

Summary
Pro-neural transcription factors and small molecules can induce the reprogramming of fibroblasts into functional neurons; however, the immediate-early molecular events that catalyze this conversion have not been well defined. We previously demonstrated that neurogenin 2 (NEUROG2), forskolin (F), and dorsomorphin (D) can reprogram fibroblasts into functional neurons with high efficiency. Here, we used this model to define the genetic and epigenetic events that initiate an acquisition of neuronal identity. We demonstrate that NEUROG2 is a pioneer factor, FD enhances chromatin accessibility and H3K27 acetylation, and synergistic transcription activated by these factors is essential to successful reprogramming. CREB1 promotes neuron survival and acts with NEUROG2 to upregulate SOX4, which co-activates NEUROD1 and NEUROD4. In addition, SOX4 targets SWI/SNF subunits and SOX4 knockdown results in extensive loss of open chromatin and abolishes reprogramming. Applying these insights, adult human glioblastoma cell and skin fibroblast reprogramming can be improved using SOX4 or chromatin-modifying chemicals.
Keywords: NEUROG2, SOX4, SWI/SNF, CREB1, small molecules, reprogramming, transdifferentiation, ATAC-seq, ChIP-seq, RNA-seq
Graphical Abstract
Highlights
-
•
NEUROG2 acts as a pioneer factor to drive neuronal reprogramming
-
•
ATAC-, ChIP-, and RNA-seq profiling reveals genome-wide mechanisms for reprogramming
-
•
SOX4 is a critical mediator of chromatin remodeling during reprogramming
-
•
SOX4 or FK228 can enhance adult human glioblastoma and skin fibroblast reprogramming
In this article, Zhang and colleagues show that NEUROG2 and two small molecules activate transcription and chromatin remodeling programs that drive fibroblast-to-neuron reprogramming in diverse human cell types. Strikingly, the transcription factor SOX4 mediates genome-wide chromatin remodeling processes critical to efficient conversion, while BCL2L1 and the chemical FK228 can improve reprogramming of adult human cell types.
Introduction
Fibroblast-to-neuron reprogramming has been demonstrated using transcription factors (Vierbuchen et al., 2010, Pang et al., 2011) and signaling molecules (Hu et al., 2015, Li et al., 2015). These factors jump-start transcriptional programs that redefine cellular identity; however, the potency of each reprogramming factor is cell age and lineage dependent (Liu et al., 2013, Masserdotti et al., 2015, Mertens et al., 2015). Therefore, mechanistically, the success of neuronal reprogramming depends upon the cell-of-origin genetic and epigenetic environment, as well as the ability of reprogramming factors to navigate that environment and induce reprogramming. For instance, the pioneer activity of ASCL1 and the permissible chromatin structures of mouse embryonic fibroblasts facilitate rapid neuronal reprogramming (Wapinski et al., 2013). Likewise, direct competition for pro-neural genetic elements between endogenous repressor complexes and overexpressed neurogenin 2 (NEUROG2) regulates reprogramming of mouse astroglia in short versus prolonged culture (Masserdotti et al., 2015).
NEUROG2 is a basic helix-loop-helix transcription factor that promotes early neurogenesis (Ma et al., 1996, Fode et al., 1998, Scardigli et al., 2001). NEUROG2 overexpression in mouse embryonic stem cells and cultured mouse cortical astroglia catalyzes conversion into functional, synapse-forming glutamatergic neurons (Berninger et al., 2007, Heinrich et al., 2010, Thoma et al., 2012). While NEUROG2 is sufficient to induce reprogramming in neural lineage cells, this factor is unable to independently reprogram somatic fibroblasts (Liu et al., 2013, Chanda et al., 2014). The high-efficiency reprogramming of human fetal fibroblasts (MRC-5) into functional cholinergic neurons by NEUROG2 requires simultaneous exposure to the small molecules forskolin, a cyclic AMP (cAMP) synthesis activator, and dorsomorphin, an inhibitor of AMP-activated protein kinase and bone morphogenetic protein type 1 receptors (Liu et al., 2013). Furthermore, the inclusion of SOX11 is required to rapidly convert adult human skin fibroblasts into neurons (Liu et al., 2013). These findings demonstrate that age- and lineage-specific genetic and epigenetic factors directly affect the ability of NEUROG2 to catalyze reprogramming.
Here, we perform a systematic analysis of the individual and synergistic actions of NEUROG2 and small molecules during reprogramming. We define critical genetic interactions, hierarchical transcription programs, epigenetic histone signatures, and open chromatin conformations that drive the immediate-early stage of reprogramming, then apply these discoveries to adult human skin and glioblastoma cells refractory to reprogramming.
Results
Small Molecules Synergize with NEUROG2 to Enhance Chromatin Occupancy and Redefine the Transcriptional Profile of Fibroblasts
MRC-5 fibroblasts transduced with NEUROG2-encoding lentivirus exhibit limited capacity for neuronal reprogramming; however, exposure to forskolin and dorsomorphin (FD) triggers adoption of class III β-tubulin (TUBB3)+ and microtubule-associated protein 2 (MAP2)+ neuronal identity with greater than 90% efficiency in 14 days (Liu et al., 2013). To investigate the genetic mechanisms that catalyze reprogramming, we performed chromatin immunoprecipitation-sequencing (ChIP-seq) and RNA-sequencing (RNA-seq) analyses at 0.5, 1, and 2 days post FD treatment (DPT) (Figure 1A).
Figure 1.

Small Molecules Enhance NEUROG2 Chromatin Occupancy and Activate Synergistic Transcription
(A) Stepwise timeline for fibroblast-to-neuron reprogramming with ATAC-seq, ChIP, ChIP-seq, and RNA-seq experiments annotated. DPI, days post infection; DPT, days post treatment with FD.
(B) Diagram representing the effects of FD exposure on NEUROG2 peak score for binding events detected at 0.5, 1, and 2 DPT. The change in peak intensity is represented as a ratio of the peak score in the presence versus absence of FD with the Venn diagram depicting the number of NFD-unique events at 0.5, 1, and 2 DPT (biological replicates, n = 3 for each time point and condition).
(C) Hierarchical clustering and heatmap of changes in transcription detected by RNA-seq relative to control fibroblasts. FD, fibroblasts exposed to FD; N, NEUROG2-transduced fibroblasts; NFD, NEUROG2-transduced fibroblasts exposed to FD (biological replicates, n = 3 for each time point and condition).
(D) Pie diagrams representing the respective number of genes induced by NEUROG2, FD, and NEUROG2-FD synergistic action at 2 DPT. Synergy represents genes only induced in the NFD condition.
(E) Time-course representation of the change in transcript expression intensity for genes regulated by FD, NEUROG2, or NFD synergy.
(F) Sunburst diagram depicting the upregulated and downregulated genes at each time point, the intensity of expression change at 2 DPT, and corresponding NEUROG2 ChIP-seq annotation data. Each segment represents one collection of time points. For example, the section with pink, red, and deep-red segments represents genes with significant differential expression at all time points, while the section with only a pink segment represents genes differentially expressed only at 0.5 DPT. The gray outer gradient represents the expression change observed for the genes in each section. NEUROG2 binding events are annotated with a blue line segment.
A ChIP-seq time-course analysis was performed to define the ordered genomic targets of NEUROG2 and identify whether FD significantly modify occupancy at these sites. NEUROG2 was immunoprecipitated from MRC-5 in both the absence and presence of FD at 0.5, 1, and 2 DPT. Replicates exhibited strong enrichment of pro-neural genetic targets, as well as the enhancer box motif (5′-CANNTG-3′). A condition-dependent comparison of NEUROG2 occupancy profiles revealed that FD both enhance NEUROG2 binding intensity at shared sites and induce binding at novel targets undetected in the absence of FD (Figure 1B). NEUROG2 was detected at up to 4-fold higher levels for both time-stable and dynamic binding events following FD treatment, indicating that FD affect NEUROG2 chromatin affinity or accessibility (Figure S1A).
The distribution of NEUROG2-bound sites is biased toward intergenic and intronic regions (Figure S1B). Although FD modestly enhanced promoter and transcription start site (TSS) occupancy, NEUROG2 seems to dominantly regulate transcription via distal regulatory elements. To define how occupancy at primary and FD-enhanced sites affects the transcription, we isolated RNA from FD-treated, NEUROG2-transduced, and NEUROG2-transduced FD-treated (NFD) fibroblasts at 0.5, 1, and 2 DPT. Sequence reads were normalized to time-point-specific GFP-transduced MRC-5 for differential expression analysis (Table S1).
NEUROG2 immediately induced an upward shift in the expression of neural genes such as DLL3 and HES6. While FD independently activated relatively few neural-specific genes, these small molecules did significantly upregulate a small subset of diverse genes including C6ORF176, C11ORF96, DNER, NR4A1, NR4A2, NRG1, and PPARGC1A (Figure 1C). In combination with NEUROG2, FD induced the expression of hundreds of genes unaltered by individual factors (Figure 1D). A temporal analysis of gene expression underscores the rapid rate of change induced by NFD (Figure 1E). Synergistic regulation accounted for 53% of the total change in gene expression at 2 DPT and the total number of genes exhibiting ≥5-fold change increased from 4% to 10% within the 36-hr window from 0.5 to 2 DPT. The synergistic induction of NEUROD1, NEUROD4, and SOX4, as well as ontological classification of immediately repressed and early-induced gene sets, highlight the functional significance of these targets (Figure S2A). Furthermore, upregulation of the basal forebrain markers NTRK1 and SSTR2 as well as the cholinergic receptor CHRNA3 and vesicular transporter SLC18A3 suggests that these early neurons are initially biased toward a cholinergic fate (Figure S2B).
To define how NEUROG2 binding correlates to the changes detected by RNA-seq, we used proximity-based peak-to-gene annotation to identify differentially expressed genes targeted by NEUROG2 (Figure 1F). These results revealed direct overlap between ChIP-seq and RNA-seq datasets ranging from high-level pan-neuronal genes to single genes such as DLL3 and SOX4. This analysis demonstrates that NEUROG2 is the primary activator of pro-neural gene expression, and FD complement this neurogenic program by enhancing transcription in a NEUROG2-sensitive manner.
Small Molecules Induce Genome-wide Chromatin Remodeling
The relative accessibility of genetic elements is cell lineage dependent and directly regulates the reprogramming potency of transcription factors. To define the binding properties of NEUROG2 in MRC-5 fibroblasts, we used the assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) to generate an atlas of accessible and modified chromatin sites.
Initially, chromatin conformation was probed at locations corresponding to the 1,000 highest score peaks from 2-DPT NEUROG2 ChIP-seq datasets. Enrichment at these sites was evaluated using ATAC-seq datasets collected from MRC-5, NEUROG2-transduced MRC-5, and NFD-treated MRC-5. The majority of these binding events occurred in the absence of ATAC-seq signal, indicating that NEUROG2 functions effectively as a pioneer factor to access closed chromatin (Figure 2A). While only a modest increase in open chromatin is detected upon NEUROG2 expression, FD significantly enhances accessible chromatin at NEUROG2 binding sites and other pro-neural genetic regulatory elements. For instance, NEUROG2 binds within closed chromatin in the INSRR-NTRK1, LRRC10B-SYT7, and GADD45G loci, but a significant increase in relaxed chromatin is observed only in the presence of FD (Figure 2B). This demonstrates a role for FD in modulating accessibility at targets relevant to neuronal reprogramming.
Figure 2.

NEUROG2 Acts as a Pioneer Transcription Factor and Small Molecules Promote Chromatin Accessibility at Pro-neural Genetic Elements
(A) Clustered heatmaps representing open chromatin events detected at NEUROG2 binding sites (2-DPT ChIP-seq, leftmost black) in fibroblasts (MRC-5, black), NEUROG2-transduced fibroblasts (N, blue), and NFD-treated fibroblasts (NFD, rightmost red). Input-normalized tag densities are plotted in log2 scale and centered in a 4-kb window around each peak.
(B) NEUROG2 ChIP-seq (2 DPT) and ATAC-seq tracks for INSRR/NTRK1, LRRC10B/SYT7, and GADD45G genomic loci. Gray shading highlights regions of NEUROG2 pioneer activity or FD-induced chromatin remodeling.
(C) Super-enhancers identified using H3K27AC and 2-DPT NEUROG2 ChIP-seq datasets with a Venn diagram depicting the distribution of super-enhancers in open versus closed chromatin.
(D) Genomic distribution of ATAC-seq peaks newly detected upon NFD treatment is biased toward intergenic and intronic genetic elements.
(E) Gene ontology analysis of open chromatin peaks newly detected following NFD treatment reveals an enrichment of neurogenesis- and differentiation-related classifications. Additionally the enhancer box, CRE half-site, and HMG-box motifs are diversely represented within these sites.
(F) Pairwise scatter plot representing the change in expression of genes annotated to ATAC-seq open chromatin peaks newly detected upon NFD treatment. Upregulated genes (red) were enriched for pro-neuronal identity, while downregulated genes (blue) had primarily non-neural function. Black dots indicate genes without significant changes.
Lineage-determinative transcription factors can bind within localized regions of chromatin containing high densities of histone modifications such as H3K27 acetylation, known as super-enhancers, to regulate gene expression (Hnisz et al., 2013). An analysis of H3K27AC super-enhancers in NFD-treated fibroblasts identified 454 potential H3K27AC super-enhancers containing a NEUROG2 bound site, of which 277 NEUROG2 binding sites were localized to regions with closed chromatin signatures (Figure 2C). While FD promoted an increase in accessibility from 50 to 118 sites, the vast majority of NEUROG2 binding sites in super-enhancers remained localized to regions of transposase-inaccessible chromatin (Figure 2C). As histone acetylation is generally considered a marker of active transcription, these dichotomous acetylation and chromatin accessibility results suggest that these features might not be easily correlated during the earliest stage of reprogramming. This also highlights the functional importance of NEUROG2 pioneer activity as a complement to FD-induced chromatin remodeling.
An analysis of accessible chromatin sites uniquely detected upon NFD treatment revealed that newly open chromatin was primarily localized to intergenic and intronic regions (Figure 2D). A functional classification of the genes annotated to these newly detected sites revealed strong enrichment of differentiation and neurogenesis-related factors (Figure 2E). A scan for transcription factor motifs that might respond to FD identified 50.1% of peaks as containing the NEUROG2-targeted enhancer box motif, as well as 4,672 peaks and 2,075 peaks containing cAMP response element (CRE) half-site and high-mobility group (HMG)-box motifs, respectively (Figure 2E). This motif-based approach hints that FD might improve the accessibility of genetic regulatory elements via activation of other transcription factors critical to reprogramming.
A pairwise analysis of RNA-seq datasets was performed for genes annotated to ATAC-seq peaks uniquely detected in the NFD condition to identify how changes in chromatin structure affect gene expression (Figure 2F). This integrated analysis identified pro-neural genes involved in neurogenic transcription, Notch and Wnt signaling, and chromatin remodeling. Collectively, these datasets demonstrate that NEUROG2 functions as a pioneer transcription factor, while FD promote chromatin remodeling at pro-neural genes upregulated during reprogramming.
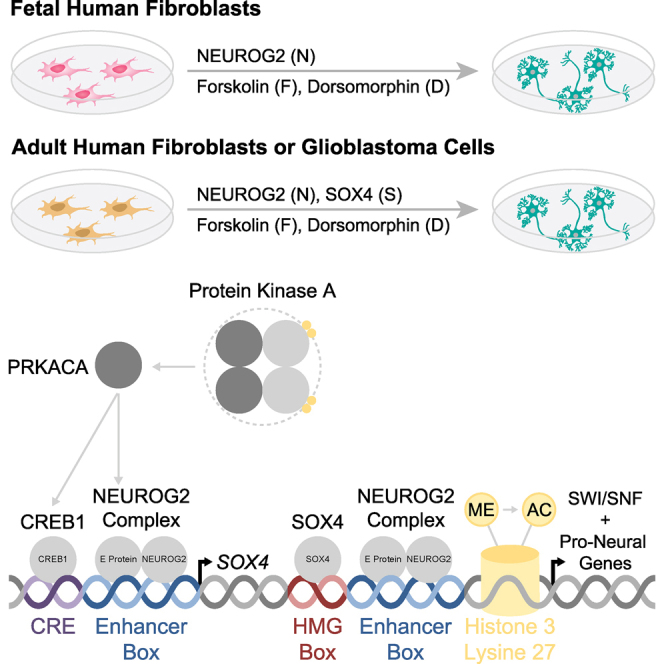
PRKACA Can Phosphorylate NEUROG2 Heterodimers to Modify DNA Affinity
Forskolin is a well-established activator of cAMP synthesis and PRKACA (protein kinase cAMP-activated catalytic subunit α) kinase activity. To demonstrate that increased intracellular cAMP is fundamental to reprogramming, we successfully replaced forskolin with cAMP or dibutyryl cAMP during reprogramming (Figure S3A). Increased intracellular cAMP catalyzes the release of PRKACA from its regulatory subunit and stimulates protein kinase A activity. Remarkably, fibroblasts co-transduced with NEUROG2 and PRKACA rapidly outgrow TUBB3+ and MAP2+ neurites within 12 days (Figure S3B).
From these findings, we hypothesized four mechanisms by which FD might enhance NEUROG2 binding and promote chromatin remodeling (Figure 3A). The simplest mechanism is an FD-induced post-translational phosphorylation event that modulates NEUROG2 function. Cyclin-dependent kinases and glycogen synthase kinase 3 have been shown to regulate NEUROG2 DNA affinity, neuron differentiation, and motor neuron specification through site-specific phosphorylation (Ali et al., 2011, Ma et al., 2008). To identify whether increased NEUROG2 phosphorylation by PRKACA or another kinase is a primary function of FD during reprogramming, we performed introduced phosphodeficient and phosphomimetic amino acids at serine residues with a high probability for phosphorylation. Surprisingly, the replacement of S24, S91, S193, S207, S209, S219, S232, S239, or S242 had no effect on reprogramming efficiency, speed of conversion, or neuron morphology (Figures S3C and S3D). Even a NEUROG2 construct carrying substitutions at seven of these residues exhibited no effect on reprogramming. Although not an exhaustive analysis, this screen suggests that FD might act via another mechanism to drive the changes observed in ChIP-seq and ATAC-seq datasets.
Figure 3.

Small Molecules Activate CREB1 to Promote Neuron Survival and SOX4 Upregulation
(A) Four hypothetical mechanisms that might underlie NEUROG2 and FD synergy.
(B) Immunoblot of shRNA-mediated CREB1 knockdown.
(C) CREB1 knockdown significantly reduces the viability of reprogrammed neurons (mean ± SD; n = 10 random image fields from biological triplicates). Relative survival represents (GFP+ and Hoechst 33342+ cells at each time point)/(total GFP+ and Hoechst 33342+ cells at 1 DPT).
(D) Phospho-CREB1 (pS133) ChIP-qPCR at CREB1 targets (NR4A1, SIK1), NEUROG2 targets with FD-enhanced expression (GADD45G, SOX4), and controls (NEUROG2 target, DLL3; within gene, NR4A1 open reading frame) for NEUROG2-transduced fibroblasts (blue) and NFD-treated fibroblasts (red) at 1 DPT (mean ± SD; biological triplicates).
(E) NEUROG2 occupancy around the SOX4 locus. NEUROG2 occupancy is represented as an overlay of 0.5-DPT (light), 1-DPT (medium), and 2 DPT (dark) NEUROG2 ChIP-seq traces from NEUROG2-transduced fibroblasts (blue) and NFD-treated fibroblasts (red). Purple hashes indicate CRE motif sequences.
(F) Condition-dependent SOX4 expression from RNA-seq results at 2 DPT (mean values from biological triplicate RNA-seq data).
(G) NEUROG2 and CREB1 ChIP from control and NFD-treated fibroblasts reveals chromatin co-occupancy in NFD-treated fibroblasts.
To evaluate the second hypothetical mechanism underlying enhanced NEUROG2 chromatin occupancy, we investigated whether FD promote the phosphorylation of heterodimeric NEUROG2 complexes to enhance chromatin affinity. NEUROG2 heterodimers have been shown to exhibit variable chromatin affinity and functional stability in developing and mature neurons (Bronicki et al., 2012, Li et al., 2012). As a proof of principle, fibroblasts transduced with a minimal NEUROG2 construct (NEUROG2110–228) containing only the DNA binding domain and a single transactivation domain exhibited significant deficits in reprogramming efficiency (Figure S3E). Importantly, this minimal structure did not entirely abolish the generation of neuron-like MAP2+ cells, indicating that NEUROG2110–228 is a functional but inefficient transcriptional activator. Next, mass spectrometry was used to identify NEUROG2 co-factors (Figure S3F). This revealed direct interaction with transcription factor 3 (TCF3), which has been shown to modulate NEUROG2 chromatin binding dynamics in a phosphorylation-dependent manner (Li et al., 2012).
To investigate whether PRKACA phosphorylates TCF3 and related co-factors that heterodimerize with NEUROG2, we co-expressed PRKACA with TCF3 isoform E12, TCF3 isoform E47, TCF4, and TCF12. A denaturing electrophoretic analysis revealed that TCF3, TCF4, and TCF12 are phosphorylated by PRKACA (Figure S3G). Next, we assessed the functional roles for this phosphorylation. Genomic sequences extracted from NEUROG2 ChIP-seq occupancy sites annotated to the 100 most upregulated and downregulated genes identified by RNA-seq were directionally searched to identify the distribution of enhancer box sequences at these sites (Figure S4A). Three unique sequences, 5′-CAGCTG-3′, 5′-CAGATG-3′, and 5′-CATCTG-3′, were strongly enriched within this subset of binding sites. Phosphorylation-dependent binding of NEUROG2 heterodimers to each of these enhancer box sequences was assayed using microscale thermophoresis (Figures S4B–S4D) (Wienken et al., 2010).
DNA binding-induced changes in the thermophoresis of recombinant GFP-labeled NEUROG2 enabled us to quantitatively define the effects of differential phosphorylation on heterodimer affinity for sequence-specific enhancer box motifs. A preliminary electrophoretic mobility shift assay demonstrated that NEUROG2 homodimers do not bind DNA (Bronicki et al., 2012), GFP fusion does not affect NEUROG2 association with TCF3, and NEUROG2 efficiently binds a ChIP-identified DLL3 enhancer box in vitro (Figure S4E). Thermophoretic traces were collected using differentially phosphorylated NEUROG2-TCF3 heterodimers and a DLL3 5′-CAGCTG-3′ native enhancer box or mutagenized internal dinucleotide pair to encode 5′-CAGATG-3′ and 5′-CATCTG-3′. Surprisingly, phosphorylation caused sequence-specific reductions in enhancer box affinity (Figures S4B–S4D). Among several possibilities, this result suggests that TCF3 phosphorylation might be a mechanism for enhancing the specificity and resolution of NEUROG2 binding in a sequence-dependent manner, but is not likely to be the primary mechanism underlying enhanced chromatin accessibility or occupancy.
CREB1 Promotes Neuron Survival
In contrast to direct post-translational modifications, FD might mediate synergistic changes in gene expression and chromatin remodeling through PRKACA-targeted transcription factors that complement the NEUROG2 transcription program. To investigate our third proposed mechanism, we performed sequence-based discovery of regulons using RNA-seq datasets for upregulated genes without NEUROG2 annotation (Janky et al., 2014). This enabled the identification of 79 FD-induced genes containing at least one CRE motif within 5 kb of the target TSS (Table S2). CRE binding protein 1 (CREB1) directly interacts with this enriched motif and is an established target of PRKACA phosphorylation (Gonzalez and Montminy, 1989). To investigate whether FD-activated CREB1 is essential to reprogramming, we performed CREB1 knockdown using a small hairpin RNA (shRNA) co-expressed with NEUROG2 (Figure 3B). CREB1 knockdown significantly reduced the survival of NFD-treated fibroblasts, indicating that CREB1 is critical to induced neuron survival (Figure 3C). This survival effect during reprogramming is consistent with its known role in endogenous neurons (Finkbeiner, 2000).
Next, an antibody specific for PRKACA-mediated CREB1 phosphorylation was used to perform ChIP-qPCR (Figure 3D). ChIP specificity was validated using established target genes (NR4A1 and SIK1), which both exhibited significant FD-induced upregulation in RNA-seq datasets. Interestingly, FD-enhanced phospho-CREB1 binding at SOX4 compared with binding in the absence of FD (Figure 3D). A genomic search revealed that several consensus CRE sites are located adjacent to NEUROG2 occupancy sites in the SOX4 enhancer (Figure 3E). Supporting a cooperative role for active CREB1 and NEUROG2 in SOX4 regulation, upregulation of SOX4 by FD could be further enhanced by NEUROG2 (Figure 3F). To evaluate whether co-binding might occur at numerous locations, we performed co-immunoprecipitation (coIP) on NEUROG2 and CREB1 chromatin (Figure 3G). NEUROG2-bound chromatin exposed positive for CREB1 and CREB1-bound chromatin validated this result, with NEUROG2 detected in NFD-treated cells but not control cells. These results demonstrate that FD enhances CREB1 enrichment at chromatin locations co-bound by NEUROG2.
SOX4 Promotes Chromatin Remodeling during Early Reprogramming
SOX4 is an HMG transcription factor with roles in pan-neuronal gene expression networks and the maturation of differentiating neurons (Bergsland et al., 2006, Shim et al., 2012). To investigate whether SOX4 upregulation is critical during early reprogramming, we performed SOX4 knockdown in NFD-treated fibroblasts (Figure 4A). Remarkably, disrupted SOX4 expression resulted in morphological defects and nearly abolished reprogramming (Figure 4B). ChIP-seq was next used to define the genetic targets of this factor. Alignment of SOX4 and NEUROG2 profiles revealed significant overlap with 413 of 1,119 total SOX4 binding events directly shared with NEUROG2 (Figure 4C).
Figure 4.

SOX4 Promotes Open Chromatin Patterns during Reprogramming
(A) Immunoblot validation of shRNA-mediated SOX4 knockdown.
(B) SOX4 knockdown significantly reduces neuronal conversion (mean ± SD; n = 10 random image fields from biological triplicates). Reprogramming efficiency represents (GFP+ and MAP2+ cells)/(total GFP+ cells).
(C) Area-proportional Venn diagram depicting the total number of SOX4 binding events and the proportion shared with NEUROG2. The adjacent pie diagram represents the relative fold increase in normalized transcript number for co-bound genes in NEUROG2-transduced fibroblasts in the presence versus absence of FD from RNA-seq datasets.
(D) NEUROG2 ChIP-seq track (2 DPT) and ATAC-seq tracks generated from fibroblasts (MRC-5), NEUROG2-transduced fibroblasts (N), NFD-treated fibroblasts (NFD), and NFD-treated fibroblasts co-expressing SOX4 shRNA (NFD + SOX4 shRNA) around the NEUROD1 and NEUROD4 loci. Gray shading highlights representative regions of FD-enhanced chromatin accessibility lost upon SOX4 knockdown.
(E) Area-proportional Venn diagram depicting the total number of ATAC-seq events in NFD-treated fibroblasts relative to the number of events detected at the same locations upon SOX4 knockdown. The adjacent pie diagram represents the relative peak score change for shared sites (NFD peak score)/(NFD + SOX4 shRNA peak score).
(F) NEUROD1 and NEUROD4 ChIP-qPCR at sites of newly open chromatin or NFD-induced genes (mean ± SD; biological triplicates).
Peak-to-gene annotation next enabled an analysis of how co-binding effects gene expression. Relative to NEUROG2 alone, NFD-treated cells exhibited enhanced transcription for 81% of co-bound genes including NEUROD1 and NEUROD4 (Figure 4C). In addition, these datasets demonstrated that SOX4 also targets components of the SWI/SNF nucleosome remodeling complex including ARID1A, ARID1B, SMARCA2, SMARCAD1, SMARCC1, SMARCC2, SMARCD1, SMARCD2, and SMARCE1 (Kwon et al., 1994, Son and Crabtree, 2014). This suggests that SOX4 not only functions to regulate transcription via direct chromatin binding but might indirectly promote chromatin remodeling to increase accessibility.
A comprehensive analysis of this indirect chromatin remodeling activity was performed using SOX4 knockdown coupled with ATAC-seq in NFD-treated fibroblasts. FD-induced chromatin decondensation at NEUROG2 binding sites and adjacent genetic elements in the NEUROD1 and NEUROD4 loci is significantly reduced following SOX4 depletion (Figure 4D). As both of these genes are significantly upregulated only in the NFD condition, this analysis demonstrates how the synergistic co-binding of NEUROG2 and SOX4 accompanied by a transition to open chromatin patterns is fundamental to the expression of genes that promote reprogramming. Broadening this to a genome-wide analysis, SOX4 knockdown results in an extensive loss of approximately 59% of total open chromatin sites in NFD-treated fibroblasts (Figure 4E). A simultaneous decrease in ATAC score is also observed for 22% of the remaining peaks following knockdown.
NEUROD1 is required to promote human fibroblast-to-neuron reprogramming (Pang et al., 2011). As they represent direct downstream targets of NEUROG2 and SOX4, ChIP for NEUROD1 and NEUROD4 was used to investigate the occupancy of these factors within regions of newly open chromatin (Figure 4F). Publicly available ChIP-seq datasets from mouse cortical tissue were analyzed for potential target sites that directly overlap with human NEUROG2 ChIP and ATAC-seq sites (Pataskar et al., 2016). NEUROD1 was enriched at NEUROG2 bound sites in regions of DLL3, HES6, and NHLH1, while NEUROD4 was significantly less enriched within these loci in reprogrammed human fibroblasts. Furthermore, NEUROD1 exhibited modest enrichment at the NEUROD4 promoter within an open chromatin site lost following SOX4 depletion. These findings collectively implicate FD-induced upregulation of SOX4 in pro-neural chromatin remodeling activity.
NEUROG2 and SOX4 Induce H3K27 Acetylation at Pro-neural Genetic Elements
We next performed a condition-dependent analysis of H3K27AC and H3K27 trimethylation (H3K27ME3) to define the native epigenetic landscape of fibroblasts and factor-specific changes triggered during reprogramming. First, a quantitative analysis of H3K27AC ChIP-seq read densities around NEUROG2 ChIP-seq peaks revealed that FD only modestly enhanced acetylation at these sites. In contrast, FD significantly enhanced H3K27AC enrichment surrounding SOX4-targeted genomic sites (Figure 5A). Second, this FD-enhanced acetylation is even more pronounced at occupancy sites annotated to genes targeted by both NEUROG2 and SOX4 (Figure 5B). This further demonstrates that the synergistic genetic and epigenetic reprogramming induced by NEUROG2 and FD is, at least in part, mediated by SOX4.
Figure 5.

Small Molecules Enhance H3K27 Acetylation at NEUROG2- and SOX4-Targeted Genetic Elements
(A and B) Distribution of H3K27 acetylation (H3K27AC) ChIP-seq reads from untreated fibroblasts (black), NEUROG2-transduced fibroblasts (blue), and NFD-treated fibroblasts (red) centered around (A) SOX4 ChIP-seq peaks and (B) NEUROG2 and SOX4 co-bound peaks within a 4-kb window.
(C and D) ChIP-seq tracks for SOX4, NEUROG2, H3K27 acetylation, and H3K27 trimethylation generated from fibroblasts (MRC-5), NEUROG2-transduced fibroblasts (N), and NFD-treated fibroblasts (NFD) around the (C) DLL3 and (D) SMARCC2 loci.
In-depth analysis of transcription factor binding profiles and histone marks at DLL3 and SMARCC2 provided insights into both the mechanism of reprogramming and the epigenetic environment of MRC-5 fibroblasts. For instance, binding sites for NEUROG2 and SOX4 directly overlap upstream of DLL3, and H3K27AC enrichment significantly increases at this site upon FD treatment (Figure 5C). A simultaneous loss of H3K27ME3 is observed throughout this region. In contrast, the SWI/SNF subunit SMARCC2 is targeted only by SOX4, and the H3K27ME3 signal is relatively low throughout this locus (Figure 5D). This indicates that MRC-5 fibroblasts might be more epigenetically poised for reprogramming than other lineage-committed adult skin fibroblasts.
Induced Neuronal Identity in Cell Types Refractory to NEUROG2 Reprogramming
The success of neuronal reprogramming depends upon the ability of exogenous reprogramming factors and downstream induced transcription factors to traverse the cell-of-origin epigenetic environment. Unlike the high-efficiency conversion of fetal MRC-5 fibroblasts, NEUROG2 and FD do not functionally or morphologically reprogram adult skin fibroblast lines without additional SOX11 overexpression (Liu et al., 2013). Likewise, the addition of FGF2 and kenpaullone to this reprogramming cocktail promote the survival of reprogrammed neurons (Liu et al., 2013, Liu et al., 2015). As SOX4 and SOX11 exhibit redundant functions in the developing nervous system (Cheung et al., 2000, Miller et al., 2013), we investigated whether an inability to activate this factor hinders adult cell reprogramming efficiency.
An initial assessment of SOX4 expression in adult skin fibroblast lines revealed that adult fibroblasts express SOX4 at comparatively lower levels than MRC-5 (Figure 6A). Next, we evaluated whether SOX4 upregulation is beneficial to reprogramming by transducing adult fibroblasts with NEUROG2- and SOX4-encoding lentivirus in combination with FD and FGF2. These factors induced TUBB3 expression and modest neurite outgrowth in all four lines (Figure 6B). However, the progressive maturation of these neurons is significantly slower and less complex than neurons derived from fetal cells (Figure 6C).
Figure 6.

Increased Chromatin Accessibility Promotes Human Adult Fibroblast and Glioblastoma Cell Reprogramming
(A) Expression of SOX4 RNA and protein in adult fibroblast lines relative to fetal MRC-5 fibroblasts (mean ± SD; biological quadruple samples for qPCR).
(B) Percentage of rounded adult fibroblasts expressing TUBB3 when treated with NFD, FGF2, and mouse Sox4 (red) or human SOX4 (gray) (mean ± SD; n = 10 random image fields from biological triplicates).
(C) Examples of progressive neuronal conversion using adult fibroblasts treated with reprogramming factors. Fibroblasts adopt a rounded morphology (5–10 DPT), outgrow low-complexity neurites (10–20 DPT), and infrequently adopt complex neuronal morphology (≥21 DPT). Scale bars, 20 μm.
(D) SOX4 ChIP-qPCR time course demonstrating SOX4 chromatin occupancy at 1, 2, and 4 DPT in adult fibroblasts treated with reprogramming factors (mean ± SD; biological triplicates for each ChIP).
(E) Screen for chromatin-modifying chemicals and SWI/SNF factors that promote neuronal reprogramming. The presence (+) or absence (−) of morphological change, soma rounding, neurite outgrowth, and TUBB3 expression at 8 DPT was evaluated.
(F) Exposure to FK228 for 20–24 hr induces rapid neuron-like morphology. Arrows indicate cells with advanced morphology, while partially converted cells at the bottom left are still undergoing morphological restructuring.
(G) BCL2L1 significantly enhances the survival of FK228 treated cells, enables the adoption of complex morphology, and promotes expression of neuronal markers such as TUBB3, MAP2, and RBFOX3. Scale bars, 50 μm.
(H) Percentage of adult AG05811 fibroblasts overexpressing NEUROG2 and BCL2L1 that reprogram into TUBB3+ neurons at 10 DPT. Each time point represents FK228 exposure during the 24-hr window from 2–3, 3–4, or 4–5 days after initial lentiviral transduction (mean ± SD; n = 10 random image fields from biological triplicates for two virus conditions at each time point).
(I) Percentage of reprogrammed U-251 glioblastoma cells expressing TUBB3 (red) and MAP2 (gray) when treated with NFD or NFD with SOX4. MAP2 and TUBB3 immunocytochemical staining of neurons generated at 10 DPT (mean ± SD; n > 10 independent experiments with biological triplicates for each experiment). Scale bar, 50 μm.
A SOX4 ChIP time course using AG05811 adult fibroblasts provided insight into the varied genetic landscapes of adult and fetal fibroblast lines. SOX4 enrichment at sites identified by ChIP-seq in 1-DPT MRC-5 cells exhibited significantly delayed occupancy in adult cells through 4 DPT (Figure 6D). This suggests that condensed chromatin might occlude the binding sites of SOX4 and other downstream NEUROG2-driven transcription factors to inhibit the neurogenic transcription programs critical to adult fibroblast reprogramming.
Under conditions of chromatin accessibility, NEUROG2, FD, and the endogenous upregulation of SOX4 should be sufficient to drive reprogramming of adult fibroblasts. Since SOX4 targets numerous subunits of the SWI/SNF complex (Figure 4C), SMARCA4 and NEUROG2 were co-expressed to determine whether chromatin remodeling could potentiate reprogramming (Figure 6E). A fraction of transduced fibroblasts rounded, outgrew neurites, and exhibited modest TUBB3 expression but, similar to SOX4 overexpression, these fibroblasts did not adopt highly complex neuronal morphology. The co-expression of SMARCB1 and SMARCC2 with NEUROG2 and SMARCA4 failed to further improve conversion.
A chemical screen targeting chromatin modifiers was then performed to identify whether compounds that enhance chromatin accessibility could improve adult fibroblast reprogramming (Figure 6E). We identified several chemicals sufficient to induce rounding and modest neurite outgrowth, but only the histone deacetylase inhibitor FK228 was sufficient to rapidly induce a large cell population with neuron-like morphology (Figure 6F). Remarkably, this morphological conversion occurred within 20–24 hr of exposure; however, cells treated with 0.5 μM FK228 failed to survive long term in culture.
In addition to histone deacetylase inhibition, FK228 suppresses the RAS-MAP kinase signaling pathway to induce apoptosis (Kobayashi et al., 2006). To abrogate these pro-apoptotic functions while retaining histone deacetylase inhibition, we pre-treated adult skin fibroblasts with the pan-caspase inhibitor Z-VAD-FMK (Wang et al., 2012), the necrosis inhibitor necrosulfonamide, and the ferroptosis inhibitor liproxstatin-1. Fibroblasts were continuously treated starting at 1 day before FK228 treatment and 2 days following FK228 exposure. Due to the rapid adoption of neuronal morphology, we also included the brain-derived neurotrophic factor BDNF, the glial cell line-derived neurotrophic factor GDNF, and neurotrophin-3 at the outset of reprogramming. Fibroblasts treated with FK228 for 24 hr starting 2 days after infection exhibited modest improvements in cell survival upon Z-VAD-FMK treatment. Liproxstatin-1 and necrosulfonamide exhibited no effect on cell survival or reprogramming efficiency. While Z-VAD-FMK increased the survival of induced neurons compared with FK228 alone, a significant fraction of cells still died 5–7 days after initial transduction.
FK228-induced apoptosis is highly sensitive to BCL2L1 activity, but remains unaffected by BCL2 (Newbold et al., 2008). We then co-expressed BCL2L1 with NEUROG2 to determine whether cell survival could be enhanced following FK228 treatment. Interestingly, adult fibroblasts co-expressing NEUROG2 and BCL2L1 for 4 days in neuron induction medium followed by a brief 24-hr exposure to FK228 from days 4–5 generated morphologically complex neurons that stained TUBB3+, MAP2+, and RBFOX3+ after 12 days in culture (Figures 6G and 6H). These results show the neuronal reprogramming capacity of FK228 when apoptotic mechanisms are controlled during conversion. This chemical-based approach to chromatin modification further demonstrates that NEUROG2 and small molecules are sufficient to induce early neurogenic programs in fibroblasts refractory to reprogramming in the absence of epigenetic barriers to activity. The toxicity of FK228 to fibroblasts not transduced with BCL2L1 resulted in fluorescent cell debris that occluded an extensive immunochemical analysis of neuronal maturation. Further optimization is needed for long-term survival, maturation, and subtype identity analyses.
Distinct cell types exhibit disparate heterochromatin condensation patterns. To investigate whether a more permissive chromatin state would facilitate rapid reprogramming, we reprogrammed adult human U-251 glioblastoma cells. In the absence of SOX4 overexpression, NFD-treated cells adopt TUBB3+, and MAP2+ neuron-like morphology with 21% ± 3% efficiency (Figure 6I). However, inclusion of SOX4 endows these cells with complex morphology and conversion efficiency of greater than 90% (Figure 6I). This alternative cellular context provides independent confirmation of the fundamental roles played by SOX4 and SOX4-targeted downstream actors in neuronal reprogramming.
Collectively, these findings represent a stepwise molecular mechanism for the synergistic events underlying early fibroblast-to-neuron reprogramming (Figure 7). The hierarchical activation of NEUROG2, CREB1, and SOX4 transcriptional programs, as well as, FD-induced chromatin remodeling, can potentiate reprogramming in a cell age- and lineage-dependent manner.
Figure 7.

Simplified Molecular Mechanisms Underlying Fibroblast-to-Neuron Reprogramming
NEUROG2, a pioneer transcription factor, targets thousands of pro-neural genetic elements. Small molecules simultaneously activate signaling cascades that promote NEUROG2 and CREB1 co-transcription, induce SOX4 expression, enhance H3K27 acetylation, and promote SOX4-dependent chromatin remodeling. NEUROG2 and SOX4 synergize to enhance the expression of diverse pro-neural transcription factors and drive neuron identity in diverse fibroblast and glioblastoma cell types.
Discussion
Here we performed genome-wide ATAC-seq, ChIP-seq, and RNA-seq profiling to define the immediate-early molecular events that catalyze fibroblast-to-neuron reprogramming by NEUROG2 and small molecules. While NEUROG2 is sufficient to reprogram glial cells into neurons, this transcription factor fails to reprogram somatic fibroblasts independent of the small molecules F and D (Liu et al., 2013). This synergistic reprogramming activity is the result of extensive SOX4-mediated chromatin remodeling that enhances NEUROG2 occupancy at pro-neural genetic elements and, in coordination with downstream factors, redefines the fibroblast transcriptome. These findings enabled us to reprogram adult fibroblasts and glioblastoma cells refractory to conversion by NEUROG2 alone.
NEUROG2 Functions as a Pioneer Factor during Early Reprogramming
Transcription factors in both pluripotent and direct reprogramming systems utilize either off-target cooperative chromatin binding (Soufi et al., 2012) or on-target pioneer binding mechanisms to activate gene expression (Wapinski et al., 2013). Similar to ASCL1 function in mouse embryonic fibroblasts, NEUROG2 exhibits on-target pioneer binding in heterochromatin regions when expressed in human fetus. Unexpectedly, however, FD-induced chromatin remodeling dramatically enhances NEUROG2 chromatin occupancy.
This enhanced occupancy demonstrates that FD act synergistically with NEUROG2 to promote reprogramming rather than function as complementary, but independent transcriptional activators. We investigated four mechanisms by which FD might enhance NEUROG2 activity in a cell lineage-dependent manner. This revealed that FD enhance chromatin accessibility to promote hierarchical associations with CREB1 and SOX4 to initiate secondary pro-neural transcription programs. These observations validated an earlier computational model indicating that CREB1 acts as a co-activator of NEUROG2 during mouse telencephalon development (Gohlke et al., 2008), as well as roles for SOX4 in establishing a cross-regulatory neurogenic network involving SWI/SNF complex factors (Ninkovic et al., 2013). The genome-wide interplay of CREB1, NEUROG2, and SOX4 during reprogramming highlights how tightly interwoven genetic regulatory networks require coordinated multi-factor occupancy to effectively enhance the expression of pro-neural downstream actors previously implicated in reprogramming such as NEUROD1 (Pang et al., 2011, Guo et al., 2014) and NEUROD4 (Masserdotti et al., 2015).
SOX4 Indirectly Enhances Neuronal Identity through Chromatin Remodeling
Sox4 and Sox11 exhibit similar expression patterns in the developing nervous system, and simultaneous, but not independent, knockout of these factors results in reduced neuron survival (Cheung et al., 2000, Miller et al., 2013). CHD7 activates a neuronal differentiation program in neural stem cells by remodeling the Sox4 and Sox11 promoters to upregulate expression (Feng et al., 2013). Of these transcription factors, only SOX4 is significantly expressed in human MRC-5 fibroblasts, and transient knockdown of SOX4 abolishes reprogramming. Importantly, NEUROG2 outcompetes REST to drive Sox4 and promote neuronal differentiation in post-mitotic neural precursor cells (Bergsland et al., 2006). In parallel with this mechanism, NEUROG2 co-binds with CREB1 to upregulate SOX4 in MRC-5 fibroblasts, demonstrating similar regulatory mechanisms during development and reprogramming.
An analysis of SOX4 targets revealed extensive coordination with NEUROG2 at factors with FD-enhanced or synergistically induced expression such as HES6, NEUROD1, NEUROD4, and WNT10A. In addition to synergistic regulation, SOX4 targets epigenetic remodeling factors such as EZH2 (Tiwari et al., 2013) and numerous SWI/SNF subunits.
In parallel with this epigenetic function, the inclusion of either SOX4 or SOX11 is required to induce adult fibroblasts and glioblastoma cells to adopt TUBB3+ identity (Liu et al., 2013, Su et al., 2014). The morphological complexity of these neurons can be significantly enhanced with ISL1 and LHX3 overexpression (Liu et al., 2015). As adult fibroblast lineages exhibit highly defined epigenetic signatures that make these cells less poised for reprogramming, mechanistic studies have predominantly relied on embryonic or fetal fibroblasts (Masserdotti et al., 2015, Wapinski et al., 2013, Gascón et al., 2016). Our proposed mechanism suggests that SOX4 reduces epigenetic barriers to reprogramming to facilitate fate conversion of adult cell lineages.
Mature mammalian neurons exhibit distinct epigenomic signatures that reflect subtype-specific functions. Based on our ATAC-seq and SOX4 ChIP-seq datasets, we hypothesized that modulating the epigenome of fibroblasts with chromatin remodeling complexes might enhance reprogramming efficiency as observed with induced pluripotent stem cells (Singhal et al., 2010). While a subset of adult fibroblasts transduced with both SMARCA4 and NEUROG2 expressed TUBB3 and low-complexity neurites, the addition of other SWI/SNF subunits such as SMARCB1 and SMARCC2 failed to improve reprogramming, likely due to imbalances in subunit stoichiometric required for a minimally functional SWI/SNF complex.
Targeting Epigenetic Modifiers Enhances Adult Human Fibroblast Reprogramming
We next turned to a small-molecule-based strategy for epigenetic remodeling inspired by recently described chemical reprogramming systems (Hu et al., 2015, Li et al., 2015, Zhang et al., 2015). This led to the replacement of SOX4 by FK228, which rapidly induces neuron morphology but confers poor survival. Upon co-expression of NEUROG2 and BCL2L1 with FK228 we observed morphologically complex neurons. While we were not able to directly image MAP-tau expression due to high background, these cells did become TUBB3+, MAP2+, and RBFOX3+. Collectively, these results highlight multiple roles for SOX4 in early reprogramming as well as the functional impact of epigenetic remodeling on reprogramming efficiency in adult cells.
Chromatin Accessibility and Histone Signatures Regulate Reprogramming Efficiency
Distinct histone acetylation and methylation signatures regulate transcription factor activity and gene expression. Patterned post-translational modifications direct transcription factors to specific genomic regulatory regions. H3K27AC enrichment was observed at both NEUROG2 and SOX4 bound sites in reprogrammed fibroblasts. Furthermore, FD significantly increased this enrichment at cooperatively bound sites.
CREB1 phosphorylation promotes the recruitment of histone acetyltransferases that induce local nucleosome remodeling (Michael et al., 2000). This activity likely mediates, in part, the FD-enhanced acetylation observed at NEUROG2 occupancy sites. Histone acetyltransferase inhibitors such as valproic acid (Niu et al., 2013) and FK228 drive the adoption of neuronal identity, underscoring the significance of persistent acetylation at neurogenic promoters during early conversion.
Collectively, this work presents a stepwise mechanism detailing the early events critical to fibroblast-to-neuron reprogramming and highlights how changes to the cell-of-origin chromatin landscape measurably affect conversion efficiency.
Experimental Procedures
ATAC-Seq
Fluorescent-sorted cells were lysed, resuspended in transposition mix, incubated for 30 min at 37°C, and amplified with barcoded primers. Single-end 50-base length reads were generated and aligned to GRCh37/hg19 for analysis.
ChIP, ChIP CoIP, and ChIP-Seq
ChIP and ChIP-seq used fibroblasts transduced with HA-NEUROG2 or NEUROG2+HA-SOX4. Cells were fixed and lysed, and nuclei suspended in shearing buffer. Chromatin was sheared, quantified, and combined with target-specific antibodies. Chromatin was captured with magnetic beads, washed, eluted, and reverse crosslinked. Single-end 50-base length reads were generated and aligned to GRCh37/hg19 for analysis. ChIP coIP followed the same protocol using denaturing buffer elution. Detailed protocols are provided in Supplemental Experimental Procedures and Table S3.
Neuron Induction
MRC-5 fibroblasts were seeded on standard 24-well glass coverslips, transduced with NEUROG2-encoding lentivirus, and supplemented with 10 μM forskolin and 1 μM dorsomorphin 48 hr after infection. Medium was changed every 2 days for the duration of culture.
RNA-Seq
Total RNA was isolated from control or treated MRC-5 fibroblasts and quantified for library synthesis. Single-end 50-base length sequencing reads were aligned to GRCh37/hg19 for differential expression analysis.
shRNA-Mediated Gene Knockdown
PCR was used to generate a lentiviral construct containing miR30 regulatory sequences and shRNAs (Table S3; Fellmann et al., 2013).
Author Contributions
D.K.S., conceptualization, data curation, formal analysis, investigation (Figures 1, 2, 3, 4, 5, 6A–6H, 7, and S1–S4), methodology, software, supervision, validation, visualization, writing – original draft, and writing – review and editing; J.Y., investigation (Figures 6G–6I); M.-L.L., supervision; C.-L.Z., conceptualization, funding acquisition, resources, and supervision.
Acknowledgments
We thank Mark Borromeo, Bradford Casey, Jane E. Johnson, Stephen Johnson, Taekyung Kim, Zhigao Wang, and members of the Zhang laboratory. This work was supported by the Decherd Foundation, the Mobility Foundation, the NIH (NS070981, NS092616, NS093502, and NS088095), the Texas Institute for Brain Injury and Repair, and the Welch Foundation (I-1724).
Published: October 27, 2016
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.stemcr.2016.09.013.
Accession Numbers
The accession number for bioinformatics datasets is GEO: GSE75912.
Supplemental Information
References
- Ali F., Hindley C., McDowell G., Deibler R., Jones A., Kirschner M., Guillemot F., Philpott A. Cell cycle-regulated multi-site phosphorylation of Neurogenin 2 coordinates cell cycling with differentiation during neurogenesis. Development. 2011;138:4267–4277. doi: 10.1242/dev.067900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsland M., Werme M., Malewicz M., Perlmann T., Muhr J. The establishment of neuronal properties is controlled by Sox4 and Sox11. Genes Dev. 2006;20:3475–3486. doi: 10.1101/gad.403406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berninger B., Costa M.R., Koch U., Schroeder T., Sutor B., Grothe B., Götz M. Functional properties of neurons derived from in vitro reprogrammed postnatal astroglia. J. Neurosci. 2007;27:8654–8664. doi: 10.1523/JNEUROSCI.1615-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronicki L.M., Bélanger G., Jasmin B.J. Characterization of multiple exon 1 variants in mammalian HuD mRNA and neuron-specific transcriptional control via neurogenin 2. J. Neurosci. 2012;32:11164–11175. doi: 10.1523/JNEUROSCI.2247-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S., Ang C.E., Davila J., Pak C., Mall M., Lee Q.Y., Ahlenius H., Jung S.W., Südhof T.C., Wernig M. Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Rep. 2014;3:282–296. doi: 10.1016/j.stemcr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung M., Abu-Elmagd M., Clevers H., Scotting P.J. Roles of Sox4 in central nervous system development. Mol. Brain Res. 2000;79:180–191. doi: 10.1016/s0169-328x(00)00109-1. [DOI] [PubMed] [Google Scholar]
- Fellmann C., Hoffmann T., Sridhar V., Hopfgartner B., Muhar M., Roth M., Lai D.Y., Barbosa I.A.M., Kwon J.S., Guan Y. An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 2013;5:1704–1713. doi: 10.1016/j.celrep.2013.11.020. [DOI] [PubMed] [Google Scholar]
- Feng W., Khan M.A., Bellvis P., Zhu Z., Bernhardt O., Herold-Mende C., Liu H.K. The chromatin remodeler CHD7 regulate adult neurogenesis via activation of SoxC transcription factors. Cell Stem Cell. 2013;13:62–72. doi: 10.1016/j.stem.2013.05.002. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25:11–14. doi: 10.1016/s0896-6273(00)80866-1. [DOI] [PubMed] [Google Scholar]
- Fode C., Gradwohl G., Morin X., Dierich A., LeMeur M., Goridis C., Guillemot F. The bHLH protein Neurogenin 2 is a determinant factor for epibranchial placode-derived sensory neurons. Neuron. 1998;20:483–494. doi: 10.1016/s0896-6273(00)80989-7. [DOI] [PubMed] [Google Scholar]
- Gascón S., Murenu E., Masserdotti G., Ortega F., Russo G.L., Petrik D., Deshpande A., Heinrich C., Karow M., Robertson S.P. Identification and successful negotiation of a metabolic checkpoint in direct neuronal reprogramming. Cell Stem Cell. 2016;18:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- Gohlke J.M., Armant O., Parham F.M., Smith M.V., Zimmer C., Castro D.S., Nguyen L., Parker J.S., Gradwohl G., Portier C.J., Guillemot F. Characterization of the proneural gene regulatory network during mouse telencephalon development. BMC Biol. 2008;6:15. doi: 10.1186/1741-7007-6-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez G.A., Montminy M.R. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Guo Z., Zhang L., Wu Z., Chen Y., Wang F., Chen G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer's disease model. Cell Stem Cell. 2014;14:188–202. doi: 10.1016/j.stem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich C., Blum R., Gascon S., Masserdotti G., Tripathi P., Sanchez R., Tiedt S., Schroeder T., Götz M., Berninger B. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8:e1000373. doi: 10.1371/journal.pbio.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W., Qiu B., Guan W., Wang Q., Wang M., Li W., Gao L., Shen L., Huang Y., Xie G. Direct conversion of normal and Alzheimer's disease human fibroblasts into neuronal cells by small molecules. Cell Stem Cell. 2015;17:204–212. doi: 10.1016/j.stem.2015.07.006. [DOI] [PubMed] [Google Scholar]
- Janky R., Verfaillie A., Imrichová H., Van de Sande B., Standaert L., Christiaens V., Hulselmans G., Herten K., Sanchez M.N., Potier D. iRegulon: from a gene list to a gene regulatory network using large motif and track collections. PLoS Comput. Biol. 2014;10:e1003731. doi: 10.1371/journal.pcbi.1003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi Y., Ohtsuki M., Murakami T., Kobayashi T., Sutheesophon K., Kitayama H., Kano Y., Kusano E., Nakagawa H., Furukawa Y. Histone deacetylase inhibitor FK228 suppresses the Ras-MAP kinase signaling pathway by upregulating Rap1 and induces apoptosis in maglinant melanoma. Oncogene. 2006;25:512–524. doi: 10.1038/sj.onc.1209072. [DOI] [PubMed] [Google Scholar]
- Kwon H., Imbalzano A.N., Khavari P.A., Kingston R.E., Green M.R. Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 1994;370:477–481. doi: 10.1038/370477a0. [DOI] [PubMed] [Google Scholar]
- Li S., Mattar P., Zinyk D., Singh K., Chaturvedi C.-P., Kovach C., Dixit R., Kurrasch D.M., Ma Y.-C., Chan J.A. GSK3 temporally regulates Neurogenin 2 proneural activity in the neocortex. J. Neurosci. 2012;32:7791–7805. doi: 10.1523/JNEUROSCI.1309-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Zuo X., Jing J., Ma Y., Wang J., Liu D., Zhu J., Du X., Xiong L., Du Y. Small-molecule-derived direct reprogramming of mouse fibroblasts into functional neurons. Cell Stem Cell. 2015;17:195–203. doi: 10.1016/j.stem.2015.06.003. [DOI] [PubMed] [Google Scholar]
- Liu M.-L., Zang T., Zou Y., Chang J.C., Gibson J.R., Huber K.M., Zhang C.-L. Small molecules enable Neurogenin 2 to efficiently convert human fibroblasts into cholinergic neurons. Nat. Commun. 2013;4:2183. doi: 10.1038/ncomms3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M.-L., Zang T., Zhang C.-L. Direct lineage reprogramming reveals disease-specific phenotypes of motor neurons from human ALS patients. Cell Rep. 2015;14:115–128. doi: 10.1016/j.celrep.2015.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Kintner C., Anderson D.J. Identification of neurogenin, a vertebrate neuronal determination gene. Cell. 1996;87:43–52. doi: 10.1016/s0092-8674(00)81321-5. [DOI] [PubMed] [Google Scholar]
- Ma Y.-C., Song M.-R., Park J.P., Ho H.-Y.H., Hu L., Kurtev M.V., Zieg J., Ma Q., Pfaff S.L., Greenberg M.E. Regulation of motor neuron specification by phosphorylation of Neurogenin 2. Neuron. 2008;58:65–77. doi: 10.1016/j.neuron.2008.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masserdotti G., Gillotin S., Sutor B., Drechsel D., Irmler M., Jørgensen H.F., Sass S., Theis F.J., Beckers J., Berninger B. Transcriptional mechanisms of proneural factors and REST in regulating neuronal reprogramming of astrocytes. Cell Stem Cell. 2015;17:74–88. doi: 10.1016/j.stem.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J., Paquola A.C.M., Ku M., Hatch E., Böhnke L., Ladjevardi S., McGrath S., Campbell B., Lee H., Herdy J.R. Directly reprogrammed human neurons retain aging-associated transcriptomic signatures and reveal age-related nucleocytoplasmic defects. Cell Stem Cell. 2015;17:705–718. doi: 10.1016/j.stem.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael L.F., Asahara H., Shulman A.I., Kraus W.L., Montminy M. The phosphorylation status of a cyclic AMP-responsive activator is modulated via a chromatin-dependent mechanism. Mol. Cell Biol. 2000;20:1596–1603. doi: 10.1128/mcb.20.5.1596-1603.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.A., Nathanson J., Franjic D., Shim S., Dalley R.A., Shapouri S., Smith K.A., Sunkin S.M., Bernard A., Bennett J.L. Conserved molecular signatures of neurogenesis in the hippocampal subgranular zone of rodents and primates. Development. 2013;140:4633–4644. doi: 10.1242/dev.097212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbold A., Lindermann R.K., Cluse L.A., Whitecross K.F., Dear A.E., Johnstone R.W. Characterisation of the novel apoptotic and therapeutic activities of the histone deacetylase inhibitor romidepsin. Mol. Cancer Ther. 2008;7:1066–1079. doi: 10.1158/1535-7163.MCT-07-2256. [DOI] [PubMed] [Google Scholar]
- Ninkovic J., Steiner-Mexxadri A., Jawerka M., Akinci U., Masserdotti G., Petricca S., Fischer J., von Holst A., Beckers J., Lie C.D. Essential role for BAF complex interacting with Pax6 in establishment of a core cross-regulatory neurogenic network. Cell Stem Cell. 2013;13:403–418. doi: 10.1016/j.stem.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu W., Zang T., Zou Y., Fang S., Smith D.K., Bachoo R., Zhang C.-L. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 2013;15:1164–1175. doi: 10.1038/ncb2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z.P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D.R., Yang T.Q., Citri A., Sebastiano V., Marro S., Südhof T.C. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pataskar A., Jung J., Smialowski P., Noack F., Calegari F., Straub T., Tiwari V.K. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. 2016;35:24–45. doi: 10.15252/embj.201591206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim S., Kwan K.Y., Li M., Lefebvre V., Šestan N. Cis-regulatory control of corticospinal system development and evolution. Nature. 2012;486:74–79. doi: 10.1038/nature11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scardigli R., Schuurmans C., Gradwohl G., Guillemot F. Crossregulation between neurogenin 2 and pathways specifying neuronal identity in the spinal cord. Neuron. 2001;31:203–217. doi: 10.1016/s0896-6273(01)00358-0. [DOI] [PubMed] [Google Scholar]
- Singhal N., Graumann J., Wu G., Araúzo-Bravo M.J., Han D.W., Greber B., Gentile L., Mann M., Schöler H.R. Chromatin-remodeling components of the BAF complex facilitate reprogramming. Cell. 2010;141:943–955. doi: 10.1016/j.cell.2010.04.037. [DOI] [PubMed] [Google Scholar]
- Son E.Y., Crabtree G.R. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. 2014;166C:333–349. doi: 10.1002/ajmg.c.31416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A., Donahue G., Zaret K.S. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;15:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z., Zang T., Liu M.L., Wang L.L., Niu W., Zhang C.-L. Reprogramming the fate of human glioma cells to impede brain tumor development. Cell Death Dis. 2014;5:e1463. doi: 10.1038/cddis.2014.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma E.C., Wischmeyer E., Offen N., Maurus K., Sirén A.-L., Schartl M., Wgner T.U. Ectopic expression of neurogenin 2 alone is sufficient to induce differentiation of embryonic stem cells into mature neurons. PLoS One. 2012;7:e38651. doi: 10.1371/journal.pone.0038651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari N., Tiwari V.K., Waldmeier L., Balwierz P.J., Arnold P., Pachkov M., Meyer-Schaller N., Schübeler D., van Nimwegen E., Christofori G. Sox4 is a mater regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23:768–783. doi: 10.1016/j.ccr.2013.04.020. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T., Ostermeier A., Pang Z.P., Kokubu Y., Südhof T.C., Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Jiang H., Chen S., Du F., Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- Wapinski O.L., Vierbuchen T., Qu K., Lee Q.Y., Chanda S., Fuentes D.R., Giresi P.G., Ng Y.H., Marro S., Neff N.F. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell. 2013;155:621–635. doi: 10.1016/j.cell.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken C.J., Baaske P., Rothbauer U., Braun D., Duhr S. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 2010;1:100. doi: 10.1038/ncomms1093. [DOI] [PubMed] [Google Scholar]
- Zhang L., Yin J.C., Yeh H., Ma N.X., Lee G., Chen X.A., Wang Y., Lin L., Chen L., Jin P. Small molecules efficiently reprogram human astroglial cells into functional neurons. Cell Stem Cell. 2015;17:735–747. doi: 10.1016/j.stem.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.