmazie 2013; 68:274-81; PMID:23700794 [PubMed] [Google Scholar]
- [33].Pines J. Cell cycle. p21 inhibits cyclin shock. Nature 1994; 369:520-1; PMID:7911227; http://dx.doi.org/ 10.1038/369520a0 [DOI] [PubMed] [Google Scholar]
- [34].Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/ 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- [35].Gorospe M, Wang X, Guyton KZ, Holbrook NJ. Protective role of p21(Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol Cell Biol 1996; 16:6654-60; PMID:8943319; http://dx.doi.org/ 10.1128/MCB.16.12.6654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wei J, Zhao J, Long M, Han Y, Wang X, Lin F, Ren J, He T, Zhang H. p21WAF1/CIP1 gene transcriptional activation exerts cell growth inhibition and enhances chemosensitivity to cisplatin in lung carcinoma cell. BMC Cancer 2010; 10:632; PMID:21087528; http://dx.doi.org/ 10.1186/1471-2407-10-632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A 2000; 97:9042-6; PMID:10908655; http://dx.doi.org/ 10.1073/pnas.160016897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu Z, Sun M, Lu K, Liu J, Zhang M, Wu W, De W, Wang Z, Wang R. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregualtion of p21(WAF1/CIP1) expression. PLoS One 2013; 8:e77293; PMID:24155936; http://dx.doi.org/ 10.1371/journal.pone.0077293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zheng L, Zhang Y, Liu Y, Zhou M, Lu Y, Yuan L, Zhang C, Hong M, Wang S, Li X. MiR-106b induces cell radioresistance via the PTEN/PI3K/AKT pathways and p21 in colorectal cancer. J Transl Med 2015; 13:252; PMID:26238857; http://dx.doi.org/ 10.1186/s12967-015-0592-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zhang Y, Geng L, Talmon G, Wang J. MicroRNA-520g confers drug resistance by regulating p21 expression in colorectal cancer. J Biol Chem 2015; 290:6215-25; PMID:25616665; http://dx.doi.org/ 10.1074/jbc.M114.620252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wang H, Zhu LJ, Yang YC, Wang ZX, Wang R. MiR-224 promotes the chemoresistance of human lung adenocarcinoma cells to cisplatin via regulating G(1)/S transition and apoptosis by targeting p21(WAF1/CIP1). Br J Cancer 2014; 111:339-54; PMID:24921914; http://dx.doi.org/ 10.1038/bjc.2014.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Elhanati S, Kanfi Y, Varvak A, Roichman A, Carmel-Gross I, Barth S, Gibor G, Cohen HY. Multiple regulatory layers of SREBP1/2 by SIRT6. Cell Rep 2013; 4:905-12; PMID:24012758; http://dx.doi.org/ 10.1016/j.celrep.2013.08.006 [DOI] [PubMed] [Google Scholar]
- [43].Kawahara TL, Rapicavoli NA, Wu AR, Qu K, Quake SR, Chang HY. Dynamic chromatin localization of Sirt6 shapes stress- and aging-related transcriptional networks. PLoS Genet 2011; 7:e1002153; PMID:21738489; http://dx.doi.org/ 10.1371/journal.pgen.1002153 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.