

Abstract
Endogenous Plastic Somatic (ePS) cells isolated from adult human tissues exhibit extensive lineage plasticity in vitro and in vivo. Here we visualize these rare ePS cells in a latent state, i.e. lacking SOX2, OCT3/4 and NANOG (SON) expression, in non-diseased breast specimens through immunohistochemical analysis of previously identified ePS-specific biomarkers (CD73+, EpCAM+ and CD90−). We also report a novel mechanism by which these latent ePS cells acquire SON expression and plasticity in vitro. Four extracellular factors are necessary for the acquisition of SON expression and lineage plasticity in ePS cells: adenosine (which is produced by the 5’ ectonucleotidase CD73 and activates in turn the PKA-dependent IL6/STAT3 pathway through the adenosine receptor ADORA2b), IL-6, FGF2 and ACTIVIN A. Blocking any pathway component renders ePS cells incapable of SON expression and lineage plasticity. Notably, hESCs do not use adenosine or IL-6 nor they express CD73 or ADORA2b and inhibition of adenosine signaling does not ablate their plasticity. Therefore, the data presented here delineate novel circuitry and physiological signals for accessing SON expression in rare, undifferentiated human cells.
Graphical abstract
1. Introduction
Until recently, it was thought that cellular plasticity, i.e. the ability to form multiple somatic lineages, was restricted to a transient population of pluripotent cells within the blastocyst (embryonic stem cells) and an enduring population in the gonads (germ cells) [1]. A self-regulating positive feedback network, that includes expression of SON proteins, maintains a plastic state in these cells. However, we now know that acquisition of cell plasticity or a pluripotent state is not restricted to the above cells. Exogenous expression of key factors has been shown to induce the transition of fully differentiated cells to a pluripotent state [2]. Additionally, rare cells within adult tissues have also been documented to express SON and generate some, or all, somatic lineages [3-10]. In some instances, such cells are isolated from stressed tissues generating “multi-lineage differentiating stress-enduring” (MUSE) cells [11]. In other instances, such rare cells (endogenous Plastic Somatic (ePS) cells) have been found in a latent state in non-damaged tissues that can subsequently be activated to express SON [12]. The latter cells provide a unique opportunity to study the activation of SON and acquisition of cellular plasticity.
The recently described ePS cells exhibit three coordinated phenotypes that are regulated by repressed p16INK4a: they (a) lack activation of cell cycle arrest when stressed, (b) express distinctive surface proteins whose expression (high CD73 and lack of CD90) enables their prospective enrichment and (c) exhibit enhanced expression of chromatin remodeling proteins [13], a state associated with acquisition of cellular plasticity and loss of differentiation properties [13-15]. The unexpected extensive lineage plasticity of ePS cells was characterized according to strict criteria previously outlined within the stem cell field [16]. In vitro, in vivo, teratoma assays and forensic analysis verified that ePS cells can give rise to a population of cells that generates all somatic lineages and retains a diploid karyotype [12]. Importantly, in contrast to other plastic cells that have the ability to differentiate into all three lineages, such as human embryonic stem cells (hESC) or induced pluripotent stem cells (iPSC), ePS cells are mortal, and retain low telomerase expression while expanding for an extensive, but finite, number of cell doublings before arresting in G1 (Table 1).
Table 1.
Phenotypes that distinguish ePS cells from pluripotent ESC and iPSC as well as cancer cells
| CHARACTERISTICS | CELL TYPES | ||||
|---|---|---|---|---|---|
| ePS | hESC | iPSC | pre- cancer |
cancer | |
| Diploid karyotype | + | + | + | +/− | − |
| Benign teratomas | + | + | + | − | − |
| Pluripotency markers | + | + | + | − | +/− |
| Functional breast tissue |
+ | not determined |
not determined |
− | − |
| Immortality | − | + | + | +/− | + |
| High telomerase activity |
− | + | + | 50% | + |
| Malignant lesions | − | +/− | +/− | − | + |
Expression of the surface marker CD73, a glycosyl phosphatidyl-inositol (GPI)-anchored ecto-5’-nucleotidase present in cell membrane lipid rafts [17, 18], is a key distinguishing marker of ePS cells. Expression of CD73, regulated by many stress-associated factors [19-21], has been widely implicated in cellular responses to stress and activating plasticity through catalyzing the generation of extracellular adenosine and control of adenosine signaling [22-24]. CD73 has been detected in many cell types where it participates in multiple functions including immune response and protection from ischemia and tissue damage [17]. Recently, CD73 has been identified as a critical reprogramming landmark in the generation of iPS cells [25]. This study examines the cellular and molecular mechanisms by which ePS cells attain a plastic state. We demonstrate that activation of SON is necessary for ePS cells to exhibit plasticity and we identify novel CD73-dependent signaling pathways that are key to the regulation of SON in these cells. In identifying these key stress-signaling cascades, we identify novel circuitry for accessing plasticity in rare human cells within the adult body and clues as to where activated ePS cells may be found in vivo.
2. Materials and methods
Information about reagents and tissue samples is shown in Tables S1 and S2.
2.1. Isolation and culture of ePS cells from human breast tissues
Acquisition of breast tissues from healthy women undergoing reduction mammoplasty after informed consent and the described experiments were approved by the UCSF Committee on Human Research under protocol 10-01532. Obtained information met Health Insurance Portability and Accountability Act guidelines. Tissues were devoid of visible disease, bacterial, fungal or viral contamination. ePS cells were isolated and cultured as described in [12]. Briefly, sorted CD73+CD90− cells (104 cells/well) were cultured on irradiated fibroblast cells (feeders) from 6.2-6.6 week placentas or expanded in a feeder-free medium (F-FM) [12]. The number of colonies over 100 μm or 400 μm in size growing on feeders was recorded. ePS cells within the R1 sorted subfraction were expanded in F-FM (α-MEM medium with glutamine supplemented with 15% (vol/vol) ES-FBS (Omega Scientific; FB-05), 18% (vol/vol) Chang B, and 2% (vol/vol) Chang C (Irvine Scientific; C-100 and C-106, respectively)) and passaged by trypsinization.
2.2. Cell culture and drug treatment
T47D breast cancer cells were maintained in DMEM+10%FBS, CD73−CD90+ cells in MEGM and ePS cells growing on feeders in mammary stem cell medium [12]. After a 3 day recovery post-sorting, ePS cells were treated with various drugs as described in the Figure legends and fed fresh medium every other day for 7 days. Cell growth was determined by daily cell counting for 4 days after initial plating of 1×105 cells in triplicate. hESCs were propagated either on irradiated MEF feeders in Knockout Serum Replacement (KSR) medium or in feeder-free medium (F-FM) used to expand ePS cells.
2.3. Immunoblotting and Real-Time qPCR
Immunoblotting was carried out as described in [12]. Total RNA was isolated from cells and cDNA synthesized as previously described [26]. qPCR (Taqman) was performed in technical triplicates using the standard curve method with gene-specific primer/probe sets and monitoring of glucuronidase B (GUSB) expression to normalize for variances in input cDNA.
2.4. FACS analysis
After epitope regeneration in MEGM + 2%FBS for 1-2 hours on ice, the single cell suspension was stained with a FITC-labeled anti-EpCAM antibody, fixed, permeabilized with a transcription factor buffer set, stained with an anti-OCT3/4 primary antibody and an Alexa 647 goat anti-rabbit secondary antibody and analyzed using a FACS Aria II cell sorter.
2.5. Apoptotic analysis
ePS cells growing on feeders were harvested and stained with a FITC-conjugated antibody against EpCAM and Alexa 647 conjugated Annexin V (Life Technologies; A23204) prior to FACS analysis.
2.6. Adipogenesis and osteogenesis differentiation assays
Adipogenesis: ePS cells or MSCs were differentiated using an adipogenesis differentiation kit. After 14 days, cells were stained with Oil Red O [12] or LipidTox following manufacturer’s instructions (Life Technology; H34350) or analyzed by qRT-PCR. Osteogenesis: 104 cells seeded into 4-well Permanox slides were treated with or without 400 μm adenosine in the presence or absence of 12 μm APCP for 48h before applying osteogenic conditions (StemPro Osteogenesis Differentiation Kit; Gibco, Inc) for 21 days. Osteogenesis was assessed after cell fixation in 4% paraformaldehyde for 15 min and staining with 2% alizarin red solution (pH 4.1-4.3) for 20 min. Stained monolayers were visualized and recorded by phase microscopy.
2.7. Embryoid body formation, cardiomyocyte differentiation and video documentation
ePS cells grown on feeders were manually dissected for embryoid body formation, subjected to cardiomyocyte differentiation and video-taped [12]. All videos time lapses were 10-15 s.
2.8. Plasmids and Retroviral Gene Transfer
Lentiviral plasmids expressing CD73-Flag or short hairpins for OCT3/4, SOX2, NANOG, STAT3 or CD73 and matched control plasmids were used to transfect packaging 293 cells. Lentiviral suspensions were used to transduce ePS cells cultured in F-FM as described in [26]. 72 h after initial transduction, cells were selected for 48 h with 1.5 μg/mL puromycin, expanded and induced with 1μg/ml doxycycline for 48 h to express the short hairpins.
2.9. Immunohistochemistry and Multiplex analysis
Serial 4 μm paraffin-embedded RM tissue sections were deparaffinized and rehydrated. One section was stained with hematoxylin and eosin (H&E) for histological evaluation and other sections subjected to multiplex immunostaining. All steps were performed at room temperature unless mentioned otherwise. Briefly, to reduce non-specific background staining due to endogenous peroxidase, sections were treated with 3% hydrogen peroxide for 10 min. For antigen retrieval, sections were microwaved for 10 min in citrate buffer (pH=6.0). For double staining, sections were incubated simultaneously for 60 min with a mouse monoclonal anti-CD73 antibody diluted 1:2,000 and either a rabbit monoclonal anti-CD90 antibody diluted 1:100 or a rabbit monoclonal anti-EpCAM antibody diluted 1:100. Sections were washed then incubated for 30 min with anti-mouse IgGs coupled to horseradish peroxidase (HRP) and anti-rabbit IgGs coupled to alkaline phosphatase (AP) (Power-StainTM 1.0 double stain kit I). Sections were washed and developed for 5 min using diaminobenzidine (DAB) and Vector blue as HRP and AP substrates, respectively. For triple staining of SON, sections were incubated first with a rabbit monoclonal anti-SOX2 antibody diluted 1:50, then with anti-rabbit IgGs coupled to AP and finally revealed with Vector blue chromogen. Sections were microwaved for 5 min in citrate buffer (pH=6.0) and incubated simultaneously with a rabbit monoclonal anti-OCT 3/4 antibody diluted 1:800 and a mouse monoclonal anti-NANOG antibody diluted 1:6,000. Sections were washed then incubated for 30 min with Power-StainTM 1.0 double stain kit I. After 2 washes, sections were developed for 5 min using DAB and Vector Red as HRP and AP substrates, respectively, and counterstained with methyl green or nuclear fast red to visualize the nuclei. Multiplex analysis was carried out after image acquisition using a multispectral Nuance FX camera and unmixing of color images using the Nuance software (Perkin-Elmer). The analysis generated black and white images showing staining pattern for each marker separately as well as a merged composite image with pseudocolors assigned to each marker to facilitate visualization of multiplex staining pattern.
2.10. Statistical analysis
Two-sided t test assuming unequal variance was used to test the relations between mRNA expression of OCT3/4, NANOG, SOX2, ADORA1, ADORA2a, ADORA2b, ADORA3, LEPTIN, FABP4, and PPARγ or EpCAM+OCT3/4+ cell percentages in ePS cells. For all comparisons, P ≤ 0.05.
3. Results
3.1. Multiplex immunohistochemical analysis of CD73 and CD90 visualizes latent ePS in adult human breast tissue
We previously reported that ePS cells isolated on the basis of the CD73+/CD90− immunophenotype constitute a small fraction (~0.15%) of cells within human breast tissue (~3% of the CD73+/CD90− population). Significant evidence differentiates ePS cells from breast tissue-specific stem cells (documented to be EpCAM negative), as well as mesenchymal stem cells, adipocytes and fibroblasts (documented to be EpCAM negative and CD90 positive) [27, 28] (Table 2). Additionally, ePS cells express EpCAM and CD49f [12], indicating that they are not mature luminal or myoepithelial breast cells since these differentiated cells are CD49f negative and EpCAM negative, respectively [29, 30]. Notably, ePS cells, either when observed in vivo in healthy tissues or when initially isolated from those tissues, do not express SON proteins. However, when these cells are placed in feeder-free or feeder conditions, SON proteins are induced to levels equivalent to those observed in hESCs. Thus, ePS cells exist as undifferentiated cells in a latent state in healthy tissues and are activated by our culture conditions, providing us with an assay to study their acquisition of SON expression and plasticity.
Table 2.
Markers that distinguish ePS cells from differentiated cells
|
MARKER
S |
CELL TYPES | |||||||
|---|---|---|---|---|---|---|---|---|
| eP S |
lumin al |
myo- epitheli al |
MSC s |
adipocyte s |
fibroblast s |
M a S C |
bone marrow- derived SC |
|
| CD90 | − | − | − | + | + | + | + | + |
| EpCAM | + | + | − | − | − | − | − | not determine d |
| CD49f | + | − | + | + | − | − | + | + |
| CD73 | + | − | − | + | − | − | − | not determine d |
Here, we sought to visualize the latent rare cells in situ by multiplex analysis of the CD73+CD90− immunomarkers. Some of the reduction mammoplasty (RM) specimens utilized were characterized in our previous study for ePS cell lineage plasticity both in vitro and in vivo [12]. Multiplex immunohistochemical analysis allowed visualization of the four subpopulations (R1-R4) of cells based on their expression of CD73 and CD90 (Figs. 1A-B and S1A-B). In agreement with our reported flow cytometric analysis [12], the majority of breast cells were negative for both CD73 and CD90 expression (R3 population). Based on our analysis, latent ePS cells were expected to be rare and enriched in a population expressing CD73 and lacking CD90 and SON. CD73+CD90− cells were more prevalent in the large ducts than in terminal alveoli (Fig. 1), with infrequent cells dispersed within the surrounding stroma. The frequency of CD73+CD90− cells was ~ 3% (136/4610 cells) consistent with the frequency (~3%) of these cells measured by flow cytometry using the same markers [12]. A similar frequency was confirmed in 11 independent RM specimens (Table S1). Multiplex analysis of CD73 and EpCAM, an additional marker expressed on ePS cells, demonstrated that a subset of CD73+ cells also express EpCAM. On the basis of the CD73+CD90− EpCAM+ immunophenotype, ePS cells can be differentiated from other cell types within the adult mammary gland (Table 2). Consistent with the proposed latent state of ePS cells, multiplex analysis of healthy breast tissue also failed to identify cells exhibiting coincident nuclear expression of SON (Fig. S1A-B), contrasting with a seminoma sample used as positive control (Fig. S1C).
Fig. 1. Detection of candidate ePS cells in vivo. A.

RM085 stained simultaneously with a monoclonal anti-CD73 antibody detected with a secondary antibody coupled to horseradish peroxidase and the chromogen, 3,3’-Diaminobenzidine (DAB) (brown) and with a polyclonal anti-CD90 antibody detected with a secondary antibody coupled to alkaline phosphatase and the chromogen, Vector blue (blue). Insets: examples of CD73+CD90− (R1), CD73+CD90+ (R2), CD73− CD90− (R3) and CD73−CD90+ (R4) cells. B. RM179 stained as described for RM085 in (A). Scale bars: 20μm for A and B.
3.2. SON signaling is necessary for ePS cell plasticity
Latent ePS cells can be activated to coincidently express nuclear SON and display lineage plasticity, providing a distinct state to allow us to investigate molecular mechanisms for induction of these two phenotypes in these rare cells. Once CD73+CD90− (R1) populations (enriched for ePS cells) are directly sorted from human breast tissues and placed on irradiated placental fibroblast feeders, the ePS cells attach and form colonies. After 9-12 days in culture, they express pluripotency-associated proteins such as SON, Lin28 [12], Tra-1-60 and Tra-1-81 (Fig. S2A) at levels comparable to hESCs cells grown on feeders. CD73+CD90− (R1) cells were also placed in F-FM conditions (Fig. S2A). These expanded ePS cells were genomic stable displaying a normal diploid phenotype (Fig. S1A). This complementary feeder-free approach allows us to specifically and sufficiently expand ePS cells to facilitate mRNA and biochemical analysis as well as perform genetic manipulations. Immunocytochemistry, qPCR, and Western blot (WB) analyses confirmed that the expression of SON was comparable in ePS cells and hESCs grown in F-FM conditions (Fig. S2B-D).
The pluripotent state of hESCs is dependent upon the expression and activity of SON [31-33]. Therefore, we next asked if the expression of each component of SON within this mutually regulated feedback circuit was also necessary for ePS cell plasticity [34-36]. ePS cells, freshly isolated from breast tissue, were cultured in F-FM conditions and transduced with a shRNA construct against OCT3/4, SOX2 or NANOG. Both qPCR (Fig. S3A) and WB (Fig. S3B) analyses confirmed efficient knockdown of each gene compared to control cells transduced with a scrambled shRNA vector. Additionally, we observed that knockdown of any one of the three SON factors promoted the downregulation of the other two untargeted SON factors (Fig. S3A and B). ePS cells silenced for OCT3/4, SOX2 or NANOG were all similarly defective in the ability to differentiate into adipocytes compared to control ePS cells, as demonstrated by the lack of accumulation of Oil Red O-positive (top panels) or LipidTox-positive (bottom panels) lipid droplets (Fig. S3C). Consistent with the functional staining, ePS cells silenced for OCT3/4, SOX2 or NANOG all failed to express molecular markers of adipocyte differentiation fatty acid binding protein 4 (FABP4), LEPTIN and the transcription factor PPARγ, (Fig. S3D). Together, these data indicate that SON expression is required for ePS cell plasticity, just as it is for hESCs.
3.3. CD73-mediated generation of adenosine is necessary for SON expression and plasticity in ePS cells
We sought to determine whether CD73, a key immunomarker used to enrich for ePS cells [12], plays a mechanistic role in the acquisition of SON expression and plasticity. Exposure of ePS cells cultured on feeders to 6 μM of adenosine 5'-(α, β-methylene) diphosphate (APCP), an inhibitor of CD73 enzymatic activity, for 1 or 3 weeks dramatically decreased the size and number of ePS colonies (Fig. 2A). Immunocytochemical analysis of ePS colonies treated with APCP revealed a dramatic reduction in SON expression compared to vehicle-treated cells and hESCs (Figs. 2B and S4A). Consistent with this, exposure of SON-expressing ePS cells cultured in F-FM to APCP for 48 hours resulted in a 40-60% reduction of SON mRNA expression (Fig. 2C) and a dramatic decrease in protein expression (Fig. S4B) compared to vehicle-treated cells. As expected, APCP treatment did not affect CD73 protein expression (Fig. S4B). Doxycycline-inducible shRNA knockdown of CD73 in ePS cells cultured in F-FM validated our results with APCP. Upon doxycycline addition, ePS cells exhibited a 3-fold reduction in CD73 mRNA (Fig. S4C) as well as a reduction in SON mRNA (Fig. 2D) and protein (Fig. S4D) compared to ePS cells transduced with scrambled shRNA. When cultured under differentiation conditions conducive to adipogenesis, both APCP-treated and shCD73-knockdown (+doxycycline) cells failed to accumulate Oil Red O-positive lipid droplets (Fig. 2E and S4E) and expressed lower levels of FABP4, LEPTIN and PPARγ (Fig. S4F) compared to control cells. Additionally, APCP-exposed ePS colonies failed to generate embryoid bodies (EB) and beating cardiomyocytes compared to control cells (Fig. 2F and Movies S1-2). APCP-mediated inhibition of CD73 activity did not affect ePS cell growth or survival in F-FM (Fig. S4G). However, upon APCP treatment, ePS colonies cultured on feeders exhibited an increase in apoptosis over time as demonstrated by EpCAM+ANNEXIN V+ staining (Fig. S4H).
Fig. 2. CD73 is necessary for the plastic state of ePS cells.

ePS colonies from RM122 and RM128 were treated with vehicle control (DMSO) or APCP and assessed for A. colony size (outlined in red dashed lines) and morphology (top panel) and number (bottom panel) after 1 week (w) or 3 w of treatment, n=2 and n=3, respectively; B. SON protein expression after 48h of treatment by immunocytochemistry along with hESC colonies (outlined in red dashed lines in corresponding bright field images) Scale bars: 10μm (n=2); Nuclei were stained with DAPI. Nuclear localization of SON in the areas outlined in white squares is shown at a higher magnification. Scale bars: 40μm. ePS cells cultured in F-FM and treated or not with APCP for 48 h were assessed for C. SON transcripts by qRT-PCR (n=2). Lentivirus carrying an inducible pTripZ shCD73 or scrambled shRNA were transduced into ePS cells from RM 122 or RM128 expanded for 21 days. Infected cells, selected with puromycin prior to and after doxycycline (Dox) induction, were assessed for D. SON transcripts by qRT-PCR. (n=3). E. Vehicle (DMSO)-treated, APCP-treated, CD73 knock-down (+Dox) and control (−Dox) ePS cells from RM122 or RM128 were subjected to adipogenic differentiation for 18 days and stained with Oil Red O (n=2). Scale bars: 10μm. F. Generation of embryoid bodies in ePS cells treated or not with APCP for 7 days (n=1 each). Scale bars: 10μm. Data from RM122 are shown in B, E and F.
Having established a role for CD73 in SON expression and ePS plasticity, we next investigated the role of adenosine (the enzymatic product of CD73 activity). Exposure of shCD73-ePS cells in F-FM conditions to 200 μM adenosine rescued OCT3/4 mRNA expression, which was increased 2-fold compared to untreated shCD73-ePS cells (Fig. S4I). More importantly, addition of adenosine was sufficient to rescue both adipogenic and osteogenic differentiation in APCP-treated ePS cells without affecting differentiation of ePS cells in the absence of APCP (Fig. S4J). As an additional control, we investigated whether adenosine was sufficient to induce SON expression in the CD73−/CD90+ subpopulation lacking ePS cells, the majority of which were of myoepithelial lineage. CD73−/CD90+ cells failed to express SON when exposed to 200 μM or 500 μM adenosine for up to 96 h (Fig. S4K), even in conditions that supported both myoepithelial (MEGM) and ePS (F-FM) cell expansion. Furthermore, ectopic expression of CD73 in T47D breast cancer cells, which do not normally express CD73, failed to upregulate OCT3/4 protein (Fig. S4L). These data indicate that CD73-mediated adenosine production is required but not sufficient for ePS colony growth on feeders, SON expression and cellular plasticity.
3.4. Adenosine receptor ADORA2b activates an intracellular signaling cascade necessary for SON expression in ePS cells
Adenosine signaling is dependent on four G protein-coupled receptors (GPCR), ADORA1, ADORA2a, ADORA2b, and ADORA3 [37]. Only ADORA2b expression was observed in ePS cells by qPCR analysis (Fig. 3A). Inhibition of ADORA2b, either specifically (MRS1754 (MRS)) or in addition to ADORA2a (DMPX, 3,7-Dimethyl-1-propargylxanthine), decreased ePS colony formation on feeders in comparison to vehicle-treated cells, similar to the effect of APCP (Fig. 3B). Treatment of ePS cells grown under F-FM conditions with DMPX or MRS resulted in a decrease in the percentage of cells expressing OCT3/4 protein (Fig. 3C) and SON mRNA (Figs. 3D and S5A). In contrast, cells exposed to inhibitors of ADORA1 or ADORA3 (8-phenyltheophylline (8-PT) and VUF-5574 (VUF), respectively) formed ePS colonies on feeders (Fig. 3B) and expressed SON mRNA in F-FM culture conditions (Figs. 3D and S5A). ePS cells cultured in MRS failed to form EBs and to differentiate into beating cardiomyocytes compared to vehicle-treated cells or cells cultured in the presence of 8-PT or VUF (Fig. 3E and Movies S3-5). In a complementary approach, ePS cells were transduced with a doxycycline-inducible shRNA against ADORA2b in F-FM conditions. Silencing ADORA2b (+doxycycline) (Fig. S5B) resulted in decreased SON mRNA (Fig. S5C) and prevented adipogenesis, as demonstrated by reduced expression of FABP4, LEPTIN, and PPARγ (Fig. 3F). These results indicate that ADORA2b receptor function is necessary for ePS cells to regulate SON expression and cellular plasticity.
Fig. 3. ADORA2b receptor signaling mediates adenosine functions in ePS cells.

ePS cells from RM136 and RM142, grown in F-FM (panels A, D and F) or on feeders (panels B, C and E), were assessed for A. ADORA1, ADORA2a, ADORA2b and ADORA3 transcript expression by qRT-PCR (n=3 each); B. size and morphology of ePS colonies (outlined with red dashed lines) treated or not with APCP and/or adenosine receptor-specific inhibitors (8-PT, MRS, DMPX or VUF; 10μm) for 7 days (n=2 each). Data from RM136 are shown. Scale bars: 10μm.; C. EpCAM+ OCT3/4+ cell fractions by FACS analysis. (n=2 each); D. OCT3/4 transcript expression by qRT-PCR in ePS cells treated or not with 8-PT, MRS, DMPX and VUF for 48 h (n=3 and n=6, respectively); E. generation of embryoid bodies treated or not with 8-PT, MRS and VUF for 7 days (n=2). Scale bars: 10μm.; F. adipogenesis at 14 days in ePS cells transduced with or without shADORA2b and induced or not with Doxycycline. Comparison of transcript expression of adipogenic differentiation markers FABP4, LEPTIN and PPARγ by qRT-PCR (n=3 each). Panels B and E show data from RM136.
3.5. Identification of PKA/STAT3 signaling downstream of ADORA2b in regulating SON expression in ePS cells
In ESCs, GPCR activity has been hypothesized to regulate SON expression and pluripotency through activation of cAMP/cAMP-dependent protein kinase (PKA) and downstream STAT3 signaling [38, 39]. ADORA2b is coupled to the stimulatory G-protein subunit that promotes cAMP production and PKA activation [40]. To determine if ADORA2b regulates SON expression through the downstream activation of the PKA and STAT3 signaling pathways, ePS cells were incubated with increasing concentrations of H89, a PKA inhibitor, or Stattic, a STAT3 inhibitor. As previously demonstrated, adenosine (200μM) enhanced SON expression in ePS cells at both mRNA (Fig 4A and S6A) and protein (Fig. 4B) levels. In this experiment, both H89 and Stattic reduced SON mRNA (Fig 4A and S6A) and protein (Fig. 4B) levels in the absence and presence of adenosine. Validating this result, ePS cells silenced for STAT3 with inducible shRNAs in F-FM conditions (Fig. 4C) also exhibited a decrease in SON protein (Fig. 4C) and mRNA (Fig. S6B).
Fig. 4. The PKA/STAT3 signaling pathway mediates ADORA2b function in ePS cells.

ePS cells from RM136 and RM159, grown in F-FM, were assessed for A. OCT3/4 transcripts by qRT-PCR in ePS cells treated or not with adenosine (Ado) in presence or absence of the PKA inhibitor H89 or the STAT3 inhibitor Stattic. (n=3 each). B. SON and STAT3 protein expression and phosphorylation status of STAT3 (p-STAT3) by WB in ePS cells with or without knock down of ADORA2b (shDORA2b), or treated with or without APCP for 48 h or treated with Ado for 48 h, in presence or absence of H89 (1 or 5μM) or Stattic (0.2 and 1μM) (n=1 and n=2, respectively). C. SON and STAT3 protein expression by WB after transduction of lentivirus carrying inducible shRNAs against STAT3 (#16 or #17) or a matched scrambled shRNA and induction by Doxycyclin for 48h (n=2 each). D. OCT3/4 transcripts by qRT-PCR in untreated ePS cells and ePS cells treated with Ado in presence or absence of H89 or Stattic, alone or various inhibitor combinations. Red dashed line: OCT3/4 expression in presence of H89, Stattic alone or their combination (n=3 each). E. OCT3/4 transcripts by qRT-PCR in MRS-treated or untreated ePS cells in presence or absence of the PKA activator 8-Br-cAMP (n=3 each). F. OCT3/4 protein expression and STAT3 phosphorylation status (phospho-STAT3) by WB in ePS cells treated or not with different concentrations of MRS-treated in presence or absence of different concentrations of 8-Br-cAMP (n=1 and n=2, respectively). G. IL6 and LIF transcripts by qRT-PCR in untreated or adenosine-treated ePS cells in presence or absence of H89 (n=3 each). H. IL6 transcripts by qRT-PCR in ePS cells treated or not with 10μm of 8-PT, MRS, DMPX and VUF (top panel). IL6 protein concentration by ELISA in CM collected from ePS cells with or without knock-down of ADORA2b (bottom panel) (n=2 each) I. IL6 transcripts by qRT-PCR in ePS cells treated or not with APCP in presence or absence of Ado or in ePS cells transduced with a CD73 shRNA or a scrambled shRNA (n=3 each). In Panels B, C and F loading control: ACTIN and data from RM136 are shown.
We next examined if the PKA and STAT3 signaling pathways were epistatic or parallel downstream of adenosine signaling. ePS cells were treated with H89 and Stattic, either alone or together in the presence of 200μM adenosine. Both the single inhibitor and combination groups exhibited a 40% reduction in OCT3/4 expression suggesting that PKA and STAT3 were part of a single pathway downstream of adenosine-induced ADORA2b activation (Fig. 4D). Consistent with this, inhibition of PKA resulted in a dramatic decrease in STAT3 phosphorylation (Fig. 4B), suggesting that PKA was upstream of STAT3 signaling.
To validate a functional link between PKA activity and SON expression, we activated PKA by treating ePS cells with 8-Br-cAMP (a non-hydrolyzable analog of cAMP) in the absence and presence of the ADORA2b-specific inhibitor MRS1754. Treatment with 8-Br-cAMP led to a 70% increase in SON mRNA (Figs. 4E and S6C) as well as increased OCT3/4 protein and STAT3 phosphorylation (Fig. 4F) compared to ePS cells treated with MRS1754 alone. These results demonstrated that in ePS cells with compromised ADORA2b activity, stimulation of the PKA/STAT3 pathway was sufficient to rescue SON expression.
We next probed the role of JAK and JNK, two kinases responsible for STAT3 activation in many cellular systems, in the response of ePS cells to adenosine. ePS cells, grown in F-FM conditions, were treated with the stable adenosine analogue 5′-(N-Ethylcarboxamido) adenosine (NECA). NECA increased JAK2 phosphorylation, but not that of JNK (Fig. S6D). Notably, phosphorylation of JAK2 in ePS cells treated with NECA was blocked by increasing concentrations of the PKA inhibitor H89 (Fig. S6D). Treatment of ePS cells with Forskolin (a PKA activator) also increased STAT3 phosphorylation and SON protein to the levels observed in NECA-treated cells. Treatment with Ruxolitinib (a specific inhibitor of JAK kinase) reduced both STAT3 phosphorylation and SON expression (Fig. S6E). Thus, in ePS cells, adenosine signaling through ADORA2b activates SON expression and cellular plasticity via an intracellular signaling cascade characterized by sequential activation of PKA, JAK and STAT3.
3.6. Increased IL6 production in response to adenosine signaling promotes autocrine activation of SON and plasticity in ePS cells
Interleukin 6 (IL6) potently activates JAK/STAT3 signaling through the cell surface type I cytokine receptor [41]. Mining of the ALEXA-Seq database [42] revealed that IL6 transcripts were expressed at high levels in sorted breast CD73+ (R1 plus R2) cells compared to other mammary cell populations and hESCs (Fig. S6F). In F-FM conditions, treatment of ePS cells with adenosine increased IL6 mRNA (Fig. 4G), while abrogation of the intracellular signaling pathway downstream of adenosine signaling in ePS cells, by treatment with the PKA inhibitor H89, prevented IL6 upregulation by adenosine (Fig. 4G). The expression of LIF, another member of the IL6 superfamily widely involved in regulating SON expression in pluripotent stem cells [43, 44], was unaltered in response to adenosine treatment (Fig. 4G). Our results suggest that adenosine signaling increased IL6 expression in a PKA-dependent manner. To validate this observation, we treated ePS cells cultured in F-FM with specific ADORA inhibitors and assessed IL6 expression by qPCR analysis. Pharmacological inhibition of ADORA2b by MRS and DMPX reduced IL6 expression whereas inhibition of ADORA1 and ADORA3 by 8-PT and VUF, respectively, did not (Fig. 4H). As described earlier, ADORA2b expression was downregulated using an inducible shRNA vector targeting ADORA2b. This knockdown of ADORA2b (+doxycycline) similarly decreased IL6 levels in the conditioned medium (CM) when measured by ELISA 72 hours after addition of fresh medium (Fig. 4H).
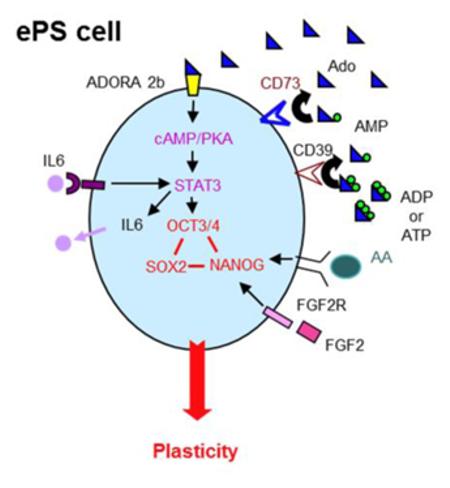
We next examined if CD73 was required for IL-6 expression. In ePS cells cultured in F-FM conditions, pharmacological inhibition of CD73 activity by APCP reduced IL6 mRNA, an effect that was blunted upon addition of adenosine (Fig. 4I). Knockdown of CD73 (Fig. 4I) and STAT3 (Fig. S6G) also decreased IL6 mRNA. These results strongly support that STAT3 activation downstream of adenosine signaling drives an autocrine positive-feedback loop through IL6. Consistent with a role for IL-6 in ePS cells, addition of exogenous IL6 to the medium of ePS cells cultured on feeders increased both the size and frequency of ePS colonies (Fig. S6H). Flow cytometry analysis revealed an increased expression of OCT3/4 protein in EpCAM-positive ePS cells (Fig. S6H-I). Reinforcing the importance of the identified positive feedback loop in control of SON expression, we observed that the addition of IL6 rescued SON protein expression in APCP-treated cells in which CD73 activity was compromised (Fig. S6J). Our observations demonstrate that generation of adenosine by CD73 contributes to SON expression in ePS cells through the autocrine activation of STAT3 by IL6 (Fig. 6D).
Fig. 6. Differential role of CD73 and ADORA2b in ePS cells compared to hESCs.

hESCs (H7), grown on feeders, were assessed for A. OCT3/4 (x-axis) and CD73 (y-axis) expression by FACS analysis (n=3). B. adenosine receptor ADORA1, 2a, 2b and 3 transcript expression in ePS cells from RM142 or RM177 (n=3 each) by qRT-PCR. C. SON protein expression in ePS cells from RM142 or RM177 (n=3 each) by WB,in the presence or absence of increasing concentrations of the CD73 enzymatic activity inhibitor APCP. Data from RM142 are shown. D. Schematic representation summarizing the four extracellular factors (adenosine, IL6, STAT3 and FGF2) essential for SON expression and cell plasticity in ePS cells.
3.7. The canonical FGF2 and ACTIVIN A signaling pathways are also essential for SON expression in ePS cells
Several extracellular factors critical to the maintenance of SON expression have been documented in hESCs, including FGF2 and ACTIVIN A, a member of the TGF-β superfamily [45-47]. The medium utilized for culturing human ePS cells on irradiated placental fibroblasts contains supplemental FGF2, suggesting that this factor may also be important for SON expression and plasticity in ePS cells. To test this hypothesis, ePS cells cultured under F-FM conditions were treated with increasing concentrations of the FGFR-specific inhibitor PD173074 and probed for SON protein expression. This treatment significantly reduced SON expression (Fig. 5A), suggesting a role for FGF2 signaling in ePS cell plasticity. Additionally, in F-FM conditions, treatment of ePS cells with Follistatin, a specific inhibitor for ACTIVIN A, reduced SON mRNA (Fig. S7A). Conversely, treatment of ePS cells with 5 ng of ACTIVIN A enhanced SON mRNA (Figs. 5B and S7B) and protein (Fig. 5C). In contrast, treatment of ePS cells either with TGF-β or with a neutralizing antibody against TGF-β did not affect SON expression (Fig. S7A). Our results are consistent with previous reports that ACTIVIN A and TGF-β fulfill different functions despite their structural similarities and ability to activate SMAD signaling [48, 49]. These data demonstrate that FGF2 and ACTIVIN A, identified as important for maintaining expression of SON in hESCs, also contributed to expression of SON in ePS cells.
Fig. 5. FGF2 and ACTIVIN A pathways are essential for SON expression in ePS cells.
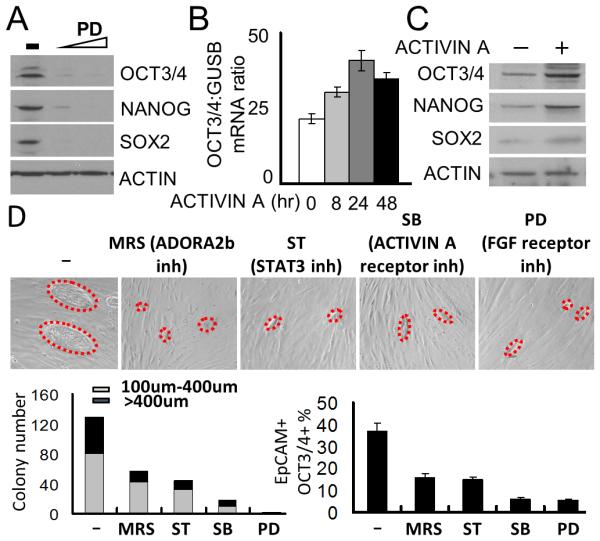
ePS cells from RM165 or RM209, grown in F-FM, were assessed for A. SON protein expression by WB in ePS cells treated with PD173074 (FGFR inh, 50nM and 500nM) for 48h. (n=1 and n=2, respectively). Loading control: ACTIN. Data from RM209 are shown; B.OCT3/4 transcript expression by qRT-PCR (B) or protein expression by WB (C) in presence or absence of 5 ng ACTIVIN A for the indicated times (n=2 and n=1, respectively). Data from RM165 are shown. ePS colonies from RM172 and RM183, grown on feeders and treated or not with the indicated inhibitors for 7 days starting on day 3 after plating, were assessed for: D. size and number of ePS colonies (outlined with red dashed lines in corresponding bright field images - bottom left). Scale bars: 10μm. Data from RM172 shown. EpCAM+OCT3/4+ cell fractions by FACS analysis (bottom right) (n=2 and n=2, respectively).
3.8. The signaling circuitry identified in ePS cells in feeder-free conditions is recapitulated in feeder conditions
The detailed characterization of signaling pathways regulating SON expression in ePS cells described above often necessitated their growth under feeder-free conditions (F-FM). We validated these findings using the feeder culture system (Fig. S2A), an important tool to access ePS cell plasticity. The CD73+/CD90-subpopulation was freshly isolated from RM tissues and seeded on irradiated placental fibroblast feeders. The ePS cells were allowed to attach and grow over 3 days before being exposed to selected pathway inhibitors for 7 days. Inhibitors targeting specifically ADORA2b, STAT3, ACTIVIN A or the FGF2 receptor all dramatically reduced formation of ePS colonies and the percentage of cells (EpCAM+) expressing OCT3/4 compared to vehicle-treated control cells (Fig. 5D). Thus, the molecular circuitry identified in ePS cells grown in F-FM is also operable in ePS cells growing as colonies on feeders.
3.9. CD73 and ADORA2b are required for SON expression in ePS cells, but not hESCs
In this study, we demonstrated that CD73 and ADORA2b signaling are required for the expression of SON and cellular plasticity in ePS cells. To date, neither CD73 nor ADORA2b have been reported to play a role in the maintenance of SON expression in hESCs. Flow cytometric analysis of CD73 and OCT3/4 revealed that <1% of hESCs express CD73 protein, while the vast majority of hESCs (>97%) expressed the core pluripotency factor OCT3/4 (Fig. 2B and 6A). Additionally, qPCR analysis revealed that hESCs expressed low levels of ADORA family adenosine receptors. Notably, ADORA2b expression in hESCs was 15-fold lower than in ePS cells (Fig. 6B). Importantly, whereas SON expression was downregulated at the protein level in ePS cells treated with the CD73 inhibitor APCP for 48 hours in F-FM conditions, APCP had no effect on SON expression in hESCs, even after 7 days of treatment (Fig. 6C). Additionally, when hESCs were cultured on feeders and exposed to inhibitors targeting CD73, ADORA1, ADORA2 or AdDORA3 activity for 7 days, immunocytochemical analysis revealed no significant change in OCT3/4 or NANOG expression in all treated groups (Fig. S8A), further demonstrating that CD73 and adenosine signaling were dispensable for maintenance of SON expression in hESCs. Similarly, exposure of hESC to either H89 or Stattic (inhibitors of the PKA and STAT3 pathways, respectively) did not significantly alter OCT3/4 or NANOG expression when measured by immunocytochemistry (Fig. S8B) or the percentage of cells (EpCAM+) expressing OCT3/4 when measured by flow cytometry (Fig. S8C). This demonstrates that the PKA/STAT3-dependent signaling pathway necessary for SON expression in ePS cells is not required for SON expression in hESCs. Exposure of hESCs cultured on feeders to either PD173014 or Follistatin (inhibitors of the FGF receptor and ACTIVIN A, respectively) for 7 days resulted in a significant reduction in OCT3/4 or NANOG expression when measured by immunocytochemistry (Fig. S8B) and in the percentage of cells (EpCAM+) expressing OCT3/4 when measured by flow cytometry (Fig. S8C). Similarly we found that both PD173014 (Fig. 5A) and Follistatin (Fig. S7A) inhibited SON expression in ePS cells, demonstrating that the ACTIVIN A and FGF2 pathways are required for SON expression in both ePS cells and hESC. In summary, our study reveals that ePS cells utilize a unique combination of signaling pathways to access SON expression when compared to hESCs.
4. Discussion
In the present study, we identified four extracellular factors (adenosine, IL-6, FGF2 and ACTIVIN A) that are required for SON expression and the extensive lineage plasticity characteristic of ePS cells. The activated ePS cell state we have described shares many characteristics with pluripotent hESCs. Both ePS cells and hESCs express high levels of canonical pluripotency markers [SON, Lin28 [12], TRA-1-60 and TRA-1-81 (Fig. S2A)], respond to similar cues for differentiation into functional ectodermal, mesodermal and endodermal derivatives, and require stable expression of SON for plasticity (Fig. S3 and [34-36]). However, for ePS colonies to access the SON machinery, they require the expression and activity of the cell surface protein CD73. CD73 is required for the production of extracellular adenosine, which binds to the adenosine receptor ADORA2b in ePS cells and activates STAT3 and IL6. The importance of STAT3 signaling and its complex relationships with IL6 and LIF signaling in control of SON expression have been previously documented in various human and mouse stem cell models [50, 51]. However, the present study identifies for the first time a key functional link between upstream CD73/adenosine/ADORA2b signaling and this downstream signaling cascade independent of LIF in ePS cells. hESCs do not express CD73 and, unlike FGF2 and ACTIVIN A, adenosine signaling is not required for the maintenance of SON expression in hESCs. The data presented here indicate that CD73, working through adenosine, is necessary, but not sufficient, for SON expression and ePS plasticity.
The canonical mechanisms controlling SON expression in embryonic stem cells (ESCs) have been extensively investigated [33, 52]. Although some aspects of this control are not fully elucidated, it is clear that environmental signals play a key role in ESC biology through action of specific growth factors [53, 54]. ESCs can be cultured either on feeder layers or in feeder-free systems. In their most potent state, the ground state, mouse ESCs require Leukemia Inhibitory Factor (LIF)/STAT3 and Bone Morphogenic Protein (BMP4) signals either secreted by feeder cells or present in the culture medium [43, 55]. As mouse ESCs acquire distinct epigenetic marks of a primed and more restricted state, equivalent to the state exhibited by human ESCs, now ACTIVIN A and FGF2 signals are necessary to maintain a primed state of pluripotency [46, 47]. These environmental signals have been shown to directly control the expression of SON and maintain the proper expression of genes required for self-renewal and repression of differentiation of these cells [31-33] or for cell reprogramming [56-58].
The requirement for CD73 signaling, and the fact that CD73 expression is controlled by stress signals, suggests that, in rare cells, activation of SON and plasticity may be linked to physiological conditions of cellular damage [19-21]. Recent studies in several tissues indicate that two distinct stem cell populations exist: one population is responsible for maintaining tissue homeostasis while the other is responsible for wound healing [59-61]. For instance, the intestine differentially uses an actively cycling stem cell population for tissue homeostasis and a distinct quiescent, damaged-induced stem cell population for tissue damage repair [61], each generating the same range of tissue-specific differentiated cells. Additionally, cells with stem-like properties and multi-lineage potential have been reported to participate in stress responses and have been described within human breast tissue and milk [62, 63], skin [64] and at multiple other tissue sites. These multiple cell types are easily distinguished from ePS cells by several criteria. MUSE (Multilineage-differentiating stress-enduring) cells, MSC (mesenchymal stem cells), VSEL (Very Small Embryonic-Like) cells, MIAMI (Marrow-Isolated Adult Multi-lineage Inducible), ADSC (adipose-derived stem cells) and MAPC (Multipotent Adult Progenitor Cells) all express CD90, whereas ePS cells do not [3-10]. Additionally, ePS cells express EpCAM and can generate teratomas, whereas MUSE and MSCs fail to do so [12]. Lastly, knockdown of OCT3/4 in MSCs does not affect differentiation into adipocytes [8], while our data demonstrate that knockdown of OCT3/4 ablates adipocyte differentiation in ePS cells (Fig. S3). Thus, ePS cells seem to represent a novel cell state that has not been previously described. Interestingly, recent reports demonstrate the expression of CD73 as a landmark in the reprogramming of iPS cells [25], thus extending the relevance of our findings in ePS cells.
This study exhibits numerous strengths: use of multiple human tissue samples (Table S1), multiple endpoints demonstrating ePS cellular plasticity, complementary analyses under both feeder and F-FM conditions and genetic and pharmacological manipulation of signaling pathways of interest. The complementary analyses under both feeder and F-FM conditions are important since growing any cell population under different culture conditions may alter gene expression, mechano-regulatory conditions or functional phenotypes. The analysis of 10 distinct human tissue samples for analysis of SON expression assures us that we are studying a general property of these cells and not a rare mutant phenotype. Most importantly for these studies, the role of stress signaling pathways in creating a niche for the activation of cellular plasticity provides predictions of where and when these cells may be found in vivo. Additionally, the in depth biochemical characterization of SON activation and complex regulation through CD73 circuitry provides insights that will allow control of these cells. Finally, comparison with hESCs demonstrates that not all cells control access to plasticity in the same manner. We will take advantage of our identification of key signaling pathways for acquisition of cellular plasticity in ePS cells in vitro to detect these cells in vivo and to inquire if they contribute to function or dysfunction in vivo.
We hypothesize that activation of plasticity in ePS cells in damaged tissues might contribute to widespread tissue repair and regeneration of functional tissues. Physiological observations may suggest why access to these plasticity circuits exists in rare cells and why they would be conserved throughout evolution. An extensive literature describes the widespread plasticity of cellular populations exposed to adverse growth conditions or injury [65-67]. The ability of pre-existing, undifferentiated latent cells to activate non-mutagenic phenotypic plasticity and explore phenotypic space could be advantageous to accomplish wound healing, differentiation, specialization or adaptation to conditions of stress. Future experiments with genetically engineered mice and stressed human tissues will be used to test these hypotheses.
5. Conclusions
The CD73+CD90-population that enriches for ePS cells can be visualized in healthy human breast tissues by multiplex IHC.
ePS cells plasticity is controlled by SON transcriptional network.
Extracellular adenosine generated by CD73 can regulate SON expression and cellular plasticity of ePS cells by activating the PKA-dependent autocrine IL6/Stat3 pathway through the ADORA 2b receptor.
The canonical Activin A and FGF2 pathways that are important for SON expression in human ESCs are also essential for SON expression in ePS cells.
CD73/Adenosine signaling is a novel pathway that is essential for SON expression in human ePS cells but not in human ESCs.
Supplementary Material
Movie S4. Cardiomyocyte differentiation of ePS cells treated with MRS1754. Absence of spontaneous beating of cardiomyocytes after differentiation of MRS1754-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Fig. S8. hESCs utilize ACTIVIN A and FGF2/Mek pathways but not adenosine-dependent PKA and IL6 signaling to control SON expression. hESCs (H7) were assessed for: A. OCT3/4 and NANOG protein expression by immunofluorescence after treated or not with 6 μm of APCP or 10 μm the adenosine receptor inhibitors 8-PT, DMPX and VUF for 7 days. Nuclei were stained with DAPI (n=2 and n=2, respectively). Scale bars: 10μm. B. OCT3/4 and NANOG protein expression by immunofluorescence after treated or not with the indicated pathway inhibitors for 7 days. Nuclei were stained with DAPI. Scale bars: 10μm; C. Epcam+OCT3/4+ cell fractions by FACS–based quantitative analysis.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table S1. Age and ethnicity of tissue samples with additional information documenting the # of times specific assays were completed and with which tissue samples.
Movie S1. Cardiomyocyte differentiation of ePS cells. Spontaneous beating of cardiomyocytes after differentiation of ePS cells from RM122 or RM128 into the myocardial lineage (n=2).
Movie S2. Cardiomyocyte differentiation of ePS cells treated with APCP. Absence of spontaneous beating of cardiomyocytes after differentiation of APCP-treated ePS cells from RM122 or RM128 into the myocardial lineage (n=2).
Movie S5. Cardiomyocyte differentiation of ePS cells treated with VUF5574. Spontaneous beating of cardiomyocytes after differentiation of VUF5574-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Movie S3. Cardiomyocyte differentiation of ePS cells treated with 8-PT. Spontaneous beating of cardiomyocytes after differentiation of 8-PT-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Fig. S1. Multiplex analysis of reduction mammoplasty sections stained simultaneously for either CD73 and CD90 or CD73 and EpCAM: Unmixing of multiplex-stained regions. Disease-free reduction mammoplasty tissue sections (RM085: panels A and B; RM179: panels C and D) stained simultaneously with an anti-CD73 antibody and an anti-CD90 antibody (panels A and C) or with an anti-CD73 antibody and an anti-EpCAM antibody (panels B and D) were imaged with a multispectral Nuance FX camera and unmixed with the Nuance software. Black and white images corresponding to unmixed images (single staining patterns) for each marker and composite images with individual marker stainings visualized with pseudo-colors (CD73: red; CD90 and EpCAM: blue; Methyl Green counterstain: green; Nuclear Fast Red counterstain: pink) are shown. Scale bars: 20μm. CD73+CD90-population isolated from RM085 displays a normal diploid 46, XX shown in panel A.
Fig. S2 ePS cells activate SON while grown on feeders or in feeder-free media. A. Schematic representation of ePS cell isolation and treatment schedules. Single cell suspensions were isolated from a representative sample of human breast tissue and subjected to FACS sorting according to their CD73 (y-axis) and CD90 (x-axis) expression levels (left panel) generating CD73+CD90− (R1 cells)(5.2%), CD73+CD90+ (R2 cells)(2.1%), CD73−CD90− (R3 cells)(85.4%) and CD73−CD90+ (R4 cells)(7.4%) fractions (Fig. 1A). The CD73+CD90− (R1) cell population was immediately cultured either on irradiated placental fibroblast feeders or in feeder-free expansion conditions. ePS cell colonies started to appear around 9 days when grown on feeders. The typical morphology of ePS cell colonies at 2 weeks is shown in two bright field images along with corresponding staining for the pluripotency markers Tra-1-60 and Tra-1-81 (left and right top panels, respectively). Analyses were conducted in ePS cells from RM172 (n=1) or RM183 (n=1). Scale bars: 10μm. Inhibitors were applied 3 days following FACS isolation to study cell plasticity (red arrows). In feeder-free expansion medium (F-FM), ePS cells were expanded for 21 days before being passaged. These cells can usually be passaged every 3 days (as indicated by the vertical marks) at a 1:4 split for a total of 6 times before losing cell plasticity. Inhibitors or shRNAs were introduced into ePS cells grown in F-FM at passage 2 (red arrow). The typical morphology of ePS cells from RM183 grown for 3 weeks in F-FM is shown in a bright field image (bottom right panel) and is representative of all RMs. Scale bars: 10μm. B. SON transcript and protein expression levels were assessed by immunofluorescence (B), qRT-PCR (C) and WB (D) in hESC and ePS cells from RM172 (n=3) or RM183 (n=3). Scale bars: 10μm.
Fig. S3. SON signaling is necessary for the plastic state of ePS cells. ePS cells from RM159 or RM177 grown in F-FM for over 21 days were transduced with a shRNA against either OCT3/4 (shOCT3/4), NANOG (shNANOG) or SOX2 (shSOX2) or a scrambled control shRNA (Scrambled), selected with puromycin then analyzed for A. SON transcript expression levels by qRT-PCR (n=3 for each RM); B. SON protein expression levels by WB (n=2). Data from RM159 are shown. Loading control: ACTIN.; C. adipogenic differentiation for 14 days and stained with the lipophilic dye Oil Red O (top panels) or LipidTox (bottom panels) for functional assessment of plasticity (n=2). Data from RM159 are shown. Scale bars: 10μm.; D. expression of three markers of adipogenic differentiation, FABP4, Leptin and PPARγ, by qRT-PCR (n=3 for each RM) normalized to expression of the housekeeping gene glucuronidase B (GUSB)
Fig. S4. CD73 dependency of ePS cell plasticity. A. ePS cells from RM128 treated with or without APCP for 1w were assessed for SON protein expression by immunocytochemistry along with hESC colonies (outlined with red dashed lines in corresponding bright field images) Scale bars: 10μm. (n=2); Nuclei were stained with DAPI. Nuclear localization of SON in the areas outlined with white squares is shown at a higher magnification. Scale bars: 40μm. Data from RM128 are shown. B. SON protein expression was analyzed by WB in ePS cells from RM122 or RM128 treated with or without APCP for 48h (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM122 are shown. ePS cells from RM122 or RM128 transduced with a doxycycline-inducible pTripZ shCD73 (vector) or a matched scrambled pTripZ shRNA (scrambled) and selected with puromycin prior to (−Dox) and after (+Dox) doxycycline induction for 48h were accessed for C. CD73 transcript by qRT-PCR. CD73 transcript expression was normalized to GUSB expression. (n=3 and n=3, respectively). D. SON and CD73 protein expression by WB. Loading control: ACTIN. (n=2). E. growth by plotting growth curves as a function of cell numbers over time for ePS cells grown with (dashed line) or without (solid line) knock-down of CD73 in F-FM (n=3 and n=3, respectively). F. ePS cells from RM122 or RM128 transduced with a matched scrambled pTripZ shRNA (scrambled) were subjected to adipocyte differentiation for 18 days and stained with Oil Red O (n=2). Data from RM122 are shown. Scale bars: 10μm. G. Expression of adipogenic differentiation markers FABP4, Leptin and PPARγ in ePS cells from RM122 or RM128 treated or not with APCP for 48 h and subjected to adipocyte differentiation for 18 days. FABP4, Leptin and PPARγ transcript expression was normalized to GUSB expression. (n=3 and n=3, respectively). H. apoptotic cell death in ePS colonies grown on feeders, treated with or without adenosine (Ado) or APCP for 48h and monitored for cell surface expression of EpCAM/Annexin V (n=3 and n=3, respectively). I. ePS cells from RM122 or RM128 with (+Dox) or without (−Dox) CD73 knock-down and exposed to 200μM adenosine (Ado) for 48 h were assessed for OCT3/4 transcript expression by qRT-PCR. OCT3/4 transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). J. ePS cells from RM122 or RM128 treated with or without APCP and exposed to 200μM adenosine (Ado) for 48 h were subjected to adipocyte differentiation for 18 days and stained with Lipidtox (n=2) or osteogenic differentiation for 21 days and stained with Alizarin Red (n=2). Data from RM122 are shown. Scale bars: 40μm. K. SON expression compared to CD73− cells isolated from the same RM by flow cytometry and expanded in MEGM then treated with 200μM or 500μM of Adenosine (Ado) in either MEGM or ePS cell F-FM for 48h (n=3 and n=3, respectively). L. The T47D breast cancer cell line with or without ectopic expression of flagged CD73 was assessed for OCT3/4 protein expression by WB. Loading control: ACTIN. (n=2)
Fig. S5. Involvement of Adenosine Receptor signaling in regulating ePS plasticity. A. ePS cells from RM136 or RM142, grown in F-FM were treated or not with 10 μm of 8-PT, MRS, DMPX and VUF for 48 h. NANOG and SOX2 transcript expression was accessed by qRT-PCR. Transcript expression was normalized to GUSB expression (n=3 and n=6, respectively); ePS cells from RM136 or RM142 transduced with a doxycycline-inducible shRNA against ADORA2b were assessed for: B. ADORA2b transcript expression and C. SON transcript expression by qRT-PCR prior to (−Dox) and after (+Dox) doxycycline treatment. ADORA2b and SON transcript expression was normalized to GUSB transcript expression (n=3 and n=3, respectively, for A and B).
Fig. S6. Involvement of the PKA/STAT3 pathway in expression of master regulators of cell plasticity. ePS cells from RM136 or RM159, grown in F-FM, were assessed for A. NANOG and SOX2 transcript expression by qRT-PCR in ePS cells treated or not with adenosine (Ado) in presence or absence of the PKA inhibitor H89 or the STAT3 inhibitor Stattic. Transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). B. STAT3, SON transcript expression by qRT-PCR after transduction of lentivirus carrying inducible shRNAs against STAT3 (#16 or #17) or a matched scrambled shRNA and induction by Doxycyclin for 48h. Transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). C. NANOG and SOX2 transcript expression by qRT-PCR in untreated or MRS-treated ePS cells from RM136 or RM159 in presence or absence of the PKA activator 8-Br-cAMP. NANOG and SOX2 transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). D. phosphorylation status of Jak (p-Jak) and Jnk (p-Jnk) kinase by WB in ePS cells treated with or without H89, a PKA inhibitor, for 48 hours in the presence or absence of NECA, a metabolically stable adenosine analogue. (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM136 are shown. E. SON and STAT3 protein expression and phosphorylation status of STAT3 (p-STAT3) by WB in ePS cells treated with or without Ruxolitinib, a Jak kinase inhibitor, in the presence or absence of NECA or Forskolin, a PKA activator. (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM136 are shown. F. IL6 transcript expression in hESCs, primary human luminal, myoepithelial or CD73+ mammary epithelial cells inferred from the Alexa-Seq database [42]. G. IL6 transcript expression by qRT-PCR after transduction of lentivirus carrying inducible shRNAs against STAT3 (#16 or #17) or a matched scrambled shRNA and induction by Doxycyclin for 48h. IL6 expression was normalized to GUSB expression (n=3 and n=3, respectively). H. ePS cells from RM136 or RM159, cultured on feeders for 3 days before incubation with or without 0.6ng/ml of IL6 for 10 days, were assessed for number of ePS colonies based on colony size (n=3 and n=3, respectively). I. Corresponding EpCAM+OCT3/4+ cell fractions by FACS analysis (n=3 and n=3, respectively). J. SON protein expression was assessed by WB in ePS cells from RM136 or RM159 treated with or without APCP in the presence or absence of different concentrations of IL6. Loading control: ACTIN. (n=1 and n=1, respectively)
Fig. S7. ACTIVIN A and TGFβ pathways function differently in ePS cells. A. ePS cells from RM136 or RM 159, grown in F-FM in the presence or absence of TGF-β (1ng/ml and 5 ng/ml), neutralizing antibody against TGF-β (anti-TGF-β (25ng/ml) or the ACTIVIN inhibitor, Follistatin (FST, 100ng/ml) for the indicated times, were assessed for SON transcript expression by qRT-PCR (n=3 and n=3, respectively). SON transcript expression was normalized to GUSB transcript expression. B. ePS cells from RM165 and RM209, grown in F-FM, were assessed for NANOG and SOX2 transcript expression by qRT-PCR in presence or absence of 5 ng ACTIVIN A for 24h (n=2 and n=1, respectively). Transcript expression was normalized to GUSB transcript expression.
Highlights.
Multiplex imaging identifies CD73+/CD90− (R1) cells that enrich for ePS cells in human breast tissue
The CD73/Adenosine/AdoRa2b/Stat3 circuitry is required for SON expression in ePS cells but not in hESCs
Activin A and FGF2 pathways are also essential for SON expression in ePS cells
ePS cells enter a plastic state through a novel circuitry not utilized by hESCs
Acknowledgments
We thank Dr. S. Fisher (USCF) for providing human placenta fibroblasts, Drs. N. Dumont (Merrimack Pharmaceuticals Inc. MA), S. Oakes and W. Finkbeiner (UCSF) and Tlsty laboratory members for helpful discussions and Drs. J. Tjoe (Comprehensive Breast Health Center Aurora Sinai Medical Center, WI); D. Baer, K. M. Yokoo, B. Hornik, and J. H. Kim (Kaiser Foundation Research Institute, CA) and Ms. K. Wiles (Cooperative Human Tissue Network, TN) for providing breast tissues.
Funding
This work was supported by National Cancer Institute Grants CA097214, CA143803, ES017154, and CA135626, Avon Foundation Research Grant 07-2007-074, Cancer League, Inc., and California Institute for Regenerative Medicine, Grant RS1-00444-1 (to T.D.T.). The authors have no conflict of interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None
Classification: Biological Science
References
- [1].Hanna JH, Saha K, Jaenisch R. Cell. 2010;143(4):508–525. doi: 10.1016/j.cell.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Takahashi K, Yamanaka S. Cell. 2006;126(4):663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- [3].Beltrami AP, Cesselli D, Bergamin N, Marcon P, Rigo S, Puppato E, D'Aurizio F, Verardo R, Piazza S, Pignatelli A, Poz A, Baccarani U, Damiani D, Fanin R, Mariuzzi L, Finato N, Masolini P, Burelli S, Belluzzi O, Schneider C, Beltrami CA. Blood. 2007;110(9):3438–3446. doi: 10.1182/blood-2006-11-055566. [DOI] [PubMed] [Google Scholar]
- [4].D'Ippolito G, Diabira S, Howard GA, Menei P, Roos BA, Schiller PC. J Cell Sci. 2004;117:2971–2981. doi: 10.1242/jcs.01103. Pt 14. [DOI] [PubMed] [Google Scholar]
- [5].Jiang Y, Vaessen B, Lenvik T, Blackstad M, Reyes M, Verfaillie CM. Exp Hematol. 2002;30(8):896–904. doi: 10.1016/s0301-472x(02)00869-x. [DOI] [PubMed] [Google Scholar]
- [6].Ratajczak MZ, Liu R, Ratajczak J, Kucia M, Shin DM. Differentiation. 2011;81(3):153–161. doi: 10.1016/j.diff.2011.01.006. [DOI] [PubMed] [Google Scholar]
- [7].Kuroda Y, Kitada M, Wakao S, Nishikawa K, Tanimura Y, Makinoshima H, Goda M, Akashi H, Inutsuka A, Niwa A, Shigemoto T, Nabeshima Y, Nakahata T, Nabeshima Y, Fujiyoshi Y, Dezawa M. Proc Natl Acad Sci U S A. 2010;107(19):8639–8643. doi: 10.1073/pnas.0911647107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lengner CJ, Camargo FD, Hochedlinger K, Welstead GG, Zaidi S, Gokhale S, Scholer HR, Tomilin A, Jaenisch R. Cell Stem Cell. 2007;1(4):403–415. doi: 10.1016/j.stem.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Aranguren XL, Luttun A, Clavel C, Moreno C, Abizanda G, Barajas MA, Pelacho B, Uriz M, Arana M, Echavarri A, Soriano M, Andreu EJ, Merino J, Garcia-Verdugo JM, Verfaillie CM, Prosper F. Blood. 2007;109(6):2634–2642. doi: 10.1182/blood-2006-06-030411. [DOI] [PubMed] [Google Scholar]
- [10].Watson JE, Patel NA, Carter G, Moor A, Patel R, Ghansah T, Mathur A, Murr MM, Bickford P, Gould LJ, Cooper DR. Adv Wound Care (New Rochelle) 2014;3(3):219–228. doi: 10.1089/wound.2013.0452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wakao S, Kitada M, Kuroda Y, Shigemoto T, Matsuse D, Akashi H, Tanimura Y, Tsuchiyama K, Kikuchi T, Goda M, Nakahata T, Fujiyoshi Y, Dezawa M. Proc Natl Acad Sci U S A. 2011;108(24):9875–9880. doi: 10.1073/pnas.1100816108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Roy S, Gascard P, Dumont N, Zhao J, Pan D, Petrie S, Margeta M, Tlsty TD. Proc Natl Acad Sci U S A. 2013;110(12):4598–4603. doi: 10.1073/pnas.1218682110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Reynolds PA, Sigaroudinia M, Zardo G, Wilson MB, Benton GM, Miller CJ, Hong C, Fridlyand J, Costello JF, Tlsty TD. J Biol Chem. 2006;281(34):24790–24802. doi: 10.1074/jbc.M604175200. [DOI] [PubMed] [Google Scholar]
- [14].Pajcini KV, Corbel SY, Sage J, Pomerantz JH, Blau HM. Cell Stem Cell. 2010;7(2):198–213. doi: 10.1016/j.stem.2010.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O'Brien M, Ng YH, Ang CE, Vaka D, Artandi SE, Dick FA, Brunet A, Sage J, Wernig M. Cell Stem Cell. 2014;16(1):39–50. doi: 10.1016/j.stem.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wagers AJ, Weissman IL. Cell. 2004;116(5):639–648. doi: 10.1016/s0092-8674(04)00208-9. [DOI] [PubMed] [Google Scholar]
- [17].Colgan SP, Eltzschig HK, Eckle T, Thompson LF. Purinergic Signal. 2006;2(2):351–360. doi: 10.1007/s11302-005-5302-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zimmermann H. Biochem J. 1992;285:345–365. doi: 10.1042/bj2850345. Pt 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Niemela J, Henttinen T, Yegutkin GG, Airas L, Kujari AM, Rajala P, Jalkanen S. J Immunol. 2004;172(3):1646–1653. doi: 10.4049/jimmunol.172.3.1646. [DOI] [PubMed] [Google Scholar]
- [20].Kim M, Ham A, Kim JY, Brown KM, D'Agati VD, Lee HT. Kidney Int. 2013;84(1):90–103. doi: 10.1038/ki.2013.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. J Clin Invest. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fredholm BB. Cell Death Differ. 2007;14(7):1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- [23].Ode A, Schoon J, Kurtz A, Gaetjen M, Ode JE, Geissler S, Duda GN. Eur Cell Mater. 2013;25:37–47. doi: 10.22203/ecm.v025a03. [DOI] [PubMed] [Google Scholar]
- [24].Takedachi M, Oohara H, Smith BJ, Iyama M, Kobashi M, Maeda K, Long CL, Humphrey MB, Stoecker BJ, Toyosawa S, Thompson LF, Murakami S. J Cell Physiol. 2012;227(6):2622–2631. doi: 10.1002/jcp.23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zunder ER, Lujan E, Goltsev Y, Wernig M, Nolan GP. Cell Stem Cell. 2015;16(3):323–337. doi: 10.1016/j.stem.2015.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fordyce CA, Patten KT, Fessenden TB, Defilippis R, Hwang ES, Zhao J, Tlsty TD. Breast Cancer Res. 2012;14(6):R155. doi: 10.1186/bcr3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].De Francesco F, Tirino V, Desiderio V, Ferraro G, D'Andrea F, Giuliano M, Libondi G, Pirozzi G, De Rosa A, Papaccio G. PLoS One. 2009;4(8):e6537. doi: 10.1371/journal.pone.0006537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lim E, Wu D, Pal B, Bouras T, Asselin-Labat ML, Vaillant F, Yagita H, Lindeman GJ, Smyth GK, Visvader JE. Breast Cancer Res. 2010;12(2):R21. doi: 10.1186/bcr2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sakaki-Yumoto M, Liu J, Ramalho-Santos M, Yoshida N, Derynck R. J Biol Chem. 2013;288(25):18546–18560. doi: 10.1074/jbc.M112.446591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Nature. 2006;439(7079):993–997. doi: 10.1038/nature04496. [DOI] [PubMed] [Google Scholar]
- [31].Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA. Cell. 2005;122(6):947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Cole MF, Young RA. Cold Spring Harb Symp Quant Biol. 2008;73:183–193. doi: 10.1101/sqb.2008.73.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Niwa H. Development. 2007;134(4):635–646. doi: 10.1242/dev.02787. [DOI] [PubMed] [Google Scholar]
- [34].Ivanova N, Dobrin R, Lu R, Kotenko I, Levorse J, DeCoste C, Schafer X, Lun Y, Lemischka IR. Nature. 2006;442(7102):533–538. doi: 10.1038/nature04915. [DOI] [PubMed] [Google Scholar]
- [35].Mitsui K, Tokuzawa Y, Itoh H, Segawa K, Murakami M, Takahashi K, Maruyama M, Maeda M, Yamanaka S. Cell. 2003;113(5):631–642. doi: 10.1016/s0092-8674(03)00393-3. [DOI] [PubMed] [Google Scholar]
- [36].Niwa H, Miyazaki J, Smith AG. Nat Genet. 2000;24(4):372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- [37].Eltzschig HK. Anesthesiology. 2009;111(4):904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Callihan P, Mumaw J, Machacek DW, Stice SL, Hooks SB. Pharmacol Ther. 2011;129(3):290–306. doi: 10.1016/j.pharmthera.2010.10.007. [DOI] [PubMed] [Google Scholar]
- [39].Liu AM, Lo RK, Wong CS, Morris C, Wise H, Wong YH. J Biol Chem. 2006;281(47):35812–35825. doi: 10.1074/jbc.M605288200. [DOI] [PubMed] [Google Scholar]
- [40].Stagg J, Smyth MJ. Oncogene. 2010;29(39):5346–5358. doi: 10.1038/onc.2010.292. [DOI] [PubMed] [Google Scholar]
- [41].Akira S, Nishio Y, Inoue M, Wang XJ, Wei S, Matsusaka T, Yoshida K, Sudo T, Naruto M, Kishimoto T. Cell. 1994;77(1):63–71. doi: 10.1016/0092-8674(94)90235-6. [DOI] [PubMed] [Google Scholar]
- [42].Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. Nat Biotechnol. 2010;28(10):1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Williams RL, Hilton DJ, Pease S, Willson TA, Stewart CL, Gearing DP, Wagner EF, Metcalf D, Nicola NA, Gough NM. Nature. 1988;336(6200):684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- [44].Hirai H, Karian P, Kikyo N. Biochem J. 2011;438(1):11–23. doi: 10.1042/BJ20102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li J, Wang G, Wang C, Zhao Y, Zhang H, Tan Z, Song Z, Ding M, Deng H. Differentiation. 2007;75(4):299–307. doi: 10.1111/j.1432-0436.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- [46].Vallier L, Alexander M, Pedersen RA. J Cell Sci. 2005;118:4495–4509. doi: 10.1242/jcs.02553. Pt 19. [DOI] [PubMed] [Google Scholar]
- [47].James D, Levine AJ, Besser D, Hemmati-Brivanlou A. Development. 2005;132(6):1273–1282. doi: 10.1242/dev.01706. [DOI] [PubMed] [Google Scholar]
- [48].Hinck AP. FEBS Lett. 2012;586(14):1860–1870. doi: 10.1016/j.febslet.2012.05.028. [DOI] [PubMed] [Google Scholar]
- [49].Massague J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- [50].Foshay KM, Gallicano GI. Stem Cells Dev. 2008;17(2):269–278. doi: 10.1089/scd.2007.0098. [DOI] [PubMed] [Google Scholar]
- [51].Do DV, Ueda J, Messerschmidt DM, Lorthongpanich C, Zhou Y, Feng B, Guo G, Lin PJ, Hossain MZ, Zhang W, Moh A, Wu Q, Robson P, Ng HH, Poellinger L, Knowles BB, Solter D, Fu XY. Genes Dev. 2013;27(12):1378–1390. doi: 10.1101/gad.221176.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Young RA. Cell. 2011;144(6):940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Dalton S. Curr Opin Cell Biol. 2013;25(2):241–246. doi: 10.1016/j.ceb.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Pera MF, Tam PP. Nature. 2010;465(7299):713–720. doi: 10.1038/nature09228. [DOI] [PubMed] [Google Scholar]
- [55].Ying QL, Nichols J, Chambers I, Smith A. Cell. 2003;115(3):281–292. doi: 10.1016/s0092-8674(03)00847-x. [DOI] [PubMed] [Google Scholar]
- [56].Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- [57].Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- [58].Wernig M, Meissner A, Foreman R, Brambrink T, Ku M, Hochedlinger K, Bernstein BE, Jaenisch R. Nature. 2007;448(7151):318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- [59].Ito M, Liu Y, Yang Z, Nguyen J, Liang F, Morris RJ, Cotsarelis G. Nat Med. 2005;11(12):1351–1354. doi: 10.1038/nm1328. [DOI] [PubMed] [Google Scholar]
- [60].Mascre G, Dekoninck S, Drogat B, Youssef KK, Brohee S, Sotiropoulou PA, Simons BD, Blanpain C. Nature. 2012;489(7415):257–262. doi: 10.1038/nature11393. [DOI] [PubMed] [Google Scholar]
- [61].Yan KS, Chia LA, Li X, Ootani A, Su J, Lee JY, Su N, Luo Y, Heilshorn SC, Amieva MR, Sangiorgi E, Capecchi MR, Kuo CJ. Proc Natl Acad Sci U S A. 2012;109(2):466–471. doi: 10.1073/pnas.1118857109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chang CC, Sun W, Cruz A, Saitoh M, Tai MH, Trosko JE. Radiat Res. 2001;155:201–207. doi: 10.1667/0033-7587(2001)155[0201:ahbect]2.0.co;2. 1 Pt 2. [DOI] [PubMed] [Google Scholar]
- [63].Hassiotou F, Hepworth AR, Williams TM, Twigger AJ, Perrella S, Lai CT, Filgueira L, Geddes DT, Hartmann PE. PLoS One. 2012;8(11):e78232. doi: 10.1371/journal.pone.0078232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Vega Crespo A, Awe JP, Reijo Pera R, Byrne JA. Biores Open Access. 2012;1(1):25–33. doi: 10.1089/biores.2012.0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Stewart PS, Franklin MJ. Nat Rev Microbiol. 2008;6(3):199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- [66].Reddien PW. Development. 2013;140(5):951–957. doi: 10.1242/dev.080499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Rinkevich Y, Lindau P, Ueno H, Longaker MT, Weissman IL. Nature. 2011;476(7361):409–413. doi: 10.1038/nature10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S4. Cardiomyocyte differentiation of ePS cells treated with MRS1754. Absence of spontaneous beating of cardiomyocytes after differentiation of MRS1754-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Fig. S8. hESCs utilize ACTIVIN A and FGF2/Mek pathways but not adenosine-dependent PKA and IL6 signaling to control SON expression. hESCs (H7) were assessed for: A. OCT3/4 and NANOG protein expression by immunofluorescence after treated or not with 6 μm of APCP or 10 μm the adenosine receptor inhibitors 8-PT, DMPX and VUF for 7 days. Nuclei were stained with DAPI (n=2 and n=2, respectively). Scale bars: 10μm. B. OCT3/4 and NANOG protein expression by immunofluorescence after treated or not with the indicated pathway inhibitors for 7 days. Nuclei were stained with DAPI. Scale bars: 10μm; C. Epcam+OCT3/4+ cell fractions by FACS–based quantitative analysis.
Table S1. Age and ethnicity of tissue samples with additional information documenting the # of times specific assays were completed and with which tissue samples.
Movie S1. Cardiomyocyte differentiation of ePS cells. Spontaneous beating of cardiomyocytes after differentiation of ePS cells from RM122 or RM128 into the myocardial lineage (n=2).
Movie S2. Cardiomyocyte differentiation of ePS cells treated with APCP. Absence of spontaneous beating of cardiomyocytes after differentiation of APCP-treated ePS cells from RM122 or RM128 into the myocardial lineage (n=2).
Movie S5. Cardiomyocyte differentiation of ePS cells treated with VUF5574. Spontaneous beating of cardiomyocytes after differentiation of VUF5574-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Movie S3. Cardiomyocyte differentiation of ePS cells treated with 8-PT. Spontaneous beating of cardiomyocytes after differentiation of 8-PT-treated ePS cells from RM136 or RM142 into the myocardial lineage (n=2).
Fig. S1. Multiplex analysis of reduction mammoplasty sections stained simultaneously for either CD73 and CD90 or CD73 and EpCAM: Unmixing of multiplex-stained regions. Disease-free reduction mammoplasty tissue sections (RM085: panels A and B; RM179: panels C and D) stained simultaneously with an anti-CD73 antibody and an anti-CD90 antibody (panels A and C) or with an anti-CD73 antibody and an anti-EpCAM antibody (panels B and D) were imaged with a multispectral Nuance FX camera and unmixed with the Nuance software. Black and white images corresponding to unmixed images (single staining patterns) for each marker and composite images with individual marker stainings visualized with pseudo-colors (CD73: red; CD90 and EpCAM: blue; Methyl Green counterstain: green; Nuclear Fast Red counterstain: pink) are shown. Scale bars: 20μm. CD73+CD90-population isolated from RM085 displays a normal diploid 46, XX shown in panel A.
Fig. S2 ePS cells activate SON while grown on feeders or in feeder-free media. A. Schematic representation of ePS cell isolation and treatment schedules. Single cell suspensions were isolated from a representative sample of human breast tissue and subjected to FACS sorting according to their CD73 (y-axis) and CD90 (x-axis) expression levels (left panel) generating CD73+CD90− (R1 cells)(5.2%), CD73+CD90+ (R2 cells)(2.1%), CD73−CD90− (R3 cells)(85.4%) and CD73−CD90+ (R4 cells)(7.4%) fractions (Fig. 1A). The CD73+CD90− (R1) cell population was immediately cultured either on irradiated placental fibroblast feeders or in feeder-free expansion conditions. ePS cell colonies started to appear around 9 days when grown on feeders. The typical morphology of ePS cell colonies at 2 weeks is shown in two bright field images along with corresponding staining for the pluripotency markers Tra-1-60 and Tra-1-81 (left and right top panels, respectively). Analyses were conducted in ePS cells from RM172 (n=1) or RM183 (n=1). Scale bars: 10μm. Inhibitors were applied 3 days following FACS isolation to study cell plasticity (red arrows). In feeder-free expansion medium (F-FM), ePS cells were expanded for 21 days before being passaged. These cells can usually be passaged every 3 days (as indicated by the vertical marks) at a 1:4 split for a total of 6 times before losing cell plasticity. Inhibitors or shRNAs were introduced into ePS cells grown in F-FM at passage 2 (red arrow). The typical morphology of ePS cells from RM183 grown for 3 weeks in F-FM is shown in a bright field image (bottom right panel) and is representative of all RMs. Scale bars: 10μm. B. SON transcript and protein expression levels were assessed by immunofluorescence (B), qRT-PCR (C) and WB (D) in hESC and ePS cells from RM172 (n=3) or RM183 (n=3). Scale bars: 10μm.
Fig. S3. SON signaling is necessary for the plastic state of ePS cells. ePS cells from RM159 or RM177 grown in F-FM for over 21 days were transduced with a shRNA against either OCT3/4 (shOCT3/4), NANOG (shNANOG) or SOX2 (shSOX2) or a scrambled control shRNA (Scrambled), selected with puromycin then analyzed for A. SON transcript expression levels by qRT-PCR (n=3 for each RM); B. SON protein expression levels by WB (n=2). Data from RM159 are shown. Loading control: ACTIN.; C. adipogenic differentiation for 14 days and stained with the lipophilic dye Oil Red O (top panels) or LipidTox (bottom panels) for functional assessment of plasticity (n=2). Data from RM159 are shown. Scale bars: 10μm.; D. expression of three markers of adipogenic differentiation, FABP4, Leptin and PPARγ, by qRT-PCR (n=3 for each RM) normalized to expression of the housekeeping gene glucuronidase B (GUSB)
Fig. S4. CD73 dependency of ePS cell plasticity. A. ePS cells from RM128 treated with or without APCP for 1w were assessed for SON protein expression by immunocytochemistry along with hESC colonies (outlined with red dashed lines in corresponding bright field images) Scale bars: 10μm. (n=2); Nuclei were stained with DAPI. Nuclear localization of SON in the areas outlined with white squares is shown at a higher magnification. Scale bars: 40μm. Data from RM128 are shown. B. SON protein expression was analyzed by WB in ePS cells from RM122 or RM128 treated with or without APCP for 48h (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM122 are shown. ePS cells from RM122 or RM128 transduced with a doxycycline-inducible pTripZ shCD73 (vector) or a matched scrambled pTripZ shRNA (scrambled) and selected with puromycin prior to (−Dox) and after (+Dox) doxycycline induction for 48h were accessed for C. CD73 transcript by qRT-PCR. CD73 transcript expression was normalized to GUSB expression. (n=3 and n=3, respectively). D. SON and CD73 protein expression by WB. Loading control: ACTIN. (n=2). E. growth by plotting growth curves as a function of cell numbers over time for ePS cells grown with (dashed line) or without (solid line) knock-down of CD73 in F-FM (n=3 and n=3, respectively). F. ePS cells from RM122 or RM128 transduced with a matched scrambled pTripZ shRNA (scrambled) were subjected to adipocyte differentiation for 18 days and stained with Oil Red O (n=2). Data from RM122 are shown. Scale bars: 10μm. G. Expression of adipogenic differentiation markers FABP4, Leptin and PPARγ in ePS cells from RM122 or RM128 treated or not with APCP for 48 h and subjected to adipocyte differentiation for 18 days. FABP4, Leptin and PPARγ transcript expression was normalized to GUSB expression. (n=3 and n=3, respectively). H. apoptotic cell death in ePS colonies grown on feeders, treated with or without adenosine (Ado) or APCP for 48h and monitored for cell surface expression of EpCAM/Annexin V (n=3 and n=3, respectively). I. ePS cells from RM122 or RM128 with (+Dox) or without (−Dox) CD73 knock-down and exposed to 200μM adenosine (Ado) for 48 h were assessed for OCT3/4 transcript expression by qRT-PCR. OCT3/4 transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). J. ePS cells from RM122 or RM128 treated with or without APCP and exposed to 200μM adenosine (Ado) for 48 h were subjected to adipocyte differentiation for 18 days and stained with Lipidtox (n=2) or osteogenic differentiation for 21 days and stained with Alizarin Red (n=2). Data from RM122 are shown. Scale bars: 40μm. K. SON expression compared to CD73− cells isolated from the same RM by flow cytometry and expanded in MEGM then treated with 200μM or 500μM of Adenosine (Ado) in either MEGM or ePS cell F-FM for 48h (n=3 and n=3, respectively). L. The T47D breast cancer cell line with or without ectopic expression of flagged CD73 was assessed for OCT3/4 protein expression by WB. Loading control: ACTIN. (n=2)
Fig. S5. Involvement of Adenosine Receptor signaling in regulating ePS plasticity. A. ePS cells from RM136 or RM142, grown in F-FM were treated or not with 10 μm of 8-PT, MRS, DMPX and VUF for 48 h. NANOG and SOX2 transcript expression was accessed by qRT-PCR. Transcript expression was normalized to GUSB expression (n=3 and n=6, respectively); ePS cells from RM136 or RM142 transduced with a doxycycline-inducible shRNA against ADORA2b were assessed for: B. ADORA2b transcript expression and C. SON transcript expression by qRT-PCR prior to (−Dox) and after (+Dox) doxycycline treatment. ADORA2b and SON transcript expression was normalized to GUSB transcript expression (n=3 and n=3, respectively, for A and B).
Fig. S6. Involvement of the PKA/STAT3 pathway in expression of master regulators of cell plasticity. ePS cells from RM136 or RM159, grown in F-FM, were assessed for A. NANOG and SOX2 transcript expression by qRT-PCR in ePS cells treated or not with adenosine (Ado) in presence or absence of the PKA inhibitor H89 or the STAT3 inhibitor Stattic. Transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). B. STAT3, SON transcript expression by qRT-PCR after transduction of lentivirus carrying inducible shRNAs against STAT3 (#16 or #17) or a matched scrambled shRNA and induction by Doxycyclin for 48h. Transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). C. NANOG and SOX2 transcript expression by qRT-PCR in untreated or MRS-treated ePS cells from RM136 or RM159 in presence or absence of the PKA activator 8-Br-cAMP. NANOG and SOX2 transcript expression was normalized to GUSB expression (n=3 and n=3, respectively). D. phosphorylation status of Jak (p-Jak) and Jnk (p-Jnk) kinase by WB in ePS cells treated with or without H89, a PKA inhibitor, for 48 hours in the presence or absence of NECA, a metabolically stable adenosine analogue. (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM136 are shown. E. SON and STAT3 protein expression and phosphorylation status of STAT3 (p-STAT3) by WB in ePS cells treated with or without Ruxolitinib, a Jak kinase inhibitor, in the presence or absence of NECA or Forskolin, a PKA activator. (n=2 and n=2, respectively). Loading control: ACTIN. Data from RM136 are shown. F. IL6 transcript expression in hESCs, primary human luminal, myoepithelial or CD73+ mammary epithelial cells inferred from the Alexa-Seq database [42]. G. IL6 transcript expression by qRT-PCR after transduction of lentivirus carrying inducible shRNAs against STAT3 (#16 or #17) or a matched scrambled shRNA and induction by Doxycyclin for 48h. IL6 expression was normalized to GUSB expression (n=3 and n=3, respectively). H. ePS cells from RM136 or RM159, cultured on feeders for 3 days before incubation with or without 0.6ng/ml of IL6 for 10 days, were assessed for number of ePS colonies based on colony size (n=3 and n=3, respectively). I. Corresponding EpCAM+OCT3/4+ cell fractions by FACS analysis (n=3 and n=3, respectively). J. SON protein expression was assessed by WB in ePS cells from RM136 or RM159 treated with or without APCP in the presence or absence of different concentrations of IL6. Loading control: ACTIN. (n=1 and n=1, respectively)
Fig. S7. ACTIVIN A and TGFβ pathways function differently in ePS cells. A. ePS cells from RM136 or RM 159, grown in F-FM in the presence or absence of TGF-β (1ng/ml and 5 ng/ml), neutralizing antibody against TGF-β (anti-TGF-β (25ng/ml) or the ACTIVIN inhibitor, Follistatin (FST, 100ng/ml) for the indicated times, were assessed for SON transcript expression by qRT-PCR (n=3 and n=3, respectively). SON transcript expression was normalized to GUSB transcript expression. B. ePS cells from RM165 and RM209, grown in F-FM, were assessed for NANOG and SOX2 transcript expression by qRT-PCR in presence or absence of 5 ng ACTIVIN A for 24h (n=2 and n=1, respectively). Transcript expression was normalized to GUSB transcript expression.