

Summary
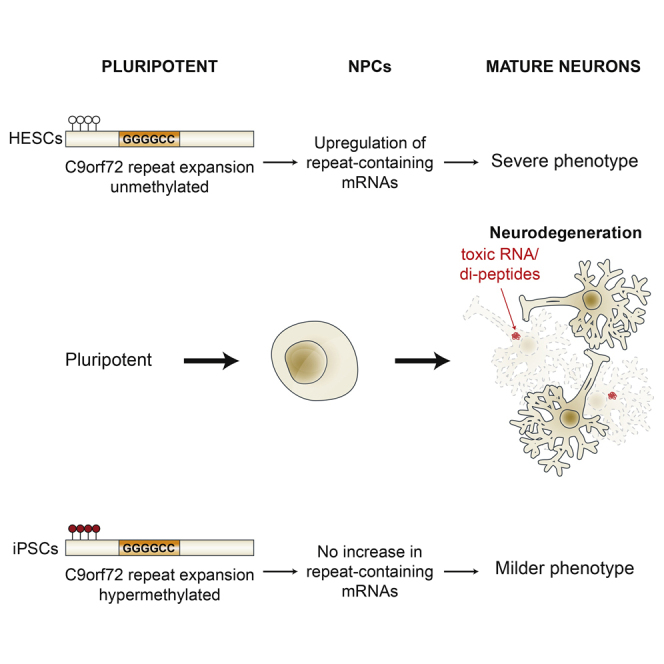
We established two human embryonic stem cell (hESC) lines with a GGGGCC expansion in the C9orf72 gene (C9), and compared them with haploidentical and unrelated C9 induced pluripotent stem cells (iPSCs). We found a marked difference in C9 methylation between the cells. hESCs and parental fibroblasts are entirely unmethylated while the iPSCs are hypermethylated. In addition, we show that the expansion alters promoter usage and interferes with the proper splicing of intron 1, eventually leading to the accumulation of repeat-containing mRNA following neural differentiation. These changes are attenuated in C9 iPSCs, presumably owing to hypermethylation. Altogether, this study highlights the importance of neural differentiation in the pathogenesis of disease and points to the potential role of hypermethylation as a neuroprotective mechanism against pathogenic mRNAs, envisaging a milder phenotype in C9 iPSCs.
Keywords: C9/ALS, unstable repeat expansions, DNA methylation, CpG islands, pluripotent stem cells, disease modelling, neurodegeneration
Graphical Abstract
Highlights
-
•
Derivation of two C9 hESC lines, haploidentical and unrelated C9 iPSCs
-
•
Striking difference in C9 methylation and transcription between C9 hESCs and iPSCs
-
•
Upregulation of repeat-containing mRNAs by differentiation, exclusively in C9 hESCs
-
•
Supports the role for C9 methylation as a neuroprotective mechanism
Eiges and colleagues characterized two C9/ALS-FTD hESC lines, and compared them with haploidentical and unrelated C9 iPSCs. They show that reprogramming excessively hypermethylates the C9 expansion, and provide evidence that when it is unmethylated the expansion enhances accumulation of repeat-containing mRNAs upon differentiation. Their study supports the potential role of C9 hypermethylation as a neuroprotective mechanism in C9-related ALS.
Introduction
Amyotrophic lateral sclerosis (ALS, OMIM #105400) is characterized by progressive muscle weakness and atrophy due to the degeneration of upper and lower motor neurons in the brain and spinal cord, while frontotemporal degeneration (FTD, OMIM #600274) affects behavior and cognition, and is caused by the preferential loss of neurons in the frontal and temporal lobe cortices. Most people who develop ALS/ALS-FTD are between the ages of 40 and 70 years, and die within 2–5 years from diagnosis. Currently there is no cure for ALS-FTD.
The leading known cause of ALS-FTD is a GGGGCC repeat expansion in the first intron of the C9orf72 gene (termed C9 mutation), between noncoding exons 1a and 1b (DeJesus-Hernandez et al., 2011, Dols-Icardo et al., 2014). This mutation can manifest as ALS, FTD, or a combined phenotype, and accounts for 20%–80% of familial and 5%–15% of sporadic ALS and FTD cases (reviewed by Cruts et al., 2013). While in most people the number of GGGGCC repeats is steady and varies between 2 and 19 units, in ALS-FTD it abnormally expands to more than 30 copies and becomes increasingly unstable (Dols-Icardo et al., 2014). The mechanism by which the C9 mutation leads to selective death of neurons is unknown, and the normal function of C9orf72 is just beginning to be defined. Multiple mechanisms for C9/ALS-FTD have been suggested, including haploinsufficiency, RNA toxicity, and abnormal translation of expanded repeat sequences by RAN translation (reviewed by Gendron et al., 2014). However, whether the C9 related neurodegeneration is initiated via a gain-of-function (toxic RNA and/or unconventional dipeptide translation) or a loss-of-function mechanism is still under investigation in animal and cellular models.
The GGGGCC repeat sequence is flanked by two CpG islands (CGIs) within a ∼1-kb region that spans from the promoter sequence into intron 1 of C9orf72. Like many CGIs in the genome, this region typically remains free of DNA methylation in subjects with 2–90 repeats. Yet large expansions lead to the formation of one large CGI with abnormal CpG methylation at the repeats (Xi et al., 2015b), which could spread to the 5′ upstream CGI in up to 37% of C9 cases (Belzil et al., 2014, Gijselinck et al., 2015, Liu et al., 2014, Russ et al., 2015, Xi et al., 2013, Xi et al., 2014, Xi et al., 2015a, Xi et al., 2015b). Hypermethylation is suggested to be coupled with the local gain of repressive histone modifications (H3K9me3 and H3K27me3) (Belzil et al., 2013, Zeier et al., 2015). It is still unclear how, or whether, hypermethylation contributes to disease pathogenesis. While some reports demonstrate an overall decrease in C9orf72 transcription, others show a change in the relative distribution between the three different mRNA isoforms, favoring transcription from exon 1a (V1 and V3, NM_145005.5 and NM_001256054.1, respectively) over exon 1b (V2, NM_018325.3) (Donnelly et al., 2013, Haeusler et al., 2014, Lee et al., 2013). While previous reports failed to detect a correlation between hypermethylation and ALS versus FTD phenotype (Xi et al., 2015b), experimental evidence demonstrates that C9orf72 haploinsufficiency affects cell morphology and function of motor neurons in zebrafish (Ciura et al., 2013). On the other hand, hypermethylation protects against the accumulation of pathogenic RNA foci and dipeptides, caused by the repeat-containing mRNA variants 1 and 3 (Bauer, 2016, Day and Roberson, 2015, Liu et al., 2014). These conflicting results warrant further investigation regarding the contribution and timing of C9orf72 hypermethylation in ALS-FTD pathogenesis, and the discrepancies may be resolved by the use of in vitro derived neurons from C9/ALS-FTD pluripotent cells. Indeed, induced pluripotent stem cells (iPSCs) from C9/ALS patient fibroblasts have already been used to generate motor neurons in culture that recapitulate the key neuropathological features of FTD-ALS (Almeida et al., 2013, Cooper-Knock et al., 2014, Cooper-Knock et al., 2015, Devlin et al., 2015, Donnelly et al., 2013, Li et al., 2015, Peters et al., 2015, Rossi et al., 2015, Sareen et al., 2013, Satoh et al., 2014, Wainger et al., 2014). Nevertheless, the epigenetic aspects of the disease have never been addressed using this model system. The aim of this study is to characterize the methylation state of the expanded region and explore its effect on C9orf72 variant transcription in C9/ALS human embryonic stem cells (hESCs), and compare them with that of their haploidentical (mother-to-child genetic identity) and unrelated C9 iPSCs before and after differentiation.
Results
Derivation and Characterization of C9/hESC Lines
We established two hESC lines with a C9 mutation (SZ-ALS1 and SZ-ALS3) from embryos, which were obtained through preimplantation genetic diagnosis (PGD) and donated for cell line derivation by a family in which the mother was an expansion carrier (patient H, 30 years old, originally diagnosed as a carrier of an expansion with >40 repeats in blood by a repeat primed PCR (rp-PCR); data not shown). Our newly established C9 hESC lines display the key features of pluripotent cells, namely unrestricted growth in culture, expression of undifferentiated cell-specific markers, and potential to differentiate into a wide range of cell types by forming teratomas (Figure S1A, B, D). Chromosome analysis by Giemsa staining demonstrated a 46(XX) karyotype for SZ-ALS1 and a 45(X0) for SZ-ALS3 (Figure S1C). Southern blot analysis identified a GGGGCC expansion of at least ∼270 repeats in both cell lines (Figure S1E).
Analysis of C9orf72 Methylation in C9 hESCs and Their Haploidentical iPSCs
Considering the accumulated data regarding hypermethylation in C9 carriers, we aimed to determine whether hypermethylation is already established in the undifferentiated state. Therefore we examined methylation levels, 200 bp upstream of the 5′ end of the GGGGCC repeats, by bisulfite DNA colony sequencing in the C9 hESCs (24 CpG sites). Interestingly, despite the presence of a large expansion, methylation was almost 0% in both cell lines (Figure 1A). To exclude the possibility that methylation had already begun, but failed to spread further upstream to the 5′ CGI, we searched for methylation at the 5′ end of the repeats by carrying out a qualitative (G4C2)n-methylation assay that is sensitive enough to detect repeat methylation in the mixture containing only 2%–5% highly methylated DNA (Xi et al., 2015b). Here again, methylation was undetectable in the mutant hESCs (Figure 1B). In contrast, the expanded allele in whole blood cells of the mother revealed methylation, although weak, in the expansion itself and the 5′ flanking region (estimated as 5% by bisulfite sequencing) (Figures 1A and 1B).
Figure 1.

Analysis of Methylation Levels in the Upstream Region of the G4C2 Repeats and in the G4C2 Repeat Itself in C9 hESCs, Maternal Blood, Primary Fibroblasts, and iPSCs Derived from Them
(A) Methylation levels upstream of the repeats were determined by bisulfite sequencing of a region 200 bp upstream of the repeats (altogether 24 CpG sites) in SZ-ALS1, SZ-ALS3, and wild-type (WT) (SZ-13) hESCs and whole blood from the mother (patient H). Each line represents a single DNA molecule (upstream region of the repeats), with methylated and unmethylated CpGs designated by black and white circles, respectively.
(B) Results of the (G4C2)n-methylation assay in WT and affected (C9/ALS) hESCs and blood cells of the mother. The left panel (blue channel) represents methylated alleles, while the right panel (green channel) represents unmethylated alleles.
(C) Methylation levels upstream to the repeats as determined by single-molecule bisulfite sequencing of the same region shown in (A) in WT iPSCs, C9 parental fibroblasts (from patient H) and iPSCs derived from them (C9-iPS#H8 and iPS#H10).
(D) Results of the (G4C2)n-methylation assay in WT and C9 affected cells described in (C).
To generate a complement model system, we established haploidentical (genetically half-identical) iPSCs (more than ten different clones) from skin fibroblasts of the currently asymptomatic mother with a C9 expansion, who donated the embryos for hESC line derivation. The primary fibroblasts, which had an unmethylated ∼700-repeat expansion (Figure S2D), were reprogrammed by standard protocol using Yamanaka's four transcription factors (Takahashi et al., 2007). The newly established C9 iPSCs with typical ESC morphology demonstrated unrestricted self-renewal, expressed undifferentiated cell-specific markers, and presented a normal karyotype (Figure S2A–C). By comparing their methylation status with that of the C9 hESCs and parental fibroblasts, we found that the C9 iPSC clones were unusual in their methylation levels. Unlike hESCs and primary fibroblasts, methylation was exceptionally high in the iPSCs. Bisulfite DNA colony sequencing of the region upstream of the expansion identified 50% fully methylated DNA copies in all iPSC clones, suggesting complete methylation of the mutant allele (Figure 1C). The (G4C2)n-methylation assay revealed hypermethylation of the repeats at the 5′ end of the expansion exclusively in C9 iPSCs (Figure 1D). However, while the upstream flanking region of the mutant allele was completely methylated based on bisulfite DNA sequencing (50%), the GGGGCC repeats themselves were not methylated in all DNA molecules (evidenced by signals of repeats beyond the normal range in both the green and blue channels in the (G4C2)n-methylation assay). Importantly, differential methylation between the iPSCs and their parental fibroblasts could not be attributed to the increase in repeat number, since expansion size remained steady following cell reprogramming, as determined by Southern blot analysis (Figure S2D). In addition, as expansion size in the C9 hESCs is well above the threshold necessary to elicit hypermethylation in C9orf72 in any other cell type thus far examined, it is very unlikely that the striking differences in hypermethylation between the C9 iPSCs and their C9 hESCs counterparts stem from a difference in expansion size. We further confirmed the fundamental difference in the epigenetic state of the C9 mutation between both cell types by demonstrating significant enrichments for the repressive histone modification H3K9me3 by chromatin immunoprecipitation (ChIP) analysis exclusively in mutant iPSCs (Figure 2A). No enrichments for H3K27me3 could be detected in wild-type (WT) or affected hESCs and iPSCs (Figure 2B). Taken together, these findings suggest that reprogramming alters the epigenetic state of this region as a consequence of the expansion in iPSCs, likely spreading from the 5′ border of the CGI toward the repeats.
Figure 2.

ChIP Analysis for H3K9me3 and H3K27me3 in WT and C9 Fibroblasts, Undifferentiated hESCs, and iPSCs
Real-time PCR ChIP analysis for (A) H3K9me3 and (B) H3K27me3 in WT and C9 affected hESCs (SZ-ALS1 and SZ-ALS3), haploidentical parental fibroblasts (Fib-H), and iPSCs derived from them (C9-iPS #H8 and #H10). APRT was used as a negative control for both modifications. Negative controls were set to 1. The data in each panel represent an average of three to five independent ChIP experiments. Error bars represent SE (paired t test, ∗∗p < 0.01, ∗∗∗p < 0.001).
Methylation Analysis in C9 iPSCs Derived from an Unrelated Symptomatic ALS Patient
To further corroborate our findings and examine whether methylation is affected by age or disease symptoms, we also generated iPSCs from a skin biopsy of a 65-year-old C9 ALS patient, 2 years following disease onset (patient M). Southern blot analysis demonstrated the presence of a ∼2,700 repeat expansion in the primary fibroblasts of the patient (Figure S2D). Reprogramming of these fibroblasts led to the establishment of more than ten different C9 iPSC clones. The newly established C9 ALS patient-derived iPSCs had the typical characteristics of hESCs (Figure S2). By comparing expansion size and methylation levels between the iPSCs and their parental fibroblasts we found that, although expansion size remained the same, methylation levels at the 5′ CGI dramatically increased from 0% in parental fibroblasts to 50% in affected iPSCs, suggesting methylation levels of 100% on the expanded allele as determined by bisulfite colony sequencing (Figure 3A). The (G4C2)n-methylation assay indicated methylation of the repeats at the 5′ end of the expansion, but not in the parental fibroblasts (Figure 3B). These results provide further evidence that somatic cell reprogramming excessively hypermethylates the 5′ UTR of the C9orf72 locus. In addition, we examined the methylation status of a different gene, SIGLEC6, as a reference locus for aberrant de novo methylation by transcription factor reprogramming, independent of the method used (integratable and nonintegratable vectors) (Huang et al., 2014). Here again, methylation was exclusively acquired in all iPSCs (WT and C9-iPSCs), but was completely absent in primary fibroblasts (patients H and M) or hESCs (SZ-ALS1 and SZ-ALS3) (Figure S3). However, unlike in C9orf72, hypermethylation in SIGLEC6 is not conditioned by a change in the DNA sequence.
Figure 3.

Analysis of Methylation Levels in the Region Upstream of the G4C2 Repeats and in the G4C2 Repeat Itself in Primary Fibroblasts and iPSCs Derived from Patient M
(A) Methylation levels upstream of the repeats were determined by bisulfite sequencing of the same region as in Figure 1A in WT and C9 affected parental fibroblasts derived from a 65-year-old C9/ALS-manifesting patient (patient M) and iPSCs derived from them (C9 iPS#M1, iPS#M9, and iPS#H10). Each line represents the upstream region of the repeats, with methylated and unmethylated CpGs designated by black and white circles, respectively.
(B) Results of the (G4C2)n-methylation assay in primary fibroblasts and iPSC clones derived from patient M. Left panel (blue channel) represents methylated alleles while the right panel (green channel) represents unmethylated alleles.
The Effect of Differentiation on the Methylation Status of C9orf72
To explore the effect of differentiation on the methylation status of the C9 mutation, we induced the unmethylated mutant hESCs (SZ-ALS1 and SZ-ALS3) and C9 hypermethylated iPSC clones (H#8 and M#9) to differentiate into disease-relevant cell types, i.e., neural precursor cells (NPCs) and neural-enriched teratomas. For NPCs, we applied a commonly used and highly efficient differentiation protocol that relies on the generation of neural rosettes by the use of two inhibitors (dorsomorphin and SB431542) (Figure S4A) (Kim et al., 2010). Differentiation efficiency into NPCs was assessed by fluorescence-activated cell sorting (FACS) analysis for NCAM1-positive cells (above 90%, Figure S4B), and by monitoring for the expression of early neural differentiation markers SOX2, Nestin, and PAX6 by RT-PCR (Figure S4C). In addition we took advantage of the teratomas, which are highly enriched for mature neurons (assessed by histological examination [H&E staining, Figures S1] and the presence of Tuj1-expressing cells [Figure S4D]), to explore the effect of differentiation on the methylation levels of the mutation. Interestingly, methylation levels remained unchanged as determined by bisulfite colony sequencing. In the NPCs and teratomas generated from C9 hESCs, methylation remained at 0%, while in the NPCs and teratomas produced from C9 iPSCs methylation remained at 50% (Figures 4A and 4C). These results were further validated by the (G4C2)n-methylation assay (Figures 4B and 4D).
Figure 4.

Analysis of Methylation Levels in Neural Precursors and Teratomas Derived from C9 and WT hESCs and iPSCs
(A) Methylation levels upstream to the repeats were determined by bisulfite sequencing in teratomas and neural precursor cells (NPC) derived from WT and C9 mutant hESCs (SZ-ALS1 and SZ-ALS3). Each line represents a single DNA molecule (upstream region of the repeats), with methylated and unmethylated CpGs designated by black and white circles, respectively.
(B) Results of the (G4C2)n-methylation assay in WT and affected (C9) teratomas and NPCs derived from hESCs. Left panel (blue channel) represents methylated alleles while the right panel (green channel) represents unmethylated alleles.
(C) Methylation levels upstream of the repeats were determined by bisulfite sequencing in teratomas and NPCs derived from WT and C9 iPSCs from patient H (C9-iPSC H#8, 700 repeats) and patient M (C9-iPSC M#9, 2,700 repeats). Each line represents a single DNA molecule (upstream region of the repeats), with methylated and unmethylated CpGs designated by black and white circles, respectively.
(D) Results of the (G4C2)n-methylation assay in WT and affected (C9/ALS) teratomas and NPCs derived from iPSCs. Left panel (blue channel) represents methylated alleles while right panel (green channel) represents unmethylated alleles.
The Effect of Hypermethylation on C9orf72 Expression
To explore whether the expansion alters C9orf72 gene expression and to examine whether it corresponds with hypermethylation, we determined the mRNA levels of the three isoforms (see schematic illustration in Figure 5A) in WT as well as C9 hESCs and iPSCs. Targeting transcript variants 1 (V1), 2 (V2), and 3 (V3) individually (amplicons spanning exon boundaries 1a-2 and 1b-2), and all together (amplicon spanning exon boundaries 2–3) with TaqMan probes, we show that while the general levels of C9orf72 (V1 + V2 + V3) as well as of V2 and V3 remain unchanged between WT and C9 cells in hESCs and iPSCs (Figure 5B), V1 alone is significantly higher in mutant versus WT undifferentiated cells (t test for equal variances, p < 0.05) (Figure 5B). Nevertheless, upregulation of V1 is at least 2.5-fold higher in C9 hESCs relative to C9 iPSCs (t test for equal variances, p < 0.05) (Figure 5B). Given the high abundance of V2 relative to the other variants (cycle threshold [Ct] values of 26–27 versus 27–34), the change in V1, even though significant, reflects minute differences overlooked when measuring overall levels of C9orf72 mRNA. We conclude that the GGGGCC expansion alters the region leading to the increased expression of C9orf72 V1 transcripts in C9 iPSCs.
Figure 5.

Analysis of C9orf72 Variant Expression Levels in Undifferentiated and Differentiated Derivatives of hESCs and iPSCs by qRT-PCR
(A) Schematic illustration of the three C9orf72 mRNA isoform variants (V1, V2, and V3).
(B and C) Mean value of qRT-PCR for C9orf72 transcription in both C9 hESC lines (SZ-ALS1 and SZ-ALS3), C9 iPSC clones derived from patients H (H8, H10) and M (M1, M9, M10), and their WT controls from the TaqMan gene expression assay for C9orf72 transcript variants 1, 2, and 3. mRNA transcription levels were determined in (B) undifferentiated (hESCs and iPSCs) and (C) differentiated cell derivatives (NPCs and teratomas) of C9 mutation carrying cells and appropriate controls (WT hESCs and WT iPSCs). Using TaqMan probes targeting transcript variants 1, 2, and 3 individually (V1, V2, V3) as well as altogether (V1 + V2 + V3), we determined the relative abundance of each transcript variant. The expression level in each cell type represents an average of three to six independent experiments. Cycle threshold (Ct) values were normalized to the corresponding Ct value of GUS. WT hESC line is SZ-13. Error bars represent SE (t test for equal variances, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
To explore whether neural differentiation contributes to the change in C9orf72 transcription, we also assayed the various isoforms upon differentiation into NPCs and teratomas. Interestingly, we found that differentiation of C9 hESCs into both cell types further enhanced the transcription of exon 1a-bearing transcripts by increasing the expression of both V3 and V1 (Figure 5C). This is in contrast to the upregulation of V2 (the most abundant transcript) in NPCs and teratomas of C9 iPSCs, contributing to the overall increase in C9orf72 mRNA levels (Figure 5C). Jointly, these findings demonstrate that the C9 mutation, together with neural differentiation, favors transcription from the upstream promoter. This effect is largely restricted in iPSCs, presumably due to hypermethylation.
Considering the central role of intron 1-retaining transcripts in the pathology of C9/ALS-FTD disease (Niblock et al., 2016), we aimed to search for the expression of repeat-containing mRNAs in C9 cells. First we generated rRNA-depleted RNA sequencing (RNA-seq) libraries from WT as well as C9-mutant undifferentiated hESCs (two cell lines) and iPSCs (five clones), and RNA deep-sequenced them utilizing next-generation sequencing (see Figures S5A and S5B for general coverage and amount of reads in intron 1 relative to exon 2 of C9orf72 and the RNA-seq data at GEO: GSE87273). As anticipated and in agreement with the report of Niblock et al. (2016), by zooming in to the 5′ part of C9orf72 we found preferential retention of intron 1 in both C9 hESCs and iPSCs (Figure 6A), indicating the propensity of the mutation to interfere with proper splicing of this region. However, quantifying this change between mutant and unaffected cells demonstrates a greater effect in hESCs (4.5-fold increase) relative to iPSCs (only 2-fold increase) (Figure 6B). Considering that C9 cells carry both the allele with expansion and a normal allele, these fold changes are predicted to reflect even greater differences in expression between WT and mutant alleles. We conclude that the shift in promoter usage is coupled with the general tendency to retain intron 1 by the expansion. Next, we aimed to explore whether this may promote accumulation of repeat-containing mRNA transcripts (exon 1a-initiating transcripts that retain repeat expansion), which form the underlying mechanism for protein/RNA gain of function in C9/ALS-FTD. cDNA sequencing at the boundary between intron 1 and exon 5 confirmed the existence of such transcripts in our cells (Figure S5). Using primers located upstream of the repeats that specifically target repeat-containing mRNAs in undifferentiated (hESCs and iPSCs, Figure 6C) and differentiated (NPCs and teratomas, Figure 6D) cells, we monitored for the existence of these unusual mRNAs by real-time RT-PCR. Importantly, while no difference could be detected between C9 and WT undifferentiated cells (hESCs and iPSCs), their level became significantly higher upon differentiation into NPCs and teratomas exclusively in C9 hESCs (7.8- and 2.6-fold change, respectively) (Figures 6D and S5), and not in C9 iPSCs. Hence, our findings provide evidence that the C9 mutation interferes with proper splicing of intron 1, thereby enhancing the formation of potentially pathogenic repeat-containing mRNAs upon differentiation. Importantly, this effect is missing in C9 iPSCs, presumably due to hypermethylation.
Figure 6.

Expression of Intron 1-Retaining C9orf72 Transcripts in Undifferentiated and Differentiated hESCs and iPSCs
(A) Genebrowser view of the first three exons (exons 1a, 1b, 2, and 3) and intronic sequences of C9orf72.
(B) Quantification of the intron 1 retention data presented in (A) in undifferentiated WT and C9 hESCs and iPSCs is based on RNA-seq data. C9 iPSC clones from patient H and M were averaged.
(C) Relative expression levels of intron 1-retaining exon 1a-initiating transcripts as determined by real-time RT-PCR in undifferentiated C9 hESCs (SZ-ALS1 and SZ-ALS3), C9 iPSC clones, and their appropriate WT controls.
(D) Relative expression levels of intron 1-retaining exon 1a-initiating transcripts as determined by real-time RT-PCR in teratomas and NPCs derived from C9 hESCs (SZ-ALS1 and SZ-ALS3), C9 iPSC clones, and their appropriate WT controls. The expression level in each cell type represents an average of three to six independent experiments.
In (C) and (D), Ct values were normalized to the corresponding Ct value of GUS. WT hESC line is SZ-13. Error bars represent SE (t test for equal variance, ∗∗p < 0.01, ∗∗∗p < 0.001).
Discussion
We report on the derivation and full characterization of two hESC lines (SZ-ALS1 and SZ-ALS3) with a GGGGCC expansion of approximately 270 repeats. Our C9 hESC lines were established from embryos obtained through PGD from a woman with a C9 mutation with >40 repeats in her peripheral blood. Interestingly, despite the sufficiently large expansion, both cell lines were completely unmethylated at the repeats (based on (G4C2)n-methylation assay [Xi et al., 2015a, Xi et al., 2015b]) and upstream of the repeats (based on bisulfite colony sequencing). In addition, we generated iPSCs clones that are haploidentical to the mutant hESCs from skin fibroblasts of the asymptomatic C9-carrier mother (700 repeats, patient H), and from an unrelated 65-year-old ALS-manifesting patient (2,700 repeats, patient M). Unexpectedly, we found a striking difference in methylation levels at the 5′ UTR of C9orf72 between the C9 iPSCs and all other cell types examined. Unlike in the C9 hESCs and parental fibroblasts, methylation was detected at the expanded repeats, and reached almost 100% at the upstream CGI in all iPSC clones. Although the difference in C9orf72 hypermethylation between the C9 hESCs and iPSCs could, in theory, result from a difference in expansion size, this is very unlikely since the number of GGGGCC repeats in the hESCs is well above the threshold for methylation to be triggered in any cell type examined thus far (Xi et al., 2015b). In addition, methylation levels in the iPSCs are much higher than those observed in somatic cells of patients (where it does not exceed 20% [Liu et al., 2014]), and is consistent with the exclusive enrichment of the repressive histone mark H3K9me3 and not H3K27me3. This is somewhat different from the data obtained by Belzil et al. (2013), who demonstrated enrichments for H3K27me3 in addition to H3K9me3, although they analyzed different cell types from those described in this study. While we used undifferentiated cells, they looked at frontal cortex and cerebellum. Indeed, our findings in undifferentiated cells cast doubt on the importance of H3K27me3 modification for setting the methylation state and transcriptionally inactive chromatin configuration at the C9 expansion.
We argue that the C9 mutation acts as a hotspot for de novo methylation by transcription factor reprogramming considering the dramatic rise in DNA methyltransferase 3B (DNMT3B) expression levels during the reprogramming procedure (Huang et al., 2014). In fragile X syndrome it was also shown that reprogramming of fibroblasts with an unmethylated full CGG expansion results in hypermethylation and complete inactivation of the FMR1 gene in mutant iPSCs (de Esch et al., 2014). Thus, perhaps hypermethylation of the GGGGCC repeats in our C9 iPSCs represents a much wider phenomenon, whereby untranslated repeat expansions that reside within CGIs provide a “sink” for DNMTs during transcription factor reprogramming (rather than first de novo methylate and then failure to demethylate). This may also explain why we and others generally fail to demethylate and reactivate the FMR1 gene by somatic cell reprogramming when producing iPSCs from cells of fragile X-affected patients (Avitzour et al., 2014, Sheridan et al., 2011, Urbach et al., 2010). It should be noted that our results contradict the report of Esanov et al. (2016), who showed demethylation (rather than de novo methylation) of the C9 mutation in C9 iPSCs. The discrepancy could result from the different cell states employed. We used primary skin fibroblasts from two unrelated patients, whereas they used an immortalized cell line from blood cells of a single patient.
To explore the effect of differentiation on the methylation status of the C9 mutation, we induced the unmethylated mutant hESCs (SZ-ALS1 and SZ-ALS3) and C9 hypermethylated iPSC clones (from C9 individuals H and M) to differentiate into disease-relevant cell types (NPCs and teratomas), which are generally enriched in neural cells. Interestingly, methylation levels remained unchanged at the repeats and upstream of them in both hESCs and iPSCs, following in vitro and in vivo differentiation (0% and 50%, respectively). Perhaps extending differentiation length to fully matured neurons, or to specific subtypes of neurons, would have elicited C9 methylation in the hESCs.
Finally, to associate hypermethylation with disease pathogenesis, we analyzed the expression of transcript variants 1, 2, and 3 individually. We show that the GGGGCC expansion alters the region to allow the enhancement of V1 transcripts, albeit with much lower levels in iPSCs relative to hESCs. In addition, we demonstrate that the C9 mutation, together with neural differentiation, favors transcription from an upstream promoter (exon 1a-initiating transcripts, V1 and V3) over a downstream promoter (exon 1b-initiating transcript, V2), and that this effect is largely restricted in iPSCs. Furthermore, we found preferential retention of intron 1 in both C9 hESCs and iPSCs by RNA deep sequencing, illustrating the propensity of the mutation to interfere with the proper splicing of this region in both exon 1a- and 1b-initiating transcripts. However, this change between WT and C9 cells demonstrates a greater effect in hESCs (4.5-fold increase) relative to iPSCs (only a 2-fold increase). More importantly, by monitoring for the expression of potentially pathogenic mRNA transcripts (i.e., exon 1a-initiating transcripts that retain intron 1), we find no difference between mutant and WT undifferentiated cells (hESCs and iPSCs). However, their level becomes significantly higher upon differentiation into disease-relevant cell types (NPCs and teratomas) exclusively in C9 hESCs, and not in C9 iPSCs. We propose that methylation counteracts the effect of the expansion by downregulating exon 1a-initiating mRNA species. In line with this idea are previous reports by Liu et al. (2014) and others (Bauer, 2016, Day and Roberson, 2015), which point to a mechanistic link between hypermethylation and reduced accumulation of RNA foci and dipeptide inclusions in patient cell lines, brain samples, and HEK293T transgenic cell lines.
Our data related to C9orf72 mRNA levels conflict with the reports of others, who demonstrate a general reduction in C9orf72 in diseased cells (Ciura et al., 2013, Cooper-Knock et al., 2013, Donnelly et al., 2013, Waite et al., 2014, Xi et al., 2013). These discrepancies may stem from the different types of samples (single versus mixed type of cells) employed. Nevertheless, even if our mutant NPCs do not reflect the physiological levels observed in fully mature disease-relevant cells, this model system could help to interpret the role of methylation in disease pathogenesis because it features both an extreme C9 hypermethylation state and an extreme C9 hypomethylation state.
To summarize, this study clearly demonstrates how reprogramming excessively hypermethylates the C9 expanded locus, and how the C9 mutation alters C9orf72 variant transcription and processing. In addition, the current work highlights the importance of neural differentiation in the pathogenesis of ALS-FTD and points to the potential role of C9 hypermethylation as a neuroprotective mechanism that attenuates the accumulation of potentially toxic repeat-containing mRNAs in C9 neurons.
Altogether, this implies that the difference between C9 hESCs and iPSCs may be crucial for investigating the neural phenotype of the C9/ALS-FTD disease, given that mutant hESCs are likely to present a more accurate and more severe phenotype than comparable iPSCs. Recognizing that C9 hypermethylation limits the increase in intron 1-retaining transcripts, it may be possible in the future (with the advent of gene editing) to modify the C9 expanded locus by specifically targeting chromatin silencing factors/DNMTs to the region. This would affect epigenetic inactivation of exon 1a-initiating variants so as to reverse/minimize the adverse effect of the C9 mutation in disease-affected tissues.
Experimental Procedures
hESC Cell Line Derivation and Characterization
The use of embryos carrying the C9 mutation, derived from PGD treatment, for hESC derivation was performed in compliance with protocols approved by the National Ethics Committee. All cell lines were established at the Shaare Zedek Medical Center (87/07). Cell line derivation was carried out as previously described (Eiges et al., 2007). All hESC lines were examined for all typical characteristics of hESCs (for primers and conditions see Supplemental Experimental Procedures).
C9 iPSC Derivation
For transcription reprogramming, Yamanaka's four retroviral vectors expressing OCT3/4, SOX2, KLF4, and c-MYC were individually packaged in 293T cells. Infectious viruses were collected 24 and 48 hr post transfection and immediately added to primary fibroblasts (88/11). Four days following infection the cells were placed on mitomycin C-treated mouse embryonic fibroblasts (MEFs) and maintained in hESC medium. Manual isolation of single clones was carried out approximately 30 days post transfection, resulting in stable cell lines with hESC-like morphology.
Teratoma Induction
Cells (2.5–5 × 106) were harvested, diluted 1:1 in medium/Matrigel, and injected subcutaneously to both sides of the back of NOD-SCID IL2Rγ−/− mice. Six to eight weeks later the mice were euthanized and tumors were isolated, sectioned, and assessed for differentiation by H&E staining.
Neuronal Precursor Differentiation of HESCs and iPSCs
The cells were grown on MEFs to similarly sized, defined colonies using hESC medium. Induced embryoid body (EB) formation was achieved by detaching colonies with collagenase IV for 30 min. The cells were centrifuged and transferred as colonies to a Petri dish containing hESC medium without basic fibroblast growth factor (bFGF) + 5 μM dorsomorphin (catalog #11967, Cayman) and 5 μM SB431542 (SB #13031, Cayman). The EBs were cultured for 4–5 days with medium replacement every other day. For the NPC expansion, the cells were plated on Matrigel-coated plates (without dissociation) and cultured with KO DMEM medium + 1XN2 supplement (stock X100) and 20 ng/mL bFGF for 8–10 days with medium replacement every other day.
Chromosome Analysis
Karyotype analysis was carried out by Giemsa staining according to standard protocol.
Southern Blot Analysis
Genomic DNAs (10–25 μg) were digested with EcoRI and HindIII (Fermentas) restriction endonucleases, separated on 0.8% agarose gels, blotted onto Hybond N+ membranes (Amersham), and hybridized with a PCR Dig-labeled 576-bp probe (primer F: TTG CGA TGA CTT TGC AGG GGA CC and primer R: CAG CGA GTA CTG TGA GAG).
RT-PCR
Total RNA was isolated from the cells by TRI Reagent extraction, then 1 μg of RNA was reverse transcribed by random hexamer priming and Multi Scribe reverse transcriptase (ABI). Amplification was performed using the primers listed in Supplemental Experimental Procedures using Super-Therm Taq DNA polymerase (Jain Biologicals).
Expression of Undifferentiated Cell-Specific Markers
Undifferentiated cell cultures were examined for the expression of undifferentiated cell-specific markers by immunostaining using monoclonal mouse OCT3/4 (Santa Cruz Biotechnology #sc-5279, 1:50 dilution) or TRA-1-60 (Santa Cruz #sc-21705, 1:50 dilution), together with Cy3-conjugated goat anti-mouse polyclonal antibodies (Jackson Immunostaining #115-035-062, 1:100 dilution). Nuclear staining was performed with Hoechst 33258 (Sigma #861405). Staining for alkaline phosphatase was carried out using an Alkaline Phosphatase Kit (Sigma Diagnostics #86R-1KT) according to the manufacturer's protocol.
Real-Time TaqMan PCR
TaqMan gene expression assays for C9orf72 were carried out according to Belzil et al. (2013). qRT-PCR experiments were conducted in triplicate using custom-made TaqMan-based expression assays for transcript variants 1 (NM_145005.5), 2 (NM_018325.3), and 3 (NM_001256054.1) individually, and altogether (adopted from DeJesus-Hernandez et al., 2011); for primers and probe sequences see Supplemental Experimental Procedures). GUS transcript (NM_000181.3) was used as housekeeping gene for normalization of ΔΔCt mean values.
Expression of Intron 1-Retaining Transcripts Upstream of the Repeats by qRT-PCR
Total RNA was isolated from cells by TRI reagent extraction. RNA (1 μg) was reverse transcribed (Multi Scribe RT, ABI) with random hexamer primers. Real-time PCR was performed using Power SYBR Green Master Mix (ABI) on an ABI 7900HT instrument. Primers are listed in Supplemental Experimental Procedures.
FACS Analysis for NCAM1-Positive Cells
NPCs were washed with PBS and dissociated to single cells using preheated (to 37°C) TrypLE Selected (Life Technologies #12563-011) for a minimal amount of time. After cell dissociation, the cells were resuspended with cold sterile PBS-FACS medium (Commercial PBS without Ca+2 Mg+2 to prevent adhesion with FCS 10%), and filtered through a mesh tube (#352235). From this point onward everything was carried out on ice or under cold centrifugation (4°C). After centrifugation 1 × 106 cells were resuspended into 200 μL of PBS-FCS. The cells were stained directly with primary anti-NCAM1 (R&D #af2408, 1:150) and incubated on ice for 1 hr. After staining the cells were washed two times by adding 5 mL of PBS-FCS and centrifuged at 150 × g for 5 min. The cells were then resuspended into the initial volume and incubated with a secondary antibody (1:200, fluorescein isothiocyanate-conjugated donkey anti-goat immunoglobulin G [IgG], #705095147) for an additional 1 hr. Control cells were stained for secondary antibody only. The cells were washed twice and assayed by FACS.
Immunostaining for Tuj1-Positive Cells in Teratoma Sections
Paraffin-embedded tissues were sectioned (4 μm) using a microtome (Leica RM2255) and transferred to silane-coated slides. After incubation in a 60°C in a dry oven for 1 hr, paraffin-embedded sections were deparaffinized in xylene and rehydrated through graded ethanol (100% to 50%). For antigen retrieval, sections were microwaved in 10 mM sodium citrate buffer (pH 6.0) for 5 min. Nonspecific antibody binding was blocked in PBS with 10% fetal bovine serum and 0.3% Triton X-100 (Sigma) for 45 min. Sections were then incubated with anti-Tuj1 antibody (rabbit IgG 1:400, Biolegend #845501) at 4°C overnight. The secondary antibody Alexa anti-rabbit (1:400, Life Technologies #A31572) was applied for 1 hr at room temperature. The sections were nuclear stained by DAPI, mounted with Vectashield (Vector Laboratories #H1000), and examined under a fluorescent microscope.
Bisulfite Sequencing
Genomic DNA (1 μg) was modified by bisulfite treatment (EZ DNA methylation Kit, Zymo Research) and amplified by FastStart DNA polymerase (Roche). Amplified products were cloned and single colonies were analyzed for CpG methylation by direct sequencing (ABI 3130). Primers are listed in Supplemental Experimental Procedures.
(G4C2)n-Methylation Assay
Methylation of the G4C2-repeat itself was studied using a reported (G4C2)n-methylation assay (Xi et al., 2015b). In brief, this assay combines rp-PCR with methylation-specific PCR, where each sample was amplified by rp-PCR using primers specific for methylated versus unmethylated DNA after bisulfite conversion. The primers were labeled by fluorescein amidite for methylated DNA amplification (blue channel) and HEX for unmethylated DNA amplification (green channel). Data were visualized by Genotyper software (version 3.6, Applied Biosystems).
Chromatin Immunoprecipitation Assay
ChIP was performed using the Upstate EZTM ChIP kit, according to the manufacturer's protocol with slight modifications. In brief, cells were harvested and then fixed, quenched, and washed in 50-mL tubes. Sonication was carried out using a Vibra Cell VCX130 with a 3-mm microtip and 30% amplitude, in five cycles of 10 s and 30 s rest on ice. Immunoprecipitation was performed using an anti-H3K27me3 (Abcam #6002) and anti-H3K9me3 (Abcam #8898) antibody. Immunoprecipitation efficiency was evaluated by HOXA9 (enriched in H3K9me3 and H3K27me3 in pluripotent stem cells). Real-time PCR was carried out on an ABI 7900HT instrument (primers are listed in Supplemental Experimental Procedures). ΔΔCt values were normalized according to a negative control (APRT) to account for histone modification enrichment.
RNA Deep Sequencing
RNA was extracted using TRI Reagent (Sigma). Depletion of rRNA was performed using the Ribominus kit (Invitrogen). RNase-R treatments were performed by adding 3 U of RNase-R (Epicenter Biotechnologies) per milligram of RNA and 15-min incubation at 37°C. cDNA libraries were generated using the TruSeq RNA sample preparation kit and protocol (Illumina), and stranded, ligation-based libraries were sequenced as previously described (Engreitz et al., 2013). RNA-seq reads were aligned to the genome (hg19) using STAR. Coverage of C9orf72 second exon (chr9:27566674-27567162) and first intron (chr9:27567163-27573426) were calculated using the samtools depth tool. We normalized the total coverage values to the length of the exon/intron to obtain average coverage per base.
Author Contributions
Y.C.-H., M.Z., E.R., and S.E.-L. contributed to the conception and design of the study, the collection and assembly of data, data analysis and interpretation, and manuscript writing. G.A., T.E-.G., E.L.-L., M.G., O.B., R.A.-F., and S.K. contributed to the collection of data. R.E. contributed to the conception and design of the study, financial support, data analysis and interpretation, and manuscript writing.
Acknowledgments
We thank the family who donated the C9 embryos for hESC line derivation, and the C9 mutation carriers (patients H and M) who made donations of skin for the generation of C9 iPSCs. We would also like to thank Dr. David Zeevi for critical reading of the manuscript and Tamar Golan-Lev for the graphic illustrations. This research was partly supported by the ALS Association initiator grant (grant no. 16-IIP-256, R.E. and S.K.), the Legacy Heritage Biomedical program of the Israel Science Foundation (grant no. 0621/02), Mirsky foundation (S.E.-L.), the Canadian Consortium on Neurodegeneration in Aging and Weston Brain Institute (E.R., M.Z.).
Published: October 20, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.09.011.
Accession Numbers
The accession number for the RNA-seq data reported in this paper is GEO: GSE87273.
Supplemental Information
References
- Almeida S., Gascon E., Tran H., Chou H.J., Gendron T.F., Degroot S., Tapper A.R., Sellier C., Charlet-Berguerand N., Karydas A. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avitzour M., Mor-Shaked H., Yanovsky-Dagan S., Aharoni S., Altarescu G., Renbaum P., Eldar-Geva T., Schonberger O., Levy-Lahad E., Epsztejn-Litman S. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014;3:699–706. doi: 10.1016/j.stemcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer P.O. Methylation of C9orf72 expansion reduces RNA foci formation and dipeptide-repeat proteins expression in cells. Neurosci. Lett. 2016;612:204–209. doi: 10.1016/j.neulet.2015.12.018. [DOI] [PubMed] [Google Scholar]
- Belzil V.V., Bauer P.O., Prudencio M., Gendron T.F., Stetler C.T., Yan I.K., Pregent L., Daughrity L., Baker M.C., Rademakers R. Reduced C9orf72 gene expression in c9FTD/ALS is caused by histone trimethylation, an epigenetic event detectable in blood. Acta Neuropathol. 2013;126:895–905. doi: 10.1007/s00401-013-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzil V.V., Bauer P.O., Gendron T.F., Murray M.E., Dickson D., Petrucelli L. Characterization of DNA hypermethylation in the cerebellum of c9FTD/ALS patients. Brain Res. 2014;1584:15–21. doi: 10.1016/j.brainres.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciura S., Lattante S., Le Ber I., Latouche M., Tostivint H., Brice A., Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013;74:180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J., Higginbottom A., Connor-Robson N., Bayatti N., Bury J.J., Kirby J., Ninkina N., Buchman V.L., Shaw P.J. C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology. 2013;81:1719–1721. doi: 10.1212/01.wnl.0000435295.41974.2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J., Walsh M.J., Higginbottom A., Robin Highley J., Dickman M.J., Edbauer D., Ince P.G., Wharton S.B., Wilson S.A., Kirby J. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J., Bury J.J., Heath P.R., Wyles M., Higginbottom A., Gelsthorpe C., Highley J.R., Hautbergue G., Rattray M., Kirby J. C9ORF72 GGGGCC Expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS One. 2015;10:e0127376. doi: 10.1371/journal.pone.0127376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., Van Langenhove T., van der Zee J., Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36:450–459. doi: 10.1016/j.tins.2013.04.010. [DOI] [PubMed] [Google Scholar]
- Day J.J., Roberson E.D. DNA methylation slows effects of C9orf72 mutations: an epigenetic brake on genetic inheritance. Neurology. 2015;84:1616–1617. doi: 10.1212/WNL.0000000000001504. [DOI] [PubMed] [Google Scholar]
- de Esch C.E., Ghazvini M., Loos F., Schelling-Kazaryan N., Widagdo W., Munshi S.T., van der Wal E., Douben H., Gunhanlar N., Kushner S.A. Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Rep. 2014;3:548–555. doi: 10.1016/j.stemcr.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devlin A.C., Burr K., Borooah S., Foster J.D., Cleary E.M., Geti I., Vallier L., Shaw C.E., Chandran S., Miles G.B. Human iPSC-derived motoneurons harbouring TARDBP or C9ORF72 ALS mutations are dysfunctional despite maintaining viability. Nat. Commun. 2015;6:5999. doi: 10.1038/ncomms6999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dols-Icardo O., Garcia-Redondo A., Rojas-Garcia R., Sanchez-Valle R., Noguera A., Gomez-Tortosa E., Pastor P., Hernandez I., Esteban-Perez J., Suarez-Calvet M. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014;23:749–754. doi: 10.1093/hmg/ddt460. [DOI] [PubMed] [Google Scholar]
- Donnelly C.J., Zhang P.W., Pham J.T., Haeusler A.R., Mistry N.A., Vidensky S., Daley E.L., Poth E.M., Hoover B., Fines D.M. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiges R., Urbach A., Malcov M., Frumkin T., Schwartz T., Amit A., Yaron Y., Eden A., Yanuka O., Benvenisty N. Developmental study of fragile X syndrome using human embryonic stem cells derived from preimplantation genetically diagnosed embryos. Cell Stem Cell. 2007;1:568–577. doi: 10.1016/j.stem.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Engreitz J.M., Pandya-Jones A., McDonel P., Shishkin A., Sirokman K., Surka C., Kadri S., Xing J., Goren A., Lander E.S. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esanov R., Belle K.C., van Blitterswijk M., Belzil V.V., Rademakers R., Dickson D.W., Petrucelli L., Boylan K.B., Dykxhoorn D.M., Wuu J. C9orf72 promoter hypermethylation is reduced while hydroxymethylation is acquired during reprogramming of ALS patient cells. Exp. Neurol. 2016;277:171–177. doi: 10.1016/j.expneurol.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron T.F., Belzil V.V., Zhang Y.J., Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta Neuropathol. 2014;127:359–376. doi: 10.1007/s00401-013-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I., Van Mossevelde S., van der Zee J., Sieben A., Engelborghs S., De Bleecker J., Ivanoiu A., Deryck O., Edbauer D., Zhang M. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry. 2015;21:1112–1124. doi: 10.1038/mp.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler A.R., Donnelly C.J., Periz G., Simko E.A., Shaw P.G., Kim M.S., Maragakis N.J., Troncoso J.C., Pandey A., Sattler R. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang K., Shen Y., Xue Z., Bibikova M., April C., Liu Z., Cheng L., Nagy A., Pellegrini M., Fan J.B. A panel of CpG methylation sites distinguishes human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014;2:36–43. doi: 10.1016/j.stemcr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.S., Lee J.S., Leem J.W., Huh Y.J., Kim J.Y., Kim H.S., Park I.H., Daley G.Q., Hwang D.Y., Kim D.W. Robust enhancement of neural differentiation from human ES and iPS cells regardless of their innate difference in differentiation propensity. Stem Cell Rev. 2010;6:270–281. doi: 10.1007/s12015-010-9138-1. [DOI] [PubMed] [Google Scholar]
- Lee Y.B., Chen H.J., Peres J.N., Gomez-Deza J., Attig J., Stalekar M., Troakes C., Nishimura A.L., Scotter E.L., Vance C. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Balasubramanian U., Cohen D., Zhang P.W., Mosmiller E., Sattler R., Maragakis N.J., Rothstein J.D. A comprehensive library of familial human amyotrophic lateral sclerosis induced pluripotent stem cells. PLoS One. 2015;10:e0118266. doi: 10.1371/journal.pone.0118266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E.Y., Russ J., Wu K., Neal D., Suh E., McNally A.G., Irwin D.J., Van Deerlin V.M., Lee E.B. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta Neuropathol. 2014;128:525–541. doi: 10.1007/s00401-014-1286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niblock M., Smith B.N., Lee Y.B., Sardone V., Topp S., Troakes C., Al-Sarraj S., Leblond C.S., Dion P.A., Rouleau G.A. Retention of hexanucleotide repeat-containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta Neuropathol. Commun. 2016;4:18. doi: 10.1186/s40478-016-0289-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters O.M., Cabrera G.T., Tran H., Gendron T.F., McKeon J.E., Metterville J., Weiss A., Wightman N., Salameh J., Kim J. Human C9ORF72 hexanucleotide expansion reproduces RNA foci and dipeptide repeat proteins but not neurodegeneration in BAC transgenic mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S., Serrano A., Gerbino V., Giorgi A., Di Francesco L., Nencini M., Bozzo F., Schinina M.E., Bagni C., Cestra G. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015;128:1787–1799. doi: 10.1242/jcs.165332. [DOI] [PubMed] [Google Scholar]
- Russ J., Liu E.Y., Wu K., Neal D., Suh E., Irwin D.J., McMillan C.T., Harms M.B., Cairns N.J., Wood E.M. Hypermethylation of repeat expanded C9orf72 is a clinical and molecular disease modifier. Acta Neuropathol. 2015;129:39–52. doi: 10.1007/s00401-014-1365-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D., O'Rourke J.G., Meera P., Muhammad A.K., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J., Yamamoto Y., Kitano S., Takitani M., Asahina N., Kino Y. Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J. Cent. Nerv Syst. Dis. 2014;6:69–78. doi: 10.4137/JCNSD.S18103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan S.D., Theriault K.M., Reis S.A., Zhou F., Madison J.M., Daheron L., Loring J.F., Haggarty S.J. Epigenetic characterization of the FMR1 gene and aberrant neurodevelopment in human induced pluripotent stem cell models of fragile X syndrome. PLoS One. 2011;6:e26203. doi: 10.1371/journal.pone.0026203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Urbach A., Bar-Nur O., Daley G.Q., Benvenisty N. Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell. 2010;6:407–411. doi: 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainger B.J., Kiskinis E., Mellin C., Wiskow O., Han S.S., Sandoe J., Perez N.P., Williams L.A., Lee S., Boulting G. Intrinsic membrane hyperexcitability of amyotrophic lateral sclerosis patient-derived motor neurons. Cell Rep. 2014;7:1–11. doi: 10.1016/j.celrep.2014.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waite A.J., Baumer D., East S., Neal J., Morris H.R., Ansorge O., Blake D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging. 2014;35:1779.e5–1779.e13. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z., Zinman L., Moreno D., Schymick J., Liang Y., Sato C., Zheng Y., Ghani M., Dib S., Keith J. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 2013;92:981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z., Rainero I., Rubino E., Pinessi L., Bruni A.C., Maletta R.G., Nacmias B., Sorbi S., Galimberti D., Surace E.I. Hypermethylation of the CpG-island near the C9orf72 G(4)C(2)-repeat expansion in FTLD patients. Hum. Mol. Genet. 2014;23:5630–5637. doi: 10.1093/hmg/ddu279. [DOI] [PubMed] [Google Scholar]
- Xi Z., van Blitterswijk M., Zhang M., McGoldrick P., McLean J.R., Yunusova Y., Knock E., Moreno D., Sato C., McKeever P.M. Jump from pre-mutation to pathologic expansion in C9orf72. Am. J. Hum. Genet. 2015;96:962–970. doi: 10.1016/j.ajhg.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Z., Zhang M., Bruni A.C., Maletta R.G., Colao R., Fratta P., Polke J.M., Sweeney M.G., Mudanohwo E., Nacmias B. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015;129:715–727. doi: 10.1007/s00401-015-1401-8. [DOI] [PubMed] [Google Scholar]
- Zeier Z., Esanov R., Belle K.C., Volmar C.H., Johnstone A.L., Halley P., DeRosa B.A., Khoury N., van Blitterswijk M., Rademakers R. Bromodomain inhibitors regulate the C9ORF72 locus in ALS. Exp. Neurol. 2015;271:241–250. doi: 10.1016/j.expneurol.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.