

Abstract
Rationale
Pulmonary arterial hypertension (PAH) is a progressive and fatal disease characterized by pulmonary arterial muscularization due to excessive pulmonary vascular cell proliferation and migration, a phenotype dependent upon growth factors and activation of receptor tyrosine kinases (RTKs). p130Cas is an adaptor protein involved in several cellular signaling pathways that control cell migration, proliferation and survival.
Objectives
We hypothesized that in experimental and human PAH p130Cas signaling is over-activated, thereby facilitating the intracellular transmission of signal induced by fibroblast growth factor (FGF)-2, epidermal growth factor (EGF), and platelet-derived growth factor (PDGF).
Measurements and Main Results
In PAH patients, levels of p130Cas protein and/or activity are higher in the serum, in walls of distal pulmonary arteries, in cultured smooth muscle (PA-SMCs) and pulmonary endothelial cells (P-ECs) than controls. These abnormalities in the p130Cas signaling were also found to be in the chronically hypoxic mice and monocrotaline-injected rats as models of human PAH. We next obtained evidence for convergence and amplification of the growth-stimulating effect of EGF, FGF2 and PDGF signaling pathways via p130Cas signaling pathway. Finally, we found that daily treatment with each of the EGF-R inhibitor gefitinib, the FGF-R inhibitor dovitinib and the PDGF-R inhibitor imatinib started 2 weeks after a subcutaneous monocrotaline injection substantially attenuate the abnormal increase in p130cas and ERK1/2 activation and regress established PH.
Conclusions
Our findings demonstrate that p130Cas signaling plays a critical role in experimental and iPAH by modulating pulmonary vascular cell migration, proliferation and by acting as an amplifier of RTKs downstream signals.
Keywords: Animals; Benzimidazoles; Mice; Monocrotaline; Myocytes, Smooth Muscle; Piperazines; Platelet-Derived Growth Factor; Protein Kinase Inhibitors; Pulmonary Artery; Pyrimidines; Quinazolines; Quinolones; Case-Control Studies; Rats; Signal Transduction; Crk-Associated Substrate Protein; Disease Models, Animal; Endothelial Cells; Epidermal Growth Factor; Fibroblast Growth Factor 2; Humans; Hypertension, Pulmonary
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a devastating disease of unknown etiology characterized by a marked and sustained increase in pulmonary artery pressure (PAP) that ultimately leads to right ventricular failure and death. Currently, there is no cure for PAH and the therapeutic options available only partially improve symptoms and increase survival. In the absence of specific treatments, patients with idiopathic PAH (iPAH) have a mean survival of 2.8 yr. Even under current therapies survival remains poor with a 15% death-rate per year in incident iPAH patients (1, 2). Irreversible remodeling of the pulmonary vasculature is the cause of increased PAP in PAH and frequently leads to progressive functional decline in patients despite treatment. This process is ascribed to the excessive proliferation, survival and migration of pulmonary vascular cells, including SMCs and ECs (3). However, the mechanisms that mediate these effects are incompletely understood.
Over recent years, there has been growing interest in the disease-promoting roles of three different growth factors: PDGF, EGF, and FGF2. Altered expression and/ or increased activities of these RTKs have been reported in human and experimental models of PH and their inhibition by specific inhibitors (TKIs) reverses experimental PH in rodents (4–8). In some cases, clinical improvements with some TKI have been documented, but partly because of their lack of selectivity, serious side-effects often limit their use in patients with PAH. A better understanding of the signaling through these RTKs may contribute to the development of new, better-tolerated and more powerful therapeutic tools.
p130Cas is an adaptor protein involved in the regulation of several cellular signaling pathways that control cell motility, proliferation and survival (9). In particular, knockdown of p130Cas causes proliferative arrest in breast cancer cell lines harboring oncogenic mutations in KRAS, BRAF, PTEN, and PIK3CA (9), implicating p130Cas as a general regulator of breast cancer cell growth induced by various oncogenes. Furthermore, p130Cas tyrosine phosphorylation (active form) has been demonstrated in response to stimulation of a wide variety of cell surface receptors including RTKs and G-protein-coupled receptors in various cell types (10–13). We investigated: 1) the p130Cas levels in the serum of iPAH patients and controls; 2) the in situ presence and activity of p130Cas within walls of iPAH and control pulmonary arteries; 3) whether or not intrinsic abnormalities in p130Cas signaling are characteristic of P-ECs and PA-SMCs from iPAH patients; 4) whether or not p130Cas is a potential target in RTK signaling pathways, particularly in the control of proliferation and/or migration of iPAH pulmonary vascular cells; 5) lung levels of p130Cas protein and activity in two rodent models of PH; 6) the efficacy of gefitinib, dovitinib and imatinib treatments on the abnormal increase in ERK1/2 and p130cas activation states and on the progression of monocrotaline-induced PH.
METHODS
Study Population
Blood samples and clinical data from 37 PAH patients (n=21 iPAH; n=16 heritable PAH) and from 40 controls were collected (Supp. Table. 1). For the in vitro and in situ studies, we studied lung specimens obtained during lung transplantation in 7 iPAH patients and during lobectomy or pneumonectomy for localized lung cancer in 7 controls (Supp. Table. 2). Preoperative echocardiological evaluation including echocardiography was performed in the controls to rule out PAH, and the lung specimens from the controls were collected at distance from tumor foci. This study was approved by the local ethics committee (CPP Ile-de-France VII, Le Kremlin-Bicêtre, France). All patients gave informed consent before the study.
Isolation, Culture and Functional Analysis of Human PA-SMCs and P-ECs
Human PA-SMCs and P-ECs were isolated and cultured as previously described (4, 14). Proliferation was assessed by 5-bromo-2-deoxyuridine (BrdU) incorporation and direct cell counting (8) and migration by a modified Boyden chamber migration assay (15).
Western blot, ELISA, Gelatin Zymography and Immunostaining
Cells were homogenized and sonicated in PBS containing protease and phosphatase inhibitors and 30 μg of protein was used to detect p130Cas (Tebu-Bio, Le Perray-en-Yvelines, France), phospho-(Y165)-p130Cas (Ozyme, Saint-Quentin-Yvelines, France), phospho-ERK1/2, ERK1 (Tebu-Bio) and β-actin (Sigma-Aldrich) as previously described (8). Circulating p130cas levels were assayed using a human p130cas enzyme-linked immunosorbent assay (ELISA) (Shanghai Boyun Biotech Co., Ltd. Shanghai, China). Gelatin zymography, immunohistochemistry and immunocytofluorescent staining were performed as previously described (8, 15, 16).
Animal Models and Hemodynamic Measurements
Young male Wistar rats (100 g) were studied 4 weeks after a single subcutaneous injection of monocrotaline (60 mg/Kg) or vehicle (14). In an additional experiment, 20 rats given monocrotaline were left untreated for 2 weeks then randomly divided into four groups and treated for 2 weeks with daily intraperitoneal injections of: imatinib (50 mg/kg/day (7)), gefitinib (50 mg/kg/day (17)), dovitinib (30 mg/kg/day (18)), and vehicle (dimetyl sulfoxide). C57BL/6 mice (12 weeks of age) were either studied in room air, or exposed to hypoxia (10% oxygen) for 3 weeks (19, 20). Animals were anesthetized with isoflurane. A polyvinyl catheter was introduced into the right jugular vein and pushed through the right ventricle into the pulmonary artery. Another polyethylene catheter was inserted into the right carotid artery. After measurement of hemodynamic parameters, the thorax was opened and the left lung immediately removed and frozen. The right lung was fixed in the distended state with formalin buffer. The right ventricular hypertrophy (RVH) index and the percentage of muscularized vessels were determined as previously described (16). Animal studies were approved by the administrative panel on animal care at Marie Lannelongue, Le Plessis-Robinson, France.
Statistical Analyses
Statistical significance was tested using the nonparametric Mann-Whitney test or two-way ANOVA with Bonferroni post hoc tests.
RESULTS
Serum p130Cas Protein Levels Are Increased in PAH Patients
Although no significant difference was noted between iPAH and hPAH (P=0.075, NS), a substantial increase in circulating p130Cas protein levels was observed in the serum of PAH patients compared with controls (1.02±0.21 vs. 0.29±0.07 ng/mL; P<0.001) (Fig. 1). In contrast, circulating p130Cas protein levels in 9 asthmatic patients without systemic corticosteroids therapy were similar to levels found in a control population (0.11 ± 0.1 versus 0.10 ± 0.10 ng/mL respectively; NS) (Supplemental Fig. S1).
Figure 1. Serum p130Cas Protein Levels.
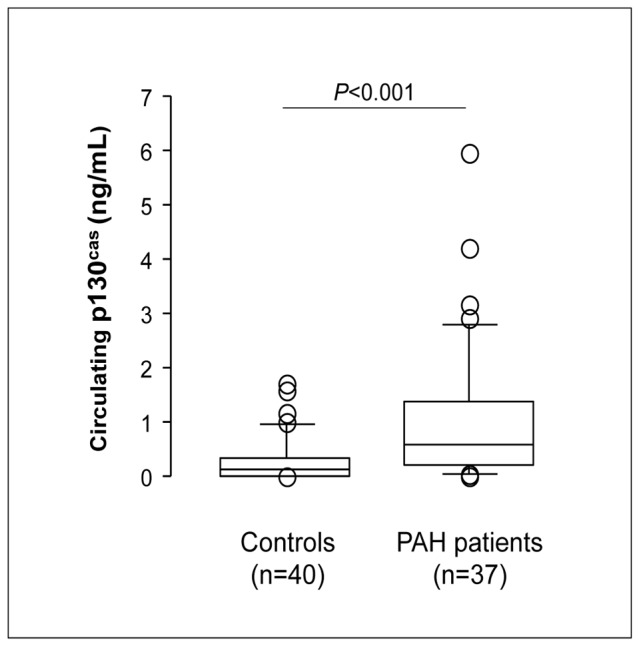
Levels of circulating p130Cas protein were markedly increased in the serum of patients with PAH compared with those from controls. Values are means±SEM.
Adapter p130Cas Protein and Activity Are Increased in Lungs and Cultured Pulmonary Vascular Cells Derived From iPAH Patients
We next quantified and localized p130Cas protein in lungs and found that phospho-p130Cas and p130Cas were abundant in walls of iPAH pulmonary arteries; in contrast, the p130Cas signal was weak in vessels from controls (Fig. 2A). SMC and EC were the major sites of p130Cas over-expression, so we investigated amounts of p130Cas and its phosphorylation state in primary PA-SMC and P-EC cultures generated from these human lung specimens. Consistent with our immunohistochemical findings, iPAH PA-SMCs (Fig. 2B) and P-ECs (Fig. 2C) contained more p130Cas and phospho-p130Cas proteins than control cells. Confocal microscopy studies confirmed the higher p130Cas activation state in iPAH than in control cells (Fig. 2D–E).
Figure 2. Over-expression of p130Cas in lungs and cultured pulmonary vascular cells from patients with iPAH.

(A) Representative photomicrographs of distal pulmonary arteries in lung sections from controls (panel a and c) and iPAH patients (panels b and d). Scale bar=100μm. Expression of p130Cas and phospho-p130Cas (B) in cultured PA-SMCs (C) and in P-ECs from iPAH patients and controls. Localization of p130Cas and phospho-p130Cas (D) in PA-SMCs and (E) in P-ECs. Scale bar= 20μm. Values are means±SEM (n=5).
p130Cas Facilitates The Proliferative and Migratory Actions of EGF, PDGF, and FGF2 in Cultured P-ECs and PA-SMCs
Next, we investigated the functional importance of p130Cas in these cells and assessed whether p130Cas may be a potential target for reducing or blocking proliferation and migration induced by RTKs. We evaluated the proliferative and migration potential of PA-SMCs with and without p130Cas inhibition by RNA-interference. Transfecting PA-SMCs with p130Cas-siRNA decreased p130Cas protein abundance by 86% with respect to transfection with scrambled-siRNA (Supplemental Fig. S2). Decreasing p130Cas similarly reduced both the proliferative and migration potentials of iPAH and control PA-SMCs under basal conditions (Fig. 3A–B and Supplemental Fig. S3). Decreasing p130Cas levels reduced the iPAH PA-SMC proliferation induced by EGF, FGF2, and PDGF by 59%, 64%, and 60%, respectively. PDGF, and in particular PDGF-BB, is one of the major stimulating migratory factors for arterial SMCs (21). We found that transfecting PA-SMCs with p130Cas-siRNA reduced the PDGF-induced migration of both iPAH and control PA-SMCs. In addition, treatments of PA-SMCs transfected with p130Cas-siRNA with either growth factor still offered significant increase in proliferation and migration compared to basal condition (Figure 3A–B). b). Since the tyrosine phosphorylation of the p130Cas substrate domain is modulated by Src, we next determined the effect of Src inhibition on PA-SMC proliferation. We found that inhibition of Src activation by the selective Src inhibitor (Src Inhibitor-1, 10−6M) substantially reduced the proliferation of iPAH and control-PA-SMCs under basal conditions or following exposure to the above mentioned growth factors (Figure 3A).
Figure 3. p130Cas Facilitates The Proliferative and Migratory Actions of EGF, PDGF, and FGF2 in Cultured Human PA-SMCs.

(A) Proliferative and (B) migration potential of PA-SMCs from iPAH- and control-patients with or without p130Cas inhibition by RNA-interference or Src inhibitor-1. (C) Representative gelatin zymography and densitometry carried out on conditioned media of human PA-SMCs. Values are means±SEM (n=5).
Matrix metalloproteinase (MMP)-2 facilitates SMC migration by degradation of the basement membrane and the extracellular matrix. We therefore determined whether the chemotactic migratory behavior of PA-SMCs was impaired by loss of p130Cas in association with reduced MMP-2 activity. Consistent with a previous report, we found that conditioned media from iPAH PA-SMCs contained more MMP-2 than that from control cells (22). Decreasing p130Cas by siRNA markedly reduced MMP-2 levels to similar levels in conditioned media from iPAH and control-PA-SMCs (Fig. 3C).
Similar to findings for PA-SMCs, we also showed that the increased p130Cas is necessary for the heightened response of iPAH P-ECs to EGF, FGF, and PDGF (Fig. 4). Decreasing p130Cas reduced iPAH P-EC proliferation by 78% and iPAH P-ECs migration by 75% under basal conditions (Fig. 4A–B).
Figure 4. p130Cas Facilitates The Proliferative and Migratory Actions of 10 ng/mL of EGF, PDGF, and FGF2 in Cultured Human P-ECs.

(A) Proliferative and (B) migration potential of P-ECs with or without p130Cas inhibition by RNA-interference. Values are means±SEM (n=5).
EGF, FGF2 and PDGF Signaling Pathways Converge to p130Cas Which Acts as an Amplifier of Downstream Signals
Then, we studied how increased p130Cas might contribute to the pathogenesis of PAH and its role in the transduction of the RTK signaling pathways. First, we analyzed its activation state in control and iPAH PA-SMCs in both basal conditions and following exposure to exogenous EGF, FGF2, and PDGF in presence or in absence of 1μM gefitinib, dovitinib, imatinib or Src Inhibitor-1, respectively. Phospho-p130Cas levels were markedly increased by stimulation with any of these three different growth factors for 15 minutes (Supplemental Fig. S4); also, the phospho-p130Cas levels were even higher in iPAH PA-SMCs than in control cells (Fig. 5A). Pre-treatment of control and iPAH PA-SMCs with either TKIs or with the Src Inhibitor-1 for 30 minutes before exposure to either exogenous growth factor substantially attenuates the increase in p130cas activation (Fig. 5A–B).
Figure 5. EGF, FGF2 and PDGF Signaling Pathways Converge to p130Cas Which Acts as an Amplifier of Downstream Signals.


(A) Representative Western blots and quantifications of the phospho-p130Cas / p130Cas and the phospho-ERK1/2/ ERK1 ratios in iPAH and control PA-SMCs in presence or in absence of EGF, FGF2 or PDGF (10ng/ml) for 15 minutes with or without a 30 minutes pretreatment with 1μM of gefitinib, dovitinib or imatinib. (B) Representative Western blots and quantifications of the phospho-p130Cas / p130Cas and the phospho-ERK1/2/ ERK1 ratios in control PA-SMCs in presence or in absence of the above mentioned growth factors for 15 minutes with or without a 30 minutes pretreatment with Src Inhibitor-1 (10−6M). (C) Representative Western blots and quantifications of the phospho-ERK1/2/ ERK1 ratio in control PA-SMCs with or without p130Cas inhibition by RNA-interference and in presence or in absence of growth factors for 15 minutes. Values are means±SEM (n=5).
To investigate whether p130Cas served as an amplifier/ transmitter of downstream signals, we conducted parallel evaluations of the ERK1/2 activation states in control and iPAH PA-SMCs exposed to either EGF, FGF2 or PDGF in absence or in presence of either TKIs, Src inhibitor-1, p130cas-siRNA or scrambled sequence. A marked increase in levels of phospho-ERK1/2 was observed following exposure to either growth factor (Fig. 5A). Inhibition of EGF-R, FGF-R, PDGF-R and Src respectively resulted in a substantial reduction in the ERK1/2 activation state in both control and iPAH PA-SMCs (Fig. 5A–B). In addition, decreasing p130Cas-signaling by RNA-interference significantly attenuate the increased levels of phospho-ERK1/2 observed following exposure to exogenous EGF, FGF2 and PDGF (Fig. 5C). Compared with PA-SMCs transfected with p130Cas-siRNA under basal conditions, treatments with these growth factors offered significant increase in levels of phospho-ERK1/2, strongly supporting that p130Cas is an amplifier of RTKs downstream signals.
Adapter p130Cas Protein and Activity Are Increased in Rat Lungs With Monocrotaline-induced PH and in Mouse Lungs With Chronic Hypoxia-induced PH
We next examined the protein expression pattern of p130Cas and phospho-p130Cas in lungs from rodent models of PH. Rats receiving a single subcutaneous injection of monocrotaline developed PH within 15 days and it further aggravated until 30 days as reflected by the significant increase in mPAP, RVH and pulmonary arterial muscularization (Supplemental Fig. S5A–B). Consistent with our findings in human lungs, we showed that p130Cas and phospho-p130Cas are substantially increased in rat lungs with monocrotaline-induced PH as well as in mouse lungs with chronic hypoxia-induced PH (Fig. 6A–B and Supplemental Fig. S6). In addition, we showed that p130Cas is also substantially increased in neointimal pulmonary vascular occlusive lesions in a pneumonectomy with monocrotaline-PH model (Supplemental Fig. S7).
Figure 6. Adapter p130Cas Protein and Activity Are Increased in Rat Lungs With Monocrotaline-induced PH and in Mouse Lungs With Chronic Hypoxia-induced PH.

Representative Western blots and quantifications of the p130Cas/ β-actin and the phospho-p130Cas/ p130Cas ratios and representative images of p130cas immunostaining (A) in lungs from monocrotaline-injected rats and controls (B) and in lungs from mice exposed to chronic hypoxia or to room air. Values are means±SEM (n=5). Scale bar = 50μm in all sections.
Efficacy of Gefitinib, Dovitinib and Imatinib Treatment on the Progression of Monocrotaline-induced PH and on the Abnormal Increase of p130Cas Activity
Daily treatment with each of the three clinically approved TKIs started 2 weeks after a subcutaneous monocrotaline injection substantially attenuate the abnormal increase in p130cas activation and regress established PH (Fig. 7A–C). In rats treated with vehicle from day-15 to day-30, a marked increase in mPAP, RVH and numbers of muscularized distal pulmonary arteries were found compared with controls. In contrast, when rats were given gefitinib, dovitinib or imatinib and compared to vehicle-treated rats, these hemodynamic parameters as well as the pulmonary vascular remodeling were substantially attenuate. Day-30 mPAP values, RVH and muscularization of pulmonary arteries in imatinib-treated rats were similar to those in controls.
Figure 7. Evaluation of the Efficacy of Gefitinib, Dovitinib and Imatinib Treatment on the Progression of Monocrotaline-induced PH and on the Abnormal Increase of p130Cas Protein and Activity.


(A) RVSP, mPAP, RVH and numbers of muscularized pulmonary arteries. (B) Representative Western blots and quantification of the phospho-p130Cas/ p130Cas ratio and the phospho-ERK1/2/ ERK1 ratio. Values are means±SEM (n=5). (C) Representative images of hematoxylin-eosin (H&E) staining, alpha-smooth muscle actin (αSM-actin), proliferating cell nuclear antigen (PCNA) and CD-68 immunostainings of monocrotaline-injected rat lungs treated or not with gefitinib, dovitinib, imatinib or vehicle. Scale bar = 50μm in all sections.
To investigate the efficacy of TKI treatments on the abnormal increase of p130Cas protein and activity, we conducted parallel evaluations of the p130Cas activation states and found that lung levels of p130Cas and phospho-p130Cas were markedly attenuate when compared to vehicle-treated rats (Fig. 7B). On day-30, the number of PCNA-positive cells (Fig. 7C) was significantly lower in pulmonary-artery walls from either TKI-treated rats than in walls from vehicle-treated rats and we found that these changes were accompanied by a marked reduction in the phosphoERK1/2:ERK1 ratio. There were no significant differences between rats given vehicle or TKIs regarding body weight, systemic blood pressure, or heart rate.
Effect of Gefitinib, Dovitinib and Imatinib on Human P-EC and PA-SMC Proliferation
EGF, FGF2 and PDGF significantly increased proliferation of cultured human PA-SMCs and P-ECs, however TKIs significantly inhibited cell proliferation. Pretreatment with gefitinib, dovitinib or imatinib (30 minutes) dose-dependently reduced the growth of iPAH and control PA-SMCs treated with EGF, FGF2 or PDGF, respectively (Fig. 8A). These growth-stimulating effects are even more reduced by TKIs in iPAH PA-SMCs than in control PA-SMCs. A similar pattern of inhibition was observed in P-ECs treated with either growth factor (Fig. 8B).
Figure 8. Effect of Gefitinib, Dovitinib and Imatinib on Human PA-SMC and P-ECs Proliferation From Patients With iPAH and Controls.

Proliferation of control- and iPAH (A) PA-SMCs and (B) P-ECs in presence or in absence of fetal calf serum (FCS), EGF, FGF2 or PDGF (10ng/ml) with or without a 30 minutes pretreatment with 1μM of gefitinib, dovitinib or imatinib (24 h). Values are means±SEM (n=5).
DISCUSSION
Migration and proliferation of ECs and SMCs constitute an essential part of the pulmonary vascular remodeling seen in PAH. Herein, we reported evidence that p130Cas and its signaling plays a critical role in the PAH pathogenesis by modulating pulmonary vascular cell migration, proliferation and by acting as an amplifier of RTKs downstream signals. We showed that the adapter p130Cas protein and activity are increased in the serum and in walls of distal pulmonary arteries and in cultured P-ECs and PA-SMCs from iPAH patients. We obtained evidence for convergence and amplification of the growth-stimulating effect of EGF, FGF2 and PDGF signaling pathways via p130Cas signaling pathway. In addition, we showed that the EGF-R inhibitor gefitinib, the FGF-R inhibitor dovitinib and the PDGF-R inhibitor imatinib dose-dependently reduced the growth-stimulating effect of EGF, FGF2 or PDGF and substantially attenuate the increase in p130cas activation. Finally, we found that p130Cas protein and activity are increased in rat lungs with monocrotaline-induced PH as well as in mouse lungs with chronic hypoxia-induced PH; daily treatment with either TKI started 2 weeks after a subcutaneous monocrotaline injection substantially attenuate the abnormal increase in p130cas activation and regress established PH, as judged on mPAP, RVH, and pulmonary arterial muscularization.
Tyrosine phosphorylation events mediated by aberrant activation of EGF-R, FGF-R and PDGF-R pathways have been proven to be involved in the development of several diseases including cancer and PAH (4–8, 23, 24). Inhibition of these specific RTK signaling decreases the abnormal proliferation and migration of pulmonary SMCs and ECs in human and experimental PH and may represent an attractive treatment for PAH. After promising phase I/II results, imatinib is moving into a phase III trial in PAH (25). All these elements support the idea that a better understanding of these signaling pathways in the pulmonary vasculature will help us understand the mechanism of pulmonary vascular remodeling and provide new or more powerful therapeutic tools/ targets for PAH.
RTK activity in resting and normal cells is tightly controlled. Upon ligand binding and activation, the intracellular catalytic domain of RTKs catalyzes receptor autophosphorylation of cytoplasmic tyrosine residues serving as docking sites for SH2- and phosphotyrosine-binding-containing molecules. Assembly of activated protein complexes triggers signaling cascades that can activate or repress genes involved in cell proliferation, migration and survival. Among these key intracellular transducers of these RTKs signals, p130Cas is a central multi-domain docking protein, with an N-terminal SH3 domain, a central “substrate domain” consisting of multiple SH2-domain binding motifs (Tyr-x-x-Pro), and a “Src-binding domain” near the C-terminus (26, 27). p130Cas is regulated in part by a phosphorylation (active form)/ dephosphorylation mechanism. The tyrosine phosphorylation of the p130Cas substrate domain is due mostly to the activity of Src and focal adhesion kinase (FAK) (28). However, little is known regarding the role of p130Cas in the aberrant activation of RTK pathways in PAH for which there is a need for development of effective targeted therapies. We found a marked increase in p130Cas protein levels in tissue and serum of iPAH patients. Although the mechanism(s) that lead to abundance of the protein in serum is not fully understood, microparticles may partly contribute to this abnormality in iPAH patients. PH patients exhibit higher circulating levels of microparticles compared to control subjects (29) and we have shown that pulmonary vascular cells exhibited high levels of p130cas. Therefore, we can speculate that the abnormal high release of these plasma membrane vesicle fragments from pulmonary vascular cells may partly contribute to this abnormal increase in levels of circulating p130Cas protein in iPAH patients. Since circulating p130Cas protein levels are not elevated in asthmatic patients without a systemic corticosteroid therapy, this abnormal high circulating p130Cas protein level may not be related to chronic inflammation. We cannot also exclude that hemodynamic forces (shear stress) could also represent another mechanisms responsible for these effects. Moreover, elevated expression of p130Cas in tissue as well as in serum of patients with either non-small-cell lung cancer or pulmonary tuberculosis has been recently reported by Bo Deng et al. (30). These in situ observations were confirmed in vitro, with cultured human PA-SMCs and P-ECs exhibiting increased expression and activity of p130Cas, providing evidence that p130Cas over-expression/ over-activity in iPAH pulmonary vascular cells is an intrinsic abnormality of these cells. The tissue sections and cells we studied were from patients with end-stage PAH receiving drug therapy, so we cannot exclude the possibility that these abnormalities represent a compensatory mechanism and are not associated with early stage vascular remodeling in PAH. Although determinations of serum p130Cas levels were also performed in less severe PAH patients, at this time we can only conclude that these abnormalities in p130Cas signaling are present at least throughout the last stages of the disease aggravation.
Over-abundance and over-activity of p130Cas signaling may contribute at multiple levels to pulmonary vascular remodeling seen in PAH. First, p130Cas signaling is known to modulate cellular proliferation. p130Cas is over-produced in many human cancers, and p130Cas is required for KRAS, BRAF, PTEN and PIK3CA oncogene-dependent proliferation (31) as well as for transformation by several oncogenes, such as Src, ERBB2, and the nucleophosmin-anaplastic lymphoma kinase (32). It has been reported that p130Cas confers resistance to anticancer drugs such as doxorubicin and tamoxifen via activation of signaling pathways mediated by various kinases, including Src, EGF-R, protein kinase B (AKT) and ERK1/2 (33, 34). In breast cancer patients, elevated p130Cas levels are associated with a decreased response to tamoxifen therapy, early disease recurrence, and lower long-term survival (35). The tamoxifen resistance conferred by p130Cas has been linked to Src-driven cell proliferation and survival pathways mediated either in complex with the estrogen receptor to promote ERK signaling and cyclin D1 induction or to an estrogen receptor-independent manner involving EGF-R and Stat5b (9, 36–38). c-Src is abundant in vascular tissue (39) and Courboulin et al. (40) showed a basal over-activation of the Src–STAT3 pathway in iPAH PA-SMCs, supporting the hypothesis that p130Cas signaling may be abnormally activated, thereby indirectly promoting cell growth and migration. Here, we showed that siRNA mediated knockdown of p130Cas or its activation by Src significantly diminished the proliferative potential of iPAH cells. In addition, we found that the growth-stimulating effect of EGF, FGF2 or PDGF is also substantially reduced in both cell types with more pronounced effects in iPAH cells. Second, a role for p130Cas signaling in migration of fibroblasts has been well documented. Impaired migration capacity of p130Cas-null mouse embryo fibroblasts can be restored by ectopic expression of wild-type p130Cas but not by mutants lacking the sites of tyrosine phosphorylation in the substrate domain (41, 42). We found that siRNA mediated knockdown of p130Cas significantly reduced the migratory potential of iPAH PA-SMCs and P-ECs. Third, we demonstrated that p130Cas acts as an amplifier of EGF-R, FGF-R and PDGF-R downstream signals. In control and iPAH PA-SMCs, we found a rapid and sustained increase in the ERK1/2 and p130Cas activation states following exposure to exogenous EGF, FGF2 and PDGF. Decreasing p130Cas-signaling by RNA-interference or by Src inhibitor-1 significantly attenuates growth factor-induced ERK activation. However, treatments of PA-SMCs transfected with p130Cas siRNA with either growth factor still offered significant increase in levels of phospho-ERK1/2, supporting the notion that p130Cas is an amplifier of RTKs downstream signals. Signaling via the p130Cas/Crk complex also promotes cell survival via activation of ERK and Rac pathways (43, 44), and it has been reported that uncoupling of p130Cas and Crk is required for c-Abl-mediated apoptosis to take place (45).
Altered bone morphogenetic protein (BMP)/TGF-β signaling is now recognized as a major factor in the PAH pathogenesis (46). Interestingly, a previous study from Kim et al. (47) demonstrates that p130Cas is also a negative modulator of the TGF-β signaling pathway by binding to smad3 and preventing its phosphorylation by the TGFβ receptor. In mammary epithelial cells, p130cas overexpression shifts TGFβ signaling from smad2 and smad3 phosphorylation to p38 MAPK activation, rendering these cells resistant to TGFβ-induced growth arrest and enhancing their metastatic potential (48). Since relationships exist between BMP and TGF-β signaling (49), further studies are therefore requested to better understand the importance of p130cas in this complex interplay.
Although the vascular changes that occur in animal models of PH do not reflect the complex pulmonary vascular lesions seen in PAH patients, our results support a pathophysiological role for p130Cas in PAH. First, we demonstrate an increase in p130Cas expression and activity within walls of remodeled pulmonary arteries of either monocrotaline- or pneumonectomy with monocrotaline-injected rats and chronically hypoxic mice. Second, we found that daily treatment with imatinib, gefitinib, or dovitinib started 2 weeks after a subcutaneous monocrotaline injection substantially attenuate the abnormal increase in p130cas activation and regress established PH. Third, we found that either TKIs in vivo and in vitro not only decreased the p130Cas and ERK1/2 activation states, but also directly inhibit pulmonary vascular cell proliferation. Further investigation with direct manipulations of p130cas in smooth muscle (SMCs) or endothelial cells (ECs) in vivo are important to demonstrate that p130cas is therapeutically targetable in PH. There are no selective p130Cas inhibitors currently available, but these results should stimulate work on the design and synthesis of such inhibitors, and encourage their evaluation in experimental PH.
In summary, our study provides the first evidence that adapter p130Cas protein and activity are abnormally increased in both pulmonary artery walls and cultured pulmonary vascular cells from PAH patients. Although further experiments are required to identify the exact mechanism underlying the over-expression of p130Cas in pulmonary vascular cells, we clearly show the critical involvement of p130Cas in regulating migration and proliferation of these cells. We report strong evidence that p130Cas acts as a nodal signaling platform on which several RTK signaling pathways converge, supporting the idea that p130Cas facilitates the intracellular transmission of the signal induced by major RTKs in iPAH.
Scientific knowledge on the subject
Increased proliferation/ migration of pulmonary vascular cells are hallmark pathogenic features of pulmonary arterial hypertension (PAH). The mechanisms underlying these abnormalities, however, remain partially unknown.
Recent discovery of selective therapies targeting crucial receptor tyrosine kinase (RTK) signaling in idiopathic PAH (iPAH) such as EGF, FGF2 and PDGF has provided encouraging experimental results, and these agents are being tested as a potential therapeutic approach to PAH
p130Cas is an adaptor protein modulating several signaling pathways that controls cell-migration and proliferation.
What this study adds to the field
Adapter p130Cas protein and activity are increased: (1) in serum and walls of pulmonary arteries and in cultured pulmonary endothelial (P-ECs) and smooth muscle cells (PA-SMCs) from iPAH-patients as compared to those from controls; (2) in experimental models of PH in rodents (chronic hypoxia-induced PH in mice; monocrotaline- and pneumonectomy with monocrotaline-induced PH in rats).
EGF, FGF2 and PDGF signaling pathways converge to p130Cas which acts as an amplifier of downstream signals
Decreasing p130Cas-signaling by RNA-interference attenuate the increase in ERK1/2 activation state and normalized migration and proliferation of PA-SMCs and P-ECs derived from iPAH patients.
These findings offer new insight into the importance of p130Cas in PAH for proliferation and migration of pulmonary vascular cells, and for the intracellular transmission of the signal induced by the major RTKs. We obtained evidence for convergence and amplification of the growth-stimulating effect of EGF, FGF2 and PDGF signaling pathways via p130Cas signaling pathway. There are no selective p130Cas inhibitors currently available, but these results should stimulate work on the design and synthesis of such inhibitors, and encourage their evaluation in experimental models of PH.
Supplementary Material
Supplemental Fig. S1: Serum p130Cas Protein Levels in Asthmatic Patients versus Controls. Values are means±SEM.
Supplemental Fig. S2: Suppression of p130Cas Protein By RNA-interference 72 Hours After Transfection of Human PA-SMCs and PECs. Representative Western immunoblots of p130Cas and β-actin loading controls and quantification of the p130Cas/ β-actin ratio. Values are means±SEM (n=5).
Supplemental Fig. S3: Proliferation of PA-SMCs and P-ECs assessed by Cell Count. Proliferative potential of PA-SMCs and P-ECs from patients with iPAH and controls in presence or absence of EGF, FGF2 and PDGF (10ng/ml; 48 hours). Values are means±SEM (n=5).
Supplemental Fig. S4: Time Course of p130Cas Activation. Representative Western blots of phospho-p130Cas and β-actin loading controls in PA-SMCs in presence or in absence of EGF, FGF2 and PDGF (10 ng/ml).
Supplemental Fig. S5: Pulmonary Hemodynamic and Vascular Remodeling in Rat Lungs With Monocrotaline-induced PH. RVSP, mPAP, RVH and percentage of muscularized pulmonary arteries (A) at day-15 and (B) day-30 in monocrotaline-injected rats and controls. Values are means±SEM (n=5).
Supplemental Fig. S6: Pulmonary Hemodynamic and Vascular Remodeling in Mouse Lungs With Chronic Hypoxia-induced PH. RVSP, RVH and percentage of muscularized pulmonary arteries in mice exposed to hypoxia (10% fiO2) or kept in room air for 3 weeks. Values are means±SEM (n=5).
Supplemental Fig. S7: p130Cas expression in the pneumonectomy with monocrotaline-induced PH in rats. Representative images of Von Willebrand factor (vWF), α-smooth muscle actin and p130Cas immunostaining in serial sections of distal pulmonary arteries from pneumonectomy-monocrotaline-injected rats:. Scale bar= 50μm.
Table 1. Characteristics Of Controls And Patients With PAH For Determination Of Serum p130cas Protein Levels:
Table 2. Characteristics Of Controls And Patients With iPAH Before Lung Transplantation:
Acknowledgments
Funding footnote: This research was supported by grants from the French National Institute for Health and Medical Research (INSERM), the French National Agency for Research: grant no P007948 ANR-GENOPATH “ATOL”, and the Legs Poix (Chancellerie des Universités de Paris). F.S.dM. (MC 1120-2009) is the recipient of a European Respiratory Society/ Marie-Curie Joint Research Fellowship
We thank Dr. Andrei Seferian and Mrs. Carole Phan for their valuable help in the revision of the manuscript as well as Dr. Elodie Gouadon for her very helpful technical assistance with use of the Confocal Microscope.
Footnotes
Authors’ contributions: Conception and design: LT, F.S.dM, DM, MH, CG; Analysis and interpretation: all; Drafting manuscript: LT, MH and CG.
“This article has an online data supplement, which is accessible from this issue’s table of content online at www.atsjournals.org”
Conflict of interest
None.
Disclosures
None.
References
- 1.Humbert M, Sitbon O, Chaouat A, Bertocchi M, Habib G, Gressin V, Yaici A, Weitzenblum E, Cordier JF, Chabot F, Dromer C, Pison C, Reynaud-Gaubert M, Haloun A, Laurent M, Hachulla E, Cottin V, Degano B, Jais X, Montani D, Souza R, Simonneau G. Survival in patients with idiopathic, familial, and anorexigen-associated pulmonary arterial hypertension in the modern management era. Circulation. 2010;122:156–163. doi: 10.1161/CIRCULATIONAHA.109.911818. [DOI] [PubMed] [Google Scholar]
- 2.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, Parent F, Savale L, Natali D, Gunther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36:549–555. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 3.Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:558–564. doi: 10.1164/rccm.200709-1369PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Izikki M, Guignabert C, Fadel E, Humbert M, Tu L, Zadigue P, Dartevelle P, Simonneau G, Adnot S, Maitre B, Raffestin B, Eddahibi S. Endothelial-derived fgf2 contributes to the progression of pulmonary hypertension in humans and rodents. J Clin Invest. 2009;119:512–523. doi: 10.1172/JCI35070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112:423–431. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 6.Perros F, Montani D, Dorfmuller P, Durand-Gasselin I, Tcherakian C, Le Pavec J, Mazmanian M, Fadel E, Mussot S, Mercier O, Herve P, Emilie D, Eddahibi S, Simonneau G, Souza R, Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 7.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, Sydykov A, Lai YJ, Weissmann N, Seeger W, Grimminger F. Reversal of experimental pulmonary hypertension by pdgf inhibition. J Clin Invest. 2005;115:2811–2821. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tu L, Dewachter L, Gore B, Fadel E, Dartevelle P, Simonneau G, Humbert M, Eddahibi S, Guignabert C. Autocrine fibroblast growth factor-2 signaling contributes to altered endothelial phenotype in pulmonary hypertension. Am J Respir Cell Mol Biol. 2011;45:311–322. doi: 10.1165/rcmb.2010-0317OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cabodi S, Tinnirello A, Di Stefano P, Bisaro B, Ambrosino E, Castellano I, Sapino A, Arisio R, Cavallo F, Forni G, Glukhova M, Silengo L, Altruda F, Turco E, Tarone G, Defilippi P. P130cas as a new regulator of mammary epithelial cell proliferation, survival, and her2-neu oncogene-dependent breast tumorigenesis. Cancer Res. 2006;66:4672–4680. doi: 10.1158/0008-5472.CAN-05-2909. [DOI] [PubMed] [Google Scholar]
- 10.Cabodi S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. Integrin signalling adaptors: Not only figurants in the cancer story. Nat Rev Cancer. 2010;10:858–870. doi: 10.1038/nrc2967. [DOI] [PubMed] [Google Scholar]
- 11.Korah R, Choi L, Barrios J, Wieder R. Expression of fgf-2 alters focal adhesion dynamics in migration-restricted mda-mb-231 breast cancer cells. Breast Cancer Res Treat. 2004;88:17–28. doi: 10.1007/s10459-004-6006-2. [DOI] [PubMed] [Google Scholar]
- 12.Ojaniemi M, Vuori K. Epidermal growth factor modulates tyrosine phosphorylation of p130cas. Involvement of phosphatidylinositol 3′-kinase and actin cytoskeleton. J Biol Chem. 1997;272:25993–25998. doi: 10.1074/jbc.272.41.25993. [DOI] [PubMed] [Google Scholar]
- 13.Pellet-Many C, Frankel P, Evans IM, Herzog B, Junemann-Ramirez M, Zachary IC. Neuropilin-1 mediates pdgf stimulation of vascular smooth muscle cell migration and signalling via p130cas. Biochem J. 2011;435:609–618. doi: 10.1042/BJ20100580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guignabert C, Raffestin B, Benferhat R, Raoul W, Zadigue P, Rideau D, Hamon M, Adnot S, Eddahibi S. Serotonin transporter inhibition prevents and reverses monocrotaline-induced pulmonary hypertension in rats. Circulation. 2005;111:2812–2819. doi: 10.1161/CIRCULATIONAHA.104.524926. [DOI] [PubMed] [Google Scholar]
- 15.El-Bizri N, Guignabert C, Wang L, Cheng A, Stankunas K, Chang CP, Mishina Y, Rabinovitch M. Sm22alpha-targeted deletion of bone morphogenetic protein receptor 1a in mice impairs cardiac and vascular development, and influences organogenesis. Development. 2008;135:2981–2991. doi: 10.1242/dev.017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guignabert C, Tu L, Izikki M, Dewachter L, Zadigue P, Humbert M, Adnot S, Fadel E, Eddahibi S. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with sm22alpha-targeted overexpression of the serotonin transporter. Faseb J. 2009;23:4135–4147. doi: 10.1096/fj.09-131664. [DOI] [PubMed] [Google Scholar]
- 17.Dahal BK, Cornitescu T, Tretyn A, Pullamsetti SS, Kosanovic D, Dumitrascu R, Ghofrani HA, Weissmann N, Voswinckel R, Banat GA, Seeger W, Grimminger F, Schermuly RT. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am J Respir Crit Care Med. 181:158–167. doi: 10.1164/rccm.200811-1682OC. [DOI] [PubMed] [Google Scholar]
- 18.Azab AK, Azab F, Quang P, Maiso P, Sacco A, Ngo HT, Liu Y, Zhang Y, Morgan BL, Roccaro AM, Ghobrial IM. Fgfr3 is overexpressed waldenstrom macroglobulinemia and its inhibition by dovitinib induces apoptosis and overcomes stroma-induced proliferation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:4389–4399. doi: 10.1158/1078-0432.CCR-10-2772. [DOI] [PubMed] [Google Scholar]
- 19.Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, Rabinovitch M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes pdgf receptor-beta-dependent pulmonary arterial muscularization. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1082–1090. doi: 10.1152/ajplung.00199.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guignabert C, Izikki M, Tu LI, Li Z, Zadigue P, Barlier-Mur AM, Hanoun N, Rodman D, Hamon M, Adnot S, Eddahibi S. Transgenic mice overexpressing the 5-hydroxytryptamine transporter gene in smooth muscle develop pulmonary hypertension. Circ Res. 2006;98:1323–1330. doi: 10.1161/01.RES.0000222546.45372.a0. [DOI] [PubMed] [Google Scholar]
- 21.Raines EW. Pdgf and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 22.Lepetit H, Eddahibi S, Fadel E, Frisdal E, Munaut C, Noel A, Humbert M, Adnot S, D’Ortho MP, Lafuma C. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Respir J. 2005;25:834–842. doi: 10.1183/09031936.05.00072504. [DOI] [PubMed] [Google Scholar]
- 23.Marmor MD, Skaria KB, Yarden Y. Signal transduction and oncogenesis by erbb/her receptors. International journal of radiation oncology, biology, physics. 2004;58:903–913. doi: 10.1016/j.ijrobp.2003.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Nam NH, Parang K. Current targets for anticancer drug discovery. Current drug targets. 2003;4:159–179. doi: 10.2174/1389450033346966. [DOI] [PubMed] [Google Scholar]
- 25.Ghofrani HA, Morrell NW, Hoeper MM, Olschewski H, Peacock AJ, Barst RJ, Shapiro S, Golpon H, Toshner M, Grimminger F, Pascoe S. Imatinib in pulmonary arterial hypertension patients with inadequate response to established therapy. Am J Respir Crit Care Med. 2010;182:1171–1177. doi: 10.1164/rccm.201001-0123OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Defilippi P, Di Stefano P, Cabodi S. P130cas: A versatile scaffold in signaling networks. Trends in cell biology. 2006;16:257–263. doi: 10.1016/j.tcb.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Sakai R, Iwamatsu A, Hirano N, Ogawa S, Tanaka T, Mano H, Yazaki Y, Hirai H. A novel signaling molecule, p130, forms stable complexes in vivo with v-crk and v-src in a tyrosine phosphorylation-dependent manner. Embo J. 1994;13:3748–3756. doi: 10.1002/j.1460-2075.1994.tb06684.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruest PJ, Shin NY, Polte TR, Zhang X, Hanks SK. Mechanisms of cas substrate domain tyrosine phosphorylation by fak and src. Mol Cell Biol. 2001;21:7641–7652. doi: 10.1128/MCB.21.22.7641-7652.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amabile N, Heiss C, Real WM, Minasi P, McGlothlin D, Rame EJ, Grossman W, De Marco T, Yeghiazarians Y. Circulating endothelial microparticle levels predict hemodynamic severity of pulmonary hypertension. Am J Respir Crit Care Med. 2008;177:1268–1275. doi: 10.1164/rccm.200710-1458OC. [DOI] [PubMed] [Google Scholar]
- 30.Deng B, Huang W, Tan QY, Fan XQ, Jiang YG, Liu L, Zhong YY, Liang YG, Wang RW. Breast cancer anti-estrogen resistance protein 1 (bcar1/p130cas) in pulmonary disease tissue and serum. Molecular diagnosis & therapy. 2011;15:31–40. doi: 10.1007/BF03257191. [DOI] [PubMed] [Google Scholar]
- 31.Pylayeva Y, Gillen KM, Gerald W, Beggs HE, Reichardt LF, Giancotti FG. Ras- and pi3k-dependent breast tumorigenesis in mice and humans requires focal adhesion kinase signaling. J Clin Invest. 2009;119:252–266. doi: 10.1172/JCI37160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cabodi S, Tinnirello A, Bisaro B, Tornillo G, del Pilar Camacho-Leal M, Forni G, Cojoca R, Iezzi M, Amici A, Montani M, Eva A, Di Stefano P, Muthuswamy SK, Tarone G, Turco E, Defilippi P. P130cas is an essential transducer element in erbb2 transformation. Faseb J. 2010;24:3796–3808. doi: 10.1096/fj.10-157347. [DOI] [PubMed] [Google Scholar]
- 33.Ta HQ, Thomas KS, Schrecengost RS, Bouton AH. A novel association between p130cas and resistance to the chemotherapeutic drug adriamycin in human breast cancer cells. Cancer Res. 2008;68:8796–8804. doi: 10.1158/0008-5472.CAN-08-2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van der Flier S, Brinkman A, Look MP, Kok EM, Meijer-van Gelder ME, Klijn JG, Dorssers LC, Foekens JA. Bcar1/p130cas protein and primary breast cancer: Prognosis and response to tamoxifen treatment. Journal of the National Cancer Institute. 2000;92:120–127. doi: 10.1093/jnci/92.2.120. [DOI] [PubMed] [Google Scholar]
- 35.Dorssers LC, Grebenchtchikov N, Brinkman A, Look MP, van Broekhoven SP, de Jong D, Peters HA, Portengen H, Meijer-van Gelder ME, Klijn JG, van Tienoven DT, Geurts-Moespot A, Span PN, Foekens JA, Sweep FC. The prognostic value of bcar1 in patients with primary breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:6194–6202. doi: 10.1158/1078-0432.CCR-04-0444. [DOI] [PubMed] [Google Scholar]
- 36.Cabodi S, Moro L, Baj G, Smeriglio M, Di Stefano P, Gippone S, Surico N, Silengo L, Turco E, Tarone G, Defilippi P. P130cas interacts with estrogen receptor alpha and modulates non-genomic estrogen signaling in breast cancer cells. Journal of cell science. 2004;117:1603–1611. doi: 10.1242/jcs.01025. [DOI] [PubMed] [Google Scholar]
- 37.Dorssers LC, van Agthoven T, Brinkman A, Veldscholte J, Smid M, Dechering KJ. Breast cancer oestrogen independence mediated by bcar1 or bcar3 genes is transmitted through mechanisms distinct from the oestrogen receptor signalling pathway or the epidermal growth factor receptor signalling pathway. Breast cancer research : BCR. 2005;7:R82–92. doi: 10.1186/bcr954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riggins RB, Thomas KS, Ta HQ, Wen J, Davis RJ, Schuh NR, Donelan SS, Owen KA, Gibson MA, Shupnik MA, Silva CM, Parsons SJ, Clarke R, Bouton AH. Physical and functional interactions between cas and c-src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res. 2006;66:7007–7015. doi: 10.1158/0008-5472.CAN-05-3952. [DOI] [PubMed] [Google Scholar]
- 39.Oda Y, Renaux B, Bjorge J, Saifeddine M, Fujita DJ, Hollenberg MD. Csrc is a major cytosolic tyrosine kinase in vascular tissue. Can J Physiol Pharmacol. 1999;77:606–617. [PubMed] [Google Scholar]
- 40.Courboulin A, Paulin R, Giguere NJ, Saksouk N, Perreault T, Meloche J, Paquet ER, Biardel S, Provencher S, Cote J, Simard MJ, Bonnet S. Role for mir-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208:535–548. doi: 10.1084/jem.20101812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Honda H, Oda H, Nakamoto T, Honda Z, Sakai R, Suzuki T, Saito T, Nakamura K, Nakao K, Ishikawa T, Katsuki M, Yazaki Y, Hirai H. Cardiovascular anomaly, impaired actin bundling and resistance to src-induced transformation in mice lacking p130cas. Nat Genet. 1998;19:361–365. doi: 10.1038/1246. [DOI] [PubMed] [Google Scholar]
- 42.Shin NY, Dise RS, Schneider-Mergener J, Ritchie MD, Kilkenny DM, Hanks SK. Subsets of the major tyrosine phosphorylation sites in crk-associated substrate (cas) are sufficient to promote cell migration. J Biol Chem. 2004;279:38331–38337. doi: 10.1074/jbc.M404675200. [DOI] [PubMed] [Google Scholar]
- 43.Cho SY, Klemke RL. Extracellular-regulated kinase activation and cas/crk coupling regulate cell migration and suppress apoptosis during invasion of the extracellular matrix. The Journal of cell biology. 2000;149:223–236. doi: 10.1083/jcb.149.1.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chodniewicz D, Klemke RL. Regulation of integrin-mediated cellular responses through assembly of a cas/crk scaffold. Biochimica et biophysica acta. 2004;1692:63–76. doi: 10.1016/j.bbamcr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 45.Holcomb M, Rufini A, Barila D, Klemke RL. Deregulation of proteasome function induces abl-mediated cell death by uncoupling p130cas and c-crkii. J Biol Chem. 2006;281:2430–2440. doi: 10.1074/jbc.M508454200. [DOI] [PubMed] [Google Scholar]
- 46.Hassoun PM, Mouthon L, Barbera JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, Newman JH, Rabinovitch M, Schermuly R, Stenmark KR, Voelkel NF, Yuan JX, Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. Journal of the American College of Cardiology. 2009;54:S10–19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 47.Kim W, Seok Kang Y, Soo Kim J, Shin NY, Hanks SK, Song WK. The integrin-coupled signaling adaptor p130cas suppresses smad3 function in transforming growth factor-beta signaling. Molecular biology of the cell. 2008;19:2135–2146. doi: 10.1091/mbc.E07-10-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wendt MK, Smith JA, Schiemann WP. P130cas is required for mammary tumor growth and transforming growth factor-beta-mediated metastasis through regulation of smad2/3 activity. J Biol Chem. 2009;284:34145–34156. doi: 10.1074/jbc.M109.023614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nasim MT, Ogo T, Chowdhury HM, Zhao L, Chen CN, Rhodes C, Trembath RC. Bmpr-ii deficiency elicits pro-proliferative and anti-apoptotic responses through the activation of tgfbeta-tak1-mapk pathways in pah. Human molecular genetics. 2012;21:2548–2558. doi: 10.1093/hmg/dds073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. S1: Serum p130Cas Protein Levels in Asthmatic Patients versus Controls. Values are means±SEM.
Supplemental Fig. S2: Suppression of p130Cas Protein By RNA-interference 72 Hours After Transfection of Human PA-SMCs and PECs. Representative Western immunoblots of p130Cas and β-actin loading controls and quantification of the p130Cas/ β-actin ratio. Values are means±SEM (n=5).
Supplemental Fig. S3: Proliferation of PA-SMCs and P-ECs assessed by Cell Count. Proliferative potential of PA-SMCs and P-ECs from patients with iPAH and controls in presence or absence of EGF, FGF2 and PDGF (10ng/ml; 48 hours). Values are means±SEM (n=5).
Supplemental Fig. S4: Time Course of p130Cas Activation. Representative Western blots of phospho-p130Cas and β-actin loading controls in PA-SMCs in presence or in absence of EGF, FGF2 and PDGF (10 ng/ml).
Supplemental Fig. S5: Pulmonary Hemodynamic and Vascular Remodeling in Rat Lungs With Monocrotaline-induced PH. RVSP, mPAP, RVH and percentage of muscularized pulmonary arteries (A) at day-15 and (B) day-30 in monocrotaline-injected rats and controls. Values are means±SEM (n=5).
Supplemental Fig. S6: Pulmonary Hemodynamic and Vascular Remodeling in Mouse Lungs With Chronic Hypoxia-induced PH. RVSP, RVH and percentage of muscularized pulmonary arteries in mice exposed to hypoxia (10% fiO2) or kept in room air for 3 weeks. Values are means±SEM (n=5).
Supplemental Fig. S7: p130Cas expression in the pneumonectomy with monocrotaline-induced PH in rats. Representative images of Von Willebrand factor (vWF), α-smooth muscle actin and p130Cas immunostaining in serial sections of distal pulmonary arteries from pneumonectomy-monocrotaline-injected rats:. Scale bar= 50μm.
Table 1. Characteristics Of Controls And Patients With PAH For Determination Of Serum p130cas Protein Levels:
Table 2. Characteristics Of Controls And Patients With iPAH Before Lung Transplantation: