

SUMMARY
Existing therapies for inflammatory bowel disease based on broad suppression of inflammation result in variable clinical benefit and unwanted side effects. A potential therapeutic approach for promoting immune tolerance is the in vivo induction of regulatory T cells (Tregs). Here we report that activation of the aryl hydrocarbon receptor using the non-toxic agonist 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) induces human Tregs in vitro that suppress effector T cells through a mechanism mediated by CD39 and Granzyme B. We then developed a humanized murine system whereby human CD4+ T cells drive colitis upon exposure to 2,4,6-trinitrobenzenesulfonic acid and assessed ITE as a potential therapeutic. ITE administration ameliorated colitis in humanized mice with increased CD39, Granzyme B, and IL-10-secreting human regulatory T cells. These results develop an experimental model to investigate human CD4+ T responses in vivo and identify the non-toxic AHR agonist ITE as a potential therapy for promoting immune tolerance in the intestine.
Graphical Abstract
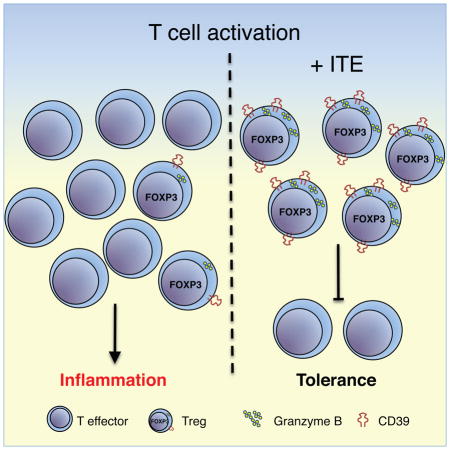
INTRODUCTION
Inflammatory bowel diseases (IBD) are complex inflammatory disorders of the intestine that are generally associated with defects in mucosal immune regulation (Khor et al., 2011). Dampening the inflammatory response to reestablish immune tolerance is a major therapeutic strategy for IBD treatment. Current clinical approaches often involve broad suppression of the immune system, resulting in limited clinical benefit and concomitant risk for opportunistic infections and other side effects (Beaugerie, 2012; Calabrese, 2006). More recently, the use of biologics such as anti-TNF antibodies have proven effective with nearly half of treated patients demonstrating a clinical response (Ben-Horin et al., 2014). However, these beneficial effects are often self-limited and highlight the need for new therapies that promote long-lasting immune tolerance.
Since the initial description that the thymus had critical immunological function (Burnet and Holmes, 1962; Miller, 1961), and subsequent work by many that the thymus had functions independent of elimination of auto-reactive T cells (Le Douarin et al., 1996), there has been a concerted effort to understand the mechanisms of immunological tolerance. Extensive experimentation has defined a group of regulatory T cells (Tregs) that are critical for both central and peripheral tolerance and loss-of-function mutations in the transcription factor, forkhead box P3 (FOXP3), causes a fatal autoimmune disorder in humans known as immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome. Similarly, Foxp3−/− mice exhibit multi-organ auto-inflammatory disease and early mortality (Bennett et al., 2001). In addition, the ability to generate or “induce” Tregs from the pool of helper T cells in the periphery in order to become tolerant to innocuous foreign antigens such as food and commensal microbes at mucosal surfaces is equally important for mucosal immune homeostasis (Atarashi et al., 2013; Hauet-Broere et al., 2003). The mechanisms by which Tregs exert their immunoregulatory function in the intestine is thought to occur via production of soluble mediators and/or direct interactions with other immune cells (Mayne and Williams, 2013). In recent years, a subpopulation of FOXP3− IL-10-secreting iTregs (termed Tr1 cells) has been implicated in the regulation of intestinal inflammation (Groux et al., 1997). This critical role for IL-10 signaling in maintaining intestinal immune homeostasis is best exemplified by the observation that loss-of-function mutations in IL10 or the IL-10 receptor cause IBD in both mice and humans (Glocker et al., 2009; Kuhn et al., 1993). Since Tregs are thought to play a central role in preventing IBD (Josefowicz et al., 2012; Mayne and Williams, 2013; Sakaguchi et al., 2010), generation or expansion of functional Tregs constitutes an attractive therapeutic approach to treat IBD (Canavan et al., 2015) and therapeutic strategies aimed at expanding Tregs in vivo have proven effective in controlling other immune mediated disorders (Koreth et al., 2011; Saadoun et al., 2011) (Desreumaux et al., 2012).
Though several polymorphisms have now been associated with altered risk for IBD, surprisingly only one third of the disease is explained by genetics, suggesting that environmental triggers play an important role. The aryl hydrocarbon receptor (AHR) is a ligand-activated transcription factor that senses certain environmental chemicals and has been shown to exert significant effects on the immune response. Previous work from our group and others demonstrates a role for AHR in the differentiation and function of Tregs and effector T cells by controlling the production of IL-10 and IL-22 (Apetoh et al., 2010; Gandhi et al., 2010; Quintana et al., 2008; Yeste et al., 2014) (Mascanfroni et al., 2015). In mice, activation of AHR suppresses experimental colitis and, although there are no current therapies that target AHR in humans, the expression of AHR is increased in IBD lesions (Arsenescu et al., 2011; Benson and Shepherd, 2011; Chinen et al., 2015; Fukumoto et al., 2014; Furumatsu et al., 2011; Monteleone et al., 2011). Given the importance of Tregs in intestinal homeostasis, coupled with the immunomodulatory effects of IL-10 and IL-22 downstream of AHR activation (Mayne and Williams, 2013; Sonnenberg et al., 2011), AHR is an attractive therapeutic target. The exogenous small molecule 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) has been shown to activate AHR; however, toxicity prevents the use of TCDD for therapeutic intervention in patients. Thus, not only is there a need for non-toxic AHR agonists but also the establishment of new experimental systems to evaluate the effects of AHR activation on human cells in the context of intestinal inflammation in vivo. In this report, we investigated the effects of the mucosal non-toxic AHR agonist 2-(1′H-indole-3′-carbonyl)-thiazole-4-carboxylic acid methyl ester (ITE) (Song et al., 2002) on human T cells in vitro and developed a new experimental model of IBD driven by human CD4+ T cells in humanized mice to assess the efficacy of AHR activation by ITE in vivo. We found that AHR activation by ITE induces suppressive human Tregs expressing of Granzyme B (GZMB) CD39, IL-10, and FOXP3 and prevents T-cell driven colitis in humanized mice. Future studies will address whether ITE is effective in treating established disease, which will be an important step to determine whether ITE would be an attractive candidate for the therapeutic induction of Tregs to treat patients with established IBD.
RESULTS
ITE induces regulatory T cells through a mechanism mediated by AHR
The induction of Tregs and the re-establishment of immune tolerance is a potential approach for the long-term treatment of IBD and other inflammatory diseases and may minimize the deleterious side effects associated with immunosuppressive approaches now in use (Chatenoud, 2015). We and others have shown that AHR activation induces Tregs that suppress the development of experimental autoimmunity and inflammation (Apetoh et al., 2010; Kerkvliet et al., 2009; Quintana et al., 2008). Although AHR activation with TCDD induces functional human Tregs, toxicity concerns exclude its use as therapeutic agent (Gandhi et al., 2010). We therefore investigated the effects of the non-toxic AHR agonist ITE isolated from mucosal tissue (Song et al., 2002). Naïve CD4+ T cells from peripheral blood mononuclear cells (PBMCs) of healthy donors were sorted based on CD4+CD62L+CD45RO− (Figure S1) and co-cultured with autologous unfractionated CD4+ T cells in the presence of anti-CD3/anti-CD28 antibodies with IL2 in the presence of ITE or vehicle control while activation in the presence of the AHR ligand TCDD was used as a positive control. ITE treatment induced suppressive activity in human T cells in vitro that was comparable to the suppressive activity observed using TCDD (Figure 1A). We confirmed the activation of AHR by ITE using quantitative real-time PCR (qPCR) to detect an increase in the expression of the AHR transcriptional target CYP1A1 (Figure 1B). To test whether the induction of suppressive T cells by ITE was mediated by AHR, we targeted AHR using siRNA that abrogated the suppressive effect of ITE and then corroborated these findings using a selective AHR antagonist CH223191 that also blocked ITE-mediated suppression in vitro (Figure 1C).
Figure 1. ITE induces suppressive human regulatory T cells through a mechanism mediated by AHR.

(A) Suppressive activity of human naïve CD4+ T cells activated with plate bound αCD3/αCD28 and IL2 in the presence of vehicle (CTL), ITE, or TCDD. Percent suppression is depicted as the mean ± SEM pooled from at least 3 independent experiments (n = 21).
(B) Relative expression of the AHR target CYP1A1 following stimulation with αCD3/αCD28 and IL2 in the presence of ITE or TCDD and compared to PBS vehicle control. Data are depicted as the mean ± SEM (n = 16).
(C) Requirement for AHR on suppressive effect of ITE. Relative suppression following siRNA-mediated knockdown of AHR compared to scrambled siRNA as the control (left). Percent T cell suppression by ITE in the presence of 10 μM CH223191, a selective antagonist of AHR, compared to DMSO as the vehicle control. Results are depicted as the mean ± SEM pooled from two independent experiments.
(D, E) qPCR analysis on RNA isolated from T cells stimulated with αCD3/αCD28 and IL2 in the presence of vehicle, ITE, or TCDD. Target fold change was calculated using vehicle control with the dashed line representing the normalized control value of 1 and the mean depicted ± SEM pooled from 3 independent experiments (n ≥ 7).
(F, G) Representative flow cytometric analysis of TBX21, GATA3, RORC, FOXP3, IL-10, GZMB, and CD39 protein expression following T cell activation using αCD3/αCD28 and IL2 in the presence or absence of ITE for 6 days. Each flow panel is representative of at least 2 independent experiments.
(H) Requirement for GZMB and CD39 on the suppressive activity of ITE using a blocking antibody against CD39 or the GZMB inhibitor AAD-CMK (left) and showing the requirement for GZMB and CD39 for TCDD-mediated suppression as a positive control (right) (n = 21).
(I) Role of IL-10 in ITE-mediated T cell suppression using 2.5 μgml−1 αIL-10 blocking antibody shown as percent suppression (left) and the ability of αIL-10 antibody to inhibit STAT3 phosphorylation in human T cells following a 20 minute stimulation with 20 ngml−1 IL-10 (right). * P < 0.05, ** P < 0.01, *** P < 0.001, n.s. = not significant
To further investigate activation of AHR by ITE in human T cells we analyzed the expression of cytokines and lineage-specific molecules and transcription factors associated with different CD4+ T cell subpopulations by qPCR. ITE treatment decreased expression of TBX21, RORC, and IL23R but not GATA3 (Figures 1D & S2), which correlates with reduced effector T cell subsets. In contrast, T cell activation in the presence of ITE led to a significant upregulation of Treg-associated gene transcripts including FOXP3, IL10, GZMB, ENTPD1, and IKZF3 (Groux et al., 1997; Quintana et al., 2012; Sakaguchi et al., 2010) (Figures 1E & S2). ITE-treatment had a modest or no effect on the expression of MAF, BCL6, BLIMP, IL12RB1, or IL12RB2 (Figure S2). Based on the transcriptional analysis we then profiled ITE-treated CD4+ T cells to assess protein expression for TBX21, RORC, GATA3, FOXP3, IL-10, GZMB, and ENTPD1 by flow cytometry and found it to be consistent with the transcriptional analysis (Figures 1F–G). While many of these markers are consistent with traditional Foxp3−IL-10+ Tr1 regulatory cells, a large percentage of ITE-treated T cell expressing CD39 were also positive for FOXP3+ suggesting that ITE induces a mixed population of regulatory cells with only some possessing a conventional Tr1 phenotype (Figure S3).
The ability of Tregs to control the activity of effector T cells can occur via several mechanisms (Josefowicz et al., 2012; Sakaguchi et al., 2010). Specifically, GZMB, CD39, and IL-10 are known to contribute to the suppressive activity of Tr1 cells (Gandhi et al., 2010; Grossman et al., 2004; Mascanfroni et al., 2015). In order to determine if the suppressive effect of ITE was mediated by GZMB, we first performed suppression assays in the presence of the GZMB inhibitor benzyloxycarbonyl-Ala-Ala-Asp-chloromethylketone (AAD-CMK) and found that ITE- and TCDD-mediated suppression was abrogated (Figure 1H). Since CD39 can also participate in the suppression of effector T cell responses, we inhibited CD39 using neutralizing antibodies in ITE-treated T cells. Similar to AAD-CMK, treatment with anti-CD39 blocked ITE- and TCDD-mediated suppressive activity in vitro (Figure 1H). Although IL-10 was upregulated by ITE by qPCR, we tested whether antibody-mediated blockade of IL-10 would inhibit suppression. While the anti-IL-10 antibody effectively blocked the phosphorylation of STAT3 in response to IL-10 (confirming a block in IL-10R receptor signaling) this had no impact on the suppressive effect of ITE (Figure 1I). Collectively, these results demonstrate that AHR activation by ITE induces functional human regulatory T cells that suppress effector T cell proliferation in a GZMB- and CD39-dependent manner.
ITE induces functional human FOXP3+ Tregs in the presence of TGFβ1
TGFβ1 is expressed by both immune and non-immune cells in the intestinal lamina propria and regulates the immune response through several mechanisms, one of which is the differentiation, maintenance, and function of FOXP3+ Tregs (Rubtsov and Rudensky, 2007). Although TGFβ1 promotes the differentiation of functional FOXP3+ Tregs in mice (Chen et al., 2003), TGFβ1-induced FOXP3+ human T cells do not exhibit suppressive activity (Tran et al., 2007). To mimic the TGFβ1 enriched microenvironment of the intestine, we previously showed that AHR activation by TCDD could cooperate with TGFβ1-induced signaling to induce suppressive FOXP3+ human Tregs in vitro (Gandhi et al., 2010). Therefore, we investigated whether human Tregs induced using TGFβ1 in the presence of ITE would mimic the suppressive effects of TCDD. Similar to our findings with TCDD, human naïve CD4+ T cells activated in the presence of TGFβ1 and ITE were suppressive in vitro (Figure 2A). Since we previously observed decreased expression of CD4+ T cell linage-specific transcription factors following ITE treatment (Figure 1D), we tested if these molecules were downregulated under Treg inducing conditions. CD4+ T cells were treated with vehicle, TGFβ1, or TGFβ1 in the presence of ITE or TCDD. While TGFβ1 significantly reduced expression of TBX21 and GATA3, no further decrease was observed if ITE or TCDD was present (Figure 2B). TGFβ1 treatment increased RORC expression that was not further enhanced in the presence of ITE and TCDD, however these data did not achieve statistical significance (Figure 2B). The expression of IL23R was elevated over control when stimulated with TGFβ1 and ITE, which was not observed in T cells treated with TGFβ1 alone or in combination with TCDD (Figure 2B). We then evaluated protein expression of these qPCR targets by flow cytometry in T cells activated in the presence of vehicle control, TGFβ1, or TGFβ1 and ITE and observed a reduction in GATA3 proteins levels and increased expression of RORC while TBX21 was slightly elevated over control only in the TGFβ1 and ITE-treated cells (Figure 2C).
Figure 2. ITE induces functional human FOXP3+ Tregs in the presence of TGFβ1.

(A) Suppressive activity of human naïve CD4+ T cells activated with αCD3/αCD28 and TGFβ1 in the presence of CTL, ITE, or TCDD. Percent suppression is depicted as the mean ± SEM pooled from at least 2 independent experiments with 10 unique donors.
(B) qPCR analysis on RNA isolated from T cells stimulated with αCD3/αCD28 and IL2 in the presence of CTL, TGFβ1, TGFβ1 + ITE, or TGFβ1 + TCDD. Target fold change was calculated using vehicle control with the dashed line representing the normalized control value of 1 with relative expression shown as the pooled mean ± SEM (n ≥ 10).
(C) Representative flow cytometric analysis of TBX21, GATA3, and RORC protein expression in T cells following activation using αCD3/αCD28 and IL2 in the presence of CTL, TGFβ1, or TGFβ1 + ITE from 3 independent experiments.
(D) qPCR analysis on RNA isolated from T cells stimulated with αCD3/αCD28 in the presence of CTL, TGFβ1, TGFβ1 + ITE, or TGFβ1 + TCDD. Target fold change was calculated using vehicle control with the dashed line representing the normalized control value of 1 with relative expression shown as the pooled mean ± SEM (n ≥ 7). ‡ P < 0.05 compared to control.
(E) Representative flow cytometric analysis of FOXP3, IL-10, CD39, and GZMB protein expression in T cells following activation using αCD3/αCD28 and IL2 in the presence of CTL, TGFβ1, or TGFβ1 + ITE from 2 independent experiments.
(F) Effect of CD39 blockade using CD39 blocking antibodies on the suppressive activity of TGF+ITE (left panel) or TGF+TCDD (right panel) with the mean depicted ± SEM (n ≥ 12). * P < 0.05, ** P < 0.01, *** P < 0.001.
Activation of AHR also upregulates the expression of the Ikaros family transcription factor AIOLOS (encoded by IKZF3) that silences the IL2 promoter and, along with IL-10 and FOXP3, is associated with suppressive human Tregs (Gandhi et al., 2010; Quintana et al., 2012; Sakaguchi et al., 2010). We analyzed Tregs induced with TGFβ1 alone or in the presence of ITE or TCDD and quantified the expression of IKZF3, IL10, and FOXP3. While FOXP3 was elevated in all treatment conditions compared to control, IL10 and IKZF3 were upregulated by TGFβ1 and ITE (Figures 2D & S4). Flow cytometric analysis largely supported the transcriptional analysis whereby increases in FOXP3, IL-10 and CD39 were also observed (Figure 2E).
Since CD39 and GZMB were required for ITE-induced AHR dependent T cell suppression in vitro in the absence of TGFβ1 (Figure 1H), we determined if GZMB and ENTPD1 were also upregulated following AHR activation with ITE or TCDD under Treg-inducing conditions with TGFβ1. While there was no increase in GZMB transcript or protein in vitro (Figures 2D–E), ENTPD1 transcript levels trended higher (Figure 2D) while cell surface expression of CD39 was increased (Figure 2E). We then tested whether CD39 was required for the suppressive effect of ITE in the presence of TGFβ1. Neutralizing CD39 abrogated the suppressive activity of T cells treated with TGFβ1 and ITE similar to that observed for TGFβ1 and TCDD and consistent with a role for CD39 in this suppressive function (Figure 2F). Taken together, these data demonstrate that AHR activation with ITE in the presence of TGFβ1 induces functional human Tregs that suppress effector T cells in a CD39-dependent manner in vitro.
A model of T-cell dependent colitis model in humanized mice
Most experimental immunotherapies successful in treating experimental autoimmunity in animal models show limited success in human clinical trials (Hay et al., 2014; Persidis, 1999). One important contributor to the limited translational application of experimental findings in mice into successful therapies for human autoimmunity is the lack of models to study the human immune system in vivo. Over the past decade immunodeficient mice have been developed that are capable of engrafting human immune cells (Rongvaux et al., 2014; Shultz et al., 2005; Traggiai et al., 2004). Although sub-optimal adaptive immune responses are often observed in many humanized murine systems, we recently described NOD.Prkdcscid.Il2rg−/− (NSG) mice that lack murine major histocompatibility complex (MHC) class II and instead express human leukocyte angtigen-DR1 (HLA-DR1) under the control of the murine MHC class II promoter (NSGAboDR1 mice) (Goettel et al., 2015). These mice intrinsically lack murine lymphocytes as well as NK cells and when made immune replete using human CD34+ hematopoietic stem cells (HSCs) the mice displayed improved human CD4+ T cell responses (Goettel et al., 2015). In order to specifically evaluate CD4+ T cell responses, we used a reductionist approach by reconstituting NSGAboDR1 mice with human CD4+ T cells isolated from allelically-matched HLA-DR1+ donors. To evaluate the effects of candidate drugs on human CD4+ T cells in vivo, we adapted an established experimental model of intestinal inflammation using the hapten 2,4,6-trinitrobenzenesulfonic acid (TNBS) that is largely mediated by T cells (Neurath et al., 1995). Reconstituted NSGAboDR1 mice were sensitized with TNBS to prime antigen-specific T cells and one week post-sensitization mice were administered a single rectal enema containing 0.25 mg TNBS in 50% ethanol (EtOH) or 50% EtOH as a vehicle control (Figure 3A). While reconstituted NSGAboDR1 mice challenged with TNBS exhibited significant weight loss 3 days post-challenge, weight loss was not readily observed in reconstituted mice challenged with EtOH or in non-reconstituted mice challenged with TNBS (Figure 3B). Consistent with these observations, blinded histological evaluation of colonic sections stained with hematoxylin and eosin (H&E) revealed extensive crypt and goblet cell loss with edema, fibrosis, and transmural inflammation in TNBS-treated NSGAboDR1 mice reconstituted with human CD4+ T but not in control mice (Figure 3C), with a histological colitis score that was significantly higher in TNBS-treated reconstituted NSGAboDR1 mice compared to controls (Figure 3D). This correlated with an increase in CD4+ T cells infiltrating in the colonic lamina propria in TNBS-treated, but not EtOH-treated, mice (Figure 3E).
Figure 3. TNBS-induced colitis mediated by human CD4+ T cells.

(A) Schematic of human CD4+ T cell reconstitution and TNBS experimental colitis in NSGAboDR1 mice.
(B) Change in body weight in NSGAboDR1 mice reconstituted with human CD4+ T cells 3 days following rectal administration of ethanol (EtOH) as a vehicle control (n = 6), TNBS (n = 9), or NSGAboDR1 mice without human T cells administered TNBS (n = 7). Each dot represents an individual animal with the mean depicted ± SEM. Data shown is pooled from 3 independent experiments and is representative of more than 6 experiments with similar results.
(C) Endoscopic images of representative mice 3 days following TNBS or EtOH challenge (top) with representative H&E stained colonic sections (bottom) from 3 independent experiments (10X magnification). Scale bar = 200 μm.
(D) Colitis scoring of H&E stained colon sections 3 days following the TNBS or EtOH rectal challenge. Scores for each individual mouse are shown with the mean for each group depicted ± SEM.
(E) Representative microscopic images of formalin-fixed paraffin-embedded colonic sections stained for human CD3 from NSGAboDR1 mice previously reconstituted with human CD4+ T cells treated with EtOH or TNBS (left) (n = 4) and quantified (right) with bars representing the mean ± SEM (20X magnification). Scale bars = 200 μm.
(F) qPCR analysis on RNA isolated from colonic tissue of reconstituted NSGAboDR1 mice treated with TNBS or EtOH (n = 6). Human cytokines were normalized to hypoxanthine phosphoribosyltransferase (HPRT) and the fold change was compared to a pooled human RNA control sample using the formula 2− (Ct(target) − Ct (HPRT)). Bars represent the mean ± SEM.
(G) Representative flow cytometric analysis of human CD4+ T cells isolated from the colonic lamina propria of NSGAboDR1 mice stimulated with phorbol myristate acetate (PMA) and ionomycin for 4 hours in the presence of GolgiStop and stained for intracellular TNF and IFNγ (top) and quantified (bottom) from 3 independent experiments (n ≥ 5). * P < 0.05, ** P < 0.01, *** P < 0.001.
In the standard model, TNBS-induced colitis leads to the upregulation of several pro-inflammatory cytokines including Tnf, Il2, Il12a, and Ifng (Hollenbach et al., 2005; Neurath et al., 1997). We analyzed the expression of human cytokines in the colons of TNBS or EtOH treated NSGAboDR1 mice by qPCR and found increased expression of TNF, IFNG, IL2, IL4, and IL17A (Figure 3F). Moreover, human T cells recovered from the colonic lamina propria of TNBS treated mice showed increased production of TNF and IFNγ following ex vivo stimulation with phorbol 12-myristate 13-acetate (PMA) and ionomycin (Figure 3G). Collectively, these data show that human CD4+ T cells mediate disease pathology in TNBS-induced colitis in humanized NSGAboDR1 mice. Moreover, these data support the use of this humanized model to assess the efficacy of therapeutics that target human CD4+ T cells to promote intestinal immune homeostasis. Consistent with our results, Neurath and colleagues recently showed that the homology of MAdCAM-1 between mice and humans permits binding of human α4β7 integrin to murine MAdCAM-1 and that human CD4+ T cells injected into DSS-treated NSG mice migrate to the colon (Fischer et al., 2015).
ITE prevents T-cell driven experimental colitis in humanized mice
To investigate the effects of ITE on human T cells in vivo we utilized the humanized mouse model described above and one-week following engraftment of human CD4+ T cells mice were administered daily injections of ITE or PBS as a vehicle control for five days. The day following the last ITE or PBS injection, mice were sensitized to TNBS and one-week later administered a single TNBS rectal challenge (Figure 4A). The administration of ITE to humanized mice followed by the TNBS challenge resulted in a trend in protection against weight loss compared to control (Figure 4B). Histological evaluation of H&E stained colonic sections showed a significant reduction in the severity of inflammation in mice receiving ITE that corresponded to a reduction in colitis score (Figures 4C). This reduction in colitis by ITE correlated with an increase in the frequency of regulatory human T cells in the colon based on increased expression of GZMB, CD39, IL-10, and FOXP3 by flow cytometry (Figures 4D). These data are highly similar to the effects of ITE on human CD4+ T cells observed in vitro (Figure 1). We further investigated the consequences of ITE treatment on human T cells in vivo by analyzing the transcriptional profile of CD4+ T cells isolated from the spleens of ITE or PBS treated mice using Nanostring nCounter arrays. ITE treatment upregulated the expression of several transcription factors and molecules linked to anti-inflammatory pathways in human T cells including IL10, IL21, IL22, IL32, GZMB, IKZF2, and IKZF3 (Figure S4A) (Evans et al., 2014; Gandhi et al., 2010; Quintana et al., 2012; Sakaguchi et al., 2010). While ITE induced cytokine transcripts known to be both pro- and anti-inflammatory, pathway analysis and functional gene clustering determined that the most affected gene sets were linked to several pathways relevant for IBD pathogenesis with significant downregulation in TNFR1, death receptor signaling, and NF-κB signaling (Figure S4B). Conversely, ITE treatment led to an upregulation of IL22 (Figure S4B), a known target of AHR linked to the protection of the intestinal epithelium (Sonnenberg et al., 2011; Yeste et al., 2014).
Figure 4. ITE prevents T cell-driven experimental colitis in humanized mice.

(A) Schematic of ITE administration in TNBS experimental colitis in NSGAboDR1 mice reconstituted with human CD4+ T cells.
(B) Body weight change 3 days following TNBS rectal challenge in NSGAboDR1 mice reconstituted with HLA-DR1 matched human CD4+ T cells treated with PBS (n = 8) or ITE (n = 13). Each dot represents and individual mouse with the mean depicted ± SEM pooled from 3 independent experiments using 2 unique HLA-DR1 matched healthy donor sources.
(C) Representative H&E stained colonic sections 3 days post-TNBS challenge of reconstituted NSGAboDR1 mice treated with PBS (n = 8) or ITE (n = 13) (left) and colitis score quantified with the mean for each group depicted ± SEM pooled from 3 independent experiments with 2 unique donor sources (right). Images are 20X magnification. Scale bars = 100 μm.
(D) Quantified flow cytometric data of human CD4+ T cells isolated from the spleen and colonic lamina propria of PBS or ITE treated NSGAboDR1 mice stained for human CD45 (hCD45), GZMB, CD39, FOXP3 (top), with representative dot plots shown below. Intracellular IL-10 staining was performed on T cells isolated from spleen and colonic lamina propria and stimulated ex vivo with phorbol myristate acetate (PMA) and ionomycin for 4 hours in the presence of GolgiStop. Bars represent the mean ± SEM pooled from 2 independent experiments using 2 unique HLA-DR1 matched healthy donors.
(E) Schematic depicting injection of in vitro generated autologous ITE-induced suppressive T cells prior to rectal challenge with TNBS in NSGAboDR1 mice previously reconstituted with matched donor CD4+ T cells.
(F) Flow cytometry dot plot showing the recovery of in vitro generated autologous Tregs (labeled with CellTrace Violet prior to intraperitoneal injection of 2×106 labeled cells) from spleens of humanized mice.
(G) Body weight change 3 days following TNBS rectal challenge in NSGAboDR1 mice reconstituted with HLA-DR1 matched human CD4+ T cells and injected with PBS (n = 9) or autologous in vitro ITE-generated suppressive cells (n = 7) 1 day prior to TNBS rectal challenge. Each dot represents and individual mouse with the mean depicted ± SEM pooled from 2 independent experiments using 2 unique HLA-DR1 matched healthy donor sources.
(H) Representative H&E stained colonic sections 3 days post-TNBS challenge of reconstituted NSGAboDR1 mice injected with PBS (n = 9) or CellTrace violet-labeled autologous ITE-induced Tregs (n = 7) (left) and colitis score quantified with the mean for each group depicted ± SEM pooled from 2 independent experiments (right). Images are 20X magnification. Scale bars = 100 μm. * P < 0.05.
Expression of AHR is not restricted to immune cells and although our data suggest that the suppressive effects of ITE in vivo were likely mediated by the effects on human CD4+ T cells, we cannot exclude the possibility that the therapeutic benefit of ITE in TNBS-induced colitis is mediated by the activation of AHR in murine cells. In order to directly test whether AHR activation by ITE in human T cells was sufficient to attenuate TNBS colitis in humanized mice, we first reconstituted NSGAboDR1 mice with HLA-matched donor CD4+ T cells. We then initiated in vitro cultures of autologous human CD4+ T cells as before to induce suppressive T cells. After 6 days of culture we labeled the autologous ITE-treated T cells with CellTrace Violet and injected 2×106 cells per mouse. We then administered the rectal TNBS challenge the next day (Figure 4E). We euthanized the mice 3 days post-TNSB challenge and confirmed the presence of the in vitro cultured Tregs based on CellTrace Violet staining (Figure 4F). Humanized mice receiving ITE-treated cells more readily recovered to their initial body weight with less colonic inflammation and a significantly improved colitis score (Figures 4G–H). Thus, autologous regulatory T cells generated in vitro were sufficient to protect against TNBS-mediated colitis in humanized mice.
DISCUSSION
Current strategies for the long-term treatment of IBD and other inflammatory diseases often depend on broad immunosuppression that can cause deleterious side effects. Unfortunately the majority of experimental immunotherapies developed using rodent models have had limited success in human clinical trials (Hay et al., 2014; Persidis, 1999). This is likely due, at least in part, to inherent physiological differences in immune cells between mice and humans. We recently described improved human T cell responses in NSGAboDR1 mice that express human, but not murine, MHC class II. Moreover, when these mice were reconstituted using HSCs isolated from an IPEX patient lacking functional FOXP3 and Tregs (Goettel et al., 2015), the mice developed a multi-organ inflammatory syndrome with the development of autoantibodies analogous to patients. We expanded upon this model and demonstrated that in the presence of normal human T cells, TNBS administration to NSGAboDR1 mice resulted in severe intestinal inflammation. Since the re-establishment of immune tolerance in immune-mediated diseases has shown early promise in the clinical setting through the induction or expansion of autologous Tregs (Desreumaux et al., 2012; Koreth et al., 2011), we investigated whether an endogenous ligand to AHR could induce human regulatory T cells and have a therapeutic effect in a humanized mouse model of intestinal inflammation. We and others have previously shown that AHR activation induces Tregs that suppress the development of inflammation and experimental autoimmunity in mice (Apetoh et al., 2010; Kerkvliet et al., 2009; Mascanfroni et al., 2015; Quintana et al., 2008; Quintana et al., 2010; Vogel et al., 2008; Wu et al., 2011; Yeste et al., 2014; Yeste et al., 2012; Zhang et al., 2009). Although, AHR activation with TCDD induces functional human Tregs in vitro, toxicity prevents the use of TCDD as a therapeutic in humans (Gandhi et al., 2010). Here we demonstrate that the non-toxic AHR agonist ITE induces functional human Tregs that suppress effector T cell proliferation in a CD39- and Granzyme B-dependent manner in vitro. Furthermore, we employed the TNBS human CD4+ T cell mouse model of colitis described above and demonstrated that ITE promoted mucosal immune homeostasis and was protective against colitis development.
For many inflammatory diseases, the effector cytokine profile of CD4+ T cells can be informative and, in some cases, indicative of the T cell subset(s) involved in disease pathogenesis. The polarization of CD4+ T cells into these specific subsets is highly regulated by transcription factors that drive Th1, Th2, and Th17 differentiation, namely TBX21, GATA3, and RORC respectively. We found that ITE, like the toxic AHR ligand TCDD (Gandhi et al., 2010), downregulated Th1 and Th17 transcription factors TBX21 and RORC, while the expression of molecules associated with Treg suppressive function including IL10, FOXP3, GZMB, and ENTPD1, were increased. This suggests that the suppression of effector T cells by ITE likely occurs by restricting differentiation of Th1 and Th17 effector subsets in addition to inducing immunoregulatory molecules associated with Tregs. Since IL-10, GZMB, and CD39 are known to participate in T cell suppression by Tregs, we tested whether the suppressive effects of ITE were mediated by any of these three molecules. ITE-mediated suppression in the absence of TGFβ1 was dependent on GZMB and CD39 as pharmacological or antibody-mediated inhibition blocked suppression. Interestingly, under Treg inducing conditions with TGFβ, ITE did not alter the expression of GZMB or ENTPD1 whereas protein levels of CD39 were increased at the cell surface. This suggests that AHR activation by ITE may mobilize intracellular pools of CD39 to the plasma membrane. Although CD39 is an important molecule in Tr1 cell differentiation and function (Mascanfroni et al., 2015) and was involved in the suppressive effect of ITE in vitro, it appears that the suppressive cells induced by ITE may be a mixed population of FOXP3− Tr1 cells expressing traditional markers CD39, GZMB, and IL-10 as well as FOXP3+ Tregs. Interestingly, we also observed a population of CD39+ cells that were also positive for FOXP3 and may constitute a “Tr1-like” cell population previously described to possess a regulatory phenotype (Borsellino et al., 2007; Deaglio et al., 2007; Moncrieffe et al., 2010).
Although IBD is a complex disorder triggered by genetic, environmental, and microbial factors, genome-wide association studies have identified over 160 polymorphisms associated with altered risk for IBD (Jostins et al., 2012). Many of these mutations are known to regulate immune responses with several being enriched in immune cells, in particular CD4+ T cells and dendritic cells (Jostins et al., 2012). Although polymorphisms in AHR have not yet been associated with IBD, AHR is known to play a central role in the regulation of intestinal inflammation and is upregulated in the inflamed gut (Arsenescu et al., 2011; Benson and Shepherd, 2011; Chinen et al., 2015; Fukumoto et al., 2014; Furumatsu et al., 2011; Huang et al., 2013; Ji et al., 2015; Mascanfroni et al., 2015; Monteleone et al., 2011; Qiu et al., 2013; Quintana et al., 2012; Singh et al., 2011; Takamura et al., 2011; Takamura et al., 2010; Yeste et al., 2014). We and others previously showed that in T cells, AHR controls the production of IL-10 (Apetoh et al., 2010; Gandhi et al., 2010; Mascanfroni et al., 2015; Wu et al., 2011) and IL22 (Quintana et al., 2008; Veldhoen et al., 2009; Veldhoen et al., 2008; Yeste et al., 2014) to modulate and promote immunological tolerance in the intestine. Consistent with these previous findings, our present data demonstrates that AHR activation by ITE induces a transcriptional program in human CD4+ T cells that promotes IL22 and IL10 expression in vivo. Moreover, ITE treatment led to an increase in GZMB, CD39, IL-10, and FOXP3 particularly in the colon of humanized mice and attenuated intestinal inflammation induced by TNBS. Although our in vivo data are consistent with in vitro findings for ITE inducing suppressive CD4+ T cells, we cannot exclude the possibility that the protective effects of ITE in vivo could also be mediated by other cells types that express AHR including intestinal epithelial cells and murine innate immune cells (Esser and Rannug, 2015). Nevertheless, we showed that in vitro generated autologous ITE-treated T cells could be administered just prior to the TNBS challenge and protect against TNBS-induced colitis in humanized mice. Further investigations on ITE using human CD4+ T cell reconstituted NSGAboDR1 mice with established TNBS-induced colitis are warranted prior to any clinical application aimed at treating IBD.
In conclusion, we have demonstrated that AHR activation with the non-toxic agonist ITE induces, in the presence or absence of TGFβ1, human regulatory T cells in vitro that can suppress effector T cell proliferation in a CD39- and GZMB-dependent manner. Given the inability to readily assess therapeutics in patients, we developed a humanized murine system to directly assess the ability of ITE, as well as other agents, to modulate human CD4+ T cell responses in vivo. We showed that TNBS-induced colitis in humanized mice requires human CD4+ T cells and that ITE attenuated colitis development, promoting immunological tolerance. This model has the advantage that it does not require full human immune reconstitution using HSCs and can be established using T cells from healthy controls as well as patient cells. The use of this model will facilitate evaluation of potential treatments for IBD and, more importantly, investigations into the inflammatory response of human CD4+ T cells from patients with mutations in loci associated with altered risk for IBD including IL10R or IL23R (Duerr et al., 2006; Glocker et al., 2009; Sarin et al., 2011). This may enable clinicians to stratify patients and distinguish responders versus non-responders leading to tailored therapeutics.
EXPERIMENTAL PROCEDURES
Antibodies and reagents
The following flow cytometry antibodies were obtained through Biolegend (San Diego, CA): FOXP3, clone: 259D; CD39, clone: A1; CD45, clone: HI30; CD4, clone: OKT4; GZMB, clone: GB11; TNF, clone: MAb11; and IFNG, clone: 4S.B3. The following flow cytometry antibodies were obtained through eBioscience (San Diego, CA): GATA3, clone: TWAJ; TBX21, clone: eBio4B10; RORC, clone: AFKJS-9. IL-10, clone: JES3-19F1 was obtained through BD Bioscience (San Jose, CA). Anti-CD3 clone: OKT3 and anti-CD28 clone CD28.6 were used for in vitro stimulation and purchased through eBioscience. TGFβ1 was obtained from R&D Systems (Minneapolis, MN). Human recombinant IL2 was obtained from the AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases (NIAID). Annexin V-PE/FITC and 7-AAD were obtained from BD Biosciences.
Isolation of naive T cells
Blood samples were collected from healthy controls upon informed consent. The institutional review board at Brigham and Women’s Hospital approved all the procedures described in this work. PBMCs were obtained by Ficoll density gradient. Total CD4+ T cells were purified using Miltenyi Biotec AutoMACS, and CD62Lhigh and CD45RO− T cells were purified by FACS sorting using FACSAria (BD Biosciences) to typically obtain 96–98% purity in post-sort analysis.
T cell differentiation
FACS-sorted naïve T cells were activated with plate bound antibodies to CD3 (1 μgml−1), soluble CD28 (1 μgml−1) and human recombinant IL2 (50 U/ml) with or without TGFβ1 (1 ngml−1), in the presence or absence of 100 nM ITE. After 6 days of differentiation, the cells were resorted to exclude dead cells and were used for suppression assays or analyzed by FACS or qPCR.
Suppression assays
Responder T cells (CD4+ T cells) were activated with beads coated with antibodies to CD3 and CD28 (1 μg/107) for 5 days in the presence of Tregs at a 2:1 (responder: regulatory) ratio. Cells were pulsed with 3H-thymidine (1 μCi/well) for 16–24 hours at the end of the incubation period. Anti-IL-10 (clone: 25209) was purchased from R&D Systems (Minneapolis, MN) and used at 2.5 μgml−1. CH223191 was purchased from Tocris (Bristol, United Kingdom) and was used at 10 μM.
Quantitative real-time PCR
RNA was extracted with Qiagne RNAeasy columns (Valencia, CA); cDNA was prepared following the manufacturer’s instructions (Applied Biosystems, Foster City, CA) and used as template for real-time PCR. All the primers and probes in this work were provided by Applied Biosystems, and were used on the GeneAmp 7500 Sequence Detection System (Applied Biosystems). Expression was normalized to the expression of GAPDH. All murine qPCR primers and reagents were obtained from Applied Biosystems.
For humanized mouse studies, qPCR was performed with SYBR Green (Bio-Rad, Hercules, CA) using a CFX96 real-time PCR (Bio-Rad) machine on cDNA generated with the iScript cDNA kit (Bio-Rad) on 1 μg total RNA isolated from whole tissue homogenized in TRIzol (Life Technologies, Carlsbad, CA). 2 μM of each human target primer was used in the reaction and quantified by normalizing the cycle threshold (Ct) of the target gene to the Ct value of HPRT and the fold change was compared to a pooled human RNA control sample using the formula 2−(Ct(target) − Ct (HPRT)).
Sequences for human targets are as follows:
IFNG For: 5′-TCGGTAACTGACTTGAATGTCCA, Rev: 5′-TCGCTTCCCTGTTTTAGCTGC,
TNF For: 5′-GAGGCCAAGCCCTGGTATG Rev: 5′-CGGGCCGATTGATCTCAGC,
IL10 For: 5′-GACTTTAAGGGTTACCTGGGTTG Rev: 5′-TCACATGCGCCTTGATGTCTG,
IL17A For: 5′-TCCCACGAAATCCAGGATGC Rev: 5′-GGATGTTCAGGTTGACCATCAC,
IL4 For: 5′-CGGCAACTTTGTCCACGGA Rev: 5′-TCTGTTACGGTCAACTCGGTG,
IL2 For: 5′-AACTCCTGTCTTGCATTGCAC Rev: 5′-GCTCCAGTTGTAGCTGTGTTT,
HPRT For: 5′-CCTGGCGTCGTGATTAGTGAT Rev: 5′-AGACGTTCAGTCCTGTCCATAA.
TNBS-induced colitis and ITE treatment in humanized mice
The generation of NOD.Cg-PrkdcscidIl2rgtm1WjlH2-Ab1tm1Doi.Tg(HLA-DRA*0101, HLA-DRB1*0101) mice (NSGAboDR1) was previously described (Goettel et al., 2015). Mice were maintained in autoclaved cages with autoclaved food and water ad libitum in the specific-pathogen free facility at Boston Children’s Hospital. All animal experiments were approved and conducted according to the institutional guidelines at Boston Children’s hospital.
6–8 week old NSGAboDR1 mice were injected intraperitoneally with 2×106 human CD4+ T cells isolated from a healthy donor that was positive for the HLA-DRB*01:01 allele were purchased from StemCell Technologies (Vancouver, BC, Canada). Two-three weeks later mice were bled and screened for human T cell reconstitution and mice exhibiting greater than 10% human chimerism in peripheral blood were selected for experimental groups. Mice were first sensitized by applying 150 microliters of a 2.5% TNBS (Sigma-Aldrich, St Louis, MO) solution in 50% ethanol to a 1 cm2 patch bare skin at the base of the neck. Seven days later, mice were anesthetized via a single intraperitoneal injection of saline containing 100 mgkg−1 ketamine and 10 mgkg−1 xylazine. Anesthetized mice were held inverted by hand and a sterile lubricated 3.5 F soft silicon catheter inserted into the colon to a distance of 3–4 cm. Mice were given a single enema containing 0.25 mg of TNBS in 50% ethanol 50% phosphate buffered saline (PBS) mixture in a volume of 50 microliters. Mice were then held inverted for 30 seconds and returned to their cage. Mice were weighed daily and colitis assessed 3 days following the TNBS rectal challenge. Histological assessment was performed and scored using a modified system previously described (Scheiffele and Fuss, 2002). For ITE treated mice, 200 μg of ITE dissolved in PBS was administered intraperitoneally starting 1 week following injection of human CD4+ T cells for five consecutive days. Mice were then sensitized and challenged as above.
Analysis of cytokine production by T cells in humanized mice
Total splenocytes and colonic lamina propria cells were plated at a concentration of 1×106 cells in RPMI 1640 (Life Technologies) supplemented with 10% FBS, 2 mM L-glutamine (Life Technologies), 1 mM sodium pyruvate (Life Technologies), 100 mM non-essential amino acids (Life Technologies), 10 mM HEPES (Life Technologies), 55 μM 2-mercaptoethanol (Life Technologies), and 100 U/ml Penicillin/Streptomycin (Life Technologies) (hereafter referred to as T cell media). Cells were stimulated with stimulated with 20 ngml−1 phorbol myristate acetate (PMA) (Sigma-Aldrich) and 1 μgml−1 ionomycin (Sigma-Aldrich) for 4 hours at 37o C in the presence of 10 μgml−1 GolgiStop (BD Biosciences). Cells were collected and washed 2X with FACS buffer (PBS supplemented with 2% FBS and 0.1% NaN3). Cell surface staining for human CD4 was performed for 30′ at RT, washed 2X with FACS buffer, then fixed using BD Cytofix/Cytoperm (BD Biosciences) following the manufacture’s protocol. Cells were then stained with IFNγ and TNF for 45′ at RT. Cells were washed 2X with FACS buffer and intracellular cytokine production detected using a 3-laser FACSCanto II (BD Biosciences) flow cytometer.
Immunohistochemistry
Histopathology was carried out on formalin-fixed paraffin-embedded colonic tissue sections stained with an anti-human CD3 antibody (cat# A0452, Dako, Carpinteria, CA). Images were acquired using an Olympus microscope mounted with an Olympus DP70 digital camera and DP-Manager software (Olympus, Melville, NY) and quantified using ImageJ software (National Institutes of Health).
Statistical analysis
Statistical analyses were performed using the Prism software (Graph Pad software, La Jolla, CA). t-test were used in Figs. 1, 2, 3E–G & 4B, C, D, G, H, & S2. ANOVA with multiple comparisons was used in Figs. 3B, D. P < 0.05 were considered significant.
Supplementary Material
Acknowledgments
We would like to thank the BIDMC Research Histology core. This work was supported by grants AI075285 and AI093903 from the National Institutes of Health (NIH) National Institute Of Allergy And Infectious Diseases and by a grant from the Harvard Institute of Translational Immunology/Helmsley Trust (F.J.Q. and S.B.S), the NIH National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) DK106311 and the Crohn’s and Colitis Foundation of America CDA 352644 (J.A.G.), NIH NIDDK DK034854 and the Helmsley Charitable Trust, and the Wolpow Family Chair in IBD Treatment and Research (S.B.S.).
Footnotes
AUTHOR CONTRIBUTIONS
J.A.G. and A.Y. performed in vivo experiments. R.G., J.E.K., S.S., D.S.S and G.M, performed in vitro expression and suppression experiments, A.E.G. performed IL10R signaling experiments, B.P. performed bioinformatics analysis, H.L.W. contributed to the initial experimental design, J.A.G., R.G., S.B.S and F.J.Q. wrote and edited the manuscript, S.B.S. and F.J.Q. supervised the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apetoh L, Quintana FJ, Pot C, Joller N, Xiao S, Kumar D, Burns EJ, Sherr DH, Weiner HL, Kuchroo VK. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat Immunol. 2010;11:854–861. doi: 10.1038/ni.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenescu R, Arsenescu V, Zhong J, Nasser M, Melinte R, Dingle RW, Swanson H, de Villiers WJ. Role of the xenobiotic receptor in inflammatory bowel disease. Inflammatory bowel diseases. 2011;17:1149–1162. doi: 10.1002/ibd.21463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- Beaugerie L. Inflammatory bowel disease therapies and cancer risk: where are we and where are we going? Gut. 2012;61:476–483. doi: 10.1136/gutjnl-2011-301133. [DOI] [PubMed] [Google Scholar]
- Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmunity reviews. 2014;13:24–30. doi: 10.1016/j.autrev.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation by TCDD reduces inflammation associated with Crohn’s disease. Toxicological sciences: an official journal of the Society of Toxicology. 2011;120:68–78. doi: 10.1093/toxsci/kfq360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsellino G, Kleinewietfeld M, Di Mitri D, Sternjak A, Diamantini A, Giometto R, Hopner S, Centonze D, Bernardi G, Dell’Acqua ML, et al. Expression of ectonucleotidase CD39 by Foxp3+ Treg cells: hydrolysis of extracellular ATP and immune suppression. Blood. 2007;110:1225–1232. doi: 10.1182/blood-2006-12-064527. [DOI] [PubMed] [Google Scholar]
- Burnet FM, Holmes MC. Immunological function of thymus and bursa of Fabricius. Thymus lesions in an auto-immune disease of mice. Nature. 1962;194:146–147. doi: 10.1038/194146a0. [DOI] [PubMed] [Google Scholar]
- Calabrese L. The yin and yang of tumor necrosis factor inhibitors. Cleve Clin J Med. 2006;73:251–256. doi: 10.3949/ccjm.73.3.251. [DOI] [PubMed] [Google Scholar]
- Canavan JB, Scotta C, Vossenkamper A, Goldberg R, Elder MJ, Shoval I, Marks E, Stolarczyk E, Lo JW, Powell N, et al. Developing in vitro expanded CD45RA+ regulatory T cells as an adoptive cell therapy for Crohn’s disease. Gut. 2015 doi: 10.1136/gutjnl-2014-306919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatenoud L. Biotherapies targeting T and B cells: from immune suppression to immune tolerance. Current opinion in pharmacology. 2015;23:92–97. doi: 10.1016/j.coph.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinen I, Nakahama T, Kimura A, Nguyen NT, Takemori H, Kumagai A, Kayama H, Takeda K, Lee S, Hanieh H, et al. The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. International immunology. 2015;27:405–415. doi: 10.1093/intimm/dxv015. [DOI] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, Chen JF, Enjyoji K, Linden J, Oukka M, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desreumaux P, Foussat A, Allez M, Beaugerie L, Hebuterne X, Bouhnik Y, Nachury M, Brun V, Bastian H, Belmonte N, et al. Safety and efficacy of antigen-specific regulatory T-cell therapy for patients with refractory Crohn’s disease. Gastroenterology. 2012;143:1207–1217. e1201–1202. doi: 10.1053/j.gastro.2012.07.116. [DOI] [PubMed] [Google Scholar]
- Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esser C, Rannug A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol Rev. 2015;67:259–279. doi: 10.1124/pr.114.009001. [DOI] [PubMed] [Google Scholar]
- Evans HG, Roostalu U, Walter GJ, Gullick NJ, Frederiksen KS, Roberts CA, Sumner J, Baeten DL, Gerwien JG, Cope AP, et al. TNF-alpha blockade induces IL-10 expression in human CD4+ T cells. Nature communications. 2014;5:3199. doi: 10.1038/ncomms4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Zundler S, Atreya R, Rath T, Voskens C, Hirschmann S, Lopez-Posadas R, Watson A, Becker C, Schuler G, et al. Differential effects of alpha4beta7 and GPR15 on homing of effector and regulatory T cells from patients with UC to the inflamed gut in vivo. Gut. 2015 doi: 10.1136/gutjnl-2015-310022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S, Toshimitsu T, Matsuoka S, Maruyama A, Oh-Oka K, Takamura T, Nakamura Y, Ishimaru K, Fujii-Kuriyama Y, Ikegami S, et al. Identification of a probiotic bacteria-derived activator of the aryl hydrocarbon receptor that inhibits colitis. Immunology and cell biology. 2014;92:460–465. doi: 10.1038/icb.2014.2. [DOI] [PubMed] [Google Scholar]
- Furumatsu K, Nishiumi S, Kawano Y, Ooi M, Yoshie T, Shiomi Y, Kutsumi H, Ashida H, Fujii-Kuriyama Y, Azuma T, et al. A role of the aryl hydrocarbon receptor in attenuation of colitis. Digestive diseases and sciences. 2011;56:2532–2544. doi: 10.1007/s10620-011-1643-9. [DOI] [PubMed] [Google Scholar]
- Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol. 2010;11:846–853. doi: 10.1038/ni.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, Perro M, Diestelhorst J, Allroth A, Murugan D, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. The New England journal of medicine. 2009;361:2033–2045. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goettel JA, Biswas S, Lexmond WS, Yeste A, Passerini L, Patel B, Yang S, Sun J, Ouahed J, Shouval DS, et al. Fatal autoimmunity in mice reconstituted with human hematopoietic stem cells encoding defective FOXP3. Blood. 2015;125:3886–3895. doi: 10.1182/blood-2014-12-618363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman WJ, Verbsky JW, Barchet W, Colonna M, Atkinson JP, Ley TJ. Human T regulatory cells can use the perforin pathway to cause autologous target cell death. Immunity. 2004;21:589–601. doi: 10.1016/j.immuni.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries JE, Roncarolo MG. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- Hauet-Broere F, Unger WW, Garssen J, Hoijer MA, Kraal G, Samsom JN. Functional CD25− and CD25+ mucosal regulatory T cells are induced in gut-draining lymphoid tissue within 48 h after oral antigen application. Eur J Immunol. 2003;33:2801–2810. doi: 10.1002/eji.200324115. [DOI] [PubMed] [Google Scholar]
- Hay M, Thomas DW, Craighead JL, Economides C, Rosenthal J. Clinical development success rates for investigational drugs. Nature biotechnology. 2014;32:40–51. doi: 10.1038/nbt.2786. [DOI] [PubMed] [Google Scholar]
- Hollenbach E, Vieth M, Roessner A, Neumann M, Malfertheiner P, Naumann M. Inhibition of RICK/nuclear factor-kappaB and p38 signaling attenuates the inflammatory response in a murine model of Crohn disease. J Biol Chem. 2005;280:14981–14988. doi: 10.1074/jbc.M500966200. [DOI] [PubMed] [Google Scholar]
- Huang Z, Jiang Y, Yang Y, Shao J, Sun X, Chen J, Dong L, Zhang J. 3,3′-Diindolylmethane alleviates oxazolone-induced colitis through Th2/Th17 suppression and Treg induction. Molecular immunology. 2013;53:335–344. doi: 10.1016/j.molimm.2012.09.007. [DOI] [PubMed] [Google Scholar]
- Ji T, Xu C, Sun L, Yu M, Peng K, Qiu Y, Xiao W, Yang H. Aryl Hydrocarbon Receptor Activation Down-Regulates IL-7 and Reduces Inflammation in a Mouse Model of DSS-Induced Colitis. Digestive diseases and sciences. 2015;60:1958–1966. doi: 10.1007/s10620-015-3632-x. [DOI] [PubMed] [Google Scholar]
- Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annual review of immunology. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet NI, Steppan LB, Vorachek W, Oda S, Farrer D, Wong CP, Pham D, Mourich DV. Activation of aryl hydrocarbon receptor by TCDD prevents diabetes in NOD mice and increases Foxp3+ T cells in pancreatic lymph nodes. Immunotherapy. 2009;1:539–547. doi: 10.2217/imt.09.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koreth J, Matsuoka K, Kim HT, McDonough SM, Bindra B, Alyea EP, 3rd, Armand P, Cutler C, Ho VT, Treister NS, et al. Interleukin-2 and regulatory T cells in graft-versus-host disease. The New England journal of medicine. 2011;365:2055–2066. doi: 10.1056/NEJMoa1108188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- Le Douarin N, Corbel C, Bandeira A, Thomas-Vaslin V, Modigliani Y, Coutinho A, Salaun J. Evidence for a thymus-dependent form of tolerance that is not based on elimination or anergy of reactive T cells. Immunol Rev. 1996;149:35–53. doi: 10.1111/j.1600-065x.1996.tb00898.x. [DOI] [PubMed] [Google Scholar]
- Mascanfroni ID, Takenaka MC, Yeste A, Patel B, Wu Y, Kenison JE, Siddiqui S, Basso AS, Otterbein LE, Pardoll DM, et al. Metabolic control of type 1 regulatory T cell differentiation by AHR and HIF1-alpha. Nat Med. 2015;21:638–646. doi: 10.1038/nm.3868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascanfroni ID, Yeste A, Vieira SM, Burns EJ, Patel B, Sloma I, Wu Y, Mayo L, Ben-Hamo R, Efroni S, et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat Immunol. 2013;14:1054–1063. doi: 10.1038/ni.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne CG, Williams CB. Induced and natural regulatory T cells in the development of inflammatory bowel disease. Inflammatory bowel diseases. 2013;19:1772–1788. doi: 10.1097/MIB.0b013e318281f5a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo L, Trauger SA, Blain M, Nadeau M, Patel B, Alvarez JI, Mascanfroni ID, Yeste A, Kivisakk P, Kallas K, et al. Regulation of astrocyte activation by glycolipids drives chronic CNS inflammation. Nat Med. 2014;20:1147–1156. doi: 10.1038/nm.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JF. Immunological function of the thymus. Lancet. 1961;2:748–749. doi: 10.1016/s0140-6736(61)90693-6. [DOI] [PubMed] [Google Scholar]
- Moncrieffe H, Nistala K, Kamhieh Y, Evans J, Eddaoudi A, Eaton S, Wedderburn LR. High expression of the ectonucleotidase CD39 on T cells from the inflamed site identifies two distinct populations, one regulatory and one memory T cell population. J Immunol. 2010;185:134–143. doi: 10.4049/jimmunol.0803474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone I, Rizzo A, Sarra M, Sica G, Sileri P, Biancone L, MacDonald TT, Pallone F, Monteleone G. Aryl hydrocarbon receptor-induced signals up-regulate IL-22 production and inhibit inflammation in the gastrointestinal tract. Gastroenterology. 2011;141:237–248. 248.e231. doi: 10.1053/j.gastro.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Kelsall BL, Stuber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath MF, Fuss I, Pasparakis M, Alexopoulou L, Haralambous S, Meyer zum Buschenfelde KH, Strober W, Kollias G. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur J Immunol. 1997;27:1743–1750. doi: 10.1002/eji.1830270722. [DOI] [PubMed] [Google Scholar]
- Persidis A. Autoimmune disease drug discovery. Nature biotechnology. 1999;17:1038. doi: 10.1038/13748. [DOI] [PubMed] [Google Scholar]
- Qiu J, Guo X, Chen ZM, He L, Sonnenberg GF, Artis D, Fu YX, Zhou L. Group 3 innate lymphoid cells inhibit T-cell-mediated intestinal inflammation through aryl hydrocarbon receptor signaling and regulation of microflora. Immunity. 2013;39:386–399. doi: 10.1016/j.immuni.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;23:23. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- Quintana FJ, Jin H, Burns EJ, Nadeau M, Yeste A, Kumar D, Rangachari M, Zhu C, Xiao S, Seavitt J, et al. Aiolos promotes T(H)17 differentiation by directly silencing Il2 expression. Nature Immunology. 2012;13:770–777. doi: 10.1038/ni.2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintana FJ, Murugaiyan G, Farez MF, Mitsdoerffer M, Tukpah AM, Burns EJ, Weiner HL. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2010;107:20768–20773. doi: 10.1073/pnas.1009201107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Willinger T, Martinek J, Strowig T, Gearty SV, Teichmann LL, Saito Y, Marches F, Halene S, Palucka AK, et al. Development and function of human innate immune cells in a humanized mouse model. Nature biotechnology. 2014;32:364–372. doi: 10.1038/nbt.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nature reviews Immunology. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- Saadoun D, Rosenzwajg M, Joly F, Six A, Carrat F, Thibault V, Sene D, Cacoub P, Klatzmann D. Regulatory T-cell responses to low-dose interleukin-2 in HCV-induced vasculitis. The New England journal of medicine. 2011;365:2067–2077. doi: 10.1056/NEJMoa1105143. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Fukuma K, Kuribayashi K, Masuda T. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I. Evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S, Miyara M, Costantino CM, Hafler DA. FOXP3+ regulatory T cells in the human immune system. Nature reviews Immunology. 2010;10:490–500. doi: 10.1038/nri2785. [DOI] [PubMed] [Google Scholar]
- Sarin R, Wu X, Abraham C. Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc Natl Acad Sci U S A. 2011;108:9560–9565. doi: 10.1073/pnas.1017854108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheiffele F, Fuss IJ. Induction of TNBS colitis in mice. Curr Protoc Immunol. 2002;Chapter 15(Unit 15):19. doi: 10.1002/0471142735.im1519s49. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- Singh NP, Singh UP, Singh B, Price RL, Nagarkatti M, Nagarkatti PS. Activation of aryl hydrocarbon receptor (AhR) leads to reciprocal epigenetic regulation of FoxP3 and IL-17 expression and amelioration of experimental colitis. PLoS One. 2011;6:e23522. doi: 10.1371/journal.pone.0023522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Clagett-Dame M, Peterson RE, Hahn ME, Westler WM, Sicinski RR, DeLuca HF. A ligand for the aryl hydrocarbon receptor isolated from lung. Proc Natl Acad Sci U S A. 2002;99:14694–14699. doi: 10.1073/pnas.232562899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg GF, Fouser LA, Artis D. Border patrol: regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat Immunol. 2011;12:383–390. doi: 10.1038/ni.2025. [DOI] [PubMed] [Google Scholar]
- Takamura T, Harama D, Fukumoto S, Nakamura Y, Shimokawa N, Ishimaru K, Ikegami S, Makino S, Kitamura M, Nakao A. Lactobacillus bulgaricus OLL1181 activates the aryl hydrocarbon receptor pathway and inhibits colitis. Immunology and cell biology. 2011;89:817–822. doi: 10.1038/icb.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamura T, Harama D, Matsuoka S, Shimokawa N, Nakamura Y, Okumura K, Ogawa H, Kitamura M, Nakao A. Activation of the aryl hydrocarbon receptor pathway may ameliorate dextran sodium sulfate-induced colitis in mice. Immunology and cell biology. 2010;88:685–689. doi: 10.1038/icb.2010.35. [DOI] [PubMed] [Google Scholar]
- Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, Lanzavecchia A, Manz MG. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304:104–107. doi: 10.1126/science.1093933. [DOI] [PubMed] [Google Scholar]
- Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Christensen J, O’Garra A, Stockinger B. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453:106–109. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- Vogel CF, Goth SR, Dong B, Pessah IN, Matsumura F. Aryl hydrocarbon receptor signaling mediates expression of indoleamine 2,3-dioxygenase. Biochem Biophys Res Commun. 2008;375:331–335. doi: 10.1016/j.bbrc.2008.07.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Quintana FJ, da Cunha AP, Dake BT, Koeglsperger T, Starossom SC, Weiner HL. In Vivo Induction of Tr1 Cells via Mucosal Dendritic Cells and AHR Signaling. PLoS One. 2011;6:e23618. doi: 10.1371/journal.pone.0023618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeste A, Mascanfroni ID, Nadeau M, Burns EJ, Tukpah AM, Santiago A, Wu C, Patel B, Kumar D, Quintana FJ. IL-21 induces IL-22 production in CD4+ T cells. Nature communications. 2014;5:3753. doi: 10.1038/ncomms4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeste A, Nadeau M, Burns EJ, Weiner HL, Quintana FJ. Nanoparticle-mediated codelivery of myelin antigen and a tolerogenic small molecule suppresses experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2012;109:11270–11275. doi: 10.1073/pnas.1120611109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ma J, Takeuchi M, Usui Y, Hattori T, Okunuki Y, Yamakawa N, Kezuka T, Kuroda M, Goto H. Activation of aryl hydrocarbon receptor suppresses experimental autoimmune uveoretinitis by inducing differentiation of regulatory T cells. Invest Ophthalmol Vis Sci. 2009 doi: 10.1167/iovs.09-3993. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.