

ABSTRACT
Prions and Amyloid beta (Aβ) peptides induce synaptic damage via complex mechanisms that include the pathological alteration of intracellular signaling cascades. The host-encoded cellular prion protein (PrPC) acts as a high-affinity cell surface receptor for both toxic species and it can modulate the endocytic trafficking of the N-methyl D-aspartate (NMDA) receptor and E-cadherin adhesive complexes via Src family kinases (SFKs). Interestingly, SFK-mediated control of endocytosis is a widespread mechanism used to regulate the activity of important transmembrane proteins, including neuroreceptors for major excitatory and inhibitory neurotransmitters. Here we discuss our recent work in zebrafish and accumulating evidence suggesting that subversion of this pleiotropic regulatory mechanism by Aβ oligomers and prions explains diverse neurotransmission deficits observed in human patients and mouse models of prion and Alzheimer's neurodegeneration. While Aβ, PrPC and SFKs constitute potential therapeutic targets on their own, drug discovery efforts might benefit significantly from aiming at protein-protein interactions that modulate the endocytosis of specific SFK targets.
KEYWORDS: Alzheimer disease, Aβ peptide, E-cadherin, endocytosis, NMDA receptor, neurotransmission, prion protein, Src family kinases, synaptic damage, zebrafish
INTRODUCTION
Prion disorders are a fascinating recurrence in medical history. They captivated the minds of scientists in the 1950s, when a bizarre neurological illness named kuru was described among the women and children of a Papua New Guinean tribe.1 They re-surfaced in the early 1980s, with the remarkable discovery that scrapie -a related condition in sheep- was caused by prions, novel infectious agents lacking nucleic acids and made exclusively of a rogue host protein, PrPSc.2 And they received massive public attention in the 1990s with the slaughtering of millions of European cattle in order to eradicate mad cow disease, a manmade epizootic which spread to humans under the name of variant Creutzfeldt-Jakob disease (vCJD).1 Almost 20 years later, we learned that PrPC might be a key piece in the puzzle of Alzheimer's disease, the most common form of age-related human dementia. Parallels between the pathogenesis of prion and Alzheimer's diseases had been recognized for some time, such as the misfolding and aggregation of neuronal membrane proteins, the formation of amyloid plaques in the brain and the disruption of synapses leading to progressive neurodegeneration.3 However, the identity of the rogue proteins involved in each case (PrPSc in prion diseases vs. the Aβ peptide and hyperphosphorylated Tau in Alzheimer's disease) suggested that these pathologies were triggered via distinct molecular pathways. The finding that PrPC mediates synaptic dysfunction induced by Aβ oligomers4 challenged this assumption and revealed that, similar to what was previously shown for prion-induced neurodegeneration,5 a PrPC-related signal contributes to Aβ neurotoxicity. In a series of compelling experiments, S. Strittmatter and coworkers revealed that PrPC is a neuronal receptor for Aβ oligomers and that this interaction activates the Src-related kinase Fyn, leading to hyper-phosphorylation and transient over-activation of the NMDA receptor at the plasma membrane. The ensuing glutamate excitotoxicity is one of the main neurotoxic mechanisms in Alzheimer's disease4,6 and certainly a prominent one in prion disorders.7 Before these studies, Aβ oligomers were known to bind neurons and trigger glutamate excitotoxicity but no mechanistic links were known. Particularly noteworthy in this Aβ-PrPC neurotoxic pathway is that Fyn effectively stabilizes the NMDA receptor at the plasma membrane by phosphorylating a C-terminal tyrosine residue (Tyr1472) of its NR2B subunit, rendering it inaccessible to the clathrin adaptor AP-2 and the endocytic machinery8 (Fig. 1). As discussed below, this mode of regulation applies to other transmembrane proteins and SFK family members, suggesting that the pleiotropic control of protein trafficking via tyrosine phosphorylation cascades is a significant role of PrPC in healthy and diseased brains.9 Here we discuss this complex scenario, its potential effects on neurotransmission, and how our recent work in zebrafish embryos adds new evidence to it.10
FIGURE 1.
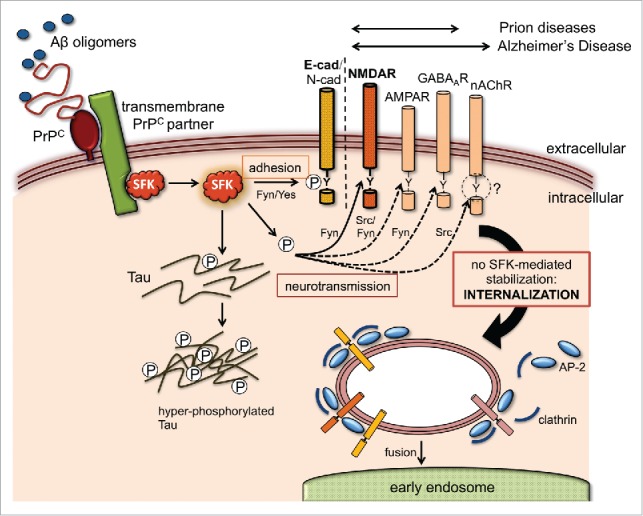
Potential effect of Aβ and PrPC on SFK-regulated trafficking of neuroreceptor and adhesion complexes. Aβ oligomers bind PrPC on neuronal cell surfaces with high affinity, triggering the activation of SFKs, often through a transmembrane PrPC partner (e.g. caveolin, mGluR5, NCAM, EGFR9). Apart from phosphorylating the disease-associated Tau protein and other cytosolic targets, SFKs can modulate the cell surface stability of numerous transmembrane proteins via endocytic tyrosine motifs at their cytoplasmic tails. Some of these, such as neuroreceptor subunits (NMDAR, AMPAR, GABAAR and nAChR) and adhesion proteins (E- and N-cadherin), are directly involved in synaptic function and can potentially explain key pathological features of prion and Alzheimer's disease. SFKs control the trafficking of these transmembrane proteins by directly phosphorylating their endocytic motifs or by targeting components of their tyrosine endocytic machinery like AP-2 and clathrin. These modifications can result in either increased stability at the plasma membrane (gain of function) or protein internalization and degradation (loss of function). To date, experimental evidence for an influence of Aβ and PrP on this mechanism is only available for NMDAR6 and AJ complexes.10 In the case of the nAChR, Src kinase activity controls its trafficking but tyrosine endocytic motifs appear dispensable (as suggested by the question mark). Double arrows above the transmembrane target proteins indicate their relevance to prion or Alzheimer's disease.
SFKs Link PrP Signaling and Cell Surface E-cadherin
The ability of PrPC to induce Fyn activation was first demonstrated by S. Mouillet-Richard and colleagues using antibody-mediated cross-linking in the 1C11 committed neuronal stem cell line.11 Although this finding strongly argued for a role of PrP in signal transduction, the apparent lack of related phenotypes in PrP knockout mice initially suggested that PrP signaling was not physiologically relevant.9 Evidence for the contrary came unexpectedly from a non-mammalian model, the zebrafish. By studying PrP loss-of-function in early fish embryos, we found that PrPC recruits Fyn at cell-cell contacts and indirectly promotes the cell surface stability of E-cadherin in vivo.12 To elucidate the molecular mechanisms behind this regulatory pathway, we recently characterized its genetic and cell biological basis.10
During gastrulation, cell-cell contacts are continuously remodeled through the controlled removal and new delivery of E-cadherin molecules at the plasma membrane, where they associate with catenins and the actin cytoskeleton to form stable multi-protein complexes known as adherens junctions (AJs).13 Our analyses of protein trafficking upon PrP gain- and loss-of-function revealed that PrPC stabilizes cell surface E-cadherin by preventing its clathrin-mediated endocytosis and subsequent degradation. Accordingly, embryos lacking PrPC accumulated E-cadherin intracellularly, lost tissue cohesion and became lethally arrested whereas embryos overexpressing fish or mouse PrP accumulated E-cadherin at the plasma membrane, underwent abnormal tissue compaction and became mechanically disrupted. Several of our observations indicate that these PrP embryonic phenotypes are mediated by Fyn and the closely related SFK Yes. First, simultaneous downregulation of Fyn and Yes mimicked PrP loss-of-function at the cellular and molecular levels, suggesting that they act along the same pathway as PrP. Second, co-expression of Fyn and Yes efficiently rescued the PrP loss-of-function phenotype, indicating that they act downstream of PrP. Third, the rescuing efficiency of exogenous Fyn and Yes correlated with their kinase activity, as constitutively active mutants performed more efficiently than wildtype ones. Moreover, the levels of activated Fyn and Yes were reduced in PrP-depleted embryos and increased following PrP overexpression. Direct activation of SFKs by PrP seems unlikely because PrP is anchored to the outer plasma membrane via a glycosylphosphatidylinositol (GPI) anchor and thus has limited access to the cytosolic space, where SFKs reside and function. In addition, SFK activity is controlled by tyrosine phosphorylation, yet PrP is neither a kinase nor a phosphatase. Our biochemical data suggest that PrP enhances the activity of SFKs indirectly, at least in 2 ways: 1) by inducing their coalescence and cross-activation or their activation via transmembrane adaptor molecules and 2) by preventing the preferential degradation of their active forms. We observed the latter phenomenon in PrP-depleted embryos, where active Fyn and Yes were significantly reduced compared to their inactive forms. The preferential ubiquitination and lysosomal degradation of active SFKs is a well-characterized mechanism whose specificity is dictated by E3 ligases like c-Cbl.14,15 Whether PrP can inhibit the targeting of SFKs by c-Cbl or other ligases is presently unknown.
The modulation of Fyn and Yes activity by PrPC provides a plausible explanation for our PrP embryonic adhesion phenotypes because SFKs are known regulators of cadherin stability at the plasma membrane.16 Classical studies in cancer cell lines established that SFK gain-of-function abolishes cell-cell adhesion mainly through the phosphorylation of key tyrosine residues in β-catenin and p120 catenin, which disrupts their physical association with E-cadherin and triggers internalization of the entire AJ complex.17,18 Interestingly, in our experiments, PrP-induced activation of embryonic SFKs triggered catenin phosphorylation but not the dissociation and internalization of AJs.10 In fact, SFK activity was required to maintain AJ stability. Although apparently paradoxical, these observations agree with accumulating evidence that SFK activity supports E-cadherin adhesion at physiological levels (such as in our in vivo experiments) but inhibits it at abnormally high levels (such as in transformed cells).16 Notably, our data from both PrP gain- and loss-of-function experiments indicate that Fyn and Yes block the endocytosis of E-cadherin independently of β-catenin phosphorylation. This strongly suggests that E-cadherin is directly stabilized by SFKs at AJs. While E-cadherin is indeed a known target of Src, it remains to be established whether it is phosphorylated upon PrP activation and whether this modulates AJ stability in vivo, similar to what has been reported in cultured cells.19 Of note, several components of the endocytic machinery such as dynamin and clathrin are also SFK targets.20,21 Particularly relevant here is the clathrin adaptor AP-2, whose phosphorylation by Src inhibits endocytosis22 and thus could potentially help explain the positive effect of PrP and SFKs on zebrafish embryonic E-cadherin. The existence of multiple SFK targets at or in the vicinity of AJs raises the question of how these simultaneous phosphorylation events are hierarchically related to one another, and what their net effect on the cell surface expression of E-cadherin might be.
Mechanistically, the modulation of zebrafish embryonic AJs by PrP bears remarkable similarities to the misregulation of the mammalian NMDA receptor complex upon binding of Aβ oligomers to PrP. In both cases, the endocytosis of key transmembrane protein components (E-cadherin and NR2B, respectively) is blocked by PrP-induced, SFK-mediated phosphorylation events at their cytoplasmic tails. This prompted us to ask if -by extension- E-cadherin would also be susceptible to misregulation by Aβ exposure. Remarkably, we found that human Aβ oligomers elicited PrP-dependent SFK activation and E-cadherin upregulation in zebrafish embryonic cells, after only one hour of incubation.10 Given the crucial roles of E-, N- and other cadherins in synapse assembly and function,23,24 this result suggests that subversion of the PrP/SFK/cadherin pathway might contribute to synaptic dysfunction in Alzheimer's disease. As shown in rat hippocampal neurons, N-cadherin is required for the broadening of the spine head that takes place upon activation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) glutamate receptor.25 Furthermore, experiments in cultured cortical neurons revealed that the inhibition of N-cadherin accelerates the Aβ-induced impairment of excitatory synapses.26 Hence, PrP- and SFK-induced alterations in cadherin adhesion could prevent the remodeling of pre- and postsynaptic membranes required for plasticity.
Potential Role of SFKs in Aβ- and Prp-Induced Neurotransmission Deficits
Fyn is undoubtedly a central component of Alzheimer's pathogenesis. Besides mediating the Aβ- and PrP-induced over-activation of the NMDA receptor, Fyn also phosphorylates Tau, an axonal microtubule-associated protein that mediates Aβ toxicity at the post-synapse, and the major constituent of neurofibrillary tangles.27 Beyond these 2 molecular events, SFKs could considerably amplify the effects of the Aβ neurotoxic cascade because this family of kinases includes several ubiquitous and tissue-specific members able to redundantly phosphorylate numerous downstream targets in neurons.28,29 Particularly relevant here is the control of endocytic trafficking via tyrosine phosphorylation signals, a widespread regulatory mechanism that influences the function of important membrane proteins like neuronal receptors, ion channels and neurotransmitter transporters.30 Hence, the modulation of E-cadherin and NMDA receptor endocytosis by PrP and SFKs is likely only part of a larger molecular network, which could be hijacked by Aβ oligomers to impair synapses and trigger neurodegeneration31 (Fig. 1). For instance, the loss of cholinergic neurons -a conspicuous event in Alzheimer's pathogenesis- is associated with reduced levels of the neurotransmitter acetylcholine (ACh) and impaired function of its nicotinic receptor (nAChR).32 Interestingly, SFKs physically interact with, phosphorylate and positively modulate the activity of the nAChR.33,34 Moreover, dynamin-independent endocytosis of the nAChR requires Src activity,35 although it does not appear to involve conventional endocytic tyrosine motifs in any of its 5 subunits.36 In contrast to excitatory glutamatergic and cholinergic pathways, inhibitory neurotransmission via gamma-aminobutyric acid (GABA) appears less affected during Alzheimer's disease and GABAergic neurons seem more resistant to neurodegeneration than their excitatory counterparts.37 Nevertheless, recent studies have found reduced levels of GABA and functionally altered GABAA receptors in Alzheimer's patients.38 Such alterations include the functional remodeling of GABAA receptors via differential up- and down-regulation of their subunits.39 Notably, one of these subunits (γ2) has a tyrosine-based endocytic motif whose phosphorylation by Fyn blocks access to AP-2 and prevents the clathrin-mediated endocytosis of GABAA receptors, thereby enhancing synaptic inhibition.40-42 This is the same mechanism that stabilizes the NR2B subunit of the NMDA receptor at neuronal cell surfaces (see above). Does this imply that Aβ oligomers can impair cholinergic and GABAergic neurotransmission via PrP and Fyn, as shown for NMDA receptor-mediated glutamate excitotoxicity? This scenario has not been tested experimentally but it would offer an alternative explanation for the concomitant increase in NMDA and GABA receptor activity in Alzheimer's patients, often interpreted as a cross-talk or compensatory mechanism to prevent and curb glutamate excitotoxicity.38
On the other hand, one might predict that if PrPC can activate SFKs, and these in turn enhance the function of NMDA, GABAA and ACh receptors, then these neuroreceptors should be affected upon PrP knockout and/or following prion infection. Some experimental evidence for this is available, although indirect and lacking mechanistic insight. For instance, electrophysiological recordings in PrP knockout mice revealed anomalies in NMDA and GABA synaptic transmission in the hippocampus and the olfactory bulb.43-45 In the absence of molecular data, it is not clear whether these phenotypes are connected to impaired SFK signaling or to altered endocytic trafficking of receptor subunits. Nevertheless, immunohistochemical analysis of cerebral, cerebellar and entorhinal cortex samples from CJD patients showed alterations in the expression levels of AMPA, NMDA and GABA receptor subunits, in partial correlation with prion protein deposition, neuronal loss and spongiform degeneration.46 Along the same lines, mice inoculated with scrapie and CJD prions underwent early, severe and selective loss of cortical GABAergic inhibitory neurons.47 In contrast to Alzheimer's disease, no significant impairment of cholinergic neurons has been reported in CJD patients or prion infected animals. However, a potential role of PrP in the modulation of cholinergic activity is likely, since it co-localizes with the nAChR at human and murine neuromuscular junctions (NMJs) and potentiates the release of ACh.48,49 Accordingly, biochemical data from human tissue and cultured cells indicate that PrPC interacts with the α7 and β4 nAChR subunits within macromolecular complexes.50,51 Furthermore, when analyzing PrP function in zebrafish embryonic trunk motor neurons, we observed strong co-localization of activated SFKs, AChR and tyrosine phosphorylated NR2B NMDA receptor subunits at PrP-positive NMJs (K. Ochs and E Málaga-Trillo, in preparation). Upon PrP downregulation, the levels of activated SFKs and neuroreceptors at these postsynaptic sites decreased and their pattern of distribution became disrupted as motor axons failed to elongate and branch properly. Thus, it seems reasonable to propose that PrP could activate SFKs within multiprotein assemblies at NJMs, thereby inducing phosphorylation of nAChRs and enhancing their activity (see above). Altogether, the evidence available suggests that the regulation of neuroreceptor cell surface stability by PrP and SFKs is a physiologically relevant mechanism with deep implications for the pathogenesis of prion and Alzheimer's disease.
Therapeutic Perspectives
The involvement of PrP and SFKs in Aβ-mediated synaptic damage has sparked renewed interest in common neurotoxic pathways and therapeutic strategies. For instance, PrPC is a targetable upstream element common to prion and Alzheimer's neurotoxicity because it can bind both PrPSc and Aβ.4,52 In fact, the use of monoclonal anti-PrPC antibodies to cure prion infection and prevent disease progression in mice is well established.53 Interestingly, one of the two PrPC sites that binds to PrPSc and mediates prion propagation (the C-PBD, at residues 100–10952) overlaps with the binding site for Aβ (residues 95–1054). Accordingly, antibody-mediated targeting of this PrPC site effectively blocks Aβ binding and Aβ-mediated disruption of synaptic plasticity.54 Subsequent work in high throughput systems led to the identification of a small drug-like molecule that can bind PrPC, block Aβ binding and interfere with prion propagation.53 As the only substrate for PrPSc replication, PrPC is a clearly undisputed therapeutic target to prevent prion-induced neurodegeneration. However, its therapeutic value in Alzheimer's disease is more difficult to assess because PrPC is not the only neuronal receptor for Aβ with the potential ability to trigger synaptic damage. Other Aβ receptors include proteins with established connections to PrPC such as the NMDA receptor and the a7 nAChR subunit as well as unrelated ones like the receptor for advanced glycation end products (RAGE), the ephrin type B receptor 2, immunoglobulin G Fc gamma receptor IIb (FccRIIb) and the paired immunoglobulin-like receptor B (PirB).55 In addition, Aβ itself can interact with cell membranes, form pores or ion channels and induce neuronal damage. Hence, targeting PrPC emerges as an encouraging strategy for the treatment of Alzheimer's neurodegeneration but its overall efficacy remains to be determined.
On the other hand, the availability of Src kinase inhibitors originally designed to treat cancer patients enables testing of SFKs as therapeutic targets to avert early stages of Aβ- and PrPC-induced neurodegeneration. In fact, inhibition of the Fyn kinase using pharmacological blockers like saracatinib and masitinib has been successfully tested in Alzheimer's mouse models and is currently undergoing clinical trials.56-58 However, to reduce the risk of undesired or off-target effects, these therapeutic strategies need to take into account the complexity of the cellular mechanisms controlled by SFKs, the diversity of their targets and their relevance to neurodegeneration. Interestingly, some of the multiple effects that SFKs exert on neuronal physiology upon PrP deregulation may be inferred from studies in PrP knockout and transgenic mice.9 These include the disturbances in ligand- and voltage-gated ion channel activity, synaptic vesicle exocytosis, and the complex genetic cascade that controls myelination. Therefore, the existence of tyrosine phosphorylation mechanisms to regulate the endocytosis of neuroreceptors and adhesion proteins at synapses provides an exciting opportunity to design or screen for new anti-prion and anti-Alzheimer's drugs that specifically target only the interaction between these molecules and SFKs in the context of PrPC-mediated signaling. Cell biological research across fish and mammalian models will help to further elucidate the complex regulatory mechanisms that impair synaptic function and how these trigger the degeneration of diverse types of neurons. Integrating this knowledge into zebrafish-based drug screening methodologies will generate physiologically relevant readouts to assess the therapeutic value of test compounds in a comprehensive manner.
ABBREVIATIONS
- PrPC
cellular prion protein
- PrPSc
infectious scrapie protein
- CJD
Creutzfeldt-Jakob disease
- Aβ
Amyloid β
- AJ
adherens junction
- NMJ
neuromuscular junction
- SFK
Src family kinase
- NMDA
N-methyl-D-aspartate
- ACh
acetylcholine
- GABA
gamma-aminobutyric acid
- GPI
glicosylphosphatidylinositol
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
DISCLOSURE OF POTENTIAL CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
ACKNOWLEDGMENTS
Our special thanks to Dora and Jack for their continued support.
Funding
Work in the Málaga-Trillo lab is funded by PRODUCE-Innóvate Perú and the Universidad Peruana Cayetano Heredia. K. Ochs is supported by a PROMOS stipend from the DAAD.
REFERENCES
- [1].Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 2001; 24:519-50; PMID:11283320; http://dx.doi.org/ 10.1146/annurev.neuro.24.1.519 [DOI] [PubMed] [Google Scholar]
- [2].Prusiner SB. Prions. Proc Natl Acad Sci U S A 1998; 95:13363-83; PMID:9811807; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Aguzzi A, Haass C. Games played by rogue proteins in prion disorders and Alzheimer disease. Science 2003; 302:814-8; PMID:14593165; http://dx.doi.org/ 10.1126/science.1087348 [DOI] [PubMed] [Google Scholar]
- [4].Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009; 457:1128-32; PMID:19242475; http://dx.doi.org/ 10.1038/nature07761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, et al.. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 2005; 308:1435-9; PMID:15933194; http://dx.doi.org/ 10.1126/science.1110837 [DOI] [PubMed] [Google Scholar]
- [6].Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 2012; 15:1227-35; PMID:22820466; http://dx.doi.org/ 10.1038/nn.3178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chiesa R. The elusive role of the prion protein and the mechanism of toxicity in prion disease. PLoS Pathog 2015; 11:e1004745; PMID:25951168; http://dx.doi.org/ 10.1371/journal.ppat.1004745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci 2004; 5:317-28; PMID:15034556; http://dx.doi.org/ 10.1038/nrn1368 [DOI] [PubMed] [Google Scholar]
- [9].Ochs K, Malaga-Trillo E. Common themes in PrP signaling: the Src remains the same. Front Cell Dev Biol 2014; 2:63; PMID:25364767; http://dx.doi.org/ 10.3389/fcell.2014.00063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sempou E, Biasini E, Pinzón-Olejua A, Harris DA, Málaga-Trillo E. Activation of zebrafish Src family kinases by the prion protein is an amyloid-β-sensitive signal that prevents the endocytosis and degradation of E-cadherin/β-catenin complexes in vivo. Mol Neurodegener 2016; 11:18; PMID:26860872; http://dx.doi.org/ 10.1186/s13024-016-0076-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science 2000; 289:1925-8; PMID:10988071; http://dx.doi.org/ 10.1126/science.289.5486.1925 [DOI] [PubMed] [Google Scholar]
- [12].Málaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol 2009; 7:e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Montero JA, Heisenberg CP. Gastrulation dynamics: cells move into focus. Trends Cell Biol 2004; 14:620-7; PMID:15519851; http://dx.doi.org/ 10.1016/j.tcb.2004.09.008 [DOI] [PubMed] [Google Scholar]
- [14].Zhimin L, Tony H. Degradation of Activated Protein Kinases by Ubiquitination. Annu Rev Biochem 2009; 78:435-75; PMID:19489726; http://dx.doi.org/ 10.1146/annurev.biochem.013008.092711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Reinecke J, Caplan S. Endocytosis and the Src family of non-receptor tyrosine kinases. Biomolecular Concepts 2014; 5(2):143-55; PMID:25372749; http://dx.doi.org/ 10.1515/bmc-2014-0003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].McLachlan RW, Yap AS. Not so simple: the complexity of phosphotyrosine signaling at cadherin adhesive contacts. J Mol Med 2007; 85:545-54; PMID:17429596; http://dx.doi.org/ 10.1007/s00109-007-0198-x [DOI] [PubMed] [Google Scholar]
- [17].Daniel JM, Reynolds AB. Tyrosine phosphorylation and cadherin/catenin function. Bioessays 1997; 19:883-91; PMID:9363682; http://dx.doi.org/ 10.1002/bies.950191008 [DOI] [PubMed] [Google Scholar]
- [18].Nelson WJ, Nusse R. Convergence of Wnt, β-catenin, and cadherin pathways. Science 2004; 303:1483-7; PMID:15001769; http://dx.doi.org/ 10.1126/science.1094291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HEM, Behrens J, Sommer T, Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol 2002; 4:222-31; PMID:11836526; http://dx.doi.org/ 10.1038/ncb758 [DOI] [PubMed] [Google Scholar]
- [20].Ahn S, Kim J, Lucaveche CL, Reedy MC, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-dependent tyrosine phosphorylation regulates dynamin self-assembly and ligand-induced endocytosis of the epidermal growth factor receptor. J Biol Chem 2002; 277:26642-51; PMID:12011079; http://dx.doi.org/ 10.1074/jbc.M201499200 [DOI] [PubMed] [Google Scholar]
- [21].Wilde A, Beattie EC, Lem L, Riethof DA, Liu SH, Mobley WC, Soriano P, Brodsky FM. EGF receptor signaling stimulates SRC kinase phosphorylation of clathrin, influencing clathrin redistribution and EGF uptake. Cell 1999; 96:677-87; PMID:10089883; http://dx.doi.org/ 10.1016/S0092-8674(00)80578-4 [DOI] [PubMed] [Google Scholar]
- [22].Zimmerman B, Simaan M, Lee MH, Luttrell LM, Laporte SA. c-Src-mediated phosphorylation of AP-2 reveals a general mechanism for receptors internalizing through the clathrin pathway. 2009; 21:103-10; PMID:18938240 [DOI] [PubMed] [Google Scholar]
- [23].Yamada S, Nelson WJ. Synapses: sites of cell recognition, adhesion, and functional specification. Ann Rev Biochem 2007; 76:267-94; PMID:17506641; http://dx.doi.org/ 10.1146/annurev.biochem.75.103004.142811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fiederling A, Ewert R, Andreyeva A, Jüngling K, Gottmann K. E-cadherin is required at GABAergic synapses in cultured cortical neurons. Neurosci Lett 2011; 501(3):167-72; http://dx.doi.org/ 10.1016/j.neulet.2011.07.009 [DOI] [PubMed] [Google Scholar]
- [25].Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N. Cadherin activity is required for activity-induced spine remodeling. J Cell Biol 2004; 167:961-72; PMID:15569714; http://dx.doi.org/ 10.1083/jcb.200406030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Andreyeva A, Nieweg K, Horstmann K, Klapper S, Muller-Schiffmann A, Korth C, Gottmann K. C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-β. Brain 2012; 135:2140-54; PMID:22637581; http://dx.doi.org/ 10.1093/brain/aws120 [DOI] [PubMed] [Google Scholar]
- [27].Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE. The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer disease. J Neurosci 2012; 32:16857-71a; PMID:23175838; http://dx.doi.org/ 10.1523/JNEUROSCI.1858-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol 1997; 13:513-609; PMID:9442882; http://dx.doi.org/ 10.1146/annurev.cellbio.13.1.513 [DOI] [PubMed] [Google Scholar]
- [29].Lau L, Huganir RL. Role of Tyrosine Phosphorylation in the Nervous System In: Siegel GJ, Agranoff BW, Alberz RW, Fischer SK, Uhler MD, eds. Basic Neurochemistry: Molecular, Cellular and Medical Aspects. Philadelphia: Lippincott-Raven, 1999 [Google Scholar]
- [30].Buckley KM, Melikian HE, Provoda CJ, Waring MT. Regulation of neuronal function by protein trafficking: a role for the endosomal pathway. J Physiol 2000; 525(Pt 1):11-9; PMID:10811720; http://dx.doi.org/ 10.1111/j.1469-7793.2000.t01-2-00011.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Crews L, Masliah E. Molecular mechanisms of neurodegeneration in Alzheimer disease. Hum Mol Genetics 2010; 19:R12-20; PMID:20413653; http://dx.doi.org/ 10.1093/hmg/ddq160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lombardo S, Maskos U. Role of the nicotinic acetylcholine receptor in Alzheimer disease pathology and treatment. Neuropharmacol: Elsevier Ltd 2015; 96:255-62 [DOI] [PubMed] [Google Scholar]
- [33].Wang K, Hackett JT, Cox ME, Van Hoek M, Lindstrom JM, Parsons SJ. Regulation of the neuronal nicotinic acetylcholine receptor by SRC family tyrosine kinases. J Biol Chem 2004; 279(10):8779-86; http://dx.doi.org/ 10.1074/jbc.M309652200 [DOI] [PubMed] [Google Scholar]
- [34].Mohamed AS, Swope SL. Phosphorylation and cytoskeletal anchoring of the acetylcholine receptor by Src class protein-tyrosine kinases. Activation by rapsyn. J Biol Chem 1999; 279(10):20529-39; http://dx.doi.org/ 10.1074/jbc.274.29.20529 [DOI] [PubMed] [Google Scholar]
- [35].Kumari S, Borroni V, Chaudhry A, Chanda B, Massol R, Mayor S, Barrantes FJ. Nicotinic acetylcholine receptor is internalized via a Rac-dependent, dynamin-independent endocytic pathway. J Cell Biol 2008; 181(7):1179-93; PMID:18591431; http://dx.doi.org/ 10.1083/jcb.200709086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rudell JC, Borges LS, Rudell JB, Beck KA, Ferns MJ. Determinants in the β and delta subunit cytoplasmic loop regulate Golgi trafficking and surface expression of the muscle acetylcholine receptor. J Biol Chem 2014; 289:203-14; PMID:24240098; http://dx.doi.org/ 10.1074/jbc.M113.502328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rissman RA, De Blas AL, Armstrong DM. GABA Areceptors in aging and Alzheimer disease. J Neurochem 2007; 103(4):1285-92; PMID:17714455; http://dx.doi.org/ 10.1111/j.1471-4159.2007.04832.x [DOI] [PubMed] [Google Scholar]
- [38].Li Y, Sun H, Chen Z, Xu H, Bu G, Zheng H. Implications of GABAergic Neurotransmission in Alzheimer Disease. Front Aging Neurosci 2016; 8:31:11; PMID:26941642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Limon A, Reyes-Ruiz JM, Miledi R. Loss of functional GABA(A) receptors in the Alzheimer diseased brain. Proc Natl Acad Sci USA 2012; 109(25):10071-6; PMID:22691495; http://dx.doi.org/ 10.1073/pnas.1204606109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Tretter V, Revilla-Sanchez R, Houston C, Terunuma M, Havekes R, Florian C, Jurd R, Vithlani M, Michels G, Couve A, et al.. Deficits in spatial memory correlate with modified {gamma}-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proc Natl Acad Sci U S A 2009; 106:20039-44; PMID:19903874; http://dx.doi.org/ 10.1073/pnas.0908840106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, Smith KR, McAinsh K, Arancibia-Carcamo IL, Saenger W, et al.. Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc Natl Acad Sci U S A 2008; 105:3616-21; PMID:18305175; http://dx.doi.org/ 10.1073/pnas.0707920105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Jurd R, Tretter V, Walker J, Brandon NJ, Moss SJ. Fyn kinase contributes to tyrosine phosphorylation of the GABAA receptor γ2 subunit. Mol Cell Neurosci: Elsevier Inc. 2010; 44(2):129-34; http://dx.doi.org/ 10.1016/j.mcn.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Carleton A, Tremblay P, Vincent JD, Lledo PM. Dose-dependent, prion protein (PrP)-mediated facilitation of excitatory synaptic transmission in the mouse hippocampus. Pflugers Arch 2001; 442:223-9; PMID:11417218; http://dx.doi.org/ 10.1007/s004240100523 [DOI] [PubMed] [Google Scholar]
- [44].Collinge J, Whittington MA, Sidle KC, Smith CJ, Palmer MS, Clarke AR, Jefferys JG. Prion protein is necessary for normal synaptic function. Nature 1994; 370:295-7; PMID:8035877; http://dx.doi.org/ 10.1038/370295a0 [DOI] [PubMed] [Google Scholar]
- [45].Le Pichon CE, Valley MT, Polymenidou M, Chesler AT, Sagdullaev BT, Aguzzi A, Firestein S. Olfactory behavior and physiology are disrupted in prion protein knockout mice. Nat Neurosci 2009; 12:60-9; PMID:19098904; http://dx.doi.org/ 10.1038/nn.2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ferrer I, Puig B. GluR2/3, NMDAepsilon1 and GABAA receptors in Creutzfeldt-Jakob disease. Acta Neuropathol 2003; 106:311-8; PMID:12835949; http://dx.doi.org/ 10.1007/s00401-003-0732-z [DOI] [PubMed] [Google Scholar]
- [47].Guentchev M, Groschup MH, Kordek R, Liberski PP, Budka H. Severe, early and selective loss of a subpopulation of GABAergic inhibitory neurons in experimental transmissible spongiform encephalopathies. Brain Pathol 1998; 8:615-23; PMID:9804371; http://dx.doi.org/ 10.1111/j.1750-3639.1998.tb00188.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Askanas V, Bilak M, Engel WK, Leclerc A, Tome F. Prion protein is strongly immunolocalized at the postsynaptic domain of human normal neuromuscular junctions. Neurosci Lett 1993; 159:111-4; PMID:8264949; http://dx.doi.org/ 10.1016/0304-3940(93)90811-X [DOI] [PubMed] [Google Scholar]
- [49].Re L, Rossini F, Re F, Bordicchia M, Mercanti A, Fernandez OS, Barocci S. Prion protein potentiates acetylcholine release at the neuromuscular junction. Pharmacological Res 2006; 53:62-8; PMID:16256362; http://dx.doi.org/ 10.1016/j.phrs.2005.09.002 [DOI] [PubMed] [Google Scholar]
- [50].Beraldo FH, Arantes CP, Santos TG, Queiroz NG, Young K, Rylett RJ, Markus RP, Prado MA, Martins VR. Role of alpha7 nicotinic acetylcholine receptor in calcium signaling induced by prion protein interaction with stress-inducible protein 1. J Biol Chem 2010; 285:36542-50; PMID:20837487; http://dx.doi.org/ 10.1074/jbc.M110.157263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Petrakis S, Irinopoulou T, Panagiotidis CH, Engelstein R, Lindstrom J, Orr-Urtreger A, Gabizon R, Grigoriadis N, Sklaviadis T. Cellular prion protein co-localizes with nAChR beta4 subunit in brain and gastrointestinal tract. Eur J Neurosci 2008; 27:612-20; PMID:18279314; http://dx.doi.org/ 10.1111/j.1460-9568.2008.06037.x [DOI] [PubMed] [Google Scholar]
- [52].Miller MB, Geoghegan JC, Supattapone S. Dissociation of infectivity from seeding ability in prions with alternate docking mechanism. PLoS Pathog 2011; 7(7):e1002128; PMID:21779169; http://dx.doi.org/ 10.1371/journal.ppat.1002128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Risse E, Nicoll AJ, Taylor WA, Wright D, Badoni M, Yang X, Farrow MA, Collinge J. Identification of a compound that disrupts binding of Amyloid-β to the prion protein using a novel fluorescence-based assay. J Biol Chem 2015; 290(27):17020-8; PMID:25995455; http://dx.doi.org/ 10.1074/jbc.M115.637124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Freir DB, Nicoll AJ, Klyubin I, Panico S, Mc Donald JM, Risse E, Asante EA, Farrow MA, Sessions RB, Saibil HR, et al.. Interaction between prion protein and toxic amyloid β assemblies can be therapeutically targeted at multiple sites. Nat Commun 2011; 2:336; PMID:21654636; http://dx.doi.org/ 10.1038/ncomms1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kam TI, Gwon Y, Jung YK. Amyloid β receptors responsible for neurotoxicity and cellular defects in Alzheimer disease. Cell Mol Life Sci 2014; 71(24):4803-13; PMID:25151011; http://dx.doi.org/ 10.1007/s00018-014-1706-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Kaufman AC, Salazar SV, Haas LT, Yang J, Kostylev MA, Jeng AT, Robinson SA, Gunther EC, van Dyck CH, Nygaard HB, et al.. Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 2015; 77:953-71; PMID:25707991; http://dx.doi.org/ 10.1002/ana.24394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Nygaard HB, van Dyck CH, Strittmatter SM. Fyn kinase inhibition as a novel therapy for Alzheimer disease. Alzheimer Res Ther 2014; 6:8; PMID:24495408; http://dx.doi.org/ 10.1186/alzrt238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nygaard HB, Wagner AF, Bowen GS, Good SP, MacAvoy MG, Strittmatter KA, Kaufman AC, Rosenberg BJ, Sekine-Konno T, Varma P, et al.. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer disease. Alzheimer Res Ther 2015; 7:35; PMID:25874001; http://dx.doi.org/ 10.1186/s13195-015-0119-0 [DOI] [PMC free article] [PubMed] [Google Scholar]