

Abstract
Over the past several years, hydrogen sulfide (H2S) has been shown to be an important player in a variety of physiological functions, including neuromodulation, vasodilation, oxidant regulation, inflammation, and angiogenesis. H2S is synthesized primarily through metabolic processes from the amino acid cysteine and homocysteine in various organ systems including neuronal, cardiovascular, gastrointestinal, and kidney. Derangement of cysteine and homocysteine metabolism and clearance, particularly in the renal vasculature, leads to H2S biosynthesis deregulation causing or contributing to existing high blood pressure. While a variety of environmental influences, such as diet can have an effect on H2S regulation and function, genetic factors, and more recently epigenetics, also have a vital role in H2S regulation and function, and therefore disease initiation and progression. In addition, new research into the role of gut microbiota in the development of hypertension has highlighted the need to further explore these microorganisms and how they influence the levels of H2S throughout the body and possibly exploiting microbiota for use of hypertension treatment. In this review, we summarize recent advances in the field of hypertension research emphasizing renal contribution and how H2S physiology can be exploited as a possible therapeutic strategy to ameliorate kidney dysfunction as well as to control blood pressure.
Keywords: Homocysteine, folic acid, MMPs, TIMPs, miRNA, epigenetics, extracellular matrix, inflammation, microbiota, polysulfides
Graphical abstract
1. Introduction
The link between mortality and hypertension is well documented and the prevalence of this disease grows each year with an estimated one-third of the population in the United States having high blood pressure and the rates steadily increase with age, accounting for 41.5% of all deaths [1]. Two of the major outcomes attributed to high blood pressure are increase risk for developing cardiovascular disease (CVD) and stroke, along with decreased life expectancy [1]. There are several risk factors associated with hypertension as the etiology and complexity of the disease is not completely understood. Genetics plays a role in determining high blood pressure and that different sets of genes affect blood pressure regulation at different stages of life [2]. Environmental factors, such as lifestyle preferences and diet can also influence blood pressure. A study in women revealed that body mass index (BMI) was the strongest indicator of predicting high blood pressure leading to hypertension, with a BMI of greater than 24.7 being the most affected group [3]. Diet, such as higher sodium, lack of fruits and vegetables intake, low physical activity, higher alcohol consumption and low folic acid (folate) supplementation were also factors related to development of hypertension [4].
The kidney is one of several organ systems that are adversely affected by hypertension and is one of the leading causes of chronic kidney diseases (CKD) resulting in kidney dysfunction [5, 6]. It is believed that CKD affects over 20 million adults in the US, with approximately 60% of these cases having associated hypertension [7]. A decrease in glomerular filtration rate (GFR) is a key feature of a hypertensive kidney [8]. Blood flow in the renal medulla is important for regulating blood pressure and excreting waste from the kidneys, such as excess sodium, and is often found to be decreased in hypertension [9]. Increased fluid retention and the activation of the renin-angiotensin system (RAS) are key characteristics of the pathophysiology in the hypertensive kidney [10]. The increased expression of angiotensin II (Ang-II) can cause endothelial damage, fibrosis, promote inflammation and mesangial cell proliferation, and vasoconstriction [11-13]. Ang-II can also cause increased reactive oxygen species (ROS), which is linked with essential hypertension, production through mitochondrial dysfunction and subsequent expression and activation of p47phox, a subunit of the NADPH-oxidase complex [14-16]. High salt intake can also stimulate NADPH-oxidase activity and oxidative stress while also decreasing superoxide dismutase scavengers, which further exacerbates renal injury [17].
Kidney, in addition to maintaining normal physiological function, also produces several gaseous molecules such as nitric oxide (NO), carbon monoxide (CO) and hydrogen sulfide (H2S) [18]. Alterations in these gaseous molecules during pathophysiological kidney remodeling impact normal kidney function. The role of NO in normal vascular function including renal vasculature in disease condition is well documented [19, 20]. Much more has yet to be studied for CO to get a comprehensive role and mechanism of its impact on kidney function. Recently, H2S has appeared a strong regulatory determinant of maintaining kidney function in normal and disease condition [21, 22]. This is primarily because of the presence of H2S production enzymes, such as cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) and 3-mercaptopyruvate sulfurtransferase (3MST) in the kidney and these enzymes are prime targets of stress in the kidney vasculature [23-25]. Diseases like hypertension diminish these enzymes expression, activity and local production of H2S [26-30]. Genes of these enzymes or their regulatory genes are also partially impacted by epigenetic modification, such as hypermethylation or homocysteinylation [31-33]. Ultimately the normal production of H2S is essentially diminished in these pathophysiological changes, which in turn affect renal functional outcomes. In this review, in light of current literature we will discuss dysregulation of H2S production in the kidney during normal and disease states leading to exaggerated renal pathophysiology of hypertension and how H2S can potentially be used to mitigate hypertension and reduced kidney function.
2. Mechanisms of renovascular hypertension
2.1 Hypertension and kidney
In general, several distinct but overlapping mechanisms of hypertension have been described in the literature involving kidney. This review is primarily focused on renovascular hypertension, however it may be worthwhile introducing a brief overview of all these mechanisms of hypertension affecting or relating to kidney for an easy distinction between these conditions, and overlapping if any, from semantic point of view. These are: 1) Primary hypertension; 2) Secondary hypertension; and 3) Other types of hypertension. Primary hypertension is also called ‘essential hypertension’ or ‘idiopathic hypertension’, which accounts 90-95% of patients diagnosed with hypertension [34, 35].There is no known cause of primary hypertension, and therefore diagnosis of this type of hypertension is predominantly made after excluding known causes of secondary hypertension. Interestingly, primary hypertension can in turn lead to ‘hypertensive nephrosclerosis’, and ‘chronic kidney disease’. Unlike primary hypertension, a number of conditions can cause secondary hypertension. The most common conditions which are related to kidney disease include diabetic nephropathy (DN), polycystic kidney diseases, glomerulosclerosis and other glomerular diseases, and renovascular hypertension [36]. Although in typical sense renovascular hypertension is caused by narrowing of one or both renal arteries, in this review article we generalized this term to mean entire renal vascular bed including glomerulus. Other types of hypertension include isolated systolic hypertension, malignant hypertension and resistant hypertension.
Besides these above described mechanisms of hypertension, there is also a topic of hypertension in primary chronic kidney disease (CKD) and its pathogenesis includes sodium retention, renin-angiotensin system (RAS) activation, enhanced activity of the sympathetic nervous system etc. [37]. It is important to note that while hypertension can cause chronic kidney disease, chronic kidney disease in turn can cause hypertension as well [38]. In all of the above-mentioned topics, which reflect the clinical setting, the role of the RAS is variable, in that it is of paramount importance in renovascular hypertension, for example. But in the other forms involvements of RAS can vary. In fact, ACE inhibitors and Angiotensin II receptor blockers (ARBs) produce an antihypertensive response in 30 to 50 percent of patients [39, 40], and ARBs itself have been found to reduce blood pressure of all ARBs about 48-55% [41]. While these topics are of great clinical importance and discussed extensively in the literature, and perhaps should be a topic of future update, in the next section of this article we focused our discussion on RAS and how H2S may modulate RAS activation.
2.2. Renin-angiotensin system and H2S
One of the primary mechanisms involved in renovascular hypertension is the renin-angiotensin system (RAS). The precise control of this system is widely recognized for its importance in regulating several physiological roles, including blood pressure and sodium homeostasis. The protease renin is the first enzyme involved in the pathway and is primarily produced in the kidney. It is responsible for converting angiotensinogen, found primarily in the liver, into angiotensin I (Ang-I), a biologically inactive precursor form to angiotensin II (Ang-II) [42]. Ang-I is converted into Ang-II by the ACE enzyme through hydrolysis and is mainly found in the lungs [43]. While Ang-II can be further converted in to other forms, such as Ang-III, Ang-IV, and Ang-(1-7), Ang-II is believed to be the most biologically active form [42, 44]. Ang-II preferentially targets the AT1-R, which is expressed in most tissue types in the kidney, and elicits a plethora of effects, including, pro-inflammatory response, release of renin, and sodium reabsorption [45, 46]. Moreover, vasoconstriction of post-glomerular arterioles leads to decreased blood flow and glomerular filtration in the kidney by increasing arteriolar resistance and contraction of mesangial cells [37]. This leads to an overall increase in arterial blood pressure. A second receptor, AT2-R, also binds Ang-II but is present in a much lower capacity. The response of Ang-II binding to AT2-R, however, appears to promote the opposite effects of AT1-R binding, through vasodilation and regulation of sodium regulation [47]. In addition, the homologue ACE2 enzyme has been shown to covert Ang-II to the Ang-(1-7) peptide, which appears to modulate the hypertensive effects elicited by Ang-II [48].
In the recent years, substantial evidences suggest a link between RAS and H2S in the kidney. In a rat model of two-kidney-one-clip (2K1C) renovascular hypertension, Lu et al demonstrated that H2S donor significantly diminished plasma renin activity and Ang-II levels compared to vehicle control without affecting plasma ACE activity [49]. Their study concluded that H2S has potential therapeutic value to treat renovascular hypertension possibly through inhibiting renin activity by decreasing its synthesis and release. In another study, Xue et al demonstrated that high glucose induced AGT, ACE and AT1 receptor and Ang-II concentration in in vitro mesangial cells [50]. While they did not measure expression or activity of H2S producing enzymes in their experiments, they observed attenuated RAS activation following H2S treatment. Similar results were also observed in a streptozotocin-induced diabetes rat model where increased RAS activity was reversed by H2S through diminished oxidative stress without affecting glucose levels [50].
2.3 Metabolic syndrome and CKD: role of H2S
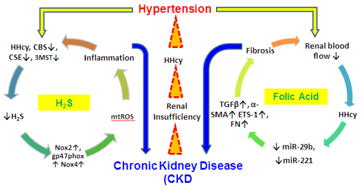
Metabolic syndrome (MetS) is defined as a clustering of cardiovascular risk factors [51]. According to the World Health Organization (WHO), diabetes mellitus, along with two or more other cardiovascular risk factors that include high blood pressure, high levels of triglycerides, low levels of HDL, obesity and increased microalbuminuria are considered as MetS. More specific information about classification of MetS is nicely summarized in a review article referenced in [52]. The association between CKD and all of these above cardiovascular risk factors are well documented in the literature. Therefore, it is obvious to infer that MetS is associated with CKD. In fact several studies have documented higher risks of CKD with increasing traits of MetS [53-55]. The importance of insulin resistance, ROS, inflammation, dyslipidemia and many other factors are proposed in contributing MetS associated CKD; however, the role of H2S in this disease mechanism is poorly addressed. We have recently shown that in diabetes, H2S deficiency contributes to unfavorable renovascular remodeling characterized by vascular constriction, increased resistive index, and diminished blood flow, and H2S treatment improves renal function [56, 57]. Others have shown inhibition of pancreatic H2S production with daily intraperitoneal injections of CSE inhibitor dl-propargylglycine (PAG) in Zucker diabetic fatty (ZDF) rats increased serum insulin levels and reduced hyperglycemia [58]. In fms-like tyrosine kinase 1-induced hypertensive Sprague-Dawley rats, Holwerda et al demonstrated that proteinuria and glomerular endotheliosis were ameliorated by H2S treatment and these changes were in part due to increased renal VEGF expression [59]. Further studies are needed to determine association of metabolic diseases and H2S in CKD, and to evaluate potential H2S therapy for ramification of renal vascular disorders. Based on the available reports, a proposed mechanism of CKD in H2S deficiency and high homocysteine levels (hyperhomocysteinemia) associated with hypertension is shown in figure 1. These are two independent, but closely related pathways. More details are explained in the legend.
Figure 1.
Schematic of CKD mechanism in hypertension depicting important roles of Hyperhomocysteinemia and H2S. Hypertension causes renal impairment of H2S producing enzymes leading to H2S deficiency and increase mitochondrial generated oxidative inflammation (on the left side). Through a different but related mechanism of H2S deficiency (since homocysteine is a precursor of H2S, homocysteine accumulation, i.e, Hyperhomocysteinemia diminishes H2S production), hypertension increases resistive index and reduces blood flow in the renal vasculature (on the right). As a result kidney impairment occurs and homocysteine clearance impairs. High levels of homocysteine in turn activate miRNAs leading to pro-fibrotic gene activation and fibrosis. These above described two vicious cycles worsen CKD in hypertension.
2.4 Epigenetic mechanism and miRNAs: is H2S a regulator?
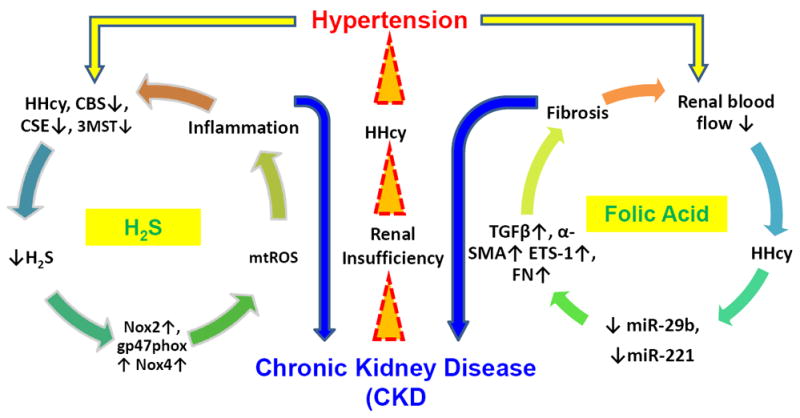
More recent studies have investigated the role of non-coding RNA involvement of hypertension. In particular, microRNAs (miRNAs) have received considerable attention in a variety of kidney diseases [60]. miRNAs are short (~22 nucleotides in length), single-stranded RNA genes that regulate post-transcriptional gene expression. miRNAs target messenger RNAs (mRNAs) by binding to complementary sequences in the 3’ untranslated region repressing their translation, and are associated with a broad spectrum of cellular and physiological processes. While there is limited data on the functional roles of miRNAs in the hypertensive kidney, a few studies have yielded promising results. Spontaneously hypertensive rats were shown to have decreased level of miR-29b in the renal cortex compared to normotensive control animals [61]. In addition, the authors reported a significant decrease in miR-29b expression in renal tubular epithelial cells treated with Ang-II. This, in turn, promoted epithelial-mesenchymal transition (EMT) through activation of key genes such as TGF-β and α-SMA, which can lead to renal fibrosis. These effects were reversed upon overexpression of miR-29b, suggesting this microRNA has an inhibitory role in Ang-II induced hypertension [61]. A similar finding was reported in the renal medulla of salt-sensitive rats, where they found miR-29b was expressed at lower levels in these rats compared to the control counterparts [62]. Transfection experiments with miR-29b showed a decrease in genes known to be involved in collagen formation, leading the authors to conclude that miR-29b plays a protective role in kidney fibrosis by inhibiting extracellular matrix (ECM) remodeling [62]. Ang-II has also been shown to downregulate the expression of miR-221 in mouse kidney and in rat renal interstitial fibroblasts, leading to an increase in the Ets-1 gene, which had been previously shown to be induced by Ang-II and result in inflammation and reactive oxygen species generation, as well as other kidney diseases [63, 64]. In a study utilizing a more global approach of microarrays, miR-638 and let-7c were found to be downregulated in the renal medulla of male hypertensive patients compared to normotensive individuals [65]. In the renal cortex, a total of 13 miRs were found to be significantly altered, including miR-181a and miR-663 which had lowered expression and led in an increase in renin mRNA expression and hypertensive effects [65]. While all these studies point towards an increasing role of miRs in the development of hypertension in the kidney, there is a lack of knowledge whether H2S is involved in the regulation of miRs or vice versa in hypertensive kidney. It was however reported that H2S donor attenuated myocardial injury through upregulation of miR-21 and suppression of inflammasome [66]. Another study reported increased expression of miR-132 and miR-212 in the heart, aortic wall and kidney of Ang-II infused hypertensive rats [67]. More interestingly the same study reported AT1R blockers decreased expression of these two miRs in surplus human mammary arterial tissue obtained from coronary bypass surgery [67]. Taken together, these above reports suggest a clear relationship between miRs and hypertensive disease mechanism. Very recently we have reported downregulation of miR-129 and miR-299b in Ang-II treated mice and normalization of these miRs following GYY4137 (a donor of H2S) treatment [68]. In contrast, we found upregulated miR-369 in Ang-II treatment which was suppressed in GYY4137 treated mice [68]. To further prove this initial finding and to elucidate the precise mode(s) of H2S action in regulating miRs in hypertensive kidney, more work needs to be completed. Here we propose a mechanism of Ang-II mediated renal inflammation and dysfunction involving epigenetic modulation of miRs, and how H2S is predicted to mitigate renal dysfunction (Figure 2).
Figure 2.
Schematic of hypertension-induced epigenetic mechanism of kidney dysfunction. Ang-II causes disruption of epigenetic mechanisms (methylation, histone modification, non-coding RNAs) leading to inflammation and increased vascular resistance causing kidney dysfunction in hypertension. GYY4137 alleviates these effects and improves kidney function. DNA methyltransferase DNMT3a and pro-inflammatory cytokine IL-17 are increased in the hypertensive kidney while miR-129 is suppressed [68]. The miR-129 is predicted to target and regulate both DNMT3a and IL-17 and its expression is increased with H2S donor GYY4137 treatment, leading to a decrease in IL-17 and DNMT3a expression [68].
3. Involvement of thiol homocysteine and H2S in renovascular hypertension
3.1 High homocysteine, low H2S and hypertension
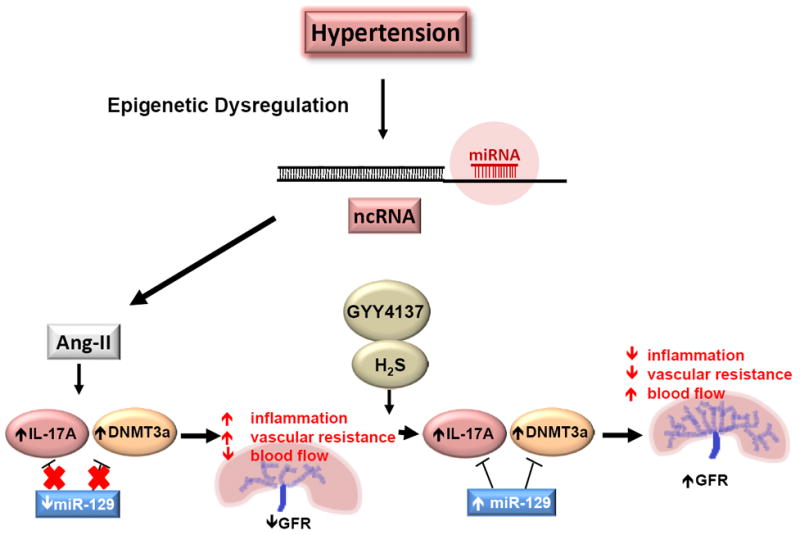
Homocysteine is an amino acid containing sulfur as one of its primary components. As part of the methionine metabolic pathway, it is an intermediate step in the processing of methionine by demethylation. After this initial step, homocysteine can either be further metabolized to cystathionine and eventually to cysteine via the transsulfuration pathway, or can undergo remethylation to once again become methionine [69] (Figure 3). There are several enzymes involved in the transsulfuration pathway, including cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE), cysteine aminotransferase (CAT), and 3-mercaptopyruvate sulfurtransferase (3MST). A substantial amount of homocysteine is metabolized in the kidney, making it a critical organ in the regulation of this amino acid as high levels of homocysteine, also known as hyperhomocysteinemia, in the plasma are a risk indicator of renal damage, hypertension and cardiovascular disease [69-72].
Figure 3.
Schematic of Homocysteine metabolism and H2S biosynthesis. H2S; hydrogen sulfide; TH4-folate, tetrahydrofolate; CBS, cystathionine β-synthase; CSE, cystathionine γ-lyase; 3MST, 3-mercaptopyruvate sulfurtransferase; AAT, aspartate aminotransferase.
There are several adverse effects that are accompanied along with increasing levels of homocysteine, one of which is increasing oxidative stress and resulting in damage to endothelial cells [73]. One mechanism that has been shown is the activation of NADH/NADPH oxidase through the increased production of ceramide by homocysteine, leading to reactive oxygen species (ROS) formation, as evidenced in rat mesangial cells [74]. Homocysteine has also been shown to decrease cellular levels of glutathione peroxidase, which is an important antioxidant enzyme in reducing hydrogen peroxide to water [75, 76]. Moreover, homocysteine can also lead to a decrease in nitric oxide (NO), a potent vasodilator, production through endothelial damage and inhibition of NO through asymmetric dimethylarginine (ADMA) [77-79]. In addition, homocysteine can be oxidized by copper while in circulation and can lead to the production of hydrogen peroxide (H2O2) [80]. Similar effects are also observed when H2S levels are low [81]. In one study where one of the key enzymes (CSE) in H2S generation was genetically eliminated, it was found that low H2S levels resulted in decreased availability of NO, resulting in oxidative stress as evidenced by increased lipid peroxidation and protein carbonyl groups [82]. It was further demonstrated that treatment with exogenous H2S restored NO levels in these animals, leading to less oxidative stress [82]. This finding was also found in a separate study utilizing mice without CSE, where endogenous H2S levels were found to be low in renal endothelial tissue, and showed increased damage due to ischemia/reperfusion injury (IR/I) in CSE-negative animals, with an increase in ROS and this was reduced in animals receiving exogenous H2S and in cells with overexpressed CSE [83].
3.2 Angiotensin-II, hyperhomocysteinemia, and CKD: possible role of folate and H2S to mitigate hypertension
Ang-II induced hypertension is known to cause renal injury by decreasing renal blood flow, glomerular filtration rates, as well as decreasing the levels of CBS and CSE enzymes, leading to elevated levels of homocysteine known as hyperhomocysteinemia [84]. In addition, Ang-II hypertension causes renal injury involving microvascular endothelial damage, which is one of the contributing factors of hypertension [85]. Renal microvascular endothelial cells express enzymes CSE and 3MST, which through the transsulfuration pathway clears homocysteine to produce the gaseous substance, H2S [86]. Ang-II mediated endothelial damage may affect these enzymes resulting in hyperhomocysteinemia and decreased production of H2S, a strong antioxidant [87-89] and vasorelaxing molecule [90, 91]. All of these effects may contribute to renovascular remodeling resulting in CKD (Figure 1).
Plasma homocysteine levels are negatively correlated with GFR and the prevalence of hyperhomocysteinemia is 85-100% among ESRD [92]. Although the precise mechanisms of hyperhomocysteinemia in kidney diseases have not been established yet, it is generally accepted that the mechanisms could be multifactorial. While increased homocysteine production or decreased excretion has been ruled out, decreased metabolism in renal or extrarenal sites seem to be the most plausible cause [93]. In fact, it has been demonstrated that folic acid supplementation does not reduce intracellular homocysteine concentration, supporting the hypothesis of negative metabolic effects that could impair intracellular homocysteine metabolism [94]. This is linked to the issue of folate therapy where “folate resistance” is present in CKD patients [95, 96]. In a recent review article, Perna and Ingrosso have creditably discussed this topic in more details with up-to-date information which are mostly based on clinical findings in patients with renal insufficiency [94]. Besides this, it has also been reported that the correction of multiple remethylation abnormalities with various other drugs in CKD patients, which are not related to folic acid supplementation, removes homocysteine from protein binding sites [97]. This information could add invaluable strategies to decrease homocysteine levels in CKD patients independent of folic acid remethylation-pathway. In this context, it is noteworthy to mention that decreased H2S and down-regulation of transsulfuration enzymes, CBS and CSE (enzymes responsible for cysteine and homocysteine metabolism) transcript have been reported in ESRD [98]. Similar results were also observed in experimental uraemic rats where malfunction of these transsulfuration enzymes were accompanied with decreased H2S levels [99]. Further supporting to these findings, in dialysis patients, levels of H2S were also shown to increase following dialysis suggesting that uraemic toxins are likely the culprits to inhibit H2S generating enzymes, and thus H2S levels [100]. Despite the progress of our scientific knowledge with these advanced researches, precise mechanism(s) of high homocysteine and low H2S levels still remain an open area of future research to discover many more unsolved mysteries of human health related to pathophysiological complexity of CKD.
3.3 Thiol metabolism and H2S production
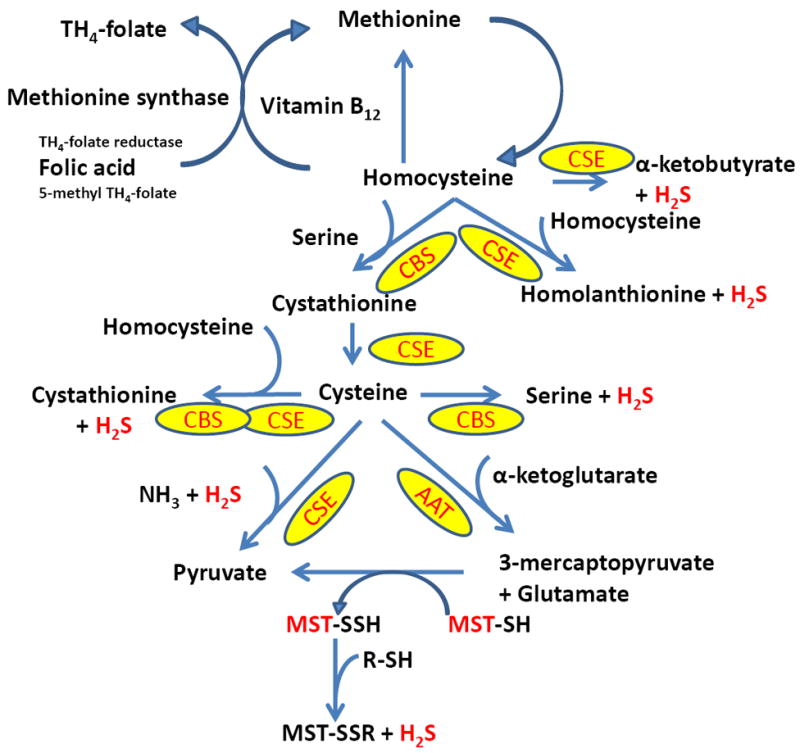
Both CBS and CSE are cytosolic enzymes and are expressed in the major organs including kidney [101-103]. While CBS produces H2S from cysteine via β-elimination / β-replacement reaction in which cysteine is condensed with homocysteine [104], CSE produces H2S via α,β-elimination of cysteine. In hyperhomocysteinemia, α,γ-elimination and γ-replacement reaction of homocysteine also produces H2S [105]. Two other enzymes, 3MST and CAT also produces H2S and this pathway known and 3MST/CAT pathway (Figure 4). First, CAT produces 3-mercaptopyruvate from cysteine and α-ketoglutarate, and then 3MST produces H2S from 3-mercaptopyruvate. Unlike CBS and CSE, 3MST is a mitochondrial enzymes and high concentration of cysteine in the mitochondria leads to 3MST/CAT pathway to produces H2S [106]. While CBS, CSE and CAT produces H2S from l-cysteine, another enzyme d-amino acid oxidase (DAO), which is localized in the peroxisome, produces H2S from d-cysteine [107, 108]. DAO metabolizes d-cysteine to achiral 3MP which further metabolizes to H2S by 3MST [109-111]. A summary of H2S production by all these above enzymes are presented in a schematic way in Figures 3 & 4.
Figure 4.
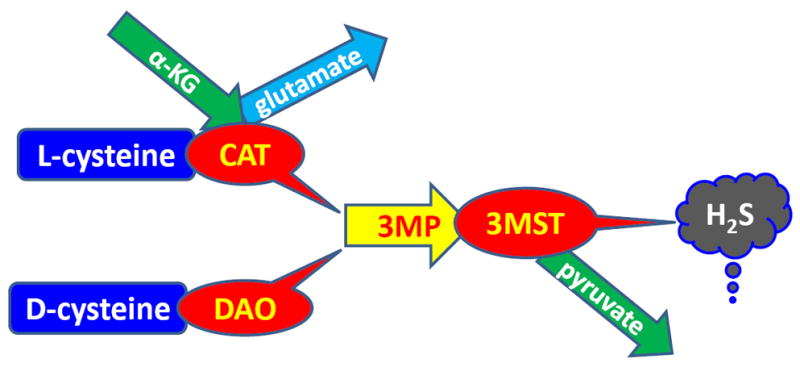
Schematic of H2S biosynthesis form cysteine. Metabolism of l-cysteine by cysteine aminotransferase (CAT) and d-cysteine by d-amino acid oxidase (DAO) produces 3-mercaptopyruvate (3MP). 3MP then metabolizes to produce H2S by 3-mercaptopyruvate sulfurtransferase (3MST).
3.4 Folic acid supplementation: regulation of blood pressure by homocysteine remethylation
Folic acid is a B-vitamin and is essential in the metabolism of homocysteine to methionine, subsequently lowering plasma levels of homocysteine through the remethylation pathway and thereby lowering oxidative and inflammatory stresses [112]. Through use of the folate cycle and B-12 vitamin, homocysteine can be remethylated back to methionine by methionine synthase and 5-methyltetrahydrofolate reductase (Figure 3). Deficits in either folate or vitamin B-12 have been linked with elevated homocysteine levels and is therefore associated with elevated blood pressure and hypertension [113]. In one study looking at end stage renal disease (ESRD), patients that were given folic acid supplement were found to have lowered plasma homocysteine levels as well as increased remethylation of homocysteine to methionine [114]. A meta-analysis conducted on individuals with end stage kidney disease (ESKD) found that folic acid treatment reduced plasma homocysteine levels 15-20% [115]. It should be taken into account that several published papers reported higher homocysteine levels without measuring corresponding H2S levels. Since homocysteine is one of the precursors of H2S, it is plausible that higher accumulation of homocysteine lowers tissue synthesis, and thus plasma levels of H2S. Using animal as experimental model, we and others have demonstrated that high levels of homocysteine in fact lower tissue and plasma levels of H2S [116-119]. However, it was not clearly demonstrated in human and in some cases it was positively related, instead of expected negative relationship. For example, it has been shown in human that in certain pathological condition hyperhomocysteinemia is associated with increased plasma and platelet H2S levels [120]. This phenomenon is unclear, but possibly linked to H2S-mediated susceptibility to thrombosis and atherosclerosis in patients with abnormal remethylation pathway [120].
Moreover, it has also been demonstrated by the same group that homocysteine is irreversibly degraded to H2S causing in vivo platelet activation via upregulation of phospholipase A2 and downstream boost of the arachidonate cascade [120, 121]. In this regard, most interventional trials focused on homocysteine lowering by folate administration did not exclude patients routinely taking arachidonate inhibitor, aspirin that could have masked this negative effect associated to H2S levels in hyperhomocysteinemia [120]. Therefore, the questions: 1) what is the most beneficial dose of folate to see reduced homocysteine levels, and 2) whether other confounding factors are eliminated for a decisive conclusion of folate therapy remains unanswered. These questions warrant future investigation [115].
Homocysteine however contributes to Ang-II mediated renovascular remodeling through NADP(H) and AT1R, in part, by decreasing vascular density and increasing oxidative, pro- inflammatory, anti-angiogenic and pro-fibrotic factors [84]. We have shown that FA ameliorate the accumulation of oxidized collagen by balancing the matrix metalloproteinase / tissue inhibitor of metalloproteinase (MMP/TIMP) axis, in particular, by decreasing MMP-2 and MMP-9 expression and elevating TIMP-1 and TIMP-2 expression levels [84]. FA supplementation also lowered systolic blood pressure, increased nitric oxide (NO) production, and lowered plasma homocysteine levels [84]. These studies suggest FA treatment can lower plasma levels of homocysteine and mitigate hypertensive effects due to elevated homocysteine through the remethylation pathway of homocysteine to methionine.
3.5 Does homocysteine cause or contribute to hypertension?
Aside from the fact of vascular complications in homocystinuria, the genetic disease where levels of plasma homocysteine are higher and hyperhomocysteinemia is one of the key clinical findings, hypertension is not described [122, 123]. The vast majority of clinical as well as experimental evidences underscore the contribution of homocysteine to hypertension is secondary to its vascular effects rather than primary cause [124, 125]. Recently, we have reported that inhibition of DNA methylation reduced homocysteine levels in CBS+/- mice, an animal model of hyperhomocysteinemia, and mitigated moderate blood pressure implicating epigenetic regulation of vascular fibrotic events involving DNA methylation [31, 126]. This is an experimental evidence of indirect cause and effect relationship between hyperhomocysteinemia and hypertension. Similar to homocystinuria patients, in CKD patients, aged and well-nourished population substantial evidences of hyperhomocysteinemia is established [127-129]. It is still not conclusive whether hyperhomocysteinemia causes hypertension in these patients [130, 131].
Nonetheless, it has been well documented in the literature that hyperhomocysteinemia exacerbates end organ damage in hypertension, if not causing hypertension, especially in ill patients including CKD population [130]. This is in part due to a vicious cycle where diseased kidney fails to properly filter uremic waste causing volume retention which in turn increases plasma homocysteine levels [132-134]. Hyperhomocysteinemia in turn causes cellular damage including endothelial lining and contributes to hypertension in CKD patients [125, 135].
The association between high homocysteine levels and isolated systolic hypertension in some older adults has been reported by Sutton-Tyrell in early nineties where the authors predicted a cause and effect relationship [136]. This association was later concluded as weak, in part, due to inaccurate blood pressure measurement and may have confounded by renal function [137]. Mechanisms that are surfaced in the literature based on animal studies include arterial stiffness, endothelial dysfunction, decreased bioavailability of nitric oxide (NO), increased oxidative stress and peroxynitrite formation [138, 139]. Interestingly, in a cross-sectional study it was estimated that higher plasma homocysteine levels at baseline were associated with an increased, but non-significant risk of incidental hypertension in men [140]. A similar result was reported in older adults where lowering homocysteine levels by B vitamins for a period of two years did not change blood pressure significantly compared to placebo control [141]. On the other hand, in a prospective study the progression of blood pressure on normotensive population over 2-years period Wang et al found that homocysteine is related to hypertension incidence at high levels, and may also increase the risk of hypertension at low levels [142]. Children with early CKD showed blood pressure abnormalities, mostly having a positive correlation with high homocysteine levels along with other factors, in a cross-sectional study [143]. Nevertheless, none of these studies provided strong evidences that the homocysteine causes human hypertension [137, 144]. Only incremental increase in blood pressure was documented in most of these studies. Thus, the contribution of homocysteine to raise blood pressure secondary to its vascular effects is generally accepted, although the direct role of homocysteine in causing hypertension is still not an established fact. Only larger case control studies can finally settle this much debated and controversial issue in future.
3.6 H2S producing gene manipulation: effect on blood pressure
In 2008, Wang et al reported that knockout of CSE gene elevated blood pressure due to lacking physiological levels of H2S production in mice [90]. This initial report was further confirmed by many other independent studies around the world using various experimental models [145-147]. In a randomized, double-blind and placebo-controlled trial, Sun et al demonstrated Taurine, a semi-essential sulfur-containing amino acid, supplementation increased plasma H2S concentration and improved endothelial-dependent and – independent vasodilation and this was negatively correlated with BP in taurine-treated prehypertensive individuals [148]. The same study also reported that taurine treatment upregulated the expression of H2S synthesizing enzymes, CBS and CSE and reduced agonist-induced vascular tone in human mouse mesenteric arteries and rodent aorta. Finally, they concluded that taurine has potential in intervening vascular tone improvement by targeting H2S-mediated inhibition of transient receptor potential channel subtype 3 (TRPC3)-mediated calcium influx [148].
3.7 H2S on pro-inflammatory molecules regulation in hyperhomocysteinemia, hypertension and macrophage differentiation
The ROS-induced cell injury initiates a pro-inflammatory response leading to further damage. We and others have previously reported that hyperhomocysteinemia can lead to elevated levels of inflammatory response molecules ICAM-1 (intercellular adhesion molecule-1) and VCAM-1 (vascular cell adhesion molecule-1), which further cause endothelial cell injury [149, 150]. It was also reported that not only hyperhomocysteinemia but also homocysteinylated proteins, especially albumin from uraemic patients have been found to increase pro-inflammatory molecules, ICAM-1 and VCAM-1 expression in in vitro endothelial cell culture. These adhesion molecules along with MCP-1 (monocyte chemoattractant protein-1) and its receptor CCR2 (C-C chemokine receptor type -2) expression in the monocyte facilitated chemokines/cytokines-mediated adhesion process and mediators of vascular remodeling including ADAM17 (a disintegrin and metallopeptidase domain 17), MCP-1 and Hsp60 (heat shock protein 60) in the endothelial cells [151]. Additionally, the mature ADAM17 in turn increased TNFα in the culture medium promoting adhesion of monocyte to the endothelial cells [151]. Interestingly, in a separate study the same group has demonstrated that H2S treatment mitigated TNFα-induced monocyte adhesion to endothelial cells by downregulating MCP-1, ADAM17 and related adhesion molecules [152]. Although the authors did not measure homocysteine levels per se in this in vitro study, they did measure homocysteine metabolizing and H2S forming enzymes, CSE and 3MST, and found that these enzymes were downregulated [152]. These findings foster the assumption of possible increase in homocysteine levels in these cells. Nevertheless, the findings clearly support the claim that H2S is protective against pro-inflammatory and pro-atherogenic response found in diseases, such as CKD [152].
Further investigation by our group and others have revealed that the exogenous H2S supplementation partially alleviated inflammatory response [153], and improved kidney function in hyperhomocysteinemia [117]. Several other cytokines and chemokines have also been shown to be upregulated in mesangial cells, leading to chronic kidney disease [154, 155]. Of note, macrophage inflammatory protein 2 (MIP-2) was found to be increased in glomerular mesangial cells after treatment with homocysteine while monocyte chemotactic protein 1 (MCP-1) has been shown to be activated in response to increased homocysteine levels through induction via NF-κB pathway [156, 157]. Both of these molecules were found to be reduced after treatment with H2S along with overexpressed CBS and CSE enzymes, concluding that H2S can regulate inflammation in response to renal injury [158].
Interleukin-6 (IL-6) has also been found to play a critical role in inflammation associated with hypertension [52, 159]. In a rat model of renal ischemia/reperfusion injury (IR/I), inhibition of H2S generation by PAG (DL-propargylglycine, an inhibitor of CSE enzyme) activated inflammatory response that include higher expression of IL-6 in renal tissue and promoted IR/I [160]. A similar study also found IR/I attenuated renal CBS and CSE mRNA and protein expression leading to increased pro-inflammatory MCP-1 and IL-6 activation. Further experiments revealed attenuation of cytokines in the renal tubular cells following H2S donor (NaHS) supplementation [161]. Another study showed Ang-II induced hypertension led to the upregulation of IL-6 in mice and loss of this cytokine drastically reduced renal injury and renal fibrosis [162]. Corroborating with animal studies, in patients with CKD, IL-6 was found to be increased in the kidney of hypertensive individuals [162]. Although the investigators did not measure H2S levels in these patients, it is possible that H2S levels may have diminished. In fact, diminished levels of H2S were confirmed by at least two other studies showing decrease levels of H2S in coronary heart disease and hypertensive patients as well as in children with hypertension [163].
Macrophages are differentiated monocytes in the damaged tissue which phagocytose foreign body and cellular debris, helps tissue regeneration and development of immunity. When monocytes enter in the injured tissue through endothelial, a process known as leukocyte extravasation, undergo a series of changes to become macrophages. Another process, known as macrophage polarization, occurs when macrophages functionally differentiate into two subtypes depending on their microenvironment signals. While classically activated M1 phenotype is for defense mechanism and can be detrimental when expressed for a long period of time, M2 phenotype is for tissue repair and regeneration. In many CKD diseases, including obstructive nephropathy, IR/I, glomerulosclerosis and diabetic nephropathy macrophage polarization plays a critical role [164, 165]. However, macrophage polarization and their differential role in hypertensive kidney have not yet been fully elucidated. Recently we have demonstrated that Ang-II-induced hypertension caused renal fibrosis by sustained activation of pro-inflammatory M1 macrophage promoting the release of inflammatory cytokines and treatment with H2S attenuated renal injury and fibrosis by promoting alternate activation of anti-inflammatory M2 macrophage [166].
Interleukin-21, which is a downstream molecule in the IL-6 signaling pathway, was also found to be upregulated in pulmonary-induced hypertension and promoted M2 macrophage polarization, which can lead to fibrosis [167]. In contrast, the anti-inflammatory cytokine interleukin-10 (IL-10) has been shown to have protective effects in response to Ang-II induced hypertension [168]. In IL-10 null mice treated with Ang-II, oxidative stress was increased as evidenced by superoxide and this was reduced in mice with functional IL-10, suggesting a protective function for this cytokine [168]. Despite involvement of interleukin signaling pathways in macrophage polarization and hypertension, the regulatory role of H2S in these pathways of hypertensive kidney has yet to be defined.
3.8 MMP/TIMP imbalance, renal matrix deposition and role of H2S in hypertension
Matrix metalloproteinases (MMPs) have a pivotal role in hypertension and extracellular matrix (ECM) remodeling. These zinc-dependent endopeptidases can cleave an array of substrates such as cell surface receptors, cytokines, chemokines, cell adhesion molecules and other proteases [169]. In addition, they are also responsible for several cellular functions, including maintaining ECM homeostasis, cell proliferation and migration, apoptotic pathways, and epithelial to mesenchymal transition (EMT) and have been associated with several diseases of the kidney, including diabetic nephropathy, glomerulonephritis, and acute renal injury [169, 170]. MMPs are regulated, in part, by a class of inhibitors known as tissue-inhibitors of metalloproteinases (TIMPs). The TIMPs are one type of regulators of MMPs through binding and subsequent inhibition [171]. The improper balance of these proteases can lead to multiple pathological consequences in the kidney. In a study utilizing spontaneous hypertensive rats, MMP-2 and MMP-9 were found to be increased in the renal cortex as well as MMP-9 in the medulla, which led to higher levels of collagen formation in the glomeruli and tubular interstitium. While TIMP-4 was found to be elevated in the medulla, the expression and activity were not sufficient to suppress MMP activity [172]. In another study using the same rodent model, MMP-2 but not MMP-9 was found to be significantly increased in the juxtamedullary cortex along with collagen expression and formation [173]. Inhibitors TIMP-1 and TIMP-2 were also found to be increased in expression, however, the lack of balance between MMPs and TIMPs highlights another example of the effects of hypertension on renal injury [173].
We have shown that in Ang-II induced hypertension, homocysteine contributes to renovascular fibrosis, in part, by disrupting MMP/TIMP balance [84]. This promotes oxidation and accumulation of collagen in the basement membrane of the glomerular capillaries [84]. In another study, we have demonstrated association of MMP-2 and MMP-9 with renal damage through induction of elevated levels of homocysteine in mice heterozygous for CBS enzyme [116]. These animals showed an increase in activity for both MMP-2 and MMP-9 accompanied with elevated superoxide production and apoptosis, but were mitigated by exogenous H2S supplementation, suggesting H2S acts as an antioxidant to alleviate and prevent renal damage caused by hypertension [116].
3.9 Polysulfides synthesis
H2S through sulfuration is stored in the tissue as bound sulfane sulfur [108, 174]. This may occur from endogenously generated H2S as well as exogenously supplied sources [175]. Endogenously bound sulfane sulfur has been reported by many workers using several tissues including brain and liver [174, 175]. However, to date it is unclear whether bound sulfane sulfur is protein specific and whether release of bound H2S under physiological condition requires a specific signal.
Very recently Bradley et al published a comprehensive review article summarizing the importance of garlic derived polysulfide synthesis and their possible role in cardiovascular protection through H2S and NO [176]. According to their hypothesis, garlic-derived polysulfides, such as, diallyl sulfide, diallyl disulfide and diallyl trisulfide are potent H2S donors that increase NO bioavailability through eNOS phosphorylation resulting in cardioprotection. Whether similar renal protection is possible in CKD by polysulfides or garlic derived H2S warrants investigation.
4. Other metabolic disorders: H2S is a modulator of renal dysfunction
4.1 Diabetes and H2S
Diabetes mellitus is linked with hypertension and is prevalent in individuals suffering from both type 1 and type 2, with the latter having a 60-80% rate of incidence [177]. Many of the hypertensive risk factors may also apply to the development of diabetes, including poor eating habits, higher salt intake and retention, lack of physical activity, arterial stiffening, and endothelial dysfunction [178]. Individuals who also are diagnosed with diabetes are at greater risk of developing congestive heart failure, stroke, myocardial infarction, periphery arterial disease and nephropathy [179]. Interestingly, accumulating evidence indicate that H2S plays a critical role in the pathogenesis of CKD including diabetic nephropathy. In streptozotocin-induced diabetic rat model, Zhou et al demonstrated plasma and renal tissue levels were diminished and intraperitoneal injection of NaHS (donor of H2S) were able to normalize H2S levels in plasma and kidney [180]. Increased glomerular basement membrane thickening, mesangial matrix deposition and renal interstitial fibrosis were evidenced in these diabetic rats resulting in declined renal function. H2S was able to alleviate diabetic nephropathy in part by attenuating matrix deposition and inflammation [180]. Similar to their study, we also reported that in genetic diabetic Akita mice, diminished tissue and plasma H2S content was associated with matrix deterioration and diminished renal function [56, 57]. Increased matrix metalloproteinse-9 (MMP-9) expression and activity was noticed in Akita diabetic kidney which was normalized by H2S supplementation. Finally, we concluded that renovascular remodeling and dysfunction in Akita kidney was in part due to oxidative stress-mediated MMP-9 activation leading to imbalance in gap junction molecules and renal functional impairment. H2S treatments attenuated oxidative stress and MMP-9 activity, and therefore ameliorated kidney ECM remodeling and function in Akita [56, 57].
The interconnectivity of diabetes and hypertension is substantial, including inflammation (IL-6, ICAM-1, VCAM-1, CRP), oxidative stress, vascular remodeling and endothelial dysfunction, all of which are induced by Ang-II [179]. Inhibition of Ang-II can lead to a decrease in proteinuria, a key feature of diabetes in urine, and slow the progression of end-stage renal disease [181]. It was reported that in streptozotocin-induced diabetic kidney angiotensin converting enzyme (ACE), AT1R and Ang-II was upregulated, and H2S in plasma and kidney tissue were diminished [180]. Direct correlation of diminished H2S and increased Ang-II was established by the fact that H2S treatment partially normalized all the above three effectors / inducers of hypertension in diabetic kidney [180]. Although this study in particular did not measure proteinuria and kidney function per se, we reported improved kidney function in Akita kidney [57]. Another study reported diuretic effect of H2S in spontaneously hypertensive diabetic rats in addition to reduction of blood pressure, restoration of H2S and plasma sodium levels compared to non-H2S treated diabetic rats [182]. In a post-hoc analysis study it was found that urinary sulfate (sulfate in the urine is the metabolic end product of H2S) was negatively associated with albuminuria suggesting the authors to conclude higher urinary sulfate concentration is associated with reduced renal risks in type 2 diabetic nephropathy [183]. Similar results were reported from another independent post-hoc prospective, randomized, controlled trial in which it was noticed that high urinary sulfate excretion was associated with slower decline of assessed GFR in diabetic nephropathy patients, and this was independent of known end-stage renal disease progression promoters [184]. All these above reports clearly establish a link between diabetes, hypertension and kidney dysfunction where H2S plays a key role to modulate hypertension and kidney dysfunction in diabetes.
4.2 Gut microbiota, CKD and H2S
An area of biomedical research receiving more attention in the recent years is the role of gut microbiota in the development of hypertension. Two main species of microbes predominately live in the gut, Firmicutes and Bacterioidetes, and the amount of these microbes can fluctuate depending on diet and physical fitness [185]. Increased population of these microbes has been shown in hypertensive animal models, including spontaneously-hypertensive rats, salt-induced models, as well as Ang-II induced hypertension [186]. Genetics and epigenetics can also influence gut microbiota. One study showed that the expression of renal olfactory receptor 78 (Olfr78) responded to propionate, a short chain fatty acid derived from gut microbiota, and regulated the amount of renin produced in the kidney. The knockout of this Olfr78 receptor, coupled with antibiotics that reduce the amount of microbiota in the gut led to an increase in blood pressure [187].
Microbiota, like many mammalian cells and tissues, also produces H2S. They exploit this gaseous molecule as an antioxidant defense mechanism, energy production, and cell cycle regulation [188-190]. In CKD patients, a close relationship between gut microbiota and kidney has often been highlighted. Deranged microbiota in the gut produces endotoxin which enters into the bloodstream and translocates to distant organs, including kidney, causing uremic pathology [191]. Due to damaged intestinal epithelial barrier it is plausible that live bacteria may even translocate to organs through the bloodstream. Therefore, it is not surprising that microbiota in the gut and/or translocated microbiome in the organ system can influence overall H2S levels in mammalian body under pathological dysbiosis condition. It was estimated that 50% of fecal H2S is derived from bacteria, since germ free mice only synthesize 50% which was observed in colonized mice [192]. Thus total plasma H2S pool varies depending on individual s health condition associated with microbiota milieu in the gut. Similarly, it is also possible that individual organs may harbor specific bacterial strain that further influence H2S synthesis and local release causing microenvironment change in microbiota-related disease condition.
To this end, currently it is not known whether microbial settlement in the kidney per se influences local H2S synthesis and release, and thus overall renal health in CKD. It is however reported that a number of uremic solutes are more prominent in plasma from dialysis patients with normal colon compared with colectomy patients [193]. Of these, five uremic toxins namely α-phenylacetyl-l-glutamine, 5-hydroxyindole, indoxyl glucuronide, p-cresol sulfate, and indoxyl sulfate were identified as colon derived uremic toxins. These important findings strongly suggest that colonic microbes produce a notable portion of uremic solutes, which otherwise absent or significantly lower in patients without colons [193]. Few years later of this aforementioned report, a similar study found even more uremic solutes in plasma of hemodialysis patients adding 48 newly identified solutes to the list of previously reported uremic toxins totaling altogether 270 [194]. Analyzing plasma from colectomy patients and normal subjects with hemodialysis the authors found that 9 solutes were colon derived, of which 6 were not previously been reported. Although they concluded their findings that many of these polyphenols are plant derived and can escape gut linings, a portion of these compounds are produced by gut microbiota [194]. In a recent review article, Vanholder and Glorieux nicely integrated how the composition of intestinal microbiota impacts overall uremic solute retention, and how these uremic status affects intestinal microbiome causing generation of more uremic solutes and pouring to the plasma [195]. It was also reported that in uremic condition colon bacteria trend to migrate or translocate to the distal parts of the body where normally they are absent including jejunum, ileum, lymph nodes, liver and spleen [196-198]. Bacterial DNAs were detected in the blood of non-dialyzed ESRD patients corroborating their specific genera overgrown in the guts [199]. Patients with bacterial DNA in the blood showed higher inflammatory marker which was indicative of direct correlation of bacterial translocation with inflammation in ESRD [199]. Using 5/6 kidney ablated rat model same research group has reported gut microbiome dysbiosis promotes bacterial translocation to the lymph and systemic circulation contributing microinflammation in experimental uremia [200]. Since gut microbiota produces considerable amount of H2S [201], it is highly possible that during dysbiosis in CKD patients translocated microbiota may also produce and add up to the local pool of H2S in their destined organ including kidney, and may affect kidney function. Research in this direction is highly warranted and in the near future we may witness some developments in this relatively unexplored area of human health.
5. Future research
At elevated levels homocysteine converts to homocysteine-thiolactone as a result of error-editing function of some aminoacyl-tRNA synthetases [202-204]. Homocysteine-thiolactone is a reactive metabolite that causes protein N-homocysteinylation through formation of amide bonds with protein lysine residues [204], which alters or impairs protein function [203]. Together, diminished CBS, CSE and 3MST enzymes in hypertension elevates homocysteine levels and reduces H2S production. Furthermore, hyperhomocysteinemia through protein homocysteinylation may contribute to renal remodeling. Whether these mechanisms are involved in hypertensive CKD, and whether H2S can modulate protein homocysteinylation, and therefore improves renal function is currently not known. Future investigation may provide some lights into these possibilities.
6. Summary and conclusion
Cardiorenovascular morbidity and mortality still remains the leading cause of death worldwide despite huge improvements in healthcare. Abnormalities in metabolic processes, such as carbohydrate, lipid and protein metabolism contribute to conditions related to vasculopathies including hypertension. While the cause of idiopathic hypertension remains to be elucidated for future therapeutic strategy, the known cause of hypertension related to dysregulated amino acid metabolism could easily be prevented or treated with available tools. H2S is such a tool that may be further exploited mechanistically either alone or in combination with available drugs to prevent or even to treat high homocysteine associated hypertensive vascular complications in patients with metabolic disorders and CKD. Towards this end, several clinical trials are ongoing to delineate safety and efficacy of H2S treatment to mitigate cardiorenovascular complications. We anticipate that the outcomes will provide us insightful information how H2S can be utilized as a future drug in treating hypertension associated with metabolic, cardiovascular and CKD.
Highlights.
H2S attenuates RAS activity in the kidney, and therefore has potential to treat renovascular hypertension.
In metabolic and epigenetic hypermethylation diseases, H2S deficiency exacerbates hypertension.
High homocysteine, and low H2S is associated with CKD-mediated renovascular hypertension
Gut dysbiosis contributes to the total plasma H2S pool, and therefore affects overall individual s health related to H2S patho/physiology.
Acknowledgments
This work was supported in part by National Institutes of Health grants HL-104103 and DK-104653.
Abbreviations
- 3MST
3-mercaptopyruvate sulfurtransferase
- ACE2
angiotensin-converting enzyme 2
- AGT
angiotensinogen
- Ang-II
angiotensin II
- AT1/T2-R
angiotensin type-1 / type-2 receptor
- BMI
body mass index
- BP
blood pressure
- CAT
cysteine aminotransferase
- CBS
cystathionine β-synthase
- CKD
chronic kidney diseases
- CO
carbon monoxide
- CRP
C-reactive protein
- CSE
cystathionine γ-lyase
- CVD
cardiovascular disease
- DAO
D-amino acid oxidase
- ECM
extracellular matrix
- EMT
epithelial-mesenchymal transition
- eNOS
endothelial nitric oxide synthase
- FA
folic acid
- GFR
glomerular filtration rate
- H2S
hydrogen sulfide
- HDL
high-density lipoproteins
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- IR/I
ischemia-reperfusion injury
- MCP-1
monocyte chemotactic protein 1
- MetS
Metabolic syndrome
- MIP-2
macrophage inflammatory protein 2
- miR
microRNA
- MMP
matrix metalloproteinase
- NADPH
nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- PAG
DL-propargylglycine
- RAS
renin-angiotensin system
- ROS
reactive oxygen species
- TGF-β
transforming growth factor-β
- TIMP
tissue inhibitor of metalloproteinase
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial growth factor
- WHO
World Health Organization
- ZDF
Zucker diabetic fatty
- α-SMA
α smooth muscle actin
Footnotes
Competing interests: The authors declare there are no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Judd SE, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Mackey RH, Magid DJ, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, 3rd, Moy CS, Mussolino ME, Neumar RW, Nichol G, Pandey DK, Paynter NP, Reeves MJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Wong ND, Woo D, Turner MB C. American Heart Association Statistics, S. Stroke Statistics. Heart disease and stroke statistics--2014 update: a report from the American Heart Association. Circulation. 2014;129(3):e28–e292. doi: 10.1161/01.cir.0000441139.02102.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kraft P, Bauman L, Yuan JY, Horvath S S. Framingham Heart. Multivariate variance-components analysis of longitudinal blood pressure measurements from the Framingham Heart Study. BMC genetics. 2003;4(Suppl 1):S55. doi: 10.1186/1471-2156-4-S1-S55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shuger SL, Sui XM, Church TS, Meriwether RA, Blair SN. Body mass index as a predictor of hypertension incidence among initially healthy normotensive women. Am J Hypertens. 2008;21(6):613–619. doi: 10.1038/ajh.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forman JP, Stampfer MJ, Curhan GC. Diet and lifestyle risk factors associated with incident hypertension in women. Jama. 2009;302(4):401–11. doi: 10.1001/jama.2009.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gargiulo R, Suhail F, Lerma EV. Hypertension and chronic kidney disease. Disease-a-month : DM. 2015;61(9):387–95. doi: 10.1016/j.disamonth.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Collins AJ, Kasiske B, Herzog C, Chavers B, Foley R, Gilbertson D, Grimm R, Liu J, Louis T, Manning W, Matas A, McBean M, Murray A, St Peter W, Xue J, Fan Q, Guo H, Li S, Li S, Roberts T, Snyder J, Solid C, Wang C, Weinhandl E, Arko C, Chen SC, Dalleska F, Daniels F, Dunning S, Ebben J, Frazier E, Johnson R, Sheets D, Forrest B, Berrini D, Constantini E, Everson S, Frederick P, Eggers P, Agodoa L S. United States Renal Data. Excerpts from the United States Renal Data System 2004 annual data report: atlas of end-stage renal disease in the United States. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2005;45(1 Suppl 1):A5–7. S1–280. doi: 10.1053/j.ajkd.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Muntner P, Anderson A, Charleston J, Chen Z, Ford V, Makos G, O’Connor A, Perumal K, Rahman M, Steigerwalt S, Teal V, Townsend R, Weir M, Wright JT, Jr I. Chronic Renal Insufficiency Cohort Study. Hypertension awareness, treatment, and control in adults with CKD: results from the Chronic Renal Insufficiency Cohort (CRIC) Study. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2010;55(3):441–51. doi: 10.1053/j.ajkd.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.James MT, Grams ME, Woodward M, Elley CR, Green JA, Wheeler DC, de Jong P, Gansevoort RT, Levey AS, Warnock DG, Sarnak MJ C.K.D.P. Consortium. A Meta-analysis of the Association of Estimated GFR, Albuminuria, Diabetes Mellitus, and Hypertension With Acute Kidney Injury. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2015;66(4):602–12. doi: 10.1053/j.ajkd.2015.02.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hamza SM, Dyck JR. Systemic and renal oxidative stress in the pathogenesis of hypertension: modulation of long-term control of arterial blood pressure by resveratrol. Frontiers in physiology. 2014;5:292. doi: 10.3389/fphys.2014.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oparil S, Zaman MA, Calhoun DA. Pathogenesis of hypertension. Annals of internal medicine. 2003;139(9):761–76. doi: 10.7326/0003-4819-139-9-200311040-00011. [DOI] [PubMed] [Google Scholar]
- 11.Wolf G, Butzmann U, Wenzel UO. The renin-angiotensin system and progression of renal disease: from hemodynamics to cell biology. Nephron Physiology. 2003;93(1):P3–13. doi: 10.1159/000066656. [DOI] [PubMed] [Google Scholar]
- 12.Liao TD, Yang XP, Liu YH, Shesely EG, Cavasin MA, Kuziel WA, Pagano PJ, Carretero OA. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008;52(2):256–63. doi: 10.1161/HYPERTENSIONAHA.108.112706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benigni A, Cassis P, Remuzzi G. Angiotensin II revisited: new roles in inflammation, immunology and aging. EMBO molecular medicine. 2010;2(7):247–57. doi: 10.1002/emmm.201000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circulation research. 2008;102(4):488–96. doi: 10.1161/CIRCRESAHA.107.162800. [DOI] [PubMed] [Google Scholar]
- 15.Kumar KV, Das UN. Are free radicals involved in the pathobiology of human essential hypertension? Free radical research communications. 1993;19(1):59–66. doi: 10.3109/10715769309056499. [DOI] [PubMed] [Google Scholar]
- 16.Chabrashvili T, Tojo A, Onozato ML, Kitiyakara C, Quinn MT, Fujita T, Welch WJ, Wilcox CS. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension. 2002;39(2):269–74. doi: 10.1161/hy0202.103264. [DOI] [PubMed] [Google Scholar]
- 17.Kitiyakara C, Chabrashvili T, Chen Y, Blau J, Karber A, Aslam S, Welch WJ, Wilcox CS. Salt intake, oxidative stress, and renal expression of NADPH oxidase and superoxide dismutase. Journal of the American Society of Nephrology : JASN. 2003;14(11):2775–82. doi: 10.1097/01.asn.0000092145.90389.65. [DOI] [PubMed] [Google Scholar]
- 18.Wesseling S, Fledderus JO, Verhaar MC, Joles JA. Beneficial effects of diminished production of hydrogen sulfide or carbon monoxide on hypertension and renal injury induced by NO withdrawal. British journal of pharmacology. 2015;172(6):1607–19. doi: 10.1111/bph.12674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veelken R, Hilgers KF, Hartner A, Haas A, Bohmer KP, Sterzel RB. Nitric oxide synthase isoforms and glomerular hyperfiltration in early diabetic nephropathy. Journal of the American Society of Nephrology : JASN. 2000;11(1):71–9. doi: 10.1681/ASN.V11171. [DOI] [PubMed] [Google Scholar]
- 20.Baylis C, Qiu C. Importance of nitric oxide in the control of renal hemodynamics. Kidney Int. 1996;49(6):1727–31. doi: 10.1038/ki.1996.256. [DOI] [PubMed] [Google Scholar]
- 21.Han SJ, Kim JI, Park JW, Park KM. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2015;30(9):1497–506. doi: 10.1093/ndt/gfv226. [DOI] [PubMed] [Google Scholar]
- 22.Kasinath BS. Hydrogen sulfide to the rescue in obstructive kidney injury. Kidney Int. 2014;85(6):1255–8. doi: 10.1038/ki.2013.529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feliers D, Lee HJ, Kasinath BS. Hydrogen Sulfide in Renal Physiology and Disease. Antioxidants & redox signaling. 2016 doi: 10.1089/ars.2015.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sen U, Sathnur PB, Kundu S, Givvimani S, Coley DM, Mishra PK, Qipshidze N, Tyagi N, Metreveli N, Tyagi SC. Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. American journal of physiology. Cell physiology. 2012;303(1):C41–51. doi: 10.1152/ajpcell.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koning AM, Frenay AR, Leuvenink HG, van Goor H. Hydrogen sulfide in renal physiology, disease and transplantation--the smell of renal protection. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2015;46:37–49. doi: 10.1016/j.niox.2015.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Yan H, Du J, Tang C. The possible role of hydrogen sulfide on the pathogenesis of spontaneous hypertension in rats. Biochemical and biophysical research communications. 2004;313(1):22–7. doi: 10.1016/j.bbrc.2003.11.081. [DOI] [PubMed] [Google Scholar]
- 27.Holwerda KM, Bos EM, Rajakumar A, Ris-Stalpers C, van Pampus MG, Timmer A, Erwich JJ, Faas MM, van Goor H, Lely AT. Hydrogen sulfide producing enzymes in pregnancy and preeclampsia. Placenta. 2012;33(6):518–21. doi: 10.1016/j.placenta.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 28.Chunyu Z, Junbao D, Dingfang B, Hui Y, Xiuying T, Chaoshu T. The regulatory effect of hydrogen sulfide on hypoxic pulmonary hypertension in rats. Biochemical and biophysical research communications. 2003;302(4):810–6. doi: 10.1016/s0006-291x(03)00256-0. [DOI] [PubMed] [Google Scholar]
- 29.Zhong G, Chen F, Cheng Y, Tang C, Du J. The role of hydrogen sulfide generation in the pathogenesis of hypertension in rats induced by inhibition of nitric oxide synthase. Journal of hypertension. 2003;21(10):1879–85. doi: 10.1097/00004872-200310000-00015. [DOI] [PubMed] [Google Scholar]
- 30.Huang P, Chen SY, Wang Y, Liu J, Yao QY, Huang YQ, Li HX, Zhu MZ, Wang SX, Li L, Tang CS, Tao YH, Yang GS, Du JB, Jin HF. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide-Biol Ch. 2015;46:192–203. doi: 10.1016/j.niox.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Pushpakumar S, Kundu S, Narayanan N, Sen U. DNA hypermethylation in hyperhomocysteinemia contributes to abnormal extracellular matrix metabolism in the kidney. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2015;29(11):4713–25. doi: 10.1096/fj.15-272443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li JJ, Li Q, Du HP, Wang YL, You SJ, Wang F, Xu XS, Cheng J, Cao YJ, Liu CF, Hu LF. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. International journal of molecular sciences. 2015;16(6):12560–77. doi: 10.3390/ijms160612560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen NC, Yang F, Capecci LM, Gu Z, Schafer AI, Durante W, Yang XF, Wang H. Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2010;24(8):2804–17. doi: 10.1096/fj.09-143651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000;101(3):329–35. doi: 10.1161/01.cir.101.3.329. [DOI] [PubMed] [Google Scholar]
- 35.Bolivar JJ. Essential hypertension: an approach to its etiology and neurogenic pathophysiology. International journal of hypertension. 2013;2013:547809. doi: 10.1155/2013/547809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viera AJ, Neutze DM. Diagnosis of secondary hypertension: an age-based approach. American family physician. 2010;82(12):1471–8. [PubMed] [Google Scholar]
- 37.Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacological reviews. 2007;59(3):251–87. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 38.Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44(5):595–601. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 39.Wolf G, Ritz E. Combination therapy with ACE inhibitors and angiotensin II receptor blockers to halt progression of chronic renal disease: pathophysiology and indications. Kidney Int. 2005;67(3):799–812. doi: 10.1111/j.1523-1755.2005.00145.x. [DOI] [PubMed] [Google Scholar]
- 40.Frank J. Managing hypertension using combination therapy. American family physician. 2008;77(9):1279–1286. [PubMed] [Google Scholar]
- 41.Barreras A, Gurk-Turner C. Angiotensin II receptor blockers. Proceedings. 2003;16(1):123–6. doi: 10.1080/08998280.2003.11927893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobori H, Ozawa Y, Suzaki Y, Prieto-Carrasquero MC, Nishiyama A, Shoji T, Cohen EP, Navar LG. Young Scholars Award Lecture: Intratubular angiotensinogen in hypertension and kidney diseases. Am J Hypertens. 2006;19(5):541–50. doi: 10.1016/j.amjhyper.2005.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Santos PC, Krieger JE, Pereira AC. Renin-angiotensin system, hypertension, and chronic kidney disease: pharmacogenetic implications. Journal of pharmacological sciences. 2012;120(2):77–88. doi: 10.1254/jphs.12r03cr. [DOI] [PubMed] [Google Scholar]
- 44.Ferrario CM, Trask AJ, Jessup JA. Advances in biochemical and functional roles of angiotensin-converting enzyme 2 and angiotensin-(1-7) in regulation of cardiovascular function. American journal of physiology. Heart and circulatory physiology. 2005;289(6):H2281–90. doi: 10.1152/ajpheart.00618.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atlas SA. The renin-angiotensin aldosterone system: pathophysiological role and pharmacologic inhibition. Journal of managed care pharmacy : JMCP. 2007;13(8 Suppl B):9–20. doi: 10.18553/jmcp.2007.13.s8-b.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siragy HM, Carey RM. Role of the intrarenal renin-angiotensin-aldosterone system in chronic kidney disease. American journal of nephrology. 2010;31(6):541–50. doi: 10.1159/000313363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carey RM, Wang ZQ, Siragy HM. Role of the angiotensin type 2 receptor in the regulation of blood pressure and renal function. Hypertension. 2000;35(1 Pt 2):155–63. doi: 10.1161/01.hyp.35.1.155. [DOI] [PubMed] [Google Scholar]
- 48.Ferrario CM. Angiotensin-converting enzyme 2 and angiotensin-(1-7): an evolving story in cardiovascular regulation. Hypertension. 2006;47(3):515–21. doi: 10.1161/01.HYP.0000196268.08909.fb. [DOI] [PubMed] [Google Scholar]
- 49.Lu M, Liu YH, Goh HS, Wang JJ, Yong QC, Wang R, Bian JS. Hydrogen sulfide inhibits plasma renin activity. Journal of the American Society of Nephrology : JASN. 2010;21(6):993–1002. doi: 10.1681/ASN.2009090949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue H, Yuan P, Ni J, Li C, Shao D, Liu J, Shen Y, Wang Z, Zhou L, Zhang W, Huang Y, Yu C, Wang R, Lu L. H(2)S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation. PloS one. 2013;8(9):e74366. doi: 10.1371/journal.pone.0074366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alberti KG, Zimmet P, Shaw J I.D.F.E.T.F.C. Group. The metabolic syndrome--a new worldwide definition. Lancet. 2005;366(9491):1059–62. doi: 10.1016/S0140-6736(05)67402-8. [DOI] [PubMed] [Google Scholar]
- 52.Singh AK, Kari JA. Metabolic syndrome and chronic kidney disease. Current opinion in nephrology and hypertension. 2013;22(2):198–203. doi: 10.1097/MNH.0b013e32835dda78. [DOI] [PubMed] [Google Scholar]
- 53.Chen J, Muntner P, Hamm LL, Jones DW, Batuman V, Fonseca V, Whelton PK, He J. The metabolic syndrome and chronic kidney disease in U.S. adults. Annals of internal medicine. 2004;140(3):167–74. doi: 10.7326/0003-4819-140-3-200402030-00007. [DOI] [PubMed] [Google Scholar]
- 54.Kurella M, Lo JC, Chertow GM. Metabolic syndrome and the risk for chronic kidney disease among nondiabetic adults. Journal of the American Society of Nephrology : JASN. 2005;16(7):2134–40. doi: 10.1681/ASN.2005010106. [DOI] [PubMed] [Google Scholar]
- 55.Palaniappan L, Carnethon M, Fortmann SP. Association between microalbuminuria and the metabolic syndrome: NHANES III. Am J Hypertens. 2003;16(11):952–958. doi: 10.1016/s0895-7061(03)01009-4. [DOI] [PubMed] [Google Scholar]
- 56.Kundu S, Pushpakumar SB, Tyagi A, Coley D, Sen U. Hydrogen sulfide deficiency and diabetic renal remodeling: role of matrix metalloproteinase-9. American journal of physiology Endocrinology and metabolism. 2013;304(12):E1365–78. doi: 10.1152/ajpendo.00604.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kundu S, Pushpakumar S, Sen U. MMP-9- and NMDA receptor-mediated mechanism of diabetic renovascular remodeling and kidney dysfunction: hydrogen sulfide is a key modulator. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2015;46:172–85. doi: 10.1016/j.niox.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu L, Yang W, Jia X, Yang G, Duridanova D, Cao K, Wang R. Pancreatic islet overproduction of H2S and suppressed insulin release in Zucker diabetic rats. Laboratory investigation; a journal of technical methods and pathology. 2009;89(1):59–67. doi: 10.1038/labinvest.2008.109. [DOI] [PubMed] [Google Scholar]
- 59.Holwerda KM, Burke SD, Faas MM, Zsengeller Z, Stillman IE, Kang PM, van Goor H, McCurley A, Jaffe IZ, Karumanchi SA, Lely AT. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. Journal of the American Society of Nephrology : JASN. 2014;25(4):717–25. doi: 10.1681/ASN.2013030291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wei Q, Mi QS, Dong Z. The regulation and function of microRNAs in kidney diseases. IUBMB life. 2013;65(7):602–14. doi: 10.1002/iub.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pan J, Zhang J, Zhang X, Zhou X, Lu S, Huang X, Shao J, Lou G, Yang D, Geng YJ. Role of microRNA-29b in angiotensin II-induced epithelial-mesenchymal transition in renal tubular epithelial cells. International journal of molecular medicine. 2014;34(5):1381–7. doi: 10.3892/ijmm.2014.1935. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, Taylor NE, Lu L, Usa K, Cowley AW, Jr, Ferreri NR, Yeo NC, Liang M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension. 2010;55(4):974–82. doi: 10.1161/HYPERTENSIONAHA.109.144428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Di J, Jiang L, Zhou Y, Cao H, Fang L, Wen P, Li X, Dai C, Yang J. Ets-1 targeted by microrna-221 regulates angiotensin II-induced renal fibroblast activation and fibrosis. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2014;34(4):1063–74. doi: 10.1159/000366321. [DOI] [PubMed] [Google Scholar]
- 64.Ebrahimian T, Li MW, Lemarie CA, Simeone SM, Pagano PJ, Gaestel M, Paradis P, Wassmann S, Schiffrin EL. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: regulation of oxidative stress. Hypertension. 2011;57(2):245–54. doi: 10.1161/HYPERTENSIONAHA.110.159889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marques FZ, Campain AE, Tomaszewski M, Zukowska-Szczechowska E, Yang YH, Charchar FJ, Morris BJ. Gene expression profiling reveals renin mRNA overexpression in human hypertensive kidneys and a role for microRNAs. Hypertension. 2011;58(6):1093–8. doi: 10.1161/HYPERTENSIONAHA.111.180729. [DOI] [PubMed] [Google Scholar]
- 66.Toldo S, Das A, Mezzaroma E, Chau VQ, Marchetti C, Durrant D, Samidurai A, Van Tassell BW, Yin C, Ockaili RA, Vigneshwar N, Mukhopadhyay ND, Kukreja RC, Abbate A, Salloum FN. Induction of microRNA-21 with exogenous hydrogen sulfide attenuates myocardial ischemic and inflammatory injury in mice. Circulation Cardiovascular genetics. 2014;7(3):311–20. doi: 10.1161/CIRCGENETICS.113.000381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eskildsen TV, Jeppesen PL, Schneider M, Nossent AY, Sandberg MB, Hansen PB, Jensen CH, Hansen ML, Marcussen N, Rasmussen LM, Bie P, Andersen DC, Sheikh SP. Angiotensin II regulates microRNA-132/-212 in hypertensive rats and humans. International journal of molecular sciences. 2013;14(6):11190–207. doi: 10.3390/ijms140611190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber G, Pushpakumar S, Mukhopadhyay M, Sen U. MicroRNA Profiling and Regulation in Hypertensive Kidney. The FASEB Journal. 2016:1028.12. [Google Scholar]
- 69.Ostrakhovitch EA, Tabibzadeh S. Homocysteine in Chronic Kidney Disease. Advances in clinical chemistry. 2015;72:77–106. doi: 10.1016/bs.acc.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 70.Selhub J. Homocysteine metabolism. Annual review of nutrition. 1999;19:217–46. doi: 10.1146/annurev.nutr.19.1.217. [DOI] [PubMed] [Google Scholar]
- 71.House JD, Brosnan ME, Brosnan JT. Characterization of homocysteine metabolism in the rat kidney. The Biochemical journal. 1997;328(Pt 1):287–92. doi: 10.1042/bj3280287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.House JD, Brosnan ME, Brosnan JT. Renal uptake and excretion of homocysteine in rats with acute hyperhomocysteinemia. Kidney Int. 1998;54(5):1601–7. doi: 10.1046/j.1523-1755.1998.00144.x. [DOI] [PubMed] [Google Scholar]
- 73.Yi F, Jin S, Zhang F, Xia M, Bao JX, Hu J, Poklis JL, Li PL. Formation of lipid raft redox signalling platforms in glomerular endothelial cells: an early event of homocysteine-induced glomerular injury. Journal of cellular and molecular medicine. 2009;13(9B):3303–14. doi: 10.1111/j.1582-4934.2009.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yi F, Zhang AY, Janscha JL, Li PL, Zou AP. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney Int. 2004;66(5):1977–87. doi: 10.1111/j.1523-1755.2004.00968.x. [DOI] [PubMed] [Google Scholar]
- 75.Handy DE, Zhang Y, Loscalzo J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. The Journal of biological chemistry. 2005;280(16):15518–25. doi: 10.1074/jbc.M501452200. [DOI] [PubMed] [Google Scholar]
- 76.Schnabel R, Lackner KJ, Rupprecht HJ, Espinola-Klein C, Torzewski M, Lubos E, Bickel C, Cambien F, Tiret L, Munzel T, Blankenberg S. Glutathione peroxidase-1 and homocysteine for cardiovascular risk prediction: results from the AtheroGene study. Journal of the American College of Cardiology. 2005;45(10):1631–7. doi: 10.1016/j.jacc.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 77.Glushchenko AV, Jacobsen DW. Molecular targeting of proteins by L-homocysteine: mechanistic implications for vascular disease. Antioxidants & redox signaling. 2007;9(11):1883–98. doi: 10.1089/ars.2007.1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boger RH, Bode-Boger SM, Szuba A, Tsao PS, Chan JR, Tangphao O, Blaschke TF, Cooke JP. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation. 1998;98(18):1842–7. doi: 10.1161/01.cir.98.18.1842. [DOI] [PubMed] [Google Scholar]
- 79.Topal G, Brunet A, Millanvoye E, Boucher JL, Rendu F, Devynck MA, David-Dufilho M. Homocysteine induces oxidative stress by uncoupling of NO synthase activity through reduction of tetrahydrobiopterin. Free radical biology & medicine. 2004;36(12):1532–41. doi: 10.1016/j.freeradbiomed.2004.03.019. [DOI] [PubMed] [Google Scholar]
- 80.Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. The Journal of clinical investigation. 1986;77(4):1370–6. doi: 10.1172/JCI112442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Perna AF, Ingrosso D. Low hydrogen sulphide and chronic kidney disease: a dangerous liaison. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27(2):486–93. doi: 10.1093/ndt/gfr737. [DOI] [PubMed] [Google Scholar]
- 82.King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, Dugas TR, Kelley EE, Elrod JW, Huang PL, Wang R, Lefer DJ. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase-nitric oxide dependent. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(8):3182–7. doi: 10.1073/pnas.1321871111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, Moser J, Hillebrands JL, Ploeg RJ, Yang G, Leuvenink HG, van Goor H. Cystathionine gamma-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. Journal of the American Society of Nephrology : JASN. 2013;24(5):759–70. doi: 10.1681/ASN.2012030268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pushpakumar SB, Kundu S, Metreveli N, Sen U. Folic acid mitigates angiotensin-II-induced blood pressure and renal remodeling. PloS one. 2013;8(12):e83813. doi: 10.1371/journal.pone.0083813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matsumoto K, Morishita R, Moriguchi A, Tomita N, Yo Y, Nishii T, Nakamura T, Higaki J, Ogihara T. Prevention of renal damage by angiotensin II blockade, accompanied by increased renal hepatocyte growth factor in experimental hypertensive rats. Hypertension. 1999;34(2):279–84. doi: 10.1161/01.hyp.34.2.279. [DOI] [PubMed] [Google Scholar]
- 86.Sen U, Mishra PK, Tyagi N, Tyagi SC. Homocysteine to hydrogen sulfide or hypertension. Cell biochemistry and biophysics. 2010;57(2-3):49–58. doi: 10.1007/s12013-010-9079-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia-reperfusion injury: role of antioxidant and antiapoptotic signaling. American journal of physiology Heart and circulatory physiology. 2008;295(2):H801–6. doi: 10.1152/ajpheart.00377.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kimura Y, Dargusch R, Schubert D, Kimura H. Hydrogen sulfide protects HT22 neuronal cells from oxidative stress. Antioxidants & redox signaling. 2006;8(3-4):661–70. doi: 10.1089/ars.2006.8.661. [DOI] [PubMed] [Google Scholar]
- 89.Kimura Y, Kimura H. Hydrogen sulfide protects neurons from oxidative stress. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2004;18(10):1165–7. doi: 10.1096/fj.04-1815fje. [DOI] [PubMed] [Google Scholar]
- 90.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–90. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. American journal of physiology Heart and circulatory physiology. 2004;287(5):H2316–23. doi: 10.1152/ajpheart.00331.2004. [DOI] [PubMed] [Google Scholar]
- 92.van Guldener C. Why is homocysteine elevated in renal failure and what can be expected from homocysteine-lowering? Nephrol Dial Transpl. 2006;21(5):1161–1166. doi: 10.1093/ndt/gfl044. [DOI] [PubMed] [Google Scholar]
- 93.Smith DE, Hornstra JM, Kok RM, Blom HJ, Smulders YM. Folic acid supplementation does not reduce intracellular homocysteine, and may disturb intracellular one-carbon metabolism. Clinical chemistry and laboratory medicine. 2013;51(8):1643–50. doi: 10.1515/cclm-2012-0694. [DOI] [PubMed] [Google Scholar]
- 94.Perna AF, Ingrosso D. Atherosclerosis determinants in renal disease: how much is homocysteine involved? Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2016;31(6):860–3. doi: 10.1093/ndt/gfv409. [DOI] [PubMed] [Google Scholar]
- 95.Massy ZA. Potential strategies to normalize the levels of homocysteine in chronic renal failure patients. Kidney international Supplement. 2003;(84):S134–6. doi: 10.1046/j.1523-1755.63.s84.28.x. [DOI] [PubMed] [Google Scholar]
- 96.Robinson K. Renal disease, homocysteine, and cardiovascular complications. Circulation. 2004;109(3):294–5. doi: 10.1161/01.CIR.0000114133.99074.96. [DOI] [PubMed] [Google Scholar]
- 97.Massy ZA. Therapy of hyperhomocysteinemia in chronic kidney disease. Semin Nephrol. 2006;26(1):24–7. doi: 10.1016/j.semnephrol.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 98.Perna AF, Luciano MG, Ingrosso D, Pulzella P, Sepe I, Lanza D, Violetti E, Capasso R, Lombardi C, De Santo NG. Hydrogen sulphide-generating pathways in haemodialysis patients: a study on relevant metabolites and transcriptional regulation of genes encoding for key enzymes. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2009;24(12):3756–63. doi: 10.1093/ndt/gfp378. [DOI] [PubMed] [Google Scholar]
- 99.Aminzadeh MA, Vaziri ND. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27(2):498–504. doi: 10.1093/ndt/gfr560. [DOI] [PubMed] [Google Scholar]
- 100.Perna AF, Sepe I, Lanza D, Ingrosso D. Hydrogen sulfide increases after a single hemodialysis session. Kidney Int. 2011;80(10):1108–9. doi: 10.1038/ki.2011.285. [DOI] [PubMed] [Google Scholar]
- 101.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochemical and biophysical research communications. 1997;237(3):527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 102.Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. The Biochemical journal. 1982;206(2):267–77. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Patel P, Vatish M, Heptinstall J, Wang R, Carson RJ. The endogenous production of hydrogen sulphide in intrauterine tissues. Reproductive biology and endocrinology : RB&E. 7(2009):10. doi: 10.1186/1477-7827-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chen X, Jhee KH, Kruger WD. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. The Journal of biological chemistry. 2004;279(50):52082–6. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 105.Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. The Journal of biological chemistry. 2009;284(17):11601–12. doi: 10.1074/jbc.M808026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Griffith OW. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free radical biology & medicine. 1999;27(9-10):922–35. doi: 10.1016/s0891-5849(99)00176-8. [DOI] [PubMed] [Google Scholar]
- 107.Gould SJ, Keller GA, Subramani S. Identification of peroxisomal targeting signals located at the carboxy terminus of four peroxisomal proteins. The Journal of cell biology. 1988;107(3):897–905. doi: 10.1083/jcb.107.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kimura H. Signaling molecules: hydrogen sulfide and polysulfide. Antioxidants & redox signaling. 2015;22(5):362–76. doi: 10.1089/ars.2014.5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hashimoto A, Oka T, Nishikawa T. Anatomical distribution and postnatal changes in endogenous free D-aspartate and D-serine in rat brain and periphery. The European journal of neuroscience. 1995;7(8):1657–63. doi: 10.1111/j.1460-9568.1995.tb00687.x. [DOI] [PubMed] [Google Scholar]
- 110.Huang J, Niknahad H, Khan S, O’Brien PJ. Hepatocyte-catalysed detoxification of cyanide by L- and D-cysteine. Biochem Pharmacol. 1998;55(12):1983–90. doi: 10.1016/s0006-2952(98)00072-0. [DOI] [PubMed] [Google Scholar]
- 111.Kimura H. The physiological role of hydrogen sulfide and beyond. Nitric oxide : biology and chemistry / official journal of the Nitric Oxide Society. 2014;41:4–10. doi: 10.1016/j.niox.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 112.Stopper H, Treutlein AT, Bahner U, Schupp N, Schmid U, Brink A, Perna A, Heidland A. Reduction of the genomic damage level in haemodialysis patients by folic acid and vitamin B12 supplementation. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2008;23(10):3272–9. doi: 10.1093/ndt/gfn254. [DOI] [PubMed] [Google Scholar]
- 113.Friedman AN, Bostom AG, Selhub J, Levey AS, Rosenberg IH. The kidney and homocysteine metabolism. Journal of the American Society of Nephrology : JASN. 2001;12(10):2181–9. doi: 10.1681/ASN.V12102181. [DOI] [PubMed] [Google Scholar]
- 114.Stam F, van Guldener C, Ter Wee PM, Jakobs C, de Meer K, Stehouwer CD. Effect of folic acid on methionine and homocysteine metabolism in end-stage renal disease. Kidney Int. 2005;67(1):259–64. doi: 10.1111/j.1523-1755.2005.00076.x. [DOI] [PubMed] [Google Scholar]
- 115.Qin X, Huo Y, Langman CB, Hou F, Chen Y, Matossian D, Xu X, Wang X. Folic acid therapy and cardiovascular disease in ESRD or advanced chronic kidney disease: a meta-analysis. Clinical journal of the American Society of Nephrology : CJASN. 2011;6(3):482–8. doi: 10.2215/CJN.05310610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sen U, Basu P, Abe OA, Givvimani S, Tyagi N, Metreveli N, Shah KS, Passmore JC, Tyagi SC. Hydrogen sulfide ameliorates hyperhomocysteinemia-associated chronic renal failure. American journal of physiology. Renal physiology. 2009;297(2):F410–9. doi: 10.1152/ajprenal.00145.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sen U, Munjal C, Qipshidze N, Abe O, Gargoum R, Tyagi SC. Hydrogen sulfide regulates homocysteine-mediated glomerulosclerosis. American journal of nephrology. 2010;31(5):442–55. doi: 10.1159/000296717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wei HJ, Xu JH, Li MH, Tang JP, Zou W, Zhang P, Wang L, Wang CY, Tang XQ. Hydrogen sulfide inhibits homocysteine-induced endoplasmic reticulum stress and neuronal apoptosis in rat hippocampus via upregulation of the BDNF-TrkB pathway. Acta pharmacologica Sinica. 2014;35(6):707–15. doi: 10.1038/aps.2013.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Flannigan KL, Agbor TA, Blackler RW, Kim JJ, Khan WI, Verdu EF, Ferraz JG, Wallace JL. Impaired hydrogen sulfide synthesis and IL-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(37):13559–64. doi: 10.1073/pnas.1413390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lupoli R, Di Minno A, Spadarella G, Franchini M, Sorrentino R, Cirino G, Di Minno G. Methylation reactions, the redox balance and atherothrombosis: the search for a link with hydrogen sulfide. Seminars in thrombosis and hemostasis. 2015;41(4):423–32. doi: 10.1055/s-0035-1549848. [DOI] [PubMed] [Google Scholar]
- 121.Bianca RDD, Coletta C, Mitidieri E, De Dominicis G, Rossi A, Sautebin L, Cirino G, Bucci M, Sorrentino R. Hydrogen sulphide induces mouse paw oedema through activation of phospholipase A(2) British journal of pharmacology. 2010;161(8):1835–1842. doi: 10.1111/j.1476-5381.2010.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pullin CH, Bonham JR, McDowell IF, Lee PJ, Powers HJ, Wilson JF, Lewis MJ, Moat SJ. Vitamin C therapy ameliorates vascular endothelial dysfunction in treated patients with homocystinuria. Journal of inherited metabolic disease. 2002;25(2):107–18. doi: 10.1023/a:1015672625913. [DOI] [PubMed] [Google Scholar]
- 123.Yap S, Naughten ER, Wilcken B, Wilcken DE, Boers GH. Vascular complications of severe hyperhomocysteinemia in patients with homocystinuria due to cystathionine beta-synthase deficiency: effects of homocysteine-lowering therapy. Seminars in thrombosis and hemostasis. 2000;26(3):335–40. doi: 10.1055/s-2000-8100. [DOI] [PubMed] [Google Scholar]
- 124.Celermajer DS, Sorensen K, Ryalls M, Robinson J, Thomas O, Leonard JV, Deanfield JE. Impaired endothelial function occurs in the systemic arteries of children with homozygous homocystinuria but not in their heterozygous parents. Journal of the American College of Cardiology. 1993;22(3):854–8. doi: 10.1016/0735-1097(93)90203-d. [DOI] [PubMed] [Google Scholar]
- 125.Chambers JC, McGregor A, Jean-Marie J, Obeid OA, Kooner JS. Demonstration of rapid onset vascular endothelial dysfunction after hyperhomocysteinemia: an effect reversible with vitamin C therapy. Circulation. 1999;99(9):1156–60. doi: 10.1161/01.cir.99.9.1156. [DOI] [PubMed] [Google Scholar]
- 126.Narayanan N, Pushpakumar SB, Givvimani S, Kundu S, Metreveli N, James D, Bratcher AP, Tyagi SC. Epigenetic regulation of aortic remodeling in hyperhomocysteinemia. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2014;28(8):3411–22. doi: 10.1096/fj.14-250183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nerbass FB, Draibe SA, Feiten SF, Chiarello PG, Vannucchi H, Cuppari L. Homocysteine and its determinants in nondialyzed chronic kidney disease patients. Journal of the American Dietetic Association. 2006;106(2):267–70. doi: 10.1016/j.jada.2005.10.035. [DOI] [PubMed] [Google Scholar]
- 128.Robinson K, Gupta A, Dennis V, Arheart K, Chaudhary D, Green R, Vigo P, Mayer EL, Selhub J, Kutner M, Jacobsen DW. Hyperhomocysteinemia confers an independent increased risk of atherosclerosis in end-stage renal disease and is closely linked to plasma folate and pyridoxine concentrations. Circulation. 1996;94(11):2743–8. doi: 10.1161/01.cir.94.11.2743. [DOI] [PubMed] [Google Scholar]
- 129.Chao MC, Hu SL, Hsu HS, Davidson LE, Lin CH, Li CI, Liu CS, Li TC, Lin CC, Lin WY. Serum homocysteine level is positively associated with chronic kidney disease in a Taiwan Chinese population. Journal of nephrology. 2014;27(3):299–305. doi: 10.1007/s40620-013-0037-9. [DOI] [PubMed] [Google Scholar]
- 130.Ye Z, Wang C, Zhang Q, Li Y, Zhang J, Ma X, Peng H, Lou T. Prevalence of Homocysteine-Related Hypertension in Patients With Chronic Kidney Disease. Journal of clinical hypertension. 2016 doi: 10.1111/jch.12881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pastore A, Noce A, Di Giovamberardino G, De Stefano A, Calla C, Zenobi R, Dessi M, Di Daniele N. Homocysteine, cysteine, folate and vitamin B(1)(2) status in type 2 diabetic patients with chronic kidney disease. Journal of nephrology. 2015;28(5):571–6. doi: 10.1007/s40620-014-0126-4. [DOI] [PubMed] [Google Scholar]
- 132.Rodriguez WE, Tyagi N, Joshua IG, Passmore JC, Fleming JT, Falcone JC, Tyagi SC. Pioglitazone mitigates renal glomerular vascular changes in high-fat, high-calorie-induced type 2 diabetes mellitus. American journal of physiology Renal physiology. 2006;291(3):F694–701. doi: 10.1152/ajprenal.00398.2005. [DOI] [PubMed] [Google Scholar]
- 133.Rodriguez WE, Sen U, Tyagi N, Kumar M, Carneal G, Aggrawal D, Newsome J, Tyagi SC. PPAR gamma agonist normalizes glomerular filtration rate, tissue levels of homocysteine, and attenuates endothelial-myocyte uncoupling in alloxan induced diabetic mice. International journal of biological sciences. 2008;4(4):236–44. doi: 10.7150/ijbs.4.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sen U, Rodriguez WE, Tyagi N, Kumar M, Kundu S, Tyagi SC. Ciglitazone, a PPARgamma agonist, ameliorates diabetic nephropathy in part through homocysteine clearance. American journal of physiology Endocrinology and metabolism. 2008;295(5):E1205–12. doi: 10.1152/ajpendo.90534.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jourde-Chiche N, Dou L, Cerini C, Dignat-George F, Brunet P. Vascular incompetence in dialysis patients--protein-bound uremic toxins and endothelial dysfunction. Seminars in dialysis. 2011;24(3):327–37. doi: 10.1111/j.1525-139X.2011.00925.x. [DOI] [PubMed] [Google Scholar]
- 136.Sutton-Tyrrell K, Bostom A, Selhub J, Zeigler-Johnson C. High homocysteine levels are independently related to isolated systolic hypertension in older adults. Circulation. 1997;96(6):1745–9. doi: 10.1161/01.cir.96.6.1745. [DOI] [PubMed] [Google Scholar]
- 137.van Guldener C, Nanayakkara PW, Stehouwer CD. Homocysteine and blood pressure. Current hypertension reports. 2003;5(1):26–31. doi: 10.1007/s11906-003-0007-z. [DOI] [PubMed] [Google Scholar]
- 138.Fischer PA, Dominguez GN, Cuniberti LA, Martinez V, Werba JP, Ramirez AJ, Masnatta LD. Hyperhomocysteinemia induces renal hemodynamic dysfunction: is nitric oxide involved? Journal of the American Society of Nephrology : JASN. 2003;14(3):653–60. doi: 10.1097/01.asn.0000053419.27133.23. [DOI] [PubMed] [Google Scholar]
- 139.Dong Y, Sun Q, Liu T, Wang H, Jiao K, Xu J, Liu X, Liu H, Wang W. Nitrative Stress Participates in Endothelial Progenitor Cell Injury in Hyperhomocysteinemia. PloS one. 2016;11(7):e0158672. doi: 10.1371/journal.pone.0158672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bowman TS, Gaziano JM, Stampfer MJ, Sesso HD. Homocysteine and risk of developing hypertension in men. J Hum Hypertens. 2006;20(8):631–4. doi: 10.1038/sj.jhh.1002052. [DOI] [PubMed] [Google Scholar]
- 141.McMahon JA, Skeaff CM, Williams SM, Green TJ. Lowering homocysteine with B vitamins has no effect on blood pressure in older adults. The Journal of nutrition. 2007;137(5):1183–7. doi: 10.1093/jn/137.5.1183. [DOI] [PubMed] [Google Scholar]
- 142.Wang Y, Chen S, Yao T, Li D, Wang Y, Li Y, Wu S, Cai J. Homocysteine as a risk factor for hypertension: a 2-year follow-up study. PloS one. 2014;9(10):e108223. doi: 10.1371/journal.pone.0108223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chien SJ, Lin IC, Hsu CN, Lo MH, Tain YL. Homocysteine and Arginine-to-Asymmetric Dimethylarginine Ratio Associated With Blood Pressure Abnormalities in Children With Early Chronic Kidney Disease. Circulation journal : official journal of the Japanese Circulation Society. 2015;79(9):2031–7. doi: 10.1253/circj.CJ-15-0412. [DOI] [PubMed] [Google Scholar]
- 144.Stehouwer CD, van Guldener C. Does homocysteine cause hypertension? Clinical chemistry and laboratory medicine. 2003;41(11):1408–11. doi: 10.1515/CCLM.2003.216. [DOI] [PubMed] [Google Scholar]
- 145.Roy A, Khan AH, Islam MT, Prieto MC, Majid DS. Interdependency of cystathione gamma-lyase and cystathione beta-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am J Hypertens. 2012;25(1):74–81. doi: 10.1038/ajh.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cindrova-Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: hydrogen sulfide as a placental vasodilator. The American journal of pathology. 2013;182(4):1448–58. doi: 10.1016/j.ajpath.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kohn C, Schleifenbaum J, Szijarto IA, Marko L, Dubrovska G, Huang Y, Gollasch M. Differential effects of cystathionine-gamma-lyase-dependent vasodilatory H2S in periadventitial vasoregulation of rat and mouse aortas. PloS one. 2012;7(8):e41951. doi: 10.1371/journal.pone.0041951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sun Q, Wang B, Li Y, Sun F, Li P, Xia W, Zhou X, Li Q, Wang X, Chen J, Zeng X, Zhao Z, He H, Liu D, Zhu Z. Taurine Supplementation Lowers Blood Pressure and Improves Vascular Function in Prehypertension: Randomized, Double-Blind, Placebo-Controlled Study. Hypertension. 2016;67(3):541–9. doi: 10.1161/HYPERTENSIONAHA.115.06624. [DOI] [PubMed] [Google Scholar]
- 149.Sen U, Tyagi N, Kumar M, Moshal KS, Rodriguez WE, Tyagi SC. Cystathionine-beta-synthase gene transfer and 3-deazaadenosine ameliorate inflammatory response in endothelial cells. American journal of physiology. Cell physiology. 2007;293(6):C1779–87. doi: 10.1152/ajpcell.00207.2007. [DOI] [PubMed] [Google Scholar]
- 150.Hofmann MA, Lalla E, Lu Y, Gleason MR, Wolf BM, Tanji N, Ferran LJ, Jr, Kohl B, Rao V, Kisiel W, Stern DM, Schmidt AM. Hyperhomocysteinemia enhances vascular inflammation and accelerates atherosclerosis in a murine model. The Journal of clinical investigation. 2001;107(6):675–83. doi: 10.1172/JCI10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Capasso R, Sambri I, Cimmino A, Salemme S, Lombardi C, Acanfora F, Satta E, Puppione DL, Perna AF, Ingrosso D. Homocysteinylated albumin promotes increased monocyte-endothelial cell adhesion and up-regulation of MCP1, Hsp60 and ADAM17. PloS one. 2012;7(2):e31388. doi: 10.1371/journal.pone.0031388. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 152.Perna AF, Sepe I, Lanza D, Capasso R, Zappavigna S, Capasso G, Caraglia M, Ingrosso D. Hydrogen sulfide reduces cell adhesion and relevant inflammatory triggering by preventing ADAM17-dependent TNF-alpha activation. Journal of cellular biochemistry. 2013;114(7):1536–48. doi: 10.1002/jcb.24495. [DOI] [PubMed] [Google Scholar]
- 153.Li JJ, Li Q, Du HP, Wang YL, You SJ, Wang F, Xu XS, Cheng J, Cao YJ, Liu CF, Hu LF. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. International journal of molecular sciences. 2015;16(6):12560–12577. doi: 10.3390/ijms160612560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Alhasson F, Dattaroy D, Das S, Chandrashekaran V, Seth RK, Schnellmann RG, Chatterjee S. NKT cell modulates NAFLD potentiation of metabolic oxidative stress-induced mesangial cell activation and proximal tubular toxicity. American journal of physiology Renal physiology. 2016;310(1):F85–F101. doi: 10.1152/ajprenal.00243.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Imaizumi T, Aizawa T, Segawa C, Shimada M, Tsuruga K, Kawaguchi S, Matsumiya T, Yoshida H, Joh K, Tanaka H. Toll-like receptor 3 signaling contributes to the expression of a neutrophil chemoattractant, CXCL1 in human mesangial cells. Clinical and experimental nephrology. 2015;19(5):761–70. doi: 10.1007/s10157-014-1060-4. [DOI] [PubMed] [Google Scholar]
- 156.Cheung GT, Siow YL, O K. Homocysteine stimulates monocyte chemoattractant protein-1 expression in mesangial cells via NF-kappaB activation. Canadian journal of physiology and pharmacology. 2008;86(3):88–96. doi: 10.1139/y08-002. [DOI] [PubMed] [Google Scholar]
- 157.Shastry S, James LR. Homocysteine-induced macrophage inflammatory protein-2 production by glomerular mesangial cells is mediated by PI3 Kinase and p38 MAPK. Journal of inflammation. 2009;6:27. doi: 10.1186/1476-9255-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sen U, Givvimani S, Abe OA, Lederer ED, Tyagi SC. Cystathionine beta-synthase and cystathionine gamma-lyase double gene transfer ameliorate homocysteine-mediated mesangial inflammation through hydrogen sulfide generation. American journal of physiology. Cell physiology. 2011;300(1):C155–63. doi: 10.1152/ajpcell.00143.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee BT, Ahmed FA, Hamm LL, Teran FJ, Chen CS, Liu Y, Shah K, Rifai N, Batuman V, Simon EE, He J, Chen J. Association of C-reactive protein, tumor necrosis factor-alpha, and interleukin-6 with chronic kidney disease. BMC nephrology. 2015;16:77. doi: 10.1186/s12882-015-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Tan Z, Shi Y, Yan Y, Liu W, Li G, Li R. Impact of endogenous hydrogen sulfide on toll-like receptor pathway in renal ischemia/reperfusion injury in rats. Renal failure. 2015;37(4):727–33. doi: 10.3109/0886022X.2015.1012983. [DOI] [PubMed] [Google Scholar]
- 161.Wang P, Isaak CK, Siow YL, O K. Downregulation of cystathionine beta-synthase and cystathionine gamma-lyase expression stimulates inflammation in kidney ischemia-reperfusion injury. Physiological reports. 2014;2(12) doi: 10.14814/phy2.12251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, Tao L, Sun H, Kellems RE, Blackburn MR, Xia Y. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59(1):136–44. doi: 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Jiang HL, Wu HC, Li ZL, Geng B, Tang CS. Changes of the new gaseous transmitter H2S in patients with coronary heart disease. Di 1 jun yi da xue xue bao = Academic journal of the first medical college of PLA. 2005;25(8):951–4. [PubMed] [Google Scholar]
- 164.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30(3):234–54. doi: 10.1016/j.semnephrol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Anders HJ, Ryu M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011;80(9):915–25. doi: 10.1038/ki.2011.217. [DOI] [PubMed] [Google Scholar]
- 166.Pushpakumar S, Kundu S, Givvimani S, Sen U. Hydrogen Sulfide Promotes Macrophage Polarization to Reduce Renal Inflammation in Angiotensin-II Induced Hypertension. Hypertension. 2014;64:A231. [Google Scholar]
- 167.Hashimoto-Kataoka T, Hosen N, Sonobe T, Arita Y, Yasui T, Masaki T, Minami M, Inagaki T, Miyagawa S, Sawa Y, Murakami M, Kumanogoh A, Yamauchi-Takihara K, Okumura M, Kishimoto T, Komuro I, Shirai M, Sakata Y, Nakaoka Y. Interleukin-6/interleukin-21 signaling axis is critical in the pathogenesis of pulmonary arterial hypertension. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(20):E2677–E2686. doi: 10.1073/pnas.1424774112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Didion SP, Kinzenbaw DA, Schrader LI, Chu Y, Faraci FM. Endogenous interleukin-10 inhibits angiotensin II-induced vascular dysfunction. Hypertension. 2009;54(3):619–24. doi: 10.1161/HYPERTENSIONAHA.109.137158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Morrison CJ, Butler GS, Rodriguez D, Overall CM. Matrix metalloproteinase proteomics: substrates, targets, and therapy. Current opinion in cell biology. 2009;21(5):645–53. doi: 10.1016/j.ceb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 170.Tan RJ, Liu Y. Matrix metalloproteinases in kidney homeostasis and diseases. American journal of physiology. Renal physiology. 2012;302(11):F1351–61. doi: 10.1152/ajprenal.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Spinale FG, Villarreal F. Targeting matrix metalloproteinases in heart disease: lessons from endogenous inhibitors. Biochem Pharmacol. 2014;90(1):7–15. doi: 10.1016/j.bcp.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Camp TM, Smiley LM, Hayden MR, Tyagi SC. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. Journal of hypertension. 2003;21(9):1719–27. doi: 10.1097/00004872-200309000-00022. [DOI] [PubMed] [Google Scholar]
- 173.Hultstrom M, Leh S, Skogstrand T, Iversen BM. Upregulation of tissue inhibitor of metalloproteases-1 (TIMP-1) and procollagen-N-peptidase in hypertension-induced renal damage. Nephrology, dialysis, transplantation : official publication of the European Dialysis and Transplant Association - European Renal Association. 2008;23(3):896–903. doi: 10.1093/ndt/gfm710. [DOI] [PubMed] [Google Scholar]
- 174.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu WT, Gazi SK, Barrow RK, Yang GD, Wang R, Snyder SH. H2S Signals Through Protein S-Sulfhydration. Science signaling. 2009;2(96) doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ishigami M, Hiraki K, Umemura K, Ogasawara Y, Ishii K, Kimura H. A Source of Hydrogen Sulfide and a Mechanism of Its Release in the Brain. Antioxidants & redox signaling. 2009;11(2):205–214. doi: 10.1089/ars.2008.2132. [DOI] [PubMed] [Google Scholar]
- 176.Bradley JM, Organ CL, Lefer DJ. Garlic-Derived Organic Polysulfides and Myocardial Protection. Journal of Nutrition. 2016;146(2):403s–409s. doi: 10.3945/jn.114.208066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Stamler J, Vaccaro O, Neaton JD, Wentworth D. Diabetes, Other Risk-Factors, and 12-Yr Cardiovascular Mortality for Men Screened in the Multiple Risk Factor Intervention Trial. Diabetes care. 1993;16(2):434–444. doi: 10.2337/diacare.16.2.434. [DOI] [PubMed] [Google Scholar]
- 178.Campbell NRC, Gilbert RE, Leiter LA, Larochelle P, Tobe S, Chockalingam A, Ward R, Morris D, Tsuyuki RT, Harris SB. Hypertension in people with type 2 diabetes Update on pharmacologic management. Can Fam Physician. 2011;57(9):997–1002. [PMC free article] [PubMed] [Google Scholar]
- 179.Volpe M, Battistoni A, Savoia C, Tocci G. Understanding and treating hypertension in diabetic populations. Cardiovasc Diagn The. 2015;5(5):353–363. doi: 10.3978/j.issn.2223-3652.2015.06.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhou X, Feng Y, Zhan ZB, Chen JC. Hydrogen Sulfide Alleviates Diabetic Nephropathy in a Streptozotocin-induced Diabetic Rat Model. Journal of Biological Chemistry. 2014;289(42):28827–28834. doi: 10.1074/jbc.M114.596593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The Effect of Angiotensin-Converting Enzyme-Inhibition on Diabetic Nephropathy. New Engl J Med. 1993;329(20):1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 182.Ahmad FUD, Sattar MA, Rathore HA, Akhter S, Jin OH, Johns EJ. Diuretic Action of Exogenous Hydrogen Sulfide in Spontaneously Hypertensive Diabetic Rats. Trop J Pharm Res. 2014;13(11):1867–1876. [Google Scholar]
- 183.van den Born JC, Frenay ARS, Bakker SJL, Pasch A, Hillebrands JL, Heerspink HJL, van Goor H. High urinary sulfate concentration is associated with reduced risk of renal disease progression in type 2 diabetes. Nitric Oxide-Biol Ch. 2016;55-56:18–24. doi: 10.1016/j.niox.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 184.Andresdottir G, Bakker SJ, Hansen HP, Parving HH, Rossing P. Urinary sulphate excretion and progression of diabetic nephropathy in Type 1 diabetes. Diabetic medicine : a journal of the British Diabetic Association. 2013;30(5):563–6. doi: 10.1111/dme.12131. [DOI] [PubMed] [Google Scholar]
- 185.Jose PA, Raj D. Gut microbiota in hypertension. Current opinion in nephrology and hypertension. 2015;24(5):403–409. doi: 10.1097/MNH.0000000000000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang T, Santisteban MM, Rodriguez V, Li E, Ahmari N, Carvajal JM, Zadeh M, Gong MH, Qi YF, Zubcevic J, Sahay B, Pepine CJ, Raizada MK, Mohamadzadeh M. Gut Dysbiosis Is Linked to Hypertension. Hypertension. 2015;65(6):1331–1340. doi: 10.1161/HYPERTENSIONAHA.115.05315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pluznick JL, Protzko RJ, Gevorgyan H, Peterlin Z, Sipos A, Han J, Brunet I, Wan LX, Rey F, Wang T, Firestein SJ, Yanagisawa M, Gordon JI, Eichmann A, Peti-Peterdi J, Caplan MJ. Olfactory receptor responding to gut microbiota-derived signals plays a role in renin secretion and blood pressure regulation. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(11):4410–4415. doi: 10.1073/pnas.1215927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lloyd D. Hydrogen sulfide: clandestine microbial messenger? Trends in microbiology. 2006;14(10):456–462. doi: 10.1016/j.tim.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 189.Ortenberg R, Beckwith J. Functions of thiol-disulfide oxidoreductases in E-coli: Redox myths, realities, and practicalities. Antioxidants & redox signaling. 2003;5(4):403–411. doi: 10.1089/152308603768295140. [DOI] [PubMed] [Google Scholar]
- 190.Poole LB. Bacterial defenses against oxidants: mechanistic features of cysteine-based peroxidases and their flavoprotein reductases. Archives of biochemistry and biophysics. 2005;433(1):240–254. doi: 10.1016/j.abb.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 191.Ramezani A, Raj DS. The Gut Microbiome, Kidney Disease, and Targeted Interventions. Journal of the American Society of Nephrology. 2014;25(4):657–670. doi: 10.1681/ASN.2013080905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Flannigan KL, McCoy KD, Wallace JL. Eukaryotic and prokaryotic contributions to colonic hydrogen sulfide synthesis. American journal of physiology. Gastrointestinal and liver physiology. 2011;301(1):G188–93. doi: 10.1152/ajpgi.00105.2011. [DOI] [PubMed] [Google Scholar]
- 193.Aronov PA, Luo FJ, Plummer NS, Quan Z, Holmes S, Hostetter TH, Meyer TW. Colonic contribution to uremic solutes. Journal of the American Society of Nephrology : JASN. 2011;22(9):1769–76. doi: 10.1681/ASN.2010121220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Tanaka H, Sirich TL, Plummer NS, Weaver DS, Meyer TW. An Enlarged Profile of Uremic Solutes. PloS one. 2015;10(8):e0135657. doi: 10.1371/journal.pone.0135657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Vanholder R, Glorieux G. The intestine and the kidneys: a bad marriage can be hazardous. Clinical kidney journal. 2015;8(2):168–79. doi: 10.1093/ckj/sfv004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Strid H, Simren M, Stotzer PO, Ringstrom G, Abrahamsson H, Bjornsson ES. Patients with chronic renal failure have abnormal small intestinal motility and a high prevalence of small intestinal bacterial overgrowth. Digestion. 2003;67(3):129–37. doi: 10.1159/000071292. [DOI] [PubMed] [Google Scholar]
- 197.de Almeida Duarte JB, de Aguilar-Nascimento JE, Nascimento M, Nochi RJ., Jr Bacterial translocation in experimental uremia. Urological research. 2004;32(4):266–70. doi: 10.1007/s00240-003-0381-7. [DOI] [PubMed] [Google Scholar]
- 198.Wei M, Wang ZG, Liu H, Jiang HL, Wang M, Liang SS, Shi KH, Feng J. Probiotic Bifidobacterium animalis subsp lactis Bi-07 alleviates bacterial translocation and ameliorates microinflammation in experimental uraemia. Nephrology. 2014;19(8):500–506. doi: 10.1111/nep.12272. [DOI] [PubMed] [Google Scholar]
- 199.Wang F, Jiang H, Shi K, Ren Y, Zhang P, Cheng S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrology. 2012;17(8):733–8. doi: 10.1111/j.1440-1797.2012.01647.x. [DOI] [PubMed] [Google Scholar]
- 200.Wang F, Zhang P, Jiang H, Cheng S. Gut bacterial translocation contributes to microinflammation in experimental uremia. Digestive diseases and sciences. 2012;57(11):2856–62. doi: 10.1007/s10620-012-2242-0. [DOI] [PubMed] [Google Scholar]
- 201.Shen X, Carlstrom M, Borniquel S, Jadert C, Kevil CG, Lundberg JO. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free radical biology & medicine. 2013;60:195–200. doi: 10.1016/j.freeradbiomed.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Jakubowski H. Metabolism of homocysteine thiolactone in human cell cultures. Possible mechanism for pathological consequences of elevated homocysteine levels. The Journal of biological chemistry. 1997;272(3):1935–42. [PubMed] [Google Scholar]
- 203.Jakubowski H. Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 1999;13(15):2277–83. [PubMed] [Google Scholar]
- 204.Jakubowski H. The pathophysiological hypothesis of homocysteine thiolactone-mediated vascular disease. Journal of physiology and pharmacology : an official journal of the Polish Physiological Society. 2008;59(Suppl 9):155–67. [PubMed] [Google Scholar]