

ABSTRACT
DNA replication initiation is a key event in the cell cycle, which is dependent on 2 kinases - CDK2 and CDC7. Here we report a novel mechanism in which p53 induces G1 checkpoint and cell cycle arrest by downregulating CDC7 kinase in response to genotoxic stress. We demonstrate that p53 controls CDC7 stability post-transcriptionally via miR-192/215 and post-translationally via Fbxw7β E3 ubiquitin ligase. The p53-dependent pathway of CDC7 downregulation is interlinked with the p53-p21-CDK2 pathway, as p21-mediated inhibition of CDK2-dependent phosphorylation of CDC7 on Thr376 is required for GSK3ß-phosphorylation and Fbxw7ß-dependent degradation of CDC7. Notably, sustained oncogenic high levels of active CDC7 exert a negative feedback onto p53, leading to unrestrained S-phase progression and accumulation of DNA damage. Thus, p53-dependent control of CDC7 levels is essential for blocking G1/S cell-cycle transition upon genotoxic stress, thereby safeguarding the genome from instability and thus representing a novel general stress response.
KEYWORDS: CDC7, CDK2, cell cycle, DNA damage, DNA replication, Fbxw7β, GSK3β, miRNA-192/215, p21, p53, protein degradation
Introduction
The threat of genomic damage to the cell arises intrinsically through errors during DNA replication, as well as through genotoxic stress from reactive oxygen metabolites and exogenous stimuli (e.g. ionising radiation, chemotherapy, ultraviolet light). The p53 tumor suppressor protein maintains genomic stability by arresting cell cycle progression following exposure to genotoxic stimuli. p53 imposes either cell cycle arrest, which prevents the replication of damaged DNA, or triggers programmed cell death (apoptosis) which reduces the risk for cell transformation.1-5 p53 executes its master regulation of cell division via transcriptional, post-transcriptional and post-translational mechanisms.6,7 A paradigm for cell cycle arrest in response to DNA damage involves p21 (a transcriptional target of p53), which blocks cell cycle progression through the inhibition of cyclin-dependent kinases (CDK2) and the DNA polymerase δ-processivity factor PCNA.8 In addition, p53-dependent upregulation of micro-RNAs (mi-RNAs)9-11 such as mi-R34a and mi-R34b/c represents a post-transcriptional mechanism for regulation of gene expression.11-13 Fbxw7β, the F-box recognition module of the Skp1-Cul1-Rbx1 E3 ubiquitin ligase complex is also a target of p53, which can modulate CDK2 activity through post-translational targeting of cyclin E.14 Inhibition of CDK2 through different p53-mediated mechanisms impacts on the Rb/E2F pathway, suppressing transcription of E2F-dependent S-phase commitment genes and therefore cell cycle progression.15
CDK2 is one of 2 critical kinases involved in the initiation of DNA replication. CDK2 governs the regulation of pre-replication complex (pre-RC) assembly through CDC6 stability, and activates replication origins through phosphorylation of MCM-proteins and Sld2-TopBP1-Sld3.16,17 However, the firing of the replication origins is strictly dependent on concerted and non-redundant MCM helicase activation in situ by both CDK2 and CDC7 (cell division cycle 7),17 with the latter considered the essential trigger.18 CDC7 is the catalytic subunit of Dbf4-dependent kinase (DDK) and acts in concert with its regulatory subunit ASK (a human homolog of yeast Dbf4).19 Notably overexpression of CDC7 has been shown to correlate significantly with the malignant phenotypes of different tumors.20-22 Moreover, CDC7 overexpression in multiple tumor cell lines and tumor specimens from the breast, ovary and lung appears to be inversely correlated with loss of p53, implying a functional connection between p53 and CDC7 kinase.23
p53 is a transcription factor, yet screening for p53-transcriptional binding sites within the human genome failed to identify CDC7 as a p53 transcriptional target.24 This implies that either an indirect transcriptional response or an alternative mechanism is responsible for the apparent p53-dependent regulation of CDC7. Recently, p53- and DNA damage-dependent miRNA-192/215 were implicated in targeting a battery of proliferation-linked transcripts including CDC7.25,26 However, the p53-dependent miRNA regulatory loop had only moderate effects on the CDC7 transcript levels (30% reduction) compared to transfection with specific CDC7 siRNAs.26 This indicates that a miRNA-based mechanism alone does not explain the inverse relationship between p53 and CDC7, pointing toward a parallel, hitherto unknown, regulatory p53-dependent pathway(s). Given the role of CDC7 as a master switch in DNA replication initiation and the association of CDC7 overexpression and malignant tumor phenotypes, CDC7 regulation by p53 might present a pathway that cooperates with p53-p21-CDK2 inhibition to prevent genomic instability.
In this study we discovered a new p53-dependent stress pathway that regulates CDC7. We demonstrate that this cell stress pathway functions through post-transcriptional and post-translational regulatory mechanisms. Moreover, the post-translational regulation of CDC7 via proteolytic degradation appears to be dependent on the p53-p21-CDK2 pathway. Furthermore, the sustained high levels of active CDC7 exert negative feedback onto p53, leading to unrestrained S-phase progression and accumulation of DNA damage. Our findings highlight the importance of p53-CDC7 cell stress response pathway for the maintenance of genomic stability and the impact deregulation of this pathway may have on tumourigenesis.
Results
Cell cycle arrest following DNA damage is dependent on p53-mediated CDC7 protein loss
We first tested whether CDC7 is targeted in response to DNA damage. IMR90 fibroblasts were released from serum starvation, exposed to irradiation (IR) or Doxorubicin (Doxo), treated with Aphidicolin, and released into a drug-free medium (Fig. 1A and Fig. S1A). In total, 95% of the irradiated cells remained in G1, while 43% of untreated cells progressed into S-phase (Fig. S1B). Furthermore, the presence of γH2A.X foci, the accumulation of Rad51 protein27 and the phosphorylation of p53 at Ser15 after treatment with either IR or Doxo confirmed the activation of DNA damage-dependent pathways (Fig. 1B and Fig. S1C and D), followed by a rise in p53 levels (Fig. 1C and D, corresponding lanes 4 and 10). Importantly, the rise in p53 levels was correlated with the down-regulation of CDC7 protein levels in both the whole cell- and nuclear extracts (Fig. 1C and D, corresponding lanes 4 and 10). The purity and lack of cross contamination between the nuclear- and cytoplasmic fractions was confirmed by immunoblotting (Fig. S1H).28 Furthermore, Aphidicolin treatment alone did not alter p53 or CDC7 protein levels (Fig. S1E), indicating that both the stabilization of p53 and downregulation of CDC7 resulted from IR or Doxo treatment.
Figure 1.

DNA Damage Induces G1 Arrest via p53-dependent Down-regulation of CDC7. (A) Schematic presentation of the synchronisation protocol (see Methods).55 p53 siRNA was introduced 12h prior and irradiation (IR) 8h after the end of serum starvation. Cells were collected for analysis 4h after release from the second Aphidicolin block. (B) Immunoblotting analysis of the nuclear extract (NE) from untreated (UT) and IR cells probed with the indicated antibodies. Orc4 was used as a loading control. (C) Immunoblotting analysis of whole cell extracts (WCE) from synchronised IMR90 cells treated with either IR (6 Gy) or with Doxorubicin (Doxo) independently or in conjunction with either CDC7 overexpression (CDC7WT) and/or p53 silencing (p53KD), probed with the indicated antibodies. β-actin was used as a loading control. (D) Immunoblotting analysis of nuclear extracts (NE) from synchronised IMR90 cells treated with either IR (6 Gy) or Doxo independently or in conjunction with either CDC7 overexpression (CDC7WT) and/or p53 silencing (p53KD), probed with the indicated antibodies. Orc4 was used as a loading control. (E) FACS analyses are shown as pie charts to demonstrate that the G1 arrest induced by IR or Doxo can be abrogated by either p53 knockdown (p53KD) or ectopic expression of CDC7 (CDC7WT), as demonstrated by the resumption of cell cycle progression into the S-phase.
To investigate whether the IR/Doxo-induced decrease in CDC7 protein levels was p53-dependent, we knocked down p53 expression in IMR90 cells prior to IR/Doxo treatment (Fig. 1C and D, corresponding lanes 9 and 12 and Fig. S1F and G). Immunoblotting analysis demonstrated that the depletion of p53 abrogated the IR/Doxo-induced down-regulation of CDC7 (Fig. 1C and D) and resulted in a significant (21–25%) increase in the number of cells in S-phase (Fig. 1E, vi and ix, compared to iv and vii respectively), while the rise in CDC7 levels after silencing of p53 or ectopic expression of CDC7WT in non-stressed cells led to a further increase of the S phase cell population by ˜6% compared to control cells (Fig. 1E, ii and iii compared to i).
Interestingly, when CDC7 protein levels were rescued by exogenous expression of CDC7 (CDC7WT) in IR/Doxo-treated cells then this resulted in a partial bypass of the cell cycle arrest (Fig. 1e, v and viii compared to iv and vii), demonstrating that the p53-mediated downregulation of CDC7 protein levels in response to stress is an integral part of the G1 cell cycle arrest. The relief of the G1 arrest upon ectopic expression of CDC7 in Doxo-treated cells was linked to CDK2 activity, as witnessed by a reduction in p21 levels and the recovery of CDK2 activity (Fig. S2A and B).
p53-dependent miR-192/215 up-regulation contributes to G1 arrest and suppression of CDC7 mRNA
We next investigated the mechanism of p53-dependent CDC7 down-regulation without employing DNA damage, thus avoiding potential side effects. To that end we used low-dose Actinomycin D (ActD) treatment, which results in highly specific activation of p53.29 Immunoblotting analysis confirmed p53 activation in ActD-treated IMR90 cells, and in keeping with our previous results, showed an inverse correlation with CDC7 protein levels and a reduction in the number of cells in S-phase compared with control cells (Fig. 2A and B). Depletion of p53 in ActD-treated cells led to the recovery of both CDC7 protein levels and S-phase initiation, while the depletion of p53 in non-stressed cells enhanced both the CDC7 levels and the proportion of cells in S phase (Fig. 2A and B), which was consistent across a number of independent experiments (Fig. S2C).
Figure 2.

p53 Down-regulates CDC7 Transcript through miR-192/215 Pathway. (A) Immunoblotting analysis of whole cell extracts (WCE) and (B) bar diagram of cell populations with different DNA content as measured by FACS (values above the bars represent the percentage of cells with S-phase DNA content) from IMR90 cells demonstrate an inverse correlation between p53 and CDC7 protein levels during stress induced with a low dose of actinomycin D (ActD; 1 nM) in the presence or absence of p53 (via knockdown with siRNA). (B) Representative of 3 independent experiments. (C) qRT-PCR quantification of relative CDC7 mRNA expression levels (-fold change) in cells treated with either ActD or DMSO, or with p53 siRNA plus control (non-targeting) siRNANT compared with siRNANT treatment alone (CTRL). p53 silencing combined with ActD treatment was compared to cells treated with ActD only. (C) Mean ± SEM from 3 independent experiments (D) IMR90 cells were transfected with either p53 siRNA for 48h or CDC7 expressing plasmid (CDC7WT), miR-192/215 siRNA alone or miR-192/215 siRNA together with miR-192/215 synthetic mimics (miR-Mimics) for 24h and together with a low dose of ActD (1 nM). Results were compared to either non-targeting control siRNA plus DMSO (CTRL) or ActD treated cells (ActD). Values above the CDC7 bands indicate CDC7 mRNA levels as measured by qRT-PCR relative to the ActD treatment (lanes 1–6) and the values above the p53 bands indicate the residual p53 mRNA after p53 silencing relative to the ActD treatment as measured by qRT-PCR (lanes 2 and 3). (E) DNA content distribution analysis of samples treated as described in (D) is presented as a bar diagram. Values above the bars indicate the percentage of S-phase cells. Representative of 3 independent experiments.
Interestingly, the loss of CDC7 protein in ActD-treated cells correlated with a strong reduction in CDC7 mRNA levels (Fig. 2C). Conversely, silencing of p53 in non-stressed cells or prior to ActD treatment resulted in a 2.5 and 3-fold increase in CDC7 mRNA, respectively, indicating that p53 activation leads to downregulation of CDC7 transcript levels (Fig. 2C and Fig. S3A).
Notably, cell treatment with ActD resulted in a 4-fold increase of known p53 targets, miR-192/215 (Fig. S3A). The depletion of miR-192/215 in ActD-treated cells resulted in a 4-fold increase in CDC7 mRNA levels, confirming their role in p53-dependent down-regulation of CDC7 mRNA (Fig. S3A). Although different control treatments (DMSO, non-targeting siRNA) have resulted in slight rise of p53 levels, only ActD treatment has significantly stabilised and activated p53 as demonstrated by the upregulation of p53 target p21 (Fig. S3B).
Importantly, the increase in CDC7 transcript levels after ActD treatment of both p53- and miR-192/215 depleted cells was reflected at the protein level only in p53-depleted cells (Fig. 2D, lane 3). Only partial recovery of CDC7 protein levels was detected in ActD-treated cells depleted of miR-192/215 (Fig. 2D, lane 5), which was negated by synthetic duplex miRNA-192/215 mimetics (Fig. 2D, lane 6). The recovery of CDC7 protein levels directly correlated with an increase in the proportion of S-phase cells in presence of ActD and after silencing of miR-192/215. The dose-response effects of synthetic miR-192/215 mimetics were first established in non-stressed cells (Fig. S3F-H) and then validated in genotoxic context (Fig. S3I-K). Both synthetic as well as endogenous miR-192/215 increased after Doxo treatment targeted specifically 3′UTR CDC7 reporter clone as revealed by reduction in luciferase activity (Fig S3H).
These data indicate that only a part of the p53-dependent downregulation of CDC7 levels is achieved via miRNA-mediated degradation of the CDC7 transcript.
p53 Targets CDC7 through SCF-Fbxw7 E3 Ubiquitin Ligase
We reasoned that p53 could also indirectly modulate CDC7 protein stability. This possibility was supported by experimental data showing that the half-life of CDC7 in ActD-treated cells was reduced ˜4-fold following cycloheximide treatment (Fig. 3A and SI Data). Furthermore, the addition of the proteasome inhibitor, MG-132, partially restored CDC7 levels, suggesting that in stressed cells CDC7 undergoes enhanced proteolysis (Fig. 3B). The recovered CDC7 levels after the addition of MG-132, and separately after the inhibition of miR-192/215 (validated in Fig. S3C-E), collectively combine to restore the CDC7 protein levels observed following p53 silencing alone, suggesting that stress-induced post-translational and post-transcriptional mechanisms co-operate in p53-dependent CDC7 down-regulation.
Figure 3.

Stress-induced Proteolysis of CDC7 is Dependent on CDC4-type Phospho-degrons (CPDs). (A) IMR90 cells were treated with cycloheximide (CHX, 10 μg/ml) in the presence of either DMSO (CTRL) or Actinomycin D (ActD; 1nM). Cells were harvested at the indicated time points and whole cell extracts (WCE) were analyzed by immunoblotting with an antibody against CDC7. (B) Immunoblotting analysis of WCE prepared from IMR90 cells that were first treated with either p53 siRNAs (48h) or miR-192/215 siRNA (24h) and then treated with ActD in the presence or absence of MG-132 (10 μM). (C) The putative Fbxw7 recognition motifs (CPDs) in the CDC7 protein sequence. The CDC4-phospho degron (CPD) consensus sequence is xT/SPxxT/S/x, where x is any amino acid residue. (D) Alignment of the phosphodegron motifs (CPDs) present in known Fbxw7 substrates with 5 CPDs in the CDC7 primary protein sequence. Positions of the key amino acid residues are highlighted in blue. (E) CDC7 structure (PDB 4f9a) shown as a ribbon with indicated positions of CPDs. Positions of CPDs 1, 2, 4 and 5 were predicted by modeling using iTasser and Phyre2 (see Methods and Fig. S4); Thr 376 in CPD3 is shown in red, ATP is shown in magenta and the Mg atom is shown as a yellow sphere. (F) CDC7 mutated on CPDs (CPDs 3, 4 and 5, CDC7CPD) and ectopically-expressed for 48h in IMR90 cells demonstrated more pronounced resistance to degradation compared to CDC7WT in the absence or presence of ActD (1 nM). CTRL represents empty vector and DMSO treatment; β-actin was used as a loading control. (G) DNA content distribution analysis of samples analyzed in (F) shown as a bar diagram. Values above the bars indicate the percentage of cells with S-phase DNA content. (G) Representative of 3 independent experiments.
The analysis of the CDC7 primary sequence has identified 5 putative degron sites, which resemble the CDC4/Fbxw7 consensus phospho-degron (CPD) motif sequence, Φ-Φ-pT/PS-P-P-X-pS/pT/E, where Φ corresponds to a hydrophobic residue and X to any amino acid (Fig. 3C and D). Protein sequence alignment analysis showed variability in CDC7 CPD conservation (Fig. S4A), with CPD3 being completely conserved in the compared species and CPDs 4 and 5 most closely resembling the consensus CDC4/Fbxw7 recognition motif that appears to prefer the Thr-Pro residue pair at its start.30 Structural information is currently available only for CPD3 motif, with other 4 CPDs located in regions missing in the available crystal structure for CDC7.31 Using structure modeling we predicted the positions of the remaining CPDs on the CDC7 structure (Fig. 3E and Fig. S4B).
The conserved threonine residues in the 3 CPD motifs were mutated to alanine (CPDs 3–5: Thr376Ala, Thr472Ala and Thr503Ala) to test whether the resulting CDC7CPD mutant protein can resist degradation via the p53-dependent pathway in cells treated with ActD. Ectopic expression of either CDC7WT or CDC7CPD proteins enhanced the S phase population of IMR90 cells by 25% and 23%, respectively. Furthermore, the CDC7CPD protein was resistant to treatment, remained at high levels, and cells harbouring CDC7CPD had unrestrained S-phase entry, despite the presence of stress. Furthermore, ectopic expression of either CDC7WT or CDC7CPD proteins enhanced S phase population of non-stressed IMR90 cells by 15% and 13% respectively (Fig. 3F and G). Likewise, expression of CDC7WT or CDC7CPD in ActD-treated IMR90 cells enhanced S phase population by 27% and 21%, respectively (Fig. 3F and G). The CDC7CPD protein was more efficient in rescuing CDC7 levels during ActD treatment compared to CDC7WT ectopic expression (Fig. 3F and Fig. S5A). Notably, levels of CDC7 in ActD-treated cells remained reduced (˜0.4-fold) compared to the non-treated control, while ActD-treated cells transfected with CDC7CPD showed ˜8-fold increase of CDC7 levels, compared to ˜5.4-fold increase measured in ActD-treated cells transfected with CDC7WT (SI Data and Fig. 3F). These data indicate that the CPD motifs are required for proteolytic degradation of CDC7 in stressed cells via the p53-dependent pathway.
Interestingly, overexpression of either CDC7WT or CDC7CPD in this experiment significantly reduced p53 levels in both non-stressed and ActD-treated cells (Fig. 3F), suggesting the existence of a negative feedback pathway by which CDC7 can influence p53 levels. We compared the p53 levels in both non-stressed and ActD-treated cells after exposure to CDC7WT or CDC7CPD for 24h and 48h (Fig. S5B). The p53 levels were unaffected after CDC7 overexpression for 24h, but significantly dropped after 48h, which was also reflected by the change in the levels of the p53 targets Mdm2 and p21, indicating that CDC7-dependent negative feedback onto p53 appears to be time-dependent and requires sustained overexpression of CDC7. Comparative transfections of cells with CDC7WT or CDC7CPD, CDC7T/A (Thr376 substituted with Ala) and CDC7T/E (Thr376 substituted to Glu) yielded similar results after 24h, such that the p53 levels were unaffected (Fig. S6).
In order to understand how CDC7 might influence the stability of p53 we expressed CDC7WT, CDC7CPD and CDC7KAD (the catalytically-inactive mutant of CDC7) for 24h and 48h in context of ActD-incurred stress and compared their effect on p53 levels. The mutants CDC7WT and CDC7CPD, but not CDC7KAD, reduced the levels of p53, indicating that CDC7 kinase activity is important for the negative feedback onto p53 (Fig. S6B). Efficient depletion of Mdm2 by siRNA (Fig. S6B and C) restored the p53 levels, indicating that CDC7 suppression of p53 levels might directly or indirectly involve Mdm2.
Given that Fbxw7β is a bona fide p53 transcriptional target32 and an E3 ubiquitin ligase, which specifically interacts with CPD motifs, we next tested whether this protein was up-regulated by p53 in ActD-treated cells. The results show that Fbxw7β protein levels were increased, whereas neither APC specific-Cdh1 and CDC20, nor β-TrCP and Skp2, demonstrated p53-dependent up-regulation (Fig. 4A). Notably, the anti-Fbxw7 antibody was specific for the Fbxw7β isoform present in the cytoplasm, as demonstrated by the subcellular fractionations (Fig. S7A and B), and this cytoplasmic Fbxw7β was upregulated upon ActD treatment (Fig. S7B). Furthermore, the depletion of Fbxw7β by siRNA led to the accumulation of CDC7 in non-stressed cells and the recovery of CDC7 protein levels in ActD-treated cells, allowing ActD-treated cells to enter into S-phase (Fig. 4B and C). Moreover, CDC7 was efficiently co-immunoprecipitated with an anti-ubiquitin antibody (and vice-versa) from IMR90 and HCT116 cell extracts but not from HCT116 Fbxw7(CDC4)−/− cells (Fig. 4D and E). Unlike IMR90 cells where ubiquitin-modified CDC7 was detected in presence or absence of stress (ActD), ubiquitination of CDC7 in non-stressed HCT116 cells was below detectable levels (compare Fig. 4D lanes-12 and 13 with Fig. 4e lanes-11 and 12) after inhibition of proteasome by MG-132, indicating that CDC7 levels are stabilised in cancer cells at the steady-state. The p53-dependent CDC7 regulation in HCT116 cells was confirmed using isogenic p53+/+ and p53−/− lines, which showed that in the absence of p53, CDC7 levels remain stable after ActD treatment (Fig. S7C).
Figure 4.

Stress-induced CDC7 Proteolysis is Mediated by Fbxw7β and GSK3β kinase. (A) Immunoblotting analysis of whole cell extracts (WCE) from IMR90 cells after treatment with Actinomycin D (ActD; 1 nM, 24h) in the presence or absence of p53 siRNA. Samples probed with anti-Fbxw7β antibody (Fbxw7β*) were prepared from cytoplasmic fractions. (B) Immunoblotting analysis of WCE with the indicated antibodies demonstrates CDC7 stabilization after Fbxw7β silencing (Fbxw7βKD) for 48h in control (CTRL) IMR90 cells or IMR90 cells treated with ActD for 24h. CTRL represents non-targeting siRNA and DMSO treatment. (C) DNA content distribution profiles for the indicated treatments as described in (B) presented as a bar diagram. Values above the bars indicate the percentage of cells with S-phase DNA content. (C) Representative of 3 independent experiments. (D and E) Immunoblotting analysis of WCE after CDC7 or ubiquitin pull-down in (d) IMR90 or (e) HCT116 Fbxw7+/+ and Fbxw7−/− cells in the presence or absence of ActD (1nM, 24h with DMSO as a negative control) and the proteasome inhibitor MG-132 (10 and 20 μM for IMR90 and HCT116, respectively) where indicated. Ubiquitinated CDC7 is revealed in presence or absence of stress (ActD) only after inhibition of the proteasome MG-132. The intense cut band on the bottom are immunoblobulins (Ig) (F) Immunoblotting analysis of the in vitro kinase assay reactions performed using the recombinant active CDC7/ASK kinase complex in the presence or absence of recombinant active GSK3β and the presence or absence of ATP. The reaction samples were analyzed with 2 different anti-phospho-threonine antibodies (pThr Ab1 and pThr Ab2), anti-pThr376 (CDC7 pT376) and an antibody recognizing all phosphorylated serines (pSer). Recombinant protein loading was controlled with anti-CDC7 and anti-GSK3 antibodies and antibodies specifically detecting the GST and His tags.
Finally, we tested whether Fbxw7β-dependent degradation of CDC7 may require phosphorylation of CDC7 CPDs by GSK3β kinase, which is also a p53 activation target.30,33 Cross-immunoprecipitations indicated the formation of complexes between CDC7 and Fbxw7β, as well as GSK3β kinase (Fig. S7D). Moreover, GSK3β phosphorylated CDC7 threonine residues in vitro (Fig. 4F), without affecting the CDC7 Thr376, the CDK2-specific phosphorylation site,18 suggesting that GSK3β phosphorylates CDC7 on threonine sites other than Thr376.
miR-192/215- and Fbxw7β dependent pathways cooperate in stress-induced p53-dependent downregulation of CDC7
We next used silencing of p53, miR-192/215, Fbxw7β and GSK3β to determine whether these novel post-transcriptional (via miR-192/215) and post-translational (via GSK3β/Fbxw7β) p53-dependent pathways act individually or cooperate in CDC7 down-regulation in IMR90 and HCT116 colorectal cancer cells (Fig. 5). First we tested the impact of down-regulating p53, miR-192/215, Fbxw7β and GSK3β in non-stressed cells and compared their impact to the ectopic expression of CDC7 alone (Fig. 5A-C). Single and combined depletions of p53, miR-192/215, Fbxw7β and GSK3β, unlike Cdh1 depletion, stabilised CDC7, indicating that the p53-dependent pathways are involved in the basal regulation of CDC7 levels. The depletions also led to a marked stimulation of cyclin A levels and DNA synthesis (Fig. 5A and B).
Figure 5.
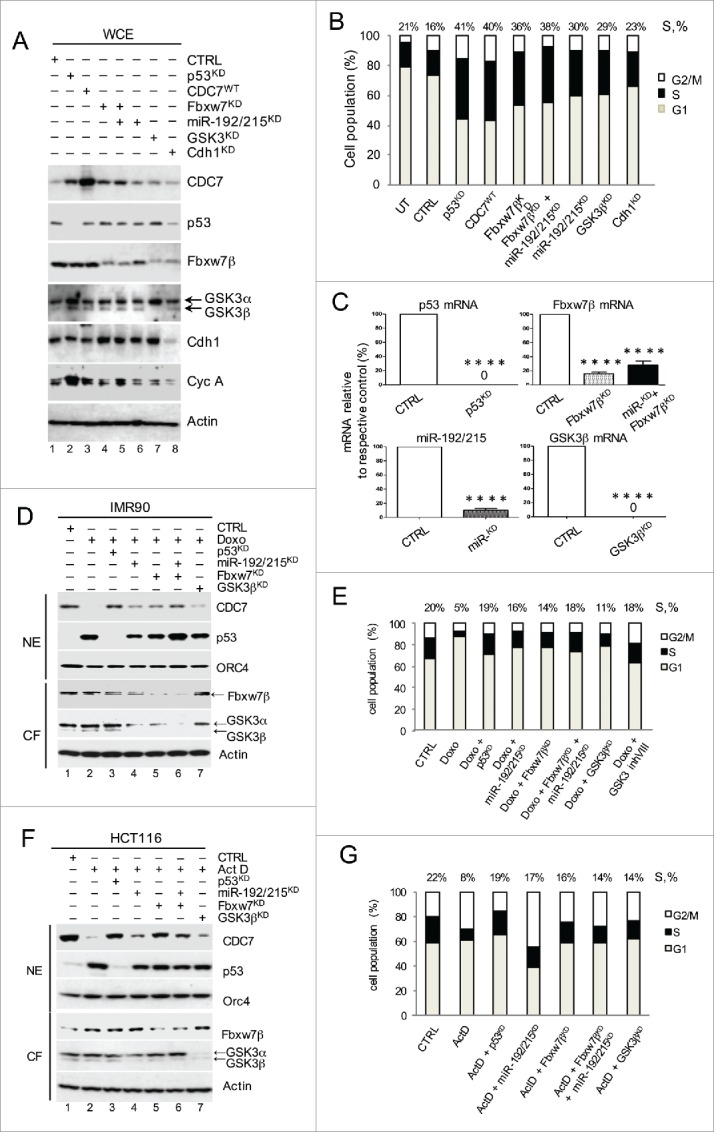
Post-transcriptional and Post-translational p53-dependend Pathways Cooperate to Down-regulate CDC7 in Cells Undergoing DNA Damage. (A) Immunoblotting analysis of whole cell extracts (WCE) from IMR90 cells treated with either p53 siRNA, miR-192/215 siRNA, Fbxw7β shRNA, a combination of miR-192/215 siRNA and Fbxw7β shRNA, GSK3β siRNA or Cdh1 siRNA. β-actin was used as a loading control. (B) DNA content distribution analysis of indicated samples as described in (A) is presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (B) Representative of 3 independent experiments. (C) Residual p53-, Fbxw7β-, miR-192/215- and GSK3β mRNA as measured by qRT-PCR in percentages (%) relative to the respective control (CTRL). (C) Mean ± SEM from 3 independent experiments (D) 2 μM Doxorubicin (Doxo) was added to IMR90 cells for 48h, after 24h pre-treatment with either p53 siRNA, miR-192/215 siRNA, Fbxw7β shRNA, a combination of miR-192/215 siRNA and Fbxw7β shRNA or GSK3β siRNA. CTRL represents the combination of non-targeting control siRNA and DMSO treatment. (E) DNA content distribution analysis of indicated samples as described in (D) is presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (E) Representative of 3 independent experiments. (F) 1 nM Actinomycin D (ActD) was added to HCT116 cells for 24h, after 24h pre-treatment with following siRNAs: either p53 siRNA, miR-192/215 siRNA, Fbxw7β shRNA, a combination of miR-192/215 siRNA and Fbxw7β shRNA or GSK3β siRNA. CTRL represents the combination of non-targeting control siRNA and DMSO treatment. (G) DNA content distribution analysis of indicated samples as described in (F) is presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (G) Representative of 3 independent experiments.
Next, we compared the impact of p53-dependent CDC7 regulators in stressed IMR90 (non-transformed) and HCT116 (cancer) cell lines (Fig. 5D-G). In IMR90 cells, Doxo treatment induced p53 upregulation, accompanied by complete downregulation of CDC7, which was partially reversed by silencing of either miR-192/215 or GSK3β/Fbxw7β (Fig. 5D and E). Combined depletion of miR-192/215 and Fbxw7β in IMR90 cells resulted in CDC7 protein recovery, reflecting the effect obtained by silencing p53 (Fig. 5D, lane 6 and lane 3). Similar effects were observed in IMR90 cells depleted of p53, miR-192/215, Fbxw7β and GSK3β in the presence or absence of ActD-induced stress (Fig. S8A and B).
Interestingly, there were differences in the recovery of CDC7 levels in normal and cancer cells when CDC7-down-regulating pathways were blocked separately or together. In IMR90 cells, the blocking of either pathway contributed to a better recovery of CDC7 levels by Fbxw7β silencing, and when both pathways were blocked the recovery of CDC7 levels was almost complete (Fig. 5D, lanes 4, 5 and 6 respectively and Fig. S8A and B). In HCT116 cells, however, blocking the Fbxw7β pathway led to a higher degree of CDC7 levels recovery than blocking the miR-192/215 pathway or combined depletion of miR-192/215 and Fbxw7β (Fig. 5F, lane 5 versus lane 4 and lane 5 vs. lane 6, respectively). In keeping with this, the S-phase population of Doxo-treated IMR90 cells was highest after combined pathway silencing (18%) and almost recovered to the level of the control cells (20%), whereas individual pathway blocking led to 14% and 16% recoveries for miR-192/215 and Fbxw7β depletion, respectively (Fig. 5E). ActD-treated HCT116 cells, however, recovered their S-phase population after both individual and combined silencing of miR-192/215 and Fbxw7β (18%, 20% and 19%, respectively, compared with 22% for the S-phase population in untreated cells) (Fig. 5G). These results showed that both p53-dependent pathways (via miR-192/215 and GSK3β/Fbxw7β) contribute to CDC7 down-regulation and act synergistically to achieve complete removal of CDC7 upon stress.
p21 complements the CDC7 downregulation under stress
We next tested whether there is any crosstalk between known p53-p21-CDK2 and novel p53-CDC7 pathways. To that end, we silenced p21 in Doxo-treated IMR90 cells and cells treated with both Doxo and miR-192/215 inhibitors (Fig. 6A and B). Silencing of p21 during Doxo-induced stress led to the recovery of CDK2 activity, which was reflected in CDC7 phosphorylation at Thr376 (CDC7 pT376) (Fig. 6A, lanes 5 and 6). CDC7 pT376 is normally present in cycling cells18 and was lost when cells were treated with Doxo and only recovered when either p53, or p21 was depleted in presence or absence of miR-192/215 (Fig. 6A lanes 3, 5 and 6). The fraction of S-phase (15%) in p21-silenced cells was similar to the S-phase population of Doxo-treated cells after miR-192/215 inhibition (16%), whereas the S-phase population following combined inhibition of miR-192/215 and p21 silencing was more pronounced (21%) and comparable with the S-phase population of Doxo-treated cells with depleted p53 (19%) (Fig. 6C).
Figure 6.

p21 complements the CDC7 downregulation under stress. (A) Levels of CDC7 and its downstream target MCM2 (phospho-Ser) pS10856 protein according to immunoblotting analysis of WCE after silencing p53 (p53KD) for 48h compared to the combination of miR-192/215 silencing and/or p21 silencing in Doxorubicin (Doxo) treated IMR90 cells. (B) Efficiency of p21 silencing (%) in cells treated with Doxo or Doxo and miR-192/215 siRNA as measured by qRT-PCR. (C) Mean ± SEM from 3 independent experiments (C) DNA content distribution profiles of indicated treatments in (A) presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (C) Representative of 3 independent experiments. (D) Immunoblotting analysis of the WCE from HCT116 p21−/− and p21+/+ cells in the presence or absence of Actinomycin D (ActD; 1 nM, 24h, DMSO as a negative control) and CDK2 silencing (CDK2KD) shows a reduction of CDC7 levels in p21−/− cells after CDK2- and CDC7 phospho-Thr376 (CDC7 pT376) inhibition. (E) DNA content distribution profiles of samples described in (D) presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (E) Representative of 3 independent experiments. (F) Effect of CDK2 inhibition by Roscovitine (Rosco, 40μM) on CDC7 recovery after p21 silencing in ActD-treated IMR90 cells as detected by immunoblotting. Phospho-Thr376 (pT376) CDC7 antibody was used to assess CDK2 inhibition by Rosco. (G) Immunofluorescence analysis of CDC7 levels in HCT116 p21−/− cells treated with either DMSO control, ActD (1 nM) or ActD (1 nM) and p21 siRNA in the presence or absence of Rosco (40 μM). Actin immunostaining (FITC) was used to visualize the cytoplasm.
In order to uncover the role of p21 in CDC7 stability, we next treated isogenic HCT116 p21+/+ and p21−/− cell lines with ActD. After 24h treatment CDC7 levels were reduced in p21+/+ cells but remained stable in p21−/− cells and were only reduced when CDK2 was silenced, which led to decreased phosphorylation of CDC7 on Thr376 (Fig. 6D). The efficiency of CDK2 silencing was confirmed by qRT-PCR (Fig. S9A). In p21−/− cells, the reduction of CDC7 levels was reflected by a drop in the proportion of S phase cells from 33% (ActD treatment only) to 12% when ActD treatment was combined with CDK2 silencing (Fig. 6E). CDK2 knockdown in IMR90 cells resulted in a reduction of CDC7 Thr376 phosphorylation similar to ActD treatment alone (Fig. S9B). These results demonstrated that CDC7 Thr376 is a CDK2 phosphorylation site and that CDK2 depletion in p21−/− cells leads to degradation of CDC7.
We next used isogenic HCT116 cancer cell lines with a genetic knockout of p53, Fbxw7 (CDC4) or p21 and treated these cells with ActD for 24h (Fig. S10A and B). In keeping with the previous observations,34 the p53 levels were up-regulated in control non-stressed p21−/− cells compared to p21+/+ cells. However, the levels of CDC7 were similar in p21−/− and p21+/+ cells and lower when compared with p53 and Fbxw7 isogenic cell lines. This low level of CDC7 was not affected by ActD treatment of p21−/− cells compared with p21+/+ cells (Fig. S10A). Consistent with our previous data, the levels of CDC7 did not decrease in cells lacking p53 and partially decreased in Fbxw7−/− cells (Fig. S10A). Unlike in HCT116 p53+/+, p21+/+ and Fbxw7+/+ cells, ActD treatment did not inflict reductions to the S-phase population in p53−/−, Fbxw7−/− and p21−/− cells (Fig. S10B).
Moreover, similar to the CDK2 knockdown, treatment with increased concentrations of the CDK2 inhibitor Roscovitine (Rosco), which mimics the presence of p21, led to a dose-dependent reduction of CDC7 levels in ActD-treated p21−/− cells, which was reflected in a reduction of CDC7 pT376 levels (Fig. S10C), confirming that CDK2 inhibition is relevant for CDC7 degradation. The combination of Rosco and ActD treatments reduced the number of cells in the S-phase (Fig. S10D) while inducing G2/M cell enrichment as previously reported.35 As expected, treatment with Rosco blocked the recovery of CDC7 levels in IMR90 cells treated with ActD and p21 siRNA (Fig. 6F, lane 5 versus lane 4 and Fig. 6G). These data indicate that p21 facilitates the p53-dependent degradation of CDC7 upon stress by inhibiting CDK2 and by negating the phosphorylation of CDC7 at Thr376 (CPD3).
CDC7 exerts negative feedback to p53 which leads to DNA damage
Prompted by the results showing CDC7-dependent negative feedback onto p53, we tested the effects of the sustained expression of CDC7WT and CDC7KAD proteins in non-transformed IMR90 cells. To achieve this, we transfected CDC7WT and CDC7KAD constructs in IMR90 cells every 48h for a total of 20 days. We observed efficient expression of both CDC7WT and CDC7KAD proteins (Fig. 7A and B). Importantly, when CDC7WT was expressed the p53 levels were depleted when compared to control expression; however, when CDC7KAD was expressed the p53 levels remained stable (Fig. 7A and B).
Figure 7.

Sustained overexpression of active CDC7 down-regulates p53, abrogates S-phase entry control and leads to DNA damage - biological implications of the p53-CDC7-regulatory pathway. Whole cell extracts (WCE) from IMR90 cells transfected every 48h with either wild type- (CDC7WT) in (A) or the kinase-inactive CDC7 (CDC7KAD) mutant in (B) over a 20 day time course and immunoblotting analysis for CDC7, p53 and γH2A.X levels (DNA damage marker). Cells transfected with an empty vector control (CTRL) acted as a negative control in both (A) and (B). β-actin was used as a loading control throughout. (C) DNA content distribution profiles of indicated treatments in (A) presented as a bar diagram. Values above the bars represent the percentage of cells with S-phase DNA content. (C) Representative of 3 independent experiments. (D) Schematic representation of the p53-dependent pathways responsible for controlling G1/S transition. The novel pathway is shown in blue. Genotoxic stress (irradiation, DNA damage and cytotoxic drugs) stabilises and activates the p53 tumor suppressor. p53 then transcriptionally up-regulates p21, which inhibits CDK2 activity and consequently prevents the CDK2-dependent phosphorylation of CDC7 at Thr376 located in CDC4-phosphodegron 3 (CPD3). Likewise, phosphorylation of Ser54, a CDK2 target in CDC6, is prevented, marking it for degradation by the APC/Cdh1 complex. Lack of CDC7 Thr376 phosphorylation facilitates CDC7 phosphorylation by stress-induced GSK3β on the other CPDs, and leads to the subsequent interaction with Fbxw7β E3 ubiquitin ligase and degradation. p53 induces both miR-192/215 which provides CDC7 transcript down-regulation, and Fbxw7β which targets CDC7 protein for degradation in response to genotoxic stress. CDC7 exerts negative feedback onto p53 through its kinase activity. The feedback loop allows CDC7 to retain its high levels and ensures cell entry into S-phase. (E) Stress-related CDC7-dependent cell cycle checkpoints. Genotoxic stress triggers (1) p53-dependent downregulation of CDC7 and (2) activation of a p53-dependent DNA origin activation checkpoint (OAC) inducing a robust G1/S cell cycle arrest.37-39 Increased CDC7 activity can down-regulate p53 levels via a negative feedback loop (3) and override the G1/S checkpoint. During S phase (4) in response to replication stress, activation of the ATR/Chk1 checkpoint results in stabilization of CDC7 that prevents replication fork collapse (40, 41). DNA-RI - DNA replication initiation.
Moreover, cells transfected with CDC7WT showed accumulation of DNA damage with the presence of γH2A.X.36 In contrast, control- or CDC7KAD-expressing cells did not accumulate DNA damage (Fig. 7A and B). In spite of the DNA damage, CDC7WT expressing cells showed a gradual rise in the S phase proportion (Fig. 7C). Thus, the sustained expression of active CDC7 kinase leads to suppression of p53, accumulation of DNA damage, and unrestrained S-phase entry despite the DNA damage.
Discussion
To maintain genomic stability, cells have to synchronise DNA damage repair and replication with the cell cycle, and the p53 tumor suppressor is central to this process. Here we have demonstrated that the p53-dependent G1 arrest following DNA damage is mediated via 2 separate signaling axes (Fig. 7D). We have shown for the first time that in addition to the p53-dependent inhibition of CDK2, blocking of the G1/S transition also requires the p53-dependent targeting of CDC7, a second key kinase involved in DNA origin firing. This pathway consists of CDC7 transcript down-regulation via miRNAs 192/215 and CDC7 protein degradation via Fbxw7β E3 ubiquitin ligase. The two arms of the novel pathway combine to reduce levels of CDC7 kinase efficiently in cells that are subject to genotoxic stress, thereby halting both cell cycle progression and origin firing. Using two separate molecular mechanisms may allow p53 to establish a temporal element of control over CDC7 levels, with Fbxw7β- providing a rapid way to eliminate large amounts of CDC7 protein, while the miR-192/215 down-regulating CDC7 transcript.
Importantly, our data shows that the 2 p53-dependent signaling pathways regulating CDK2 and CDC7 are interlinked (Fig. 7D). Here we discovered that CDK2 inhibition by p21 abrogates Thr376 phosphorylation of CDC7, which facilitates Fbxw7β-mediated CDC7 degradation, in a similar fashion to Ser54 in CDC6 which then leads to Cdc6 degradation and blocking of MCM recruitment to the origins.16 Through these interlinked pathways p53 is therefore able to mediate a powerful G1/S phase arrest in response to genotoxic insult via inhibition of pre-RC assembly and suppression of DNA origin firing. Interestingly, our data suggests the existence of further cross talk between the p53-p21-CDK2 and the p53-[miR-192/215 Fbxw7β]-CDC7 signaling axes. Thus, we observed that abrogation of G1/S arrest by CDC7 overexpression is linked to a recovery of CDK2 activity and a reduction in p21 levels. In contrast, reducing CDC7 levels in response to stress provides an important trigger of G1/S arrest by the p53-dependent DNA origin activation checkpoint (OAC)37-39 (Fig. 7E). The role of p53 in reducing CDC7 levels, and the fact that CDC7 is an integral component of the OAC, might incur a potential positive feedback loop on the G1/S arrest, thereby assuring continuous protection of the genome until DNA repair is completed. Thus, loss of p53 will not only abrogate the OAC but also will increase the levels of the CDC7.
In this context, loss of p53 will affect the basal proliferation since perturbing one of p53-dependent pathways (miR-192/215 or Fbxw7β) resulted in enhanced proportion of cells in the S phase due to stabilised CDC7. These results indicate that p53-CDC7 regulatory pathway could at least in a part determine the cellular turnover, worth addressing in the future.
The importance of CDC7 downregulation is further re-iterated in the fact that over-expression of CDC7 in cells undergoing DNA damage was sufficient to override p53-induced G1/S arrest, permitting cells to enter S-phase despite their genomes being severely damaged, in keeping with the oncogenic potential of CDC7 and CDC7-dependent malignancies.20,22
The oncogenic potential of sustained CDC7 overexpression was supported by our data revealing a novel negative feedback loop from CDC7 to p53. Notably, this feedback requires CDC7 kinase activity and might involve Mdm2 (Fig. S6B and C). The exact mechanism of Cdc7 negative feedback on p53 warrants further detailed investigation.
In the context of abrogation of cell cycle control, it appears that CDC7 overexpression has different effects on G1/S, S and G2/M transitions. Thus, in G1, high levels of CDC7 suppress the p53 pathway via negative feedback loop to override the p53-CDC7 G1/S checkpoint leading to an unrestrained entry into S-phase. In contrast, during S phase compromised DNA replication is dealt with by the ATR/Chk1 checkpoint leading to CDC7 stabilization which prevents replication fork collapse (Fig. 7E).40,41 Similar to oncogenic CDC642 and given the role of CDC7 in the ATR/Chk1 checkpoint,43,44 it is possible that oncogenic levels of CDC7 can influence the G2/M arresting pathway.
Interestingly recent work by Galanos et al.45 indicated that prolonged expression of p21 in p53-deficient cells can deregulate DNA replication leading to genomic instability further consolidating CDC7 as a potent anti-cancer target in p53 mutant tumors.37,46-48
In summary, this study shows that the p53-induced G1 growth arrest following genotoxic stress is critically dependent on the p53-[miR-192/215; Fbxw7β]-CDC7 signaling pathway, in addition to the classical p53-p21-CDK2 pathway. The parallel and synergistic action of these pathways therefore represents a novel and fundamental cell cycle mechanism, a p53-linked regulatory network that plays an important role in mediating G1 growth arrest in response to genotoxic insult.
Methods
Cell culture and cell synchronisation
Wild type and HCT116 p53−/−, HCT116 p21−/− and HCT116 Fbxw7 (Cdc4)−/− cell lines were a kind gift from Dr Bert Vogelstein. IMR90 (ATCC# CCL-186), a diploid human fibroblast cell line, was obtained from LGC Standards (Middlesex, UK) at population doubling (PD) 10. IMR90 cells were cultured at 37°C and 5% CO2 in DMEM (Invitrogen, Paisley, UK), supplemented with 10% FCS (Invitrogen), 100 U/ml penicillin and 100 μg/ml streptomycin. For the preparation of synchronous cell populations, IMR90 cells were treated with 2 consecutive blockades, for G0/G1 enrichment, by starvation with 0.1% FCS for 72 h and for G1/S enrichment, by the addition of Aphidicolin (5 μg/ml) (Sigma) for 12 h, separated by an interval of 12h in FCS-complemented medium. Cells were harvested for analysis 4 h after release from the second block.
Actinomycin D, Doxorubicin, Cycloheximide, Roscovitine and MG-132 were purchased from Sigma. GSK3β inhibitor VIII49 was purchased from Calbiochem.
Doxorubicin was used at 2 µM final concentration. Ionising irradiation of 6 Gy/min was performed using an AGO HS X-Ray System Model CP320 with a Varian NDI-321 X-ray tube of 320kV and an oil cooled stationary anode metal ceramic X-ray source.
Cell cycle analysis
Flow cytometric cell cycle analyses were performed using Beckman Coulter CyAN ADP as described elsewhere. Gating and analysis of FACS histograms were performed using FlowJo software (Tree Star, Inc. USA).
Cell fractionation and immunoprecipitation
For immunoblotting analysis, cells were harvested and whole cell extracts (WCE) and subcellular fractions were prepared in modified RIPA buffer.37 Chromatin-bound fraction (CBF) was isolated as described previously.37 Subcellular fractionation and in vitro kinase assays were performed as described previously.37 Phosphorylation of truncated Rb (CDK2 substrate; QED Bioscience, San Diego, CA) was detected with anti-Rb phospho Ser 612 antibody (Invitrogen).
The recovery of CDK2 activity after ectopic expression of CDC7 in stressed cells was assessed by in vitro kinase assay.37
Luciferase assay was obtained from Promega and performed according to manufacturer's instructions in lysates from HCT116 cells co-transfected with 3′UTR luciferase reporter clone of CDC7 (OriGene) and miR-192/215 synthetic mimetics (Qiagen, Ltd.).
Immunoblotting
Protein concentration was determined using the DC Bio-Rad protein assay kit (Bio-Rad Hemel Hempstead, UK). 15–60 μg of total protein was loaded in each lane and separated by 4% to 20% SDS-PAGE. Protein was transferred from polyacrylamide gels onto PVDF membranes (Bio-Rad) by semi-dry electroblotting. Blocking, antibody incubations and washing steps were performed as described.22 The antibodies used for immunoblotting (see below) included: Rb, CDK2, GSK3, Fbxw7, c-MYC (N-262), Skp2, Cdc-20 and Rad51 from Santa Cruz (Santa Cruz, CA); Orc4, MCM2 (BM28), p21 and β-TrCP from BD Biosciences (Oxford, UK); ubiquitin from Abcam (Cambridge, UK), p53 (Ab-6); Cdh1 from Merck (Beeston, UK); CDC7 and CDC7 phospho-Thr376 from MBL International (Woburn, MA); β-actin from Sigma (Gillingham, UK); p53 phospho-Ser-15 and phospho-histone γH2A.X (Ser-139) from New England Biolabs (Hitchin, UK); and Rb phospho-Ser 612 from Invitrogen. Affinity-purified rabbit polyclonal anti-geminin antibody G95 was generated as described.50 The immunoblot images were scanned, and densitometric analysis of the immunodetected protein bands was performed using ImageJ software (SI Data).
RNA interference
CDC7 and p53 mRNA depletion was performed with double-stranded RNA custom oligos as previously described,37 while miR-192/215 levels were silenced by custom inhibitors with targeting sequence AAUUGACAGCCAGUGCUCUCGUU for miR-192 and AAUGACCUAUGAAUUGACAGAUU for miR-215 purchased from Thermo Scientific Dharmacon. p21, GSK3β and ASK (Dbf4) SMART pool siRNAs were purchased from Thermo Scientific Dharmacon. shFbxw7β was obtained under the cloning code HNRCLO-136632 from NKI Cancer Specific pRetroSuperCam shRNA library of the Scientific Services at the Wolfson Institute for Biomedical Research (UCL, London, UK). Non-targeting siRNA (Invitrogen) was used as a negative control. Lipofectamine RNAiMAX (Invitrogen) was used in all transfections according to the manufacturer's recommendations. The transient transfections were performed (see Ref 37) using 10 nM of either CDC7, p53, GSK3 and p21 duplex, or a mixture of 10 nM miR-192 and 10 nM miR-215 (referred to as miR-192/215). siRNAs and inhibitors were used according to the manufacturer's instructions (Invitrogen).
For p53 knockdown, cells were incubated for 24 to 72h depending on the experimental setup, for miR-192/215 depletion for 48h, and for Fbxw7 knockdown for 24h. All experiments were performed at least 3 times. QRT-PCR and immunoblotting were used to confirm selective silencing of the corresponding proteins.
Cell transfections
For rescue experiments, the full 1725 bp CDC7 cDNA sequence inserted into pCMV6-AC expression vector (OriGene) was used, as described in.37 The specificity of miR-192/215 inhibition was tested with a rescue experiment using a cocktail of synthetic miR192/215 mimics (Qiagen Ltd.). pCMV6-AC CDC7CPD (CDC7 Thr376A/Thr472A/Thr503A), pCMV6-AC CDC7T/A (CDC7 Thr376A), pCMV6-AC CDC7T/E (CDC7 Thr376E) and pCMV6-AC CDC7KAD (CDC7 catalytically inactive - K90A; D196A) were generated synthetically (Geneart; Life Technologies). 3′ UTR luciferase reporter clone of CDC7 for miRNA validation was obtained from OriGene. All plasmid DNAs used for transfection were prepared with a Plasmid Maxi Prep Kit (Qiagen) following the manufacturer's protocol, and their quality and integrity was assessed by DNA sequencing. Transfection of the plasmids was performed using Lipofectamine LTX according to the manufacturer's instructions.
RNA extraction and qRT-PCR
To evaluate the efficiency of transfection with CDC7, p53, p21, CDK2, Mdm2, GSK3β, Fbxw7β and miR-192/215 mimics, mRNA transcription levels were assessed by qRT-PCR. Total RNA was isolated using a PureLink Micro-to-Midi kit (Invitrogen) according to the manufacturer's instructions. Reverse transcription reactions using 40 ng of total RNA were performed in a one step process using a SuperScript III Platinum SYBR Green One Step qRT-PCR Kit (Invitrogen). Melting temperatures (Tm) used for the annealing step were 47°C for CDC7, 50°C for p53 and 54°C for miR-192/215. Relative quantifications were obtained using the comparative Ct method with Realplex software according to the manufacturer's protocol (Eppendorf, Heidelberg, Germany). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used to normalize each of the extracts for amplifiable human DNA. Primers were provided by Eurofins MWG Operon (Ebersberg, Germany). Cycle conditions and primers are available upon request. For miR-192/215 relative quantification, first specific cDNA was synthesized using LNA-modified primers for miR-192 and miR-215 and miRCURY LNA Universal RT microRNA cDNA Synthesis Kit from Exiqon (Vedbaek, Denmark), followed by qRT-PCR using a QuantiFAST SYBR Green Kit from Qiagen. MiR-16 purchased from Exiqon (Vedbaek, Denmark) was used as the normalizing control. The PCR conditions for miR-192/215 were as follows: initial activation at 95°C for 5 min then 40 cycles of 95°C for 10s and 60°C for 30s with a ramp rate of 1.6°C/s.
CDC7 protein sequence and structure analysis
CDC7 primary sequence was analyzed using ExPASy Bioinformatics Resource portal tools
http://www.expasy.org/proteomics and ELM51 (http://elm.eu.org/). Multiple protein sequence alignment was prepared by using CLUSTALW.52
CDC7 structural modeling has been performed by using Phyre253 (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) and i-TASSER54 (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). Illustrations were generated using PyMOL (http://pymol.sourceforge.net/).
Statistics
Statistical significance was determined using a 2-tailed t test or ANOVA where appropriate. P < 0.05 was considered significant (*), P < 0.01 (**); P < 0.001 (***) and P < 0.0001 (****).
Supplementary Material
Abbreviations
- ActD
Actinomycin D
- CDC7
cell division cycle 7
- CDK2
cyclin-dependent kinase 2
- Doxo
Doxorubicine
- IR
irradiation
- miR-192/215
micro RNAs 192 and/or 215
- OAC
origin of replication activation checkpoint
- Rosco
Roscovitine
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Evelyn Gomez-Espinosa, Sofija Lazarevska, Eva Dainyte and Kenny Vongbunyong for technical help with the cell cultures.
Funding
STT was funded by Celia Abraham, Matthew Baldwin Glioblastoma Research Fund and Ernst Jung Stiftung. AO was funded by the BBSRC (BB/E019862/1) and CRDC (F175). This study has been supported by Cancer Research UK scientific program grant C428/A6263 (KS and GHW). SL was supported by Industry Research grant from REPLEK Pharm to STT.
Authors' contribution
STT, KS, AO and GHW generated the hypothesis. STT has designed and performed all the experiments with help from AD and AO. STT, PM, AO, KS and GHW analyzed the data. STT, AO, and GHW wrote the manuscript.
ORCID
Paul Mulholland http://orcid.org/0000-0002-7926-824X
References
- [1].Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell 2009; 137:413-31; PMID:19410540; http://dx.doi.org/ 10.1016/j.cell.2009.04.037 [DOI] [PubMed] [Google Scholar]
- [2].Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275-83; PMID:17380161; http://dx.doi.org/ 10.1038/nrm2147 [DOI] [PubMed] [Google Scholar]
- [3].Junttila MR, Evan GI. p53–a Jack of all trades but master of none. Nat Rev Cancer 2009; 9:821-9; PMID:19776747; http://dx.doi.org/ 10.1038/nrc2728 [DOI] [PubMed] [Google Scholar]
- [4].Lane D, Levine A. p53 Research: the past thirty years and the next thirty years. Cold Spring Harbor Perspect Biol 2010; 2:a000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Aylon Y, Oren M. New plays in the p53 theater. Curr Opin Gen Dev 2011; 21:86-92; http://dx.doi.org/ 10.1016/j.gde.2010.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer 2009; 9:724-37; PMID:19776742; http://dx.doi.org/ 10.1038/nrc2730 [DOI] [PubMed] [Google Scholar]
- [7].Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 2008; 9:702-12; PMID:18719709; http://dx.doi.org/ 10.1038/nrm2451 [DOI] [PubMed] [Google Scholar]
- [8].Waga S, Hannon GJ, Beach D, Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994; 369:574-8; PMID:7911228; http://dx.doi.org/ 10.1038/369574a0 [DOI] [PubMed] [Google Scholar]
- [9].He X, He L, Hannon GJ. The guardian's little helper: microRNAs in the p53 tumor suppressor network. Cancer Res 2007; 67:11099-101; PMID:18056431; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2672 [DOI] [PubMed] [Google Scholar]
- [10].He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al.. A microRNA component of the p53 tumour suppressor network. Nature 2007; 447:1130-4; PMID:17554337; http://dx.doi.org/ 10.1038/nature05939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].He L, He X, Lowe SW, Hannon GJ. microRNAs join the p53 network–another piece in the tumour-suppression puzzle. Nat Rev Cancer 2007; 7:819-22; PMID:17914404; http://dx.doi.org/ 10.1038/nrc2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bommer GT, Gerin I, Feng Y, Kaczorowski AJ, Kuick R, Love RE, Zhai Y, Giordano TJ, Qin ZS, Moore BB, et al.. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr Biol 2007; 17:1298-307; PMID:17656095; http://dx.doi.org/ 10.1016/j.cub.2007.06.068 [DOI] [PubMed] [Google Scholar]
- [13].Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci U S A 2007; 104:15472-7; PMID:17875987; http://dx.doi.org/ 10.1073/pnas.0707351104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 2001; 294:173-7; PMID:11533444; http://dx.doi.org/ 10.1126/science.1065203 [DOI] [PubMed] [Google Scholar]
- [15].Sala A, Nicolaides NC, Engelhard A, Bellon T, Lawe DC, Arnold A, Grana X, Giordano A, Calabretta B. Correlation between E2F-1 requirement in the S phase and E2F-1 transactivation of cell cycle-related genes in human cells. Cancer Res 1994; 54:1402-6; PMID:8137237 [PubMed] [Google Scholar]
- [16].Duursma A, Agami R. p53-Dependent regulation of Cdc6 protein stability controls cellular proliferation. Mol Cell Biol 2005; 25:6937-47; PMID:16055707; http://dx.doi.org/ 10.1128/MCB.25.16.6937-6947.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Labib K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Gen Dev 2010; 24:1208-19; http://dx.doi.org/ 10.1101/gad.1933010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Masai H, Matsui E, You Z, Ishimi Y, Tamai K, Arai K. Human Cdc7-related kinase complex. In vitro phosphorylation of MCM by concerted actions of Cdks and Cdc7 and that of a criticial threonine residue of Cdc7 bY Cdks. J Biol Chem 2000; 275:29042-52; PMID:10846177; http://dx.doi.org/ 10.1074/jbc.M002713200 [DOI] [PubMed] [Google Scholar]
- [19].Kumagai H, Sato N, Yamada M, Mahony D, Seghezzi W, Lees E, Arai K, Masai H. A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells. Mol Cell Biol 1999; 19:5083-95; PMID:10373557; http://dx.doi.org/ 10.1128/MCB.19.7.5083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rodriguez-Acebes S, Proctor I, Loddo M, Wollenschlaeger A, Rashid M, Falzon M, Prevost AT, Sainsbury R, Stoeber K, Williams GH. Targeting DNA replication before it starts: Cdc7 as a therapeutic target in p53-mutant breast cancers. Am J Pathol 2010; 177:2034-45; PMID:20724597; http://dx.doi.org/ 10.2353/ajpath.2010.100421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Loddo M, Kingsbury SR, Rashid M, Proctor I, Holt C, Young J, El-Sheikh S, Falzon M, Eward KL, Prevost T, et al.. Cell-cycle-phase progression analysis identifies unique phenotypes of major prognostic and predictive significance in breast cancer. Br J Cancer 2009; 100:959-70; PMID:19240714; http://dx.doi.org/ 10.1038/sj.bjc.6604924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kulkarni AA, Kingsbury SR, Tudzarova S, Hong HK, Loddo M, Rashid M, Rodriguez-Acebes S, Prevost AT, Ledermann JA, Stoeber K, et al.. Cdc7 kinase is a predictor of survival and a novel therapeutic target in epithelial ovarian carcinoma. Clin Cancer Res 2009; 15:2417-25; PMID:19318489; http://dx.doi.org/ 10.1158/1078-0432.CCR-08-1276 [DOI] [PubMed] [Google Scholar]
- [23].Bonte D, Lindvall C, Liu H, Dykema K, Furge K, Weinreich M. Cdc7-Dbf4 kinase overexpression in multiple cancers and tumor cell lines is correlated with p53 inactivation. Neoplasia 2008; 10:920-31; PMID:18714392; http://dx.doi.org/ 10.1593/neo.08216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, Shahab A, Yong HC, Fu Y, Weng Z, et al.. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207-19; PMID:16413492; http://dx.doi.org/ 10.1016/j.cell.2005.10.043 [DOI] [PubMed] [Google Scholar]
- [25].Georges SA, Biery MC, Kim SY, Schelter JM, Guo J, Chang AN, Jackson AL, Carleton MO, Linsley PS, Cleary MA, et al.. Coordinated regulation of cell cycle transcripts by p53-Inducible microRNAs, miR-192 and miR-215. Cancer Res 2008; 68:10105-12; PMID:19074876; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1846 [DOI] [PubMed] [Google Scholar]
- [26].Feng S, Cong S, Zhang X, Bao X, Wang W, Li H, Wang Z, Wang G, Xu J, Du B, et al.. MicroRNA-192 targeting retinoblastoma 1 inhibits cell proliferation and induces cell apoptosis in lung cancer cells. Nucl Acids Res 2011; 39:6669-78; PMID:21511813; http://dx.doi.org/ 10.1093/nar/gkr232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tashiro S, Walter J, Shinohara A, Kamada N, Cremer T. Rad51 accumulation at sites of DNA damage and in postreplicative chromatin. J Cell Biol 2000; 150:283-91; PMID:10908572; http://dx.doi.org/ 10.1083/jcb.150.2.283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Barkley LR, Hong HK, Kingsbury SR, James M, Stoeber K, Williams GH. Cdc6 is a rate-limiting factor for proliferative capacity during HL60 cell differentiation. Exp Cell Res 2007; 313:3789-99; PMID:17689530; http://dx.doi.org/ 10.1016/j.yexcr.2007.07.004 [DOI] [PubMed] [Google Scholar]
- [29].Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 2009; 8:2810-8; PMID:19657224; http://dx.doi.org/ 10.4161/cc.8.17.9503 [DOI] [PubMed] [Google Scholar]
- [30].Welcker M, Clurman BE. FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 2008; 8:83-93; PMID:18094723; http://dx.doi.org/ 10.1038/nrc2290 [DOI] [PubMed] [Google Scholar]
- [31].Hughes S, Elustondo F, Di Fonzo A, Leroux FG, Wong AC, Snijders AP, Matthews SJ, Cherepanov P. Crystal structure of human CDC7 kinase in complex with its activator DBF4. Nat Struct Mol Biol 2012; 19:1101-7; PMID:23064647; http://dx.doi.org/ 10.1038/nsmb.2404 [DOI] [PubMed] [Google Scholar]
- [32].Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004; 432:775-9; PMID:15592418; http://dx.doi.org/ 10.1038/nature03155 [DOI] [PubMed] [Google Scholar]
- [33].Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, Johnson GV, Jope RS. Direct, activating interaction between glycogen synthase kinase-3beta and p53 after DNA damage. Proc Natl Acad Sci U S A 2002; 99:7951-5; PMID:12048243; http://dx.doi.org/ 10.1073/pnas.122062299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Broude EV, Demidenko ZN, Vivo C, Swift ME, Davis BM, Blagosklonny MV, Roninson IB. p21 (CDKN1A) is a negative regulator of p53 stability. Cell Cycle 2007; 6:1468-71; PMID:17585201; http://dx.doi.org/ 10.4161/cc.6.12.4313 [DOI] [PubMed] [Google Scholar]
- [35].Wu PC, Tai MH, Hu DN, Lai CH, Chen YH, Wu YC, Tsai CL, Shin SJ, Kuo HK. Cyclin-dependent kinase inhibitor roscovitine induces cell cycle arrest and apoptosis in rabbit retinal pigment epithelial cells. J Ocular Pharmacol Therap 2008; 24:25-33; http://dx.doi.org/ 10.1089/jop.2007.0044 [DOI] [PubMed] [Google Scholar]
- [36].Ismail IH, Hendzel MJ. The gamma-H2A.X: is it just a surrogate marker of double-strand breaks or much more? Envir Mol Mutagen 2008; 49:73-82; http://dx.doi.org/ 10.1002/em.20358 [DOI] [PubMed] [Google Scholar]
- [37].Tudzarova S, Trotter MW, Wollenschlaeger A, Mulvey C, Godovac-Zimmermann J, Williams GH, Stoeber K. Molecular architecture of the DNA replication origin activation checkpoint. EMBO J 2010; 29:3381-94; PMID:20729811; http://dx.doi.org/ 10.1038/emboj.2010.201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene 2002; 21:6624-32; PMID:12242660; http://dx.doi.org/ 10.1038/sj.onc.1205910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Montagnoli A, Tenca P, Sola F, Carpani D, Brotherton D, Albanese C, Santocanale C. Cdc7 inhibition reveals a p53-dependent replication checkpoint that is defective in cancer cells. Cancer Res 2004; 64:7110-6; PMID:15466207; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-1547 [DOI] [PubMed] [Google Scholar]
- [40].Yamada M, Watanabe K, Mistrik M, Vesela E, Protivankova I, Mailand N, Lee M, Masai H, Lukas J, Bartek J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Gen Dev 2013; 27:2459-72; http://dx.doi.org/ 10.1101/gad.224568.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yamada M, Masai H, Bartek J. Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 2014; 13:1859-66; PMID:24841992; http://dx.doi.org/ 10.4161/cc.29251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Clay-Farrace L, Pelizon C, Santamaria D, Pines J, Laskey RA. Human replication protein Cdc6 prevents mitosis through a checkpoint mechanism that implicates Chk1. EMBO J 2003; 22:704-12; PMID:12554670; http://dx.doi.org/ 10.1093/emboj/cdg046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Kim JM, Kakusho N, Yamada M, Kanoh Y, Takemoto N, Masai H. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene 2008; 27:3475-82; PMID:18084324; http://dx.doi.org/ 10.1038/sj.onc.1210994 [DOI] [PubMed] [Google Scholar]
- [44].Rainey MD, Harhen B, Wang GN, Murphy PV, Santocanale C. Cdc7-dependent and -independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle 2013; 12:1560-8; PMID:23598722; http://dx.doi.org/ 10.4161/cc.24675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, et al.. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol 2016; 18:777-89; PMID:27323328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Williams GH, Stoeber K. The cell cycle and cancer. J Pathol 2012; 226:352-64; PMID:21990031; http://dx.doi.org/ 10.1002/path.3022 [DOI] [PubMed] [Google Scholar]
- [47].Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, et al.. A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 2008; 4:357-65; PMID:18469809; http://dx.doi.org/ 10.1038/nchembio.90 [DOI] [PubMed] [Google Scholar]
- [48].Swords R, Mahalingam D, O'Dwyer M, Santocanale C, Kelly K, Carew J, Giles F. Cdc7 kinase - a new target for drug development. Eur J Cancer 2010; 46:33-40; PMID:19815406; http://dx.doi.org/ 10.1016/j.ejca.2009.09.020 [DOI] [PubMed] [Google Scholar]
- [49].Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, et al.. Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem 2003; 278:45937-45; PMID:12928438; http://dx.doi.org/ 10.1074/jbc.M306268200 [DOI] [PubMed] [Google Scholar]
- [50].Wharton SB, Hibberd S, Eward KL, Crimmins D, Jellinek DA, Levy D, Stoeber K, Williams GH. DNA replication licensing and cell cycle kinetics of oligodendroglial tumours. Br J Cancer 2004; 91:262-9; PMID:15199392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Dinkel H, Van Roey K, Michael S, Davey NE, Weatheritt RJ, Born D, Speck T, Kruger D, Grebnev G, Kuban M, et al.. The eukaryotic linear motif resource ELM: 10 years and counting. Nucleic Acids Res 2014; 42:D259-66; PMID:24214962; http://dx.doi.org/ 10.1093/nar/gkt1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al.. Clustal W and Clustal X version 2.0. Bioinformatics 2007; 23:2947-8; PMID:17846036; http://dx.doi.org/ 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- [53].Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protocols 2009; 4:363-71; PMID:19247286; http://dx.doi.org/ 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- [54].Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformat 2008; 9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci U S A 2009; 106:11960-5; PMID:19617534; http://dx.doi.org/ 10.1073/pnas.0904875106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, Santocanale C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem 2006; 281:10281-90; PMID:16446360; http://dx.doi.org/ 10.1074/jbc.M512921200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.