

Abstract
Various nanodelivery systems have been designed to release therapeutic agents upon contact with specific enzymes. However, enzyme-triggered release typically takes place in the tissue interstitium, thereby resulting in the extracellular delivery of drugs. Here, we have designed an enzyme-stimulated multistage vector (ESMSV), which enables stimulus-triggered release of drug-encapsulated nanoparticles from a microparticle. Specifically, polymeric nanoparticles with a surface matrix metalloproteinase-2 (MMP2) peptide substrate were conjugated to the surface of porous silicon microparticles. In the presence of MMP2, the polymeric nanoparticles were released into the tumor interstitium. This platform can be used to attain triggered drug release, while simultaneously facilitating the cellular internalization of drugs. The results indicate that nanoparticle release was MMP2-specific and resulted in improved intracellular uptake of hydrophobic agents in the presence of MMP2. Furthermore, in a mouse model of melanoma lung metastasis, systemic delivery of ESMSVs caused a substantial increase in intracellular accumulation of agents in cancer cells in comparison to delivery with non-stimulus-responsive particles.
Keywords: enzyme-responsive release, lung metastasis, MMP2, multistage vector, porous silicon
Graphical abstract
1. Introduction
Nanotechnology has been used in medicine to improve the biodistribution of drugs, thereby increasing therapeutic efficacy and reducing adverse side effects. In particular, nanodelivery systems can prolong the blood circulation of drugs, overcome transport barriers within the body, and preferentially accumulate in pathological tissue [1-5]. Nevertheless, in the case of all nanodrugs in current clinical use, drug release is non-specific [6, 7]. Furthermore, the premature release of drugs from a nanocarrier in the circulation can increase off-target effects, while fast release in the cancer microenvironment rather than directly in target cells may reduce therapeutic efficacy. To address these issues, stimuli-responsive delivery systems that enable spatiotemporal control of drug release have been developed. Such nanocarriers respond to environments with unique properties such as increased acidity, oxidative stress, or hypoxia, which are common characteristics of tumor tissue [8-14]. For instance, polymeric nanoparticles for nucleic acid delivery have been designed to release an outer polyethylene glycol (PEG) layer when exposed to hypoxic [15] or acidic [16] environments. The removal of this stealth layer promotes the internalization of the polymeric nanoparticles, resulting in increased exposure of tumor cells to nucleic acids. Another example of stimuli-responsive carriers is nanoparticles designed to release drugs in response to reactive oxygen species (ROS). Such nanodelivery systems have utilized polymers that degrade when subjected to ROS [17] or incorporated catalase in the aqueous core of nanoparticles, consequently causing oxygen production and nanoparticle rupture in the presence of H2O2 [18].
Enzyme activity can also be used as a stimulus to trigger drug release, since tumor tissue usually displays increased levels of specific enzymes. For instance, cancer proliferation, metastasis, and angiogenesis often result in overexpression of matrix metalloproteinases (MMPs), which are a family of zinc and calcium-dependent proteolytic enzymes [19, 20]. MMPs digest several components of the extracellular matrix (ECM), including collagen, laminin, fibronectin, vitronectin, elastin, and proteoglycans [21, 22]. Especially MMP2 and MMP9 play important roles in tumorigenesis, cell migration, invasion, and metastasis [23-25]. Indeed, nanocarriers responding to MMPs have been designed for delivery of cancer therapeutics [26]. Typically, such nanocarriers are functionalized with a peptide sequence that can be cleaved by MMPs. For example, Dorresteijn et al. synthesized polylactide (PLLA)-peptide-PLLA triblock copolymer nanoparticles that displayed controlled drug release in vitro in response to MMP2 cleavage of the peptide [27]. However, since MMP-driven dissolution of nanoparticles takes place in the ECM, the released drugs may display low levels of intracellular uptake when disassociated from nanocarriers. Furthermore, once the drugs are released in the ECM, they may aggregate and degrade in this environment prior to establishing contact with cancer cells.
To address the aforementioned problems, a multistage system was utilized for drug delivery. Namely, a first-stage carrier (microparticle) was used to transport second-stage carriers (nanoparticles) to tumor tissue. Previous studies have indicated that the multistage vector system serves as a safe and effective vehicle for drug delivery [28-40]. In this study, the microparticle component consisted of a porous silicon disk (2.6 μm × 0.7 μm), while the nanoparticle component comprised poly(lactic-co-glycolic acid) (PLGA)-PEG particles. The multistage delivery system was assembled through conjugation of the polymeric nanoparticles to the surface of the silicon microdisk. Moreover, the nanoparticles were loaded with coumarin 6, a fluorescent small molecule, which served as a model for hydrophobic drugs. This multistage platform was designed to release nanoparticles from the microparticle upon contact with MMPs, as the surface of the PLGA-PEG particles was modified with a MMP2 substrate peptide. This strategy enabled intact nanocarriers to be internalized by cancer cells, thus increasing intracellular uptake and preventing extracellular aggregation/degradation of drugs. Moreover, it has been shown that nanoparticle-mediated drug uptake can decrease drug resistance by reducing the expulsion of drugs from multi-drug resistance efflux pumps [41]. The major difference between the enzyme-stimulated multistage vector (ESMSV) and the majority of previously reported enzyme-responsive drug delivery platforms is the location of drug release. While the ESMSV increases tumor-specific exposure to drugs, the drug release process does not take place in the extracellular environment, as MMPs target the bond between nanoparticles and microparticles. Previously, it has been shown that intravenously administered silicon microdisks display high accumulation in lung tissue due to geometrical features [40, 42, 43]. Indeed, the small capillaries of the lungs promote interactions between discoidal particles and endothelial cells [42]. Compared to spherical particles, microdisks have the ability to interact with endothelial cells through a much larger contact area [40]. These interactions promote particle adhesion to the vascular wall. In particular, discoidal microparticles have been shown to accumulate to a greater extent in tumor-bearing lungs compared to healthy lung tissue [41]. The main reason for enhanced particle accumulation in tumors is abnormal blood flow patterns that lead to reduced shear rates [40]. Consequently, discoid particles are able to adhere to cancer blood vessels, while they are dislodged from the endothelial wall of healthy vasculature. Based on this natural tropism of microdisks for tumor-bearing lungs, the performance of the ESMSV was evaluated in a mouse model of A375 melanoma lung metastasis.
2. Experimental Section
2.1. Materials
PLGA-PEG-COOH was prepared as previously reported [44]. The MMP2 substrate with a peptide sequence of AGFSGPLGMWSAGSFG was purchased from Peptide 2.0 (Chantilly, VA, USA). Sulfo-NHS (N-hydroxysulfosuccinimide) was acquired from Thermo Fisher Scientific, Inc. Phosphate buffered saline (PBS), fetal bovine serum (FBS), Medium 200, Low Serum Growth Supplement (LSGS), Dulbecco's Modified Eagle Medium (DMEM), trypsin, and penicillin/streptomycin solution were purchased from GE Healthcare Life Sciences (Pittsburgh, PA, USA). All other chemicals were acquired from Sigma-Aldrich.
2.2. Preparation and characterization of the ESMSV
The ESMSV was prepared by conjugating MMP2 substrate-modified PLGA-PEG nanoparticles to 3-aminopropyltriethoxysilane (APTES)-modified silicon microdisks. Photolithography and electrochemical etching were used to produce the porous silicon microdisks (2.6 μm × 0.7 μm, 50∼60 nm pores) as previously described [45]. Particles were then oxidized in a 30% solution of hydrogen peroxide for 2 h at 95 °C and aminated in 2% APTES in isopropyl alcohol for 48 h at 65 °C.
Coumarin 6-loaded PLGA-PEG-COOH nanoparticles were prepared using the nanoprecipitation method [46, 47]. The nanoparticles were then modified with the MMP2 substrate according to a previously described procedure [48]. Briefly, 1 mg of nanoparticles was dissolved in 10 mL PBS. Next, 3 mg of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 2.4 mg sulfo-NHS were added to the solution to activate the functional groups on the nanoparticle surface. The MMP2 substrate was then added to the solution at a concentration of 0.2 mg/mL and the reaction was left to proceed for 3 h. Finally, the MMP2-conjugated PLGA-PEG nanoparticles were washed with water and centrifuged three times at 10000 rpm.
To prepare the ESMSVs, 20 mg of MMP2 conjugated PLGA-PEG nanoparticles were dissolved in PBS and activated with EDC (10 mg) and sulfo-NHS (8 mg) for 30 min. APTES-modified silicon microdisks (0.2 billion) were added to the solution (final volume: 10 mL) and stirred for 3 h. After the reaction, the ESMSVs were washed with water and centrifuged three times at 4500 rpm.
The size and zeta potential of the particles were measured using the Zetasizer (Zetasizer Nano, Malvern). For each sample, three measurements were taken with ten runs each. Scanning electron microscopy (SEM) characterization was performed using an ultra-high resolution microscope (SEM 230, NovaNano). A drop of sample solution was added to a silicon wafer, vacuum dried, and coated with 5-10 nm of platinum.
2.3. Enzyme-stimulated release
Coumarin 6-loaded PLGA-PEG nanoparticles and peptide-PLGA-PEG nanoparticles were centrifuged at 1500 rpm for 5 min to discard larger particles and the supernatant containing the nanoparticles was collected. The nanoparticles were then conjugated to silicon microparticles. The micro/nano particles were centrifuged at 1500 rpm for 5 min and the pellet was collected. The micro/nano particles were then dispersed in 200 μL MMP2 reaction buffer (50 mM HEPES with 10 mM CaCl2) with or without MMP2 enzymes at the concentration of 2 μg/mL. The solutions were then placed on an orbital shaker (120 rpm, 37 °C). At designated time intervals, the solutions were centrifuged at 1500 rpm for 5 min and the supernatant was collected for analysis. Subsequently, the pellet was resuspended in fresh buffer and the drug release study was continued. The collected supernatants were added in black 96-well plates and the fluorescent intensity of coumarin 6 (Ex 430 nm/Em 485 nm) was measured using a microplate reader (Genios, Tecan, Switzerland). The experiments were performed in triplicate.
2.4. Cell culture
A375 human melanoma cells were obtained from ATCC, while HUVECs were purchased from PromoCell. A375 super-metastasis cells with luciferase (A375 SM-Luc) were obtained from Dr. Isaiah Fidler's laboratory (MD Anderosn Cancer Center Houston, TX, USA). A375 human melanoma cells were cultured in DMEM supplemented with 10% FBS (v:v) and 1% (v:v) penicillin/streptomycin solution, while HUVECs were cultured in Medium 200 supplemented with 2% (v:v) LSGS and 1% (v:v) penicillin/streptomycin solution. Cells were maintained in an incubator at 37 °C and 5% CO2. The cells were sub-cultivated at 70% confluency with 0.25% trypsin.
2.5. Cellular uptake
The cellular uptake of MSVs and ESMSVs was quantitatively measured. A375 cells were seeded in 96-well black plates (Costar, IL, USA) at a density of 5×103 cells/well (0.1 ml). When the cell confluency reached 80%, the cells were exposed to coumarin 6-loaded MSVs or ESMSVs at a concentration of 0.1 billion particles/mL. The medium was supplemented with 2 μg/mL MMP2. At various time points, the cells were washed three times with 0.1 ml PBS and 50 μL of 0.5% Triton X-100 in 0.2 N NaOH solution was added to lyse the cells. A microplate reader (Genios, Tecan, Switzerland) was then used to measure the fluorescence intensity of coumarin 6 (Ex 430 nm/Em 485 nm).
2.6. Confocal microscopy
A375 cells were seeded overnight in 4-well chamber slides (LAB-TEK, Nagle Nunc, IL, USA) at a density of 2×104 cells/well. The cells were then exposed to MSVs or ESMSVs at the concentration of 0.1 billion particles/mL. After 24 h, the cells were washed twice with pre-warmed 1× PBS and fixed with 70% ethanol for 20 min. Subsequently, the cells were washed twice with PBS and the nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole) for 30 min. Finally, the cells were washed twice with 1× PBS and visualized with confocal microscopy.
2.7. Animal model
The animal studies were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals following protocols approved by the Institutional Animal Care and Use Committee (IACUC 3/5/2014). Six-week-old female athymic nude mice were purchased from Charles River Laboratories (Boston, MA, USA). A375 SM-Luc cells were injected through the tail vein (106 cells/mouse). Lung metastasis was monitored using a bioluminescence imaging system (IVIS 200).
2.8. Expression of MMP2 in vitro and in vivo
The expression levels of MMP2 and MMP9 mRNA were measured in A375 cells and HUVECs using quantitative reverse transcription polymerase chain reaction (RT-PCR). For evaluating the expression of excreted MMP2 and MMP9, A375 cells were cultured in medium without FBS. After 72 h, the medium was collected and analyzed by silver staining and western blot. In order to evaluate the expression of MMP2 in vivo, mice were injected with A375 SM-Luc cells. After six weeks, the lungs were harvested, fixed and stained following standard immunohistochemistry protocols using a monoclonal MMP2 antibody and a peroxidase-labeled secondary antibody. Randomly selected images were captured with an optical microscope.
2.9. Accumulation of ESMSVs in cancer cells in vivo
Six weeks after injection of A375 SM-Luc cells, mice were intravenously administered with MSVs or ESMSVs (15 billion/kg). After 24 h, mice were sacrificed and the lungs were minced and digested in DMEM/F12 medium containing 300 U/mL collagenase (100 mg tissue/mL medium) for 1.5 h at 37°C. The suspension was filtered through a 40 μm mesh to attain a single-cell solution. For flow cytometry analysis (Fortessa, BD FACS), dead cells were stained with CYTOX® Blue (Life Technologies) and A375 cells were stained with the APC-Cy7 Anti-human HLA-ABC antibody (BD Bioscience).
3. Results and Discussion
3.1. Design, synthesis, and characterization of the ESMSV
The process of designing and synthesizing the ESMSV is shown in Fig. 1. A peptide with an amino acid sequence that serves as a substrate for MMP2 was used as a linker to conjugate PLGA-PEG nanoparticles to porous silicon microparticles. A hydrophobic model drug, coumarin 6, was encapsulated in the PLGA-PEG nanoparticles. The morphology of uncoated porous silicon microparticles and ESMSVs are shown in Fig. 2a and 2b. The back side of the silicon particle is characterized by a thin solid base, while the rest of the particle has a porous structure (pore diameter: 30-60 nm; porosity: 70 nm). Upon conjugation, both the front and back of the ESMSV displayed a homogenous coating of PLGA-PEG NPs (Fig. 2b). The size distribution of PLGA-PEG nanoparticles and PLGA-PEG-peptide nanoparticles was measured by dynamic light scattering. The PLGA-PEG nanoparticles displayed an average size of ∼75 nm, while peptide conjugation caused the size to increase by ∼45 nm (Fig. 2c), most likely due to peptide crosslinking. Both nanoparticles displayed a narrow size distribution. The zeta potential of PLGA-PEG nanoparticles was approximately -40 mV and was unaffected by peptide conjugation (Fig. 2d). Likewise, upon conjugation of nanoparticles to the silicon microparticle, the zeta potential remained at ∼-40 mV. Notably, a negative zeta potential value is usually an indicator of colloidal stability and increased safety, since positively charged nanoparticles have frequently been reported to have toxic effects [49, 50]. Confocal microscopy studies confirmed successful conjugation of coumarin 6-loaded PLGA-PEG nanoparticles to the microparticle surface (Fig. 2e). Indeed, in this study, coumarin 6 was used as a model for hydrophobic drugs. Previously, we have demonstrated that docetaxel loading efficiencies of ∼53 μg/mg PLGA-PEG and ∼804 μg/billion silicon particles can be achieved [43]. This drug loading capacity was sufficient to markedly suppress tumor burden in a mouse model of melanoma lung metastasis [43]. Notably, the polymeric coating does not compromise the loading of other nanoparticles into the porous interior of the microparticles, as previously demonstrated [43]. Although not a focus of this study, it is possible to incorporate additional nanoparticles, such as siRNA liposomes, in the ESMSV [43].
Fig. 1.
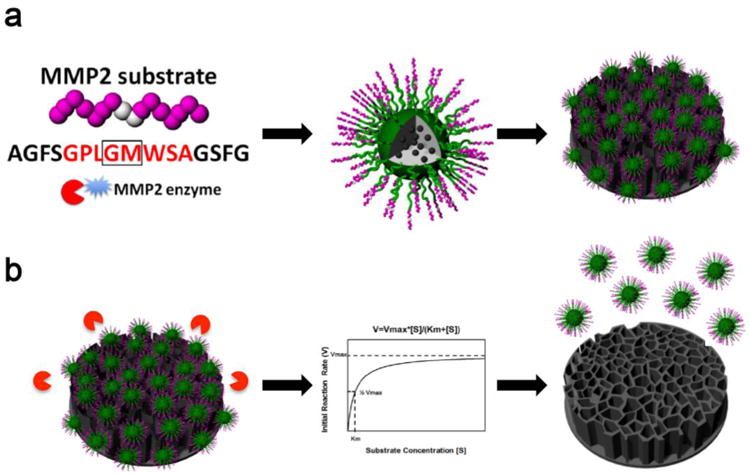
Schematic illustrating the enzyme-stimulated multistage vector (ESMSV). a) A matrix metalloproteinase-2 (MMP2) peptide substrate was conjugated to poly(lactic-co-glycolic acid) (PLGA)- polyethylene glycol (PEG) nanoparticles, which were further conjugated to the surface of porous silicon microparticles. A hydrophobic model drug, coumarin 6, was encapsulated in the polymeric nanoparticles. b) In the presence of MMP2 enzymes, the polymeric nanoparticles disassociate from the silicon microparticles.
Fig. 2.
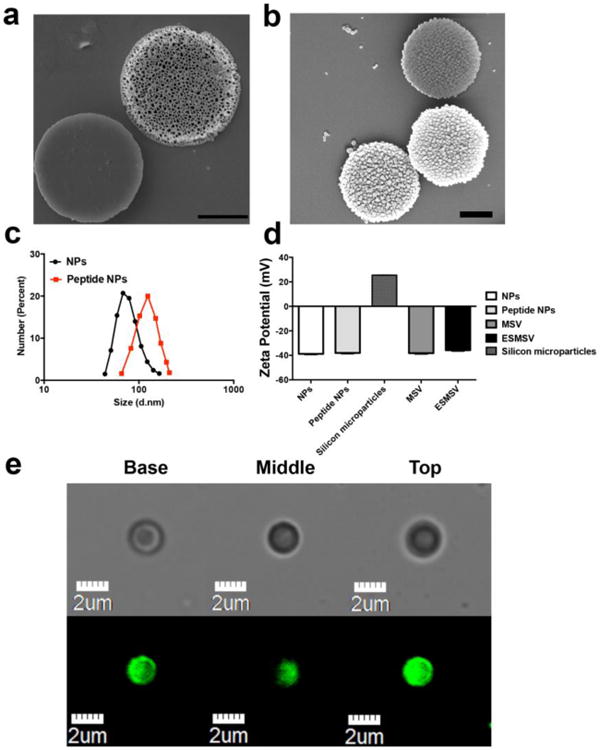
Characterization of the ESMSV. a-b) Scanning electron microscopy (SEM) images of the front and back sides of MSVs (a) and ESMSVS (b). Scale bar, 1 μm. c) Size distribution of NPs and peptide (MMP2 substrate)-NPs obtained with dynamic light scattering. d) Zeta potential of NPs, peptide-NPs, silicon microparticles, MSVs, and ESMSVs. Results are presented as mean ± SD of three measurements. e) Confocal laser scanning microscopy images. Columns from left to right show the basal, middle, and top planes of the ESMSV. The upper row was captured in the bright field channel and the lower row in the FITC channel (coumarin 6).
3.2. Release profile of the ESMSV in vitro
Enzyme-stimulated release of PLGA-PEG nanoparticles from silicon microparticles was assessed in vitro by comparing the fluorescence intensity of the release buffer in the presence or absence of MMP2 (Fig. 3). Centrifugal separation ensured that the released polymeric nanoparticles were separated from the ESMSV. In the absence of MMP2, 40% of coumarin 6 had been released from the ESMSV after 6 h, as opposed to 80% in the presence of MMP2 (Fig. 3). Furthermore, the results demonstrate that the accelerated release was due to cleavage of the peptide substrate, since MMP2 did not affect the release of PLGA-PEG nanoparticles lacking the peptide (Fig. 3). It is worth noting that some of the drug could be passively released before reaching the target tissue. However, a previous study with discoidal porous silicon particles of the same size demonstrated that the particles accumulated in high amounts in tumor-bearing lungs and were cleared from the circulation 1 h post-injection [41]. The rapid deposition of particles in lung tissue suggests that premature passive release is not a major concern for this drug delivery system.
Fig. 3.
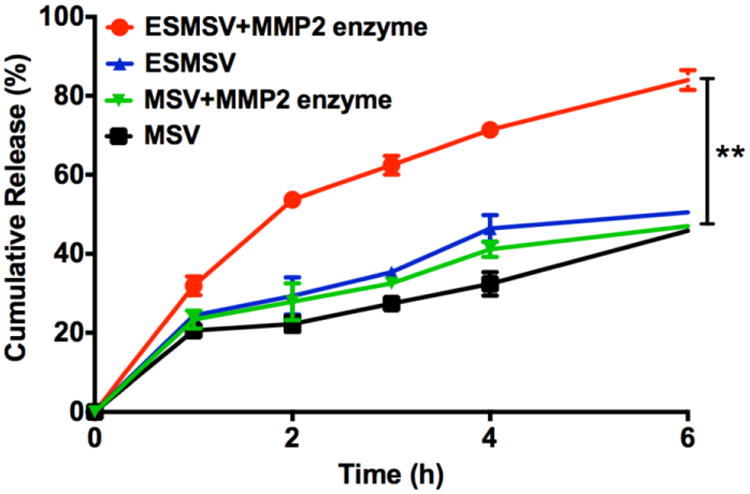
Release of polymeric nanoparticles from MSVs and ESMSVs in the presence or absence of MMP2. Experiments were performed in triplicate and results are presented as mean ± SD. **, P < 0.001.
3.3. Enhanced cellular uptake of the ESMSVs in vitro
The cellular internalization of coumarin 6 was measured in A375 human melanoma cells exposed to ESMSVs or MSVs. MSVs were identical to ESMSVs with the exception that the MSVs lacked the MMP2 peptide substrate [43]. Fluorescence intensity measurements confirmed that the loading efficiency of coumarin 6 in MSVs and ESMSVs was comparable. The cell culture media was supplemented with MMP2 to simulate the tumor microenvironment. Since cell culture media is frequently changed, levels of MMP2 are substantially lower than those that accumulate over time in tumor tissue. Compared to MSVs, treatment with ESMSVs caused a 1.2-fold and 1.5-fold increase in the cellular uptake of coumarin 6 after 6 h and 24 h, respectively (Fig. 4a). This difference in cellular internalization rates is likely due to the fact that nanoparticles are internalized more rapidly than microparticles [51, 52]. Indeed, cleavage of the peptide substrate in ESMSVs caused the cells to be exposed to a larger number of nanoparticles. Moreover, the cellular internalization of coumarin 6 in A375 melanoma cells exposed to MSVs or ESMSVs was also visualized using confocal microscopy (Fig. 4b). In accordance with the quantitative uptake studies, the fluorescence signal was substantially higher in the cells treated with ESMSVs. These results indicate that this triggered-release strategy enables preferential internalization of cargo in cancer cells. Since we have previously demonstrated that the discoidal microparticles display increased deposition in lung tumors upon intravenous injection [42, 43], the ESMSV platform embodies two complementing strategies for increasing selective exposure of drugs to cancer cells.
Fig. 4.
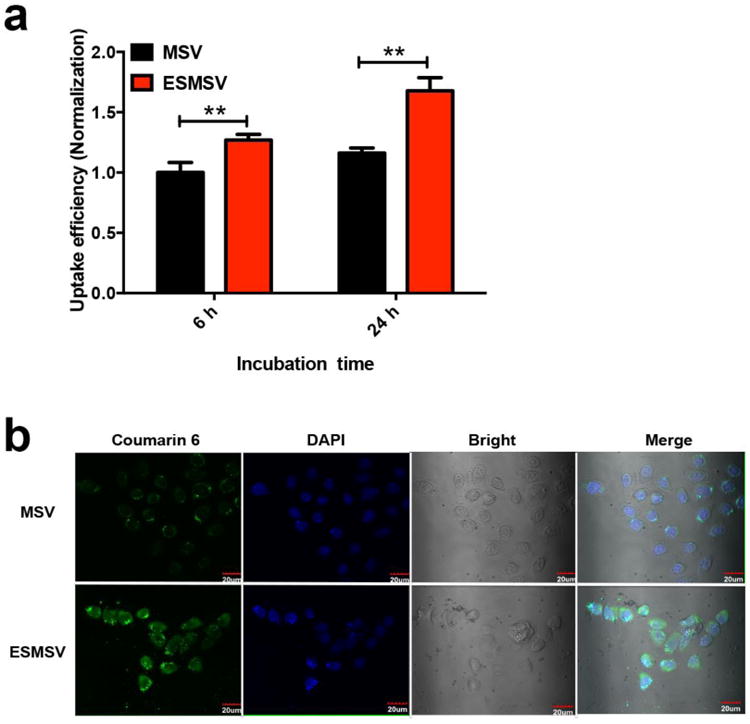
Cellular uptake of polymeric nanoparticles (loaded with coumarin 6) released from MSVs or ESMSVs in A375 human melanoma cells. The cell culture medium was supplemented with MMP2. a) Quantitative measurements of cellurar uptake. Experiments were performed in triplicate and results are presented as mean ± SD. **, P < 0.001. b) Confocal microscopy images of cellular uptake. Nuclei were stained with DAPI. Scale bar, 20 μm.
3.4. Expression of MMP2 in a melanoma lung metastasis model
Prior to assessing the benefits of the ESMSV platform in vivo, the expression of MMP2 in cancer cells and tumor tissues was evaluated. The levels of MMP2 mRNA were measured in A375 melanoma cells and human umbilical vein endothelial cells (HUVECs). The results indicate that MMP2 expression levels were three times higher in the cancer cells than in the endothelial cells (Fig. 5a). Since MMP9 also plays an important role in tumor progression, the mRNA levels of this enzyme were measured. Contrary to MMP2, the expression of MMP9 was higher in HUVECs compared to A375 cells (Fig. 5a), suggesting that MMP2 was a better target for enzyme-triggered release. Next, the excretion of MMP2 and MMP9 in cell culture medium was evaluated using silver staining and western blot. In accordance with the mRNA results, the protein levels of MMP2 were substantially higher than those of MMP9 (Fig. 5b). Moreover, an A375 melanoma lung metastasis model was employed to assess the expression of MMP2 in tumor tissue. Lung sections with metastatic nodules were stained by immunohistochemistry. As can be seen in Fig. 5c and 5d, the expression levels of MMP2 were high in lung tissue.
Fig. 5.

MMP2 expression and in vivo drug release. a-d) Expression of MMP2 and MMP9 in A375 cells and human umbilical vein endothelial cells (HUVECs). a) mRNA expression of MMP2 and MMP9. Experiments were performed in triplicate and results are presented as mean ± SD. **, P < 0.001. b) Protein levels of MMP2 and MMP9 in cell culture medium. c, d) Immunohistochemistry of MMP2 expression in lung tissues in a mouse model of melanoma lung metastasis. Scale bars: 500 μm (left); 50 μm (right). e-f) Flow cytomety analysis of the intracellular uptake of polymeric nanoparticles (loaded with Coumarin 6) following intravenous injection of MSVs or ESMSVs in an A375 melanoma lung metastasis model. e) Schematic illustrating the flow cytometry process. The mice with melanoma lung metastasis were sacrficed and lungs were collected. The NPs with fluorescence in cancer cells were quantified with flow cytometry. f) Percentage of coumarin 6-positive A375 cells. Cancer cells were labeled with the APC-Cy7 anti-human HLA-ABC antibody. SSC, side scatter.
3.5. Enhanced intracellular uptake of the ESMSV in vivo
The performance of the ESMSV in vivo was evaluated using an A375 melanoma lung metastasis model. Previous findings comparing the performance of polymeric nanoparticles and the MSV indicate that the latter results in a twofold increase in the uptake of fluorescent cargo in lung cancer cells [43]. In this study, mice received intravenous injections of the ESMSV or the MSV and lung tissues were collected, digested, and analyzed by flow cytometry (Fig. 5e). The results demonstrate that 46.8% and 64.6% of the melanoma cells were positive for coumarin 6 in mice treated with the MSV and the ESMSV, respectively (Fig. 5f). These results are consistent with the in vitro uptake studies, which showed that the ESMSV was able to enhance the intracellular uptake of coumarin 6 in A375 melanoma cells. Notably, this enzyme-triggered improvement in cellular internalization was more pronounced in vivo than in cell culture.
Overall, the ESMSV system shows promise as an effective delivery vehicle for stimulus-triggered release of nanoparticles in the tumor microenvironment. Although qualitative measurements demonstrated that peptide conjugation was successful, future experiments involving Fourier transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS), nuclear magnetic resonance (NMR) spectroscopy, or elemental analysis would be necessary to determine the peptide conjugation efficiency. Moreover, confocal microscopy images of fluorescent PLGA-PEG particles could shed light upon the mechanism of coumarin 6 cellular entry. Additionally, to provide further evidence to support the primary objective of this study, it would be useful to compare the performance of the ESMSV to state-of-the-art vehicles for extracellular drug delivery.
4. Conclusions
In this study, we have developed an enzyme-stimulated multistage platform to improve the localized delivery of drugs to cancer cells. This platform consists of a porous silicon microparticle conjugated to drug-containing polymeric nanoparticles. Specifically, this system was designed to accumulate in tumor tissue in the lungs, based on prior observations. To further improve the preferential deposition of drugs in cancer cells, an enzyme-stimulated strategy was employed to induce the release of nanoparticles in tumor tissue. Upon exposure to MMP2 in the tumor microenvironment, the linker connecting the nanoparticles to the microparticle was cleaved, consequently enabling nanoparticle entry into cancer cells. The results revealed that this enzyme-responsive strategy caused increased intracellular accumulation of cargo in A375 melanoma cells in vitro and in vivo. In particular, intravenous injection of the ESMSV in an A375 melanoma lung metastasis model resulted in increased internalization of drugs in cancer cells compared to delivery with a non-stimulus-driven multistage vehicle. Our results indicate that the ESMSV could serve as an effective drug delivery system for treating melanoma lung metastasis.
Supplementary Material
Acknowledgments
This work was supported by the Houston Methodist Research Institute, and the Ernest Cockrell Jr. Distinguished Endowed Chair (M.F.), and the US Department of Defense (W81XWH-09-1-0212) (M.F.), the National Institute of Health (U54CA143837, U54CA151668) (M.F.), Department of Defense grant W81XWH-12-1-0414 (M.F.) and the State of Texas CPRIT grant RP121071 (M.F. and H.S.). We thank Dr. Kemi Cui, Dr. Jianhua James Gu, and Dr. Xukui Wang for providing experimental facilities.
Footnotes
Conflict of interest: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 2.Peer D, Karp JM, Hong S, FaroKHzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 3.Feng SS, Chien S. Chemotherapeutic engineering: Application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci. 2003;58(18):4087–4114. [Google Scholar]
- 4.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3(5):242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 5.Wolfram J, Zhu M, Yang Y, Shen J, Gentile E, Paolino D, Fresta M, Nie G, Chen C, Shen H, Ferrari M, Zhao Y. Safety of Nanoparticles in Medicine. Curr Drug Targets. 2015;16(14):1671–1681. doi: 10.2174/1389450115666140804124808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Min Y, Caster JM, Eblan MJ, Wang AZ. Clinical Translation of Nanomedicine. Chem Rev. 2015;115(19):11147–11190. doi: 10.1021/acs.chemrev.5b00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markman JL, Rekechenetskiy A, Holler E, Ljubimova JY. Nanomedicine therapeutic approaches to overcome cancer drug resistance. Adv Drug Deliv Rev. 2013;65(13-14):1866–1879. doi: 10.1016/j.addr.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Shim MS, Levinson NS, Sung HW, Xia Y. Stimuli-Responsive Materials for Controlled Release of Theranostic Agents. Adv Funct Mater. 2014;24(27):4206–4220. doi: 10.1002/adfm.201400279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleige E, Quadir MA, Haag R. Stimuli-responsive polymeric nanocarriers for the controlled transport of active compounds: concepts and applications. Adv Drug Deliv Rev. 2012;64(9):866–884. doi: 10.1016/j.addr.2012.01.020. [DOI] [PubMed] [Google Scholar]
- 10.Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. J Control Release. 2008;126(3):187–204. doi: 10.1016/j.jconrel.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 11.Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem Soc Rev. 2013;42(17):7289–7325. doi: 10.1039/c3cs60048c. [DOI] [PubMed] [Google Scholar]
- 12.Huang X, Appelhans D, Formanek P, Simon F, Voit B. Tailored synthesis of intelligent polymer nanocapsules: an investigation of controlled permeability and pH-dependent degradability. ACS Nano. 2012;6(11):9718–9726. doi: 10.1021/nn3031723. [DOI] [PubMed] [Google Scholar]
- 13.Gu Z, Dang TT, Ma M, Tang BC, Cheng H, Jiang S, Dong Y, Zhang Y, Anderson DG. Glucose-responsive microgels integrated with enzyme nanocapsules for closed-loop insulin delivery. ACS Nano. 2013;7(8):6758–6766. doi: 10.1021/nn401617u. [DOI] [PubMed] [Google Scholar]
- 14.Mo R, Jiang T, DiSanto R, Tai W, Gu Z. ATP-triggered anticancer drug delivery. Nat Commun. 2014;5:3364. doi: 10.1038/ncomms4364. [DOI] [PubMed] [Google Scholar]
- 15.Perche F, Biswas S, Wang T, Zhu L, Torchilin VP. Hypoxia-targeted siRNA delivery. Angew Chem Int Ed Engl. 2014;53(13):3362–3366. doi: 10.1002/anie.201308368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sawant RR, Sriraman SK, Navarro G, Biswas S, Dalvi RA, Torchilin VP. Polyethyleneimine-lipid conjugate-based pH-sensitive micellar carrier for gene delivery. Biomaterials. 2012;33(15):3942–3951. doi: 10.1016/j.biomaterials.2011.11.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pu HL, Chiang WL, Maiti B, Liao ZX, Ho YC, Shim MS, Chuang EY, Xia Y, Sung HW. Nanoparticles with dual responses to oxidative stress and reduced ph for drug release and anti-inflammatory applications. ACS Nano. 2014;8(2):1213–1221. doi: 10.1021/nn4058787. [DOI] [PubMed] [Google Scholar]
- 18.Chen H, He W, Guo Z. An H(2)O(2)-responsive nanocarrier for dual-release of platinum anticancer drugs and O(2): controlled release and enhanced cytotoxicity against cisplatin resistant cancer cells. Chem Commun. 2014;50(68):9714–9717. doi: 10.1039/c4cc03385j. [DOI] [PubMed] [Google Scholar]
- 19.Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5(7):793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- 20.Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411(6835):375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 21.Jezierska A, Motyl T. Matrix metalloproteinase-2 involvement in breast cancer progression: a mini-review. Med Sci Monit. 2009;15(2):RA32–40. [PubMed] [Google Scholar]
- 22.Tayebjee MH, Lip GY, MacFadyen RJ. Matrix metalloproteinases in coronary artery disease: clinical and therapeutic implications and pathological significance. Curr Med Chem. 2005;12(8):917–925. doi: 10.2174/0929867053507270. [DOI] [PubMed] [Google Scholar]
- 23.Roomi MW, Monterrey JC, Kalinovsky T, Rath M, Niedzwiecki A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol Rep. 2009;21(5):1323–1333. doi: 10.3892/or_00000358. [DOI] [PubMed] [Google Scholar]
- 24.Hofmann UB, Westphal JR, Waas ET, Zendman AJ, Cornelissen IM, Ruiter DJ, van Muijen GN. Matrix metalloproteinases in human melanoma cell lines and xenografts: increased expression of activated matrix metalloproteinase-2 (MMP-2) correlates with melanoma progression. Br J Cancer. 1999;81(5):774–782. doi: 10.1038/sj.bjc.6690763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roh MR, Zheng Z, Kim HS, Kwon JE, Jeung HC, Rha SY, Chung KY. Differential expression patterns of MMPs and their role in the invasion of epithelial premalignant tumors and invasive cutaneous squamous cell carcinoma. Exp Mol Pathol. 2012;92(2):236–242. doi: 10.1016/j.yexmp.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Yu SS, Miteva M, Nelson CE, Werfel T, Giorgio TD, Duvall CL. Matrix Metalloproteinase Responsive, Proximity-activated Polymeric Nanoparticles for siRNA Delivery. Adv Funct Mater. 2013;23(24):3040–3052. doi: 10.1002/adfm.201202215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorresteijn R, Billecke N, Schwendy M, Putz S, Bonn M, Parekh SH, Klapper M, Mullen K. Polylactide-block-Polypeptide-block-Polylactide Copolymer Nanoparticles with Tunable Cleavage and Controlled Drug Release. Adv Funct Mater. 2014;24(26):4026–4033. [Google Scholar]
- 28.Godin B, Tasciotti E, Liu X, Serda RE, Ferrari M. Multistage nanovectors: from concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44(10):979–989. doi: 10.1021/ar200077p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferrari M. Frontiers in cancer nanomedicine: directing mass transport through biological barriers. Trends Biotechnol. 2010;28(4):181–188. doi: 10.1016/j.tibtech.2009.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasciotti E, Liu XW, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MMC, Decuzzi P, Tour JM, Robertson F, Ferrari M. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol. 2008;3(3):151–157. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 31.Ananta JS, Godin B, Sethi R, Moriggi L, Liu X, Serda RE, Krishnamurthy R, Muthupillai R, Bolskar RD, Helm L, Ferrari M, Wilson LJ, Decuzzi P. Geometrical confinement of gadolinium-based contrast agents in nanoporous particles enhances T1 contrast. Nat Nanotechnol. 2010;5(11):815–821. doi: 10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parodi A, Quattrocchi N, van de Ven AL, Chiappini C, Evangelopoulos M, Martinez JO, Brown BS, Khaled SZ, Yazdi IK, Enzo MV, Isenhart L, Ferrari M, Tasciotti E. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat Nanotechnol. 2013;8(1):61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen J, Wu X, Lee Y, Wolfram J, Yang Z, Mao ZW, Ferrari M, Shen H. Porous silicon microparticles for delivery of siRNA therapeutics. J Vis Exp. 2015;(95):52075. doi: 10.3791/52075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scavo MP, Gentile E, Wolfram J, Gu J, Barone M, Evangelopoulos M, Martinez JO, Liu X, Celia C, Tasciotti E, Vilar E, Shen H. Multistage vector delivery of sulindac and silymarin for prevention of colon cancer. Colloids Surf B Biointerfaces. 2015;136:694–703. doi: 10.1016/j.colsurfb.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 35.Shen J, Xu R, Mai J, Kim HC, Guo X, Qin G, Yang Y, Wolfram J, Mu C, Xia X, Gu J, Liu X, Mao ZW, Ferrari M, Shen H. High capacity nanoporous silicon carrier for systemic delivery of gene silencing therapeutics. ACS Nano. 2013;7(11):9867–9880. doi: 10.1021/nn4035316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mai J, Huang Y, Mu C, Zhang G, Xu R, Guo X, Xia X, Volk DE, Lokesh GL, Thiviyanathan V, Gorenstein DG, Liu X, Ferrari M, Shen H. Bone marrow endothelium-targeted therapeutics for metastatic breast cancer. J Control Release. 2014;187:22–29. doi: 10.1016/j.jconrel.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez JO, Evangelopoulos M, Bhavane R, Acciardo S, Salvatore F, Liu X, Ferrari M, Tasciotti E. Multistage Nanovectors Enhance the Delivery of Free and Encapsulated Drugs. Curr Drug Targets. 2015;16(14):1582–1590. doi: 10.2174/1389450115666141015113914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xia X, Mai J, Xu R, Perez JE, Guevara ML, Shen Q, Mu C, Tung HY, Corry DB, Evans SE, Liu X, Ferrari M, Zhang Z, Li XC, Wang RF, Shen H. Porous silicon microparticle potentiates anti-tumor immunity by enhancing cross-presentation and inducing type I interferon response. Cell Rep. 2015;11(6):957–966. doi: 10.1016/j.celrep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wolfram J, Shen H, Ferrari M. Multistage vector (MSV) therapeutics. J Control Release. 2015;219:406–415. doi: 10.1016/j.jconrel.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, Shen J, Liu H, Hu Z, Chen L, Huang Y, Koay E, Huang Y, Liu J, Ensor JE, Blanco E, Liu X, Ferrari M, Shen H. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016;34(4):414–418. doi: 10.1038/nbt.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Decuzzi P, Godin B, Tanaka T, Lee SY, Chiappini C, Liu X, Ferrari M. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141(3):320–327. doi: 10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 43.Mi Y, Mu C, Wolfram J, Deng Z, Hu TY, Liu X, Blanco E, Shen H, Ferrari M. A Micro/Nano Composite for Combination Treatment of Melanoma Lung Metastasis. Adv Healthc Mater. 2016;5:936–949. doi: 10.1002/adhm.201500910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, Levy-Nissenbaum E, Radovic-Moreno AF, Langer R, Farokhzad OC. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28(5):869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Discoidal Porous Silicon Particles: Fabrication and Biodistribution in Breast Cancer Bearing Mice. Adv Funct Mater. 2012;22(20):4225–4235. doi: 10.1002/adfm.201200869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mi Y, Zhao J, Feng SS. Targeted co-delivery of docetaxel, cisplatin and herceptin by vitamin E TPGS-cisplatin prodrug nanoparticles for multimodality treatment of cancer. J Control Release. 2013;169(3):185–192. doi: 10.1016/j.jconrel.2013.01.035. [DOI] [PubMed] [Google Scholar]
- 47.Tian X, Lara H, Wagner KT, Saripalli S, Hyder SN, Foote M, Sethi M, Wang EN, Caster JM, Zhang LZ, Wang AZ. Improving DNA double-strand repair inhibitor KU55933 therapeutic index in cancer radiotherapy using nanoparticle drug delivery. Nanoscale. 2015;7(47):20211–20219. doi: 10.1039/c5nr05869d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mi Y, Liu X, Zhao J, Ding J, Feng SS. Multimodality treatment of cancer with herceptin conjugated, thermomagnetic iron oxides and docetaxel loaded nanoparticles of biodegradable polymers. Biomaterials. 2012;33(30):7519–7529. doi: 10.1016/j.biomaterials.2012.06.100. [DOI] [PubMed] [Google Scholar]
- 49.Kedmi R, Ben-Arie N, Peer D. The systemic toxicity of positively charged lipid nanoparticles and the role of Toll-like receptor 4 in immune activation. Biomaterials. 2010;31(26):6867–6875. doi: 10.1016/j.biomaterials.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 50.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kulkarni SA, Feng SS. Effects of particle size and surface modification on cellular uptake and biodistribution of polymeric nanoparticles for drug delivery. Pharm Res. 2013;30(10):2512–2522. doi: 10.1007/s11095-012-0958-3. [DOI] [PubMed] [Google Scholar]
- 52.Win KY, Feng SS. Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials. 2005;26(15):2713–2722. doi: 10.1016/j.biomaterials.2004.07.050. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.