

Abstract
Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide. Although recent advances in therapeutic approaches for treating HCC have improved the prognoses of patients with HCC, this cancer is still associated with a poor survival rate mainly due to late diagnosis. Therefore, a diagnosis must be made sufficiently early to perform curative and effective treatments. There is a need for a deeper understanding of the molecular mechanisms underlying the initiation and progression of HCC because these mechanisms are critical for making early diagnoses and developing novel therapeutic strategies. Over the past decade, much progress has been made in elucidating the molecular mechanisms underlying hepatocarcinogenesis. In particular, recent advances in next-generation sequencing technologies have revealed numerous genetic alterations, including recurrently mutated genes and dysregulated signaling pathways in HCC. A better understanding of the genetic alterations in HCC could contribute to identifying potential driver mutations and discovering novel therapeutic targets in the future. In this article, we summarize the current advances in research on the genetic alterations, including genomic instability, single-nucleotide polymorphisms, somatic mutations and deregulated signaling pathways, implicated in the initiation and progression of HCC. We also attempt to elucidate some of the genetic mechanisms that contribute to making early diagnoses of and developing molecularly targeted therapies for HCC.
Keywords: Genetic alterations, Chromosomal instability, Somatic mutations, Signaling pathways, Hepatocellular carcinoma
Core tip: Hepatocellular carcinoma (HCC) is one of the leading causes of cancer-related deaths worldwide. The poor survival rate is mainly due to late diagnosis of HCC. Elucidating the molecular mechanisms underlying hepatocarcinogenesis is critical for making early diagnoses of and developing targeted therapies for HCC. Recent studies on HCC using deep sequencing have provided increasing lines of evidence indicating that genetic alterations play important roles in the initiation and progression of HCC, which are summarized in this article. We also attempt to elucidate some of the genetic mechanisms underlying HCC, which may help in making early diagnoses of and developing molecularly targeted therapies for this disease.
INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth most common cancer worldwide and the third leading cause of cancer-related deaths[1]. HCC has a high incidence rate, and patients with this disease have a poor prognosis. Rising incidence and mortality rates for HCC have been observed in most countries, particularly in eastern/south-eastern Asia and in Africa[2]. Currently, it is generally accepted that persistent hepatitis B virus (HBV) and hepatitis C virus (HCV) infections are the primary causes of chronic liver disease leading to liver cirrhosis and HCC. Aflatoxin B1 (AFB1) exposure and chronic alcohol abuse are also important risk factors for developing HCC[2]. Despite improved overall survival (OS) rates among patients with HCC due to advancements in surgical techniques, 5-year OS remains low at 18%[3]. The survival rate of HCC patients is poor because most patients cannot be treated by surgical resections or liver transplantation (LT), mainly due to late diagnosis. In addition, HCC is associated with a high recurrence rate, which exceeds 50% at 5 years after surgery[4]. Therefore, the early detection of HCC is urgently needed to perform curative and effective treatments and to improve long-term survival rates. There is a need for a deeper understanding of the molecular mechanisms underlying the initiation and progression of HCC because this understanding is critical to making early diagnoses and developing novel therapeutic strategies.
It is widely accepted that carcinogenesis is a multistep process triggered by the accumulation of genetic alterations that activate different signal transduction pathways and drive the progressive transformation of normal cells into malignant cells[5,6]. The precise molecular mechanisms underlying the initiation and progression of HCC remain obscure. The phenotypic (morphological and microscopic) and genetic heterogeneity of HCCs also adds a new level of complexity to our understanding of hepatocarcinogenesis. However, despite many remaining challenges, substantial progress has been made in this field. As in other solid cancers, numerous genetic alterations accumulate during the process of hepatocarcinogenesis. Genetic alterations accumulate slowly in a limited number of genes and chromosomal loci during the early preneoplastic stage and accelerate throughout dysplasia and into the development of HCC[7]. Previous studies have shown that the incidence of genetic alterations in HCC is relatively rare and limited to a subset of a few cancer-specific genes[8]. Encouragingly, functional genomic approaches that have been applied in recent years, such as array-based comparative genomic hybridization, genome-wide association studies (GWAS) and next-generation sequencing (NGS), have advanced our understanding of the genetic basis of HCC. Specifically, recent advances in NGS technologies have identified major cancer-driving genes and associated oncogenic signaling pathways that play important roles in the initiation and progression of HCC.
It is known that HCC cells are extremely resistant to almost all conventional chemotherapeutic drugs, and until now, there have been only a limited number of chemotherapeutic agents available for the treatment of patients with HCC, especially those with advanced, unresectable cancer. Currently, oncologists are testing novel, molecularly targeted agents for treating HCC. Therefore, in an era of precision cancer medicine, monitoring clinically relevant genetic alterations is important for stratifying patients for targeted therapies[9].
The molecular mechanisms leading to the development of HCC are extremely complicated and consist of prominent genetic and epigenetic alterations[10]. Although it has been widely accepted that epigenetic alterations also play a significant role in hepatocarcinogenesis, this topic is beyond the scope of this article. Instead, in this article, we focus on the current advances in understanding the genetic alterations, including genomic instability, single-nucleotide polymorphisms (SNPs), somatic mutations, and the deregulated signaling pathways implicated in the initiation and progression of HCC. We also attempt to elucidate some of the underlying genetic mechanisms, which could contribute to making early diagnoses of and developing molecularly targeted therapies for HCC. The impact of genetic alterations on hepatocarcinogenesis is presented in Figure 1.
Figure 1.
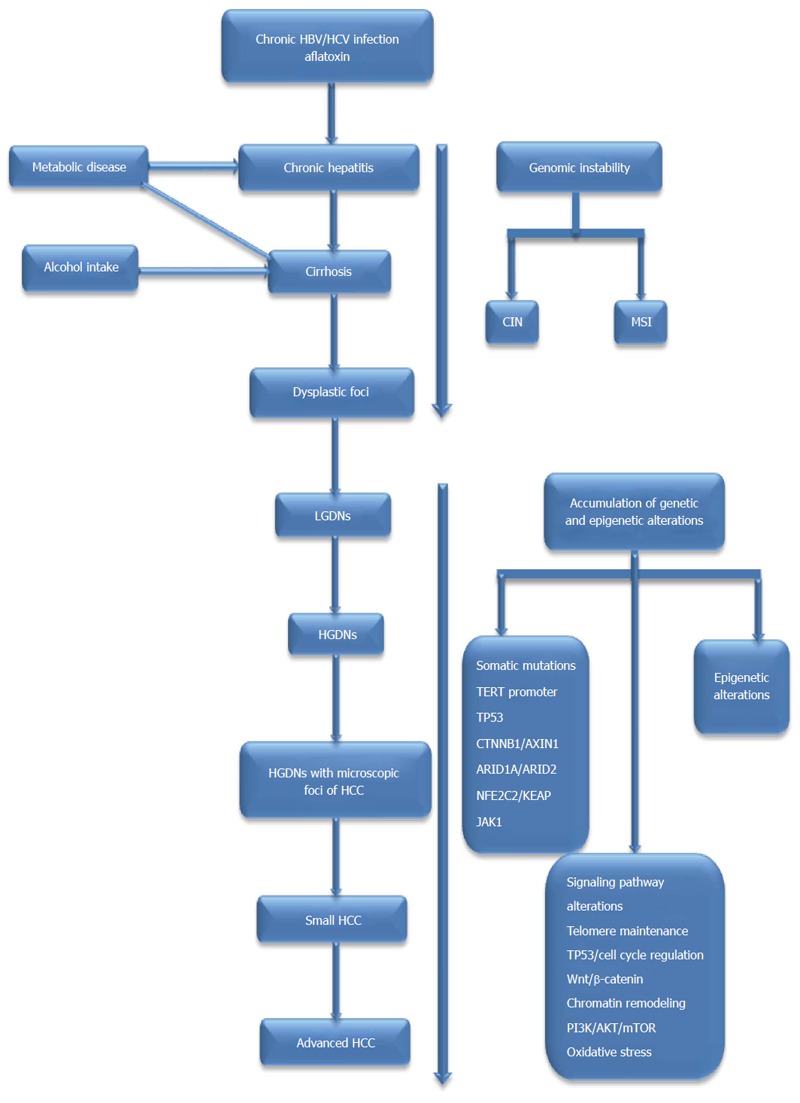
The impact of genetic alterations on hepatocarcinogenesis. Genetic alterations in hepatocarcinogenesis are connected to underlying etiologies, such as HBV, HCV, dietary AFB1 exposure and alcohol intake. Genomic instability accumulates slowly in a limited number of genes during the early preneoplastic stage, such as the development of cirrhosis, and the accumulation of genetic and epigenetic alterations accelerates throughout the formation of preneoplastic lesions, such as LGDNs and HGDNs, and into the development HCC; HBV: Hepatitis B virus; HCV: Hepatitis C virus; AFB1: Aflatoxin B1; LGDN: Low grade dysplastic nodule; HGDN: High grade dysplastic nodule; HCC: Hepatocellular carcinoma; CIN: Chromosomal instability; MSI: Microsatellite instability; TERT: Telomerase reverse-transcriptase; ARID1A: AT-rich interactive domain-containing protein 1A; ARID2: AT-rich interactive domain-containing protein 2; NFE2L2 or NRF2: Nuclear factor erythroid-derived 2-like 2; KEAP1: Kelch-like ECH-associated protein 1; JAK1: Janus kinase 1; RPS6KA3: Ribosomal protein S6 kinase polypeptide 3.
GENOMIC INSTABILITY
Genomic instability (also known as “genetic instability” or “genome instability”) is defined as a high frequency of mutations within the genome, including changes in nucleic acid sequences, chromosomal rearrangements, or aneuploidy[11]. However, it remains unclear whether genomic instability is a cause or a consequence of tumorigenesis. In recent years, accumulating evidence has strongly indicated that genomic instability could be a major driving force in tumorigenesis and the development of cancer[12-18]. In neoplasms, genomic instability can be broadly classified based on its origin as chromosomal instability (CIN) or, less commonly, microsatellite instability (MSI)[19]. Currently, there are many technologies that can be used to detect genomic instability, including karyotyping, flow cytometry, fluorescent in situ hybridization (FISH), array comparative genome hybridization (aCGH), high-density single-nucleotide polymorphism (SNP) arrays, the random amplified polymorphic DNA (RAPD) technique, and NGS technology.
Chromosomal instability
In cancer, aneuploidy is a consequence of an increased rate of whole-chromosome missegregation during mitosis, a process known as chromosomal instability (CIN)[20]. CIN usually involves both numerical and structural chromosomal changes. Numerical CIN is characterized by gross chromosomal abnormalities, such as the gain or loss of whole chromosomes, leading to an altered DNA copy number (aneuploidy)[21]. Structural CIN might involve only fractions of chromosomes, resulting in the gain or loss of chromosome fragments, translocations, inversions, amplifications, deletions and allelic loss [loss of heterozygosity (LOH)][22]. CIN is a hallmark of human cancer and is believed to contribute to tumorigenesis, tumor progression, and the development of therapy resistance[20]. In addition, it has been widely accepted that CIN is associated with clinical and pathological parameters in solid tumors, and CIN is one of the most frequent abnormalities in HCC. The characteristics of CIN and its possible correlations with clinical and pathological parameters in HCC patients are summarized in Table 1. In addition, we also review the role of micronuclei, which are indicators of CIN, and chromothripsis, which is a new class of complex catastrophic chromosomal rearrangement.
Table 1.
The characteristics of chromosomal instability and possible correlations with clinical and pathological parameters in hepatocellular carcinoma discussed in this review
| Chromosome | Type of aberration | Targeted genes | Correlations with clinical and pathological parameters | Ref. |
| 1q21 | Gain | CHD1L, CKS1B, JTB, SHC1 | Progression of HCC | Hyeon et al[43] |
| 1q21-23 | Gain | - | Early development | Yim et al[40] |
| 1q21-q22 | Gain | - | Metastasis | Wang et al[41] |
| 1q21.1-q23.2 | Gain | BCL9, ARNT, TPM3, MUC1, NTRK1 | Poorly differentiated HCV-associated HCC | Liu et al[42] |
| 1q22-23.1 | Gain | CD1d | Diagnosis and prognosis | Zhang et al[44] |
| 1q24.1-24.2 | Gain | MPZL1 | Intrahepatic metastasis | Jia et al[45] |
| 8q24.21-24.22 | Gain | MYC, DDEF1, MLZE | Prognosis (DFS and OS) | Pedica et al[47] |
| 8q21.13 | Gain | HEY1 | Proliferation | Jia et al[37] |
| 8q22.3 | Gain | CTHRC1 | Aggressive HCC | Tameda et al[48] |
| 8q24.3 | Gain | BOP1 | Advanced-stage HCC, microvascular invasion and shorter DFS | Chung et al[50] |
| 7q21.3 | Gain | SGCE, DYNC1I1, PEG10 | Hepatocarcinogenesis | Tsuji et al[51] |
| 4q34.3-35 | LOH | ING2 | Progression | Zhang et al[56] |
| 4q13.3-q35.2 | LOH | ADH4, ADH1C, ADH1A, ADH6 | HBV- and AFB1-related HCC carcinogenesis | Qi et al[58] |
| 8p | LOH | DLC1, CCDC25, ELP3, PROSC, SH2D4A, SORBS3 | Early stage of hepatocarcinogenesis, poor outcomes | Tornillo et al[59]; Roessler et al[30] |
| 8p22-p23 | LOH | MCPH1, KIAA1456, TUSC3, ZDHHC2 | Metastasis and prognosis | Peng et al[62] |
| D4S2964 | LOH | ARD1B, SEPT11 | Prognosis (OS) | Huang et al[63] |
| 6q26-q27 | LOH | M6P/IGF2R | Poor outcomes | Jang et al[64] |
HCC: Hepatocellular carcinoma; HBV: Hepatitis B virus; HCV: Hepatitis C virus; AFB1: Aflatoxin B1; DFS: Disease-free survival; OS: Overall survival; CHD1L: Chromodomain helicase/ATPase DNA binding protein 1-like; CKS1B: Cyclin-dependent kinases regulatory subunit 1; JTB: Jumping translocation breakpoint; SHC1: SHC-transforming protein 1; BCL9: B-cell CLL/lymphoma 9 protein; ARNT: Aryl hydrocarbon receptor nuclear translocator; TPM3: Tropomyosin alpha-3 chain; MUC1: Mucin 1; NTRK1: Neurotrophic tyrosine kinase receptor type 1; CD1d: Antigen-presenting glycoprotein; MPZL1: Myelin protein zero-like protein 1; MYC: Myelocytomatosis viral oncogene; DDEF1: Arf-GAP with SH3 domain, ANK repeat and PH domain-containing protein 1; MLZE: Human melanomaderived leucine zipper extra-nuclear factor; HEY1: YRPW motif protein 1; CTHRC1: Collagen triple helix repeat containing 1; BOP1: Block of proliferation 1; SGCE: Epsilon-sarcoglycan; DYNC1I1: Cytoplasmic dynein 1 intermediate chain 1; PEG10: Parternal express gene 10; ING2: Interferon regulatory factor 2; ADH4: Alcohol dehydrogenase 4; ADH1C: Alcohol dehydrogenase 1C; ADH1A: Alcohol dehydrogenase 1A; ADH6: Alcohol dehydrogenase 6; DLC1: Deleted in liver cancer 1; CCDC25: Coiled-coil domain-containing protein 25; ELP3: Longator complex protein 3P; ROSC: Proline synthetase co-transcribed bacterial homolog; SH2D4A: SH2 domain-containing protein 4A; SORBS3: Sorbin and SH3 domain containing 3; MCPH1: Microcephalin 1; KIAA1456: tRNA methyltransferase 9-like; TUSC3: Tumor suppressor candidate 3; ZDHHC2: DHHC-type containing 2; ARD1B: ARD1 homolog B (S. cerevisiae); SEPT11: Mus musculus septin 11; M6P/IGF2R: Mannose 6-phosphate/insulin-like growth factor 2 receptor.
DNA copy number alterations (CNAs) are important subclasses of somatic mutations, with aberrant chromosomal regions of amplifications or deletions commonly associated with overexpressed oncogenes or the loss of tumor suppressor genes (TSGs)[23]. Thus, CNAs can be used as an effective method for identifying driver genes with causal roles in carcinogenesis[24]. Such alterations are related to certain types of cancer, including HCC, and it is possible that the identification of driver genes by means of cancer-specific CNAs could provide new insights for understanding the molecular mechanisms underlying the initiation and progression of HCC. In particular, the elucidation of the molecular roles of CNAs could contribute to developing clinically relevant prognostic and predictive markers and novel therapeutic targets for treating HCC, which might ultimately be used in personalized therapeutics. Currently, CNAs in HCC cells are usually detected via conventional methods, such as FISH, comparative genomic hybridization, aCGH and SNP arrays. Lately, NGS technology has been used to detect CNAs in several types of tumors[25-27]. These studies have suggested that NGS has obvious advantages in sensitivity, reliability and accuracy in detecting CNAs relative to the use of aCGH and SNP arrays. However, there is currently only one study that has reported NGS-based CNAs detected in HCC[28].
Although the distribution of aberrant chromosomal arms differs among HCCs, numerous studies have shown, using aCGH data and SNP arrays, that certain regions are frequently affected in HCC, including gains in chromosomes 1q, 5p, 6p, 7q, 8q, 17q, and 20q and losses in 1p, 4q, 6q, 8p, 9p, 13q, 14q, 16p-q, 17p, 21p-q, and 22q[28-33]. These findings reflect a high degree of CIN in HCC[34], contributing to hepatocarcinogenesis. In addition, some of these regions contain CNA-associated oncogenes or TSGs, such asc-myelocytomatosis viral oncogene (c-myc) (8q), cyclin A2 (4q), cyclin D1 (11q), retinoblastoma 1 (Rb1) (13q), axis inhibition protein 1 (AXIN1) (16p), p53 (17p), mannose-6-phosphate receptor (IGFRII/M6PR) (6q), p16 (9p), epithelial cadherin (E-cadherin) (16q), suppressor of cytokine signaling (SOCS) (16p), and phosphatase and tensin homolog (PTEN) (10q), which have been identified to be associated with HCC[35,36]. These findings could provide us with information critical for understanding the genetic events involved in the pathogenesis and progression of HCC. However, studies employing unbiased genome-wide searches for HCC driver genes have been limited, particularly for genes related to cancer prognosis[30]. Hence, an integrated approach, such as a combined analysis of CNAs and gene expression, might be necessary to identify driver mutations.
A copy number gain at 1q is one of the most frequently detected alterations in HCC (58%-86%), and it has been suggested to be an early genomic event in the development of HCC[37]. Notably, the region 1q21 is the most frequent minimal amplifying region (MAR)[38]. A research group showed that 1q21 was the most frequently amplified region in chromosome 1q; its amplification was detected in 36 of 60 (60%) HCC specimens[39]. In addition, a gain in 1q21-23 was identified as a genomic event associated with the early development of HCC[40], and regional 1q21-q22 gains were found in 40% of advanced metastatic HCC cases[41]. In particular, a gain of 1q21.1-q23.2 was significantly associated with grades II-IV HCC and moderately or poorly differentiated HCV-associated HCCs. 1q21.1-q23.2 target genes encode five cancer genes: B-cell CLL/lymphoma 9 protein (BCL9), aryl hydrocarbon receptor nuclear translocator (ARNT), tropomyosin alpha-3 chain (TPM3), mucin 1 (MUC1), and neurotrophic tyrosine kinase receptor type 1 (NTRK1)[42]. These findings indicate that 1q21 might harbor many potential oncogenes, and the overexpression of these genes via amplification plays an important role in the pathogenesis of HCC[38]. In recent years, several research groups have focused on the identification and characterization of 1q21 target genes, such as chromodomain helicase/ATPase DNA binding protein 1-like (CHD1L), cyclin-dependent kinase regulatory subunit 1 (CKS1B), jumping translocation breakpoint (JTB) and SHC-transforming protein 1 (SHC1), in the progression of HCC. Of these, CHD1L was shown to be amplified and overexpressed in HCC cases[39]. A recent study found no nuclear immunoreactivity for CHD1L in normal livers or dysplastic nodules (DNs). In contrast, CHD1L expression in cases of HCC was significantly associated with microvascular invasion, major portal vein invasion, and higher American Joint Committee on Cancer (AJCC) T stage values[43], suggesting that CHD1L expression might not be an early event in hepatocarcinogenesis, whereas it is an independent predictor of lower disease-free survival (DFS) rates in HCC patients after surgical resection. Given these findings, it is vital to elucidate the roles of candidate target genes within 1q21 amplicons in the initiation and progression of HCC, which could contribute to our understanding of HCC carcinogenesis.
In addition to chromosome 1q21, a novel potential oncogene antigen-presenting glycoprotein (CD1d) amplicon at 1q22-23.1 could be a potential target for this amplicon in HCC[44]. In addition, using an integrated analysis of copy number and expression profiling data, one recent study found that the recurrent region of the 1q24.1-24.2 amplicon specifically targets the myelin protein zero-like protein 1 (MPZL1) gene in HCC; the expression levels of MPZL1 were positively correlated with the intrahepatic metastasis of the HCC specimens[45].
Chromosome 8q is the second most frequently amplified region in HCC. More specifically, 8q24.21-24.22 is the most frequently amplified region in chromosome 8q, with amplification occurring in 53.4% of samples and targeting the known oncogenes myelocytomatosis viral oncogene (MYC), Arf-GAP with SH3 domain, ANK repeat and PH domain-containing protein 1 (DDEF1), and human melanoma-derived leucine zipper extra-nuclear factor (MLZE)[45]. MYC has been identified as a central regulator of malignant transformations in early hepatocarcinogenesis[46], and c-myc gene amplification has also been found to be significantly correlated with DFS and OS in patients with HCC after surgical resection[47]. These findings suggest that c-myc gene amplification plays an important role in the pathogenesis and progression of HCC. Additionally, three other recurrent amplified regions at chromosome 8q have been found: 8q21.13, 8q22.3, and 8q24.3. The 8q21.13 region targets the hairy/enhancer-of-split related with YRPW motif protein 1 (HEY1), and functional experiments have shown that the enhanced expression of HEY1 significantly promotes the in vitro and in vivo proliferation of HCC cells[37]. The 8q22.3 region targets two genes: collagen triple helix repeat containing 1 (CTHRC1) and grainy head-like transcription factor 2 (GRHL2). CTHRC1 has the potential to be a new biomarker of aggressive HCC[48], while a gain in GRHL2 was found to be associated with an early recurrence of HCC[49]. The 8q24.3 region contains several genes that could be functionally related to HCC, including scribble (SCRIB) and block of proliferation 1 (BOP1). It has been reported that increased expression of BOP1 is associated with advanced-stage HCC, microvascular invasion and lower DFS[50].
Other amplifications include the 7q21.3 locus, which might contribute to the development or progression of HCC. Epsilon-sarcoglycan (SGCE), cytoplasmic dynein 1 intermediate chain 1 (DYNC1I1) and paternal express gene 10 (PEG10) have been identified as putative oncogenes located within the amplified 7q21.3 locus in HCC[51,52]. These results indicate that the amplification of 7q21.3 might be implicated in hepatocarcinogenesis.
The LOH is a marker of CIN that involves the loss of one of the two alleles at one or more loci in a heterozygote[53]. The LOH is one of the main mechanisms for the inactivation of TSGs, and the identification and characterization of LOHs could provide potential means for finding HCC-related TSGs. The LOH is frequently observed on chromosomes 1p, 4q, 6q, 8p, 9p, 10q, 11p, 13q, 14q, 16q, and 17p and is commonly observed in HCC patients[54,55]. Of these, losses on 4q and 8p are the most frequent chromosomal alterations in HCC.
The LOH at 4q has been reported to be strongly correlated with increased in alpha-fetoprotein (AFP) levels in HCC[56], and it has been found to be significantly more frequently in poorly differentiated HCCs[57]. These results suggest that the inactivation of TSGs on chromosome 4q might be a late progression event that occurs after malignant transformation. Using a high-throughput SNP array, 4q24-26 and 4q34.3-35 were found to be hot regions of chromosome 4q in HCC[56]. Three TSGs, including nei endonuclease VIII-like 3 (NEIL3), interferon regulatory factor 2 (IRF2) and inhibitor of growth family member 2 (ING2), are located on chromosome 4q34.3-35, but only ING2 is a potential TSG associated with HCC[48]. In addition, the loss of 4q13.3-q35.2 is related to both HBV- and AFB1-related HCC[58], suggesting that genetic abnormalities in 4q13.3-q35.2 might play a role in both HBV- and AFB1-related HCC carcinogenesis. Four TSGs, including alcohol dehydrogenase 4 (ADH4), alcohol dehydrogenase 1C (ADH1C), alcohol dehydrogenase 1A (ADH1A), and alcohol dehydrogenase 6 (ADH6), are located in this region[58].
The LOH on chromosome 8p is one of the most common alterations in HCC. A group of researchers found that allelic losses on 8p were observed in high-grade dysplastic nodules (HGDNs)[59], indicating that these losses might occur in the early stage of hepatocarcinogenesis. Chromosome 8p is rich in candidate and validated TSGs, with a cluster of six genes, including deleted in liver cancer 1 (DLC1), coiled-coil domain-containing protein 25 (CCDC25), elongator complex protein 3 (ELP3), proline synthetase co-transcribed bacterial homolog (PROSC), SH2 domain-containing protein 4A (SH2D4A), and sorbin and SH3 domain containing 3 (SORBS3), located on chromosome 8p that have been deleted in HCC samples from patients with poor outcomes[30]. Notably, numerous studies have revealed a high frequency for LOH on 8p22-p23 in HCC[60,61], and deletions of alleles on 8p22-p23 have been found to be associated with metastasis and poor prognoses for HCC patients[56]. Four specific genes - microcephalin 1 (MCPH1), tRNA methyltransferase 9-like (KIAA1456), tumor suppressor candidate 3 (TUSC3), and zinc finger, DHHC-type containing 2 (ZDHHC2) - are located in this region. Of these genes, a LOH for ZDHHC2 might contribute to the early metastatic recurrence of HCC after LT[62]. These findings suggest that 8p22-p23 harbors numerous TSGs that play important roles in the progression of HCC, which could contribute to assessing the risk of metastasis and recurrence in HCC patients.
In addition, a few recent studies have investigated the associations between LOH for new TSGs and the clinicopathological features of HCC. For example, LOH in the genes ARD1 homolog B (S. cerevisiae) (ARD1B) and Mus musculus septin 11 (SEPT11) were found to be significant prognostic factors for poor OS[63], and LOH in mannose 6-phosphate/insulin-like growth factor 2 receptor (M6P/IGF2R) was found to be predictive of poor clinical outcomes in surgically resected primary HCC patients[64].
In summary, the aforementioned findings provide valuable information that could contribute to our understanding of HCC carcinogenesis. However, there are still many important LOH regions that must be explored with regard to the genes that are involved in carcinogenesis and their biological and clinical implications[63].
MN are extra-nuclear bodies that contain damaged chromosome fragments and/or whole chromosomes that are not incorporated into the nucleus after cell division[65]. The frequency of MN is higher in tumor cells and cells with defective DNA damage repair systems or disrupted cell cycle checkpoint machinery; hence, MN could serve as indicators of CIN[66,67]. In one study, the micronucleus index was found to gradually increase along with the progression of hepatocarcinogenesis. HCCs showed the highest micronucleus index values, which were significantly greater than those of HGDNs and DNs with HCC foci[68]. In another study, a progressively increasing number of MN were also documented in the transition from cirrhotic nodules (CNs) to large regenerative nodules (LRNs), DNs and HCC; MN were significantly more frequent in DNs than in CNs or LRNs[69]. These results suggest that CIN might occur in the early stage of hepatocarcinogenesis, and HCC cells generally have acquired chromosomal abnormalities; therefore, the degree of CIN could increase during the progression of HCC.
Recently, chromothripsis has been identified using whole-genome sequencing(WGS) as a new class of complex catastrophic chromosomal rearrangement. Chromothripsis is a single cellular crisis in which a chromosome is broken and reassembled by a DNA repair mechanism, resulting in a large number of rearrangements clustered in a chromosomal region[70]. Although chromothripsis appears to be relatively rare, it can be an extreme outcome of a mutagenic mechanism that could be widespread in human cancers[71]. Furthermore, chromothripsis could affect cancer gene function and thereby have a major impact on the progression, prognosis, and therapeutic response of cancer[72]. To date, we are aware of only one study that investigated the role of chromothripsis in the incidence of HCC. In this study, chromothripsis and CIN were found to recurrently affect chromosomal arms 1q and 8q to create gene amplifications, suggesting that chromothripsis might contribute to hepatocarcinogenesis[33]. It seems that more attention should be paid to this concept.
MSI
MSI is the result of defects in mismatch repair genes that leads to the expansion and contraction of short nucleotide repeats called microsatellites[18]. Microsatellites are simple tandem repeats that are present at millions of loci in the human genome. MSI can result in the inactivation of TSGs or can disrupt other noncoding regulatory sequences, thereby playing a role in carcinogenesis[73]. MSI has been described in cirrhosis, mainly when cirrhosis is associated with an HBV infection[74,75]. Recent limited data are available on the incidence of MSI in HCCs. Several studies have suggested that MSI might play a minor role in hepatocarcinogenesis[76,77]. Furthermore, MSI is not implicated in the pathogenesis of a subset of HCCs affecting elderly patients without chronic liver disease[78]. Nevertheless, two studies have shown that high levels of MSI (MSI-H > 30%) were significantly associated with more aggressive histological tumor features and shorter median delays before recurrence[79], and the degree of MSI was significantly correlated with the poor differentiation and portal vein involvement of HCC[80]. These findings suggest that MSI could play a minor role in hepatocarcinogenesis and might be associated with the progression of HCC in patients with a background of chronic hepatitis and/or cirrhosis.
SNPS
SNPs are the most common form of human genetic polymorphisms that can contribute to an individual’s susceptibility and progression to cancer. Accumulating evidence suggests an association between SNPs in certain genes and HCC susceptibility[81]. GWAS have emerged as a new approach for identifying less penetrant cancer susceptibility alleles that might be associated with the initiation and progression of cancer.
Recent GWAS have identified numerous SNPs associated with the risk of HCC (Table 2); however, most findings have been both conflicting and inconsistent. For example, three researchers investigated whether an SNP (rs17401966) of kinesin-like factor 1 B (KIF1B) might be associated with the risk of HBV-related HCC in Chinese individuals. One study found that it was[82], but another study found that it was not[83]. A third study found that KIF1B alone was not associated with the risk but that the gene-environment interaction between the KIF1B variant and alcohol consumption was associated with the risk of HCC[84]. These inconsistent findings could be attributed to a lack of controlling for confounding variables, such as epidemiological and environmental risk factors in the first two studies. Therefore, it is important to evaluate the role of KIF1B rs17401966 in the genetic susceptibility to HCC and gene-environment interactions. Interestingly, three studies found that KIF1B rs17401966 was not associated with the development of HBV-related HCC in Thai, Japanese, and Saudi Arabian patients[85-87], and two other studies identified that KIF1B rs17401966 exerted protective effects against the susceptibility to HBV-related HCC in Chinese patients[88,89]. These inconsistencies might partly be because different ethnicities or study populations have distinct genetic architectures. In another example, three GWAS identified that MHC class I polypeptide-relatedsequence A (MICA) and DEP domain containing 5 (DEPDC5) SNPs were strongly associated with HCC in Japanese populations with chronic HCV infections[90-92]. However, two other studies found that neither DEPDC5 rs1012068 nor MICA rs2596542 was associated with HCC in Europeans with chronic HCV infections[93] or in Chinese populations with chronic HBV infections[94]. The discrepancies among these studies might be due to different study designs[93] or to differences in the different racial/ethnic groups. The inconsistent findings for HBV- and HCV-related HCC suggest that whether SNPs in the MICA and DEPDC5 loci affect the susceptibility to HCC is subject to race/ethnicity-specific differences. Undoubtedly, the same variability also applies to all the other HCC-related SNPs, which could be explained by gene-gene and gene-environment interactions contributing to the inconsistent findings in different racial or ethnic groups that have been studied[95].
Table 2.
Summary of single-nucleotide polymorphisms associated with the risk of hepatocellular carcinoma identified from genome-wide association studies
| Related gene | SNP | Etiology of HCC | Odds ratio (95%CI) | P value | Ref. |
| TPTE2 | rs2880301 | HBV/HCV, Republic of Korea | 0.27 (0.19-0.39) | 1.74 × 10-12 | Clifford et al[96] |
| KIF1B | rs17401966 | HBV, China | 0.61 (0.55-0.67) | 1.70 × 10-18 | Zhang et al[82] |
| KIF1B | rs17401966 | HBV interacting with alcohol consumption, China | 2.36 (1.49-3.74) | Chen et al[84] | |
| GRIK1 | rs455804 | HBV, China | 0.84 (0.80-0.89) | 5.24 × 10-10 | Li et al[94] |
| HLA-DQA1/DRB1 | rs9272015 | HBV, China | 1.28 (1.22-1.35) | 1.13 × 10-19 | Li et al[94] |
| MICA | rs2596542 | HCV, Japan | 1.39 (1.27-1.52) | 4.21 × 10-13 | Kumar et al[91] |
| HLA-DQ | rs9275319 | HBV, China | 1.51 (1.38-1.66) | 8.65 × 10-19 | Jiang et al[97] |
| DEPDC5 | rs1012068 | HCV, Japan | 1.75 (1.51-2.03) | 1.27 × 10-13 | Miki et al[90] |
| DDX18 | rs2551677 | HBV/HCV, Republic of Korea | 3.38 (2.07-5.53) | 1.41 × 10-10 | Clifford et al[96] |
| FasL | rs763110 | HBV/HCV, Egypt | 1.970 (1.250-3.105) | 0.003 | Khalifa et al[98] |
| DLC1 | rs3816747 | HBV, China | 0.486 (0.2450.962)/ 0.51 (0.2670.974) | 0.037/0.039 | Xie et al[99] |
| STAT4 | rs7574865 | HBV, China | 1.22 (1.15-1.29) | 1.66 × 10-11 | Jiang et al[97] |
| FOXP3 | rs3761549 | HBV, China | 1.32 (1.03-1.70) | 0.030 | Chen et al[100] |
SNP: Single-nucleotide polymorphism; HCC: Hepatocellular carcinoma; GWAS: Genome-wide association studies; HBV: Hepatitis B virus; HCV: Hepatitis C virus; TPTE2: Transmembrane phosphoinositide 3-phosphatase and tensin homolog 2; KIF1B: Kinesin-like factor 1 B; GRIK1: Glutamate receptor, ionotropic, kainate 1; HLA-DQA1/DRB1: Major histocompatibility complex, class II, DQ alpha 1, DR beta 1; MICA: MHC class I polypeptide-relatedsequence A; HLA-DQ: Major histocompatibility complex class II antigen; DEPDC5: DEP domain containing 5; DDX18: DEAD (Asp-Glu-Ala-Asp) box polypeptide 18; FasL: Fas ligand; DLC1: Deleted in liver cancer 1; STAT4: Signal transducer and activator of transcription 4; FOXP3: Forkhead box P3.
Taken together, the available results show that most findings related to the SNPs detected in GWAS on HCC can be problematic to replicate due to differences among different racial/ethnic groups, different study designs, and genetic heterogeneity. GWAS have so far identified numerous SNPs associated with HCC susceptibility[90-100]; however, most of these investigations were limited by relatively small sample sizes or the inclusion of only one racial/ethnic group. The inconsistency of these findings could be attributed to many factors, such as a lack of control for confounding variables, different study designs or the different racial/ethnic groups in the studies. Given the high variability/inconsistency in findings related to SNPs found in GWAS, at least to date, we cannot recommend the continued study of SNPs in relation to HCC as a means for identifying reliable markers of the initiation and progression of HCC. Therefore, further well-designed investigations with larger sample sizes and multiple races/ethnicities are warranted to elucidate the impact of SNPs on susceptibility to HCC.
SOMATIC MUTATIONS IN HCC
Similar to any other cancer, HCCs consist of highly heterogeneous tumors with multiple genetic alterations, particularly somatic mutations. Recent advances in NGS technologies, such as WGS or whole-exome sequencing (WES), have enabled us to identify global driver genes related to the development of HCC. In addition to confirming the high frequency of somatic mutations in tumor protein p53 (TP53), catenin beta 1 (CTNNB1) and AXIN1, recent studies applying deep-sequencing analyses have identified numerous novel mutations in genes, such as mutations in genes related to chromatin remodeling (ARID1A and ARID2), oxidative stress (NFE2L2 and KEAP1), RAS/MAPK signaling (RPS6KA3), and the janus kinase/signal transducers and activators of the transcription (JAK/STAT) pathway (JAK1)[28,101-103]. With the exception of ARID1A (10%-16%), most of these newly identified driver genes are mutated in less than 10% of HCC cases. It is encouraging that recurrent telomerase reverse transcriptase (TERT)-promoter mutations have been recently identified as the most frequent molecular alterations in HCC and as the first gene that is recurrently mutated in cirrhotic preneoplastic lesions[104,105]. There is abundant evidence to support the notion that TERT, TP53, CTNNB1, ARID1A and AXIN1 are recurrently mutated genes involved in HCC[28,102,103,106-111]. Specifically, driver mutations in TERT, TP53, and CTNNB1 are among the most frequent genetic alterations that have been defined as additive events in the development of HCC, irrespective of etiological background[28,106-108,112-115].
In this section, we briefly summarize previously well-known driver mutations and some novel gene mutations discovered in NGS studies. CTNNB1 and AXIN1 are subsequently reviewed in relation to the Wnt/β-catenin signaling pathway. The role and characteristics of frequent recurrent somatic mutations in HCC and their associations with clinical pathological parameters are summarized in Table 3.
Table 3.
The characteristics of frequent recurrent somatic mutations and their correlations with clinical and pathological parameters in hepatocellular carcinoma based on deep-sequencing analyses
| Gene | Altered pathway | Correlations with clinical and pathological parameters | Ref. |
| TERT promoter | Telomere stability | Hepatocarcinogenesis | Nault et al[104]; Yang et al[165] |
| TP53 | Cell cycle control | Under debate: an early event in the context of aflatoxin exposure and chronic HBV infection, or it might not play a role in carcinogenesis | Qi et al[136] |
| Poor prognosis | El-Din et al[117] | ||
| Cleary SP et al[138] | |||
| CTNNB1 | Wnt/β-catenin signaling | Under debate: a late event for malignant progression or earlier during hepatocarcinogenesis | Park et al[253]; Vilarinho et al[256] |
| Under debate: worse outcomes or better outcomes | Tornesello et al[263]; Wang et al[269] | ||
| AXIN1 | Wnt/β-catenin signaling | Hepatocarcinogenesis and progression | Guan et al[242] |
| ARID1A | Chromatin remodeling | Initiation and progression of HCC | Schulze et al[106] |
| ARID2 | Chromatin remodeling | Initiation and progression of HCC | Totoki et al[107] |
| NFE2L2 | Oxidative stress | Hepatocarcinogenesis and progression | Nault JC et al[105] |
| KEAP1 | Oxidative stress | Hepatocarcinogenesis and progression | Schulze et al[106] |
| JAK1 | JAK/STAT pathway | Hepatocarcinogenesis | Kan et al[28] |
| RPS6KA3 | RAS/MAPK signaling | Hepatocarcinogenesis | Guichard et al[102] |
TERT: Telomerase reverse-transcriptase; ARID1A: AT-rich interactive domain-containing protein 1A; ARID2: AT-rich interactive domain-containing protein 2; NFE2L2/NRF2: Nuclear factor erythroid-derived 2-like 2; KEAP1: Kelch-like ECH-associated protein 1; JAK1: Janus kinase 1; RPS6KA3: Ribosomal protein S6 kinase polypeptide 3.
TP53
TP53 is a key molecule in the TP53/cell cycle signaling pathway. The mutation or deletion of the p53 gene, which plays an important role in cell growth, division and apoptosis by acting as a transcription factor or by forming complexes with other proteins, is one of the most frequent genetic changes detected in HCC[116,117]. Strikingly, TP53 mutation rates in HCC vary in different geographic areas, reflecting differences in etiological agents and susceptibility factors[118]. The TP53 mutation in HCC occurs most commonly in sub-Saharan Africa and Southeast Asia, where the combination of widespread dietary AFB1 exposure and endemic hepatitis B fosters a high rate of mutagenesis in the liver[119]. In these areas, AFB1 is a particularly common mutagen of TP53, causing G:C to T:A transversions at the third base of codon 249 in TP53 (R249S), and the rate of TP53 R249S mutations can be accelerated in the presence of a viral infection[120,121]. This mutation was not detected in HCC cases from non-aflatoxin-contaminated areas[119].
Accumulating evidence shows that the HBV X (HBx) protein is a multifunctional regulator that plays a crucial role in HBV-associated hepatocarcinogenesis[122]. However, the potential synergistic effects between the HBx protein and TP53 mutations during hepatocarcinogenesis remain unclear. Several studies have suggested that the HBx protein affects the function of the P53 protein and contributes to the development of HCC. For example, complete HBx sequences were often associated with the presence of TP53 R249S mutations[123], and HBx was found to be associated with TP53 R249S mutations in HCC patients with no documented history of cirrhosis[124]. In addition, HBx mutations were found to interact with TP53 R249S mutations in altering cell proliferation and chromosome stability in hepatocytes[125]. HBx has also been shown to bind to p53 and to block p53-sequence-specific DNA-binding and p53-dependent transcription, ultimately blocking p53-mediated apoptosis[126]. HBx and TP53 mutations have been suggested to synergistically contribute to the formation of HCC in animal models[127]. These findings suggest that HBx is involved in the etiology of TP53 mutations during the molecular pathogenesis of HCC.
Persistent HCV infections could play a role in hepatocarcinogenesis; however, the mechanisms underlying this process remain unclear. A possible mechanism of HCV-induced oncogenesis seems to result from the interference of HCV proteins in the intracellular signal transduction processes via a mechanism including the dysregulation of cell cycle control[128]. In the presence of DNA damage, the P53 protein can be activated, promoting the expression of several important genes involved in cell cycle arrest, DNA repair, and apoptosis[129]. Accordingly, whether HCV infections occur concurrently with other genomic alterations, such as TP53 mutations, in hepatocarcinogenesis is of interest. Currently, several studies have provided some evidence for the direct action of HCV-related proteins on TP53. For example, HCV infection impairs the function of P53 through the overexpression of 3β-hydroxysterol delta 24-reductase (DHCR24), which up-regulates the interaction between P53 and MDM2 (mouse double minute 2 homolog, also known as HDM2, a P53-specific E3 ubiquitin ligase) in the cytoplasm and suppresses P53 acetylation in the nucleus[130]. Additionally, a novel TP53 mutation, 616ins14del1 (14-1 microindel), has been detected in a case of HCC associated with an HCV infection, providing evidence that HCCs characterized by HCV infections are typically associated with the mutational inactivation of the TP53 gene[131]. In addition, genetic changes in TP53 have been detected in non-neoplastic lesions linked to chronic HCV infections[132]. Collectively, the aforementioned findings suggest that HCV is implicated in the etiology of TP53 mutations during hepatocarcinogenesis. However, these results were obtained in vitro using cell culture models or animal models, and the synergistic effects of TP53 mutations and HCV infections in human hepatocarcinogenesis must be further investigated.
A TP53 mutation has been identified as one of the most frequent molecular alterations in HCC; however, the role of TP53 mutations in hepatocarcinogenesis remains debatable. Strikingly, a missense mutation in exon 7 (R249S) of p53 has been found specifically in HCC patients from regions with high levels of AFB1 exposure[133]. Several studies have suggested that TP53 R249S mutations are likely to occur as early events in association with aflatoxin exposure and chronic HBV infection[134-136]. A recent study showed that TP53 R249S mutations are an important factor in HCC carcinogenesis in Brazil, where aflatoxin exposure levels are high[137]. In contrast, TP53 mutations can occur as a late event in carcinogenesis without a typical mutational pattern in areas with low levels of AFB1 intake[135]. Furthermore, another study showed that TP53 R249S mutations might not play a role in the carcinogenesis of HCC in Egypt, where HCV infections are highly prevalent and are a major risk factor for the development of HCC[117]. Taken together, these findings show that TP53 mutations could play an important role in hepatocarcinogenesis in populations with chronic HBV infections, especially in those exposed to excessive levels of AFB1. It follows that these inconsistent and even conflicting results regarding the role of TP53 mutations in hepatocarcinogenesis might primarily be due to heterogeneity in the geographic and etiological backgrounds of the cases studied.
Recent reports have shown that TP53 mutations can be used to predict HCC. For example, mutations in TP53 were found to be associated with a significantly higher rate of recurrence and a lower DFS[138]. In addition, two systematic reviews concluded that TP53 mutations were associated with poor OS, relapse-free survival rates (RFS), and DFS in HCC patients, with similar results found between patients with HBV infections and HCV infections[139,140]. However, a recent study showed that TP53 mutations were associated with shorter survival time only in cases of HBV-related HCC, although R249S hot spot mutations were not associated with survival rates in patients of European origin with HBV-related HCC[141]. In contrast, another study found that TP53 mutations, particularly the hot spot mutations R249S and V157F, regardless of sample origin, were associated with poor prognoses in patients with HCC[142]. This finding was echoed by another recent study on the relationship between TP53 mutations and the recurrence of HCC in patients with HCC of various etiologies[143]. Taken together, these inconsistent and even conflicting findings might be largely due to the use of different racial and regional groups as well as other possible contributing factors, including the small sample sizes of the studies. Therefore, these confounding factors should be considered when evaluating the prognostic value of TP53 mutations in HCC.
Increasing evidence suggests that the stabilization of mutant p53 in tumors is crucial for its oncogenic activities, while the depletion of mutant p53 attenuates the malignant properties of cancer cells. Thus, mutant p53 is an attractive drug target for cancer therapies[144].
Telomerase reverse-transcriptase
The human telomerase reverse transcriptase (hTERT) gene encodes a rate-limiting catalytic subunit of telomerase, which maintains the length of telomeric DNA and chromosomal stability[145]. hTERT is the major determinant of telomerase activity, and it plays a key role in cellular immortalization and the development and progression of human cancers. The reactivation of telomerase activity is observed in approximately 90% of human cancers, enabling cells to overcome replicative senescence and to escape apoptosis, which are fundamental steps in the initiation of malignant transformation[146,147]. The precise mechanism behind the reactivation of telomerase activity in cancer remains elusive, but it likely involves multiple changes that occur during the progression of cancer, including mutations and chromosomal rearrangements[148].
In two recent studies, researchers identified mutations that created new binding sites in the TERT promoter for particular transcriptional regulators, such as E-twenty-six (ETS)/ternary complex factors (TCFs) factors, and resulted in increased transcriptional activity at the TERT promoter, which could in turn lead to the increased expression of the gene and the endless cell division characteristic of cancer cells[149,150]. These findings suggest that TERT promoter mutations could be potential mechanisms for TERT reactivation in cancer cells. In more recent studies, investigators found that two highly recurrent point mutations (G228T and G250T) in the TERT promoter might be among the fundamental mechanisms underlying telomerase reactivation/expression in several types of human cancers[149,151-154].
The molecular mechanisms involved in telomerase reactivation in HCC have been only partially elucidated, with the most important being TERT promoter mutations[104]. TERT amplification and the recurrent integration of HBx into the TERT gene promoter are alternative explanations for telomerase reactivation[107,155-157]. In particular, TERT promoter mutations were found to be associated with CTNNB1 mutations in HCC[104,106,107,158], suggesting that TERT promoter mutations and the deregulation of the Wnt/β-catenin pathway could interact in the malignant transformation of hepatocytes. Overall, the identification of TERT promoter mutations in association with HCC has provided new insights into telomerase reactivation and telomere maintenance in hepatocarcinogenesis[148]. Despite these compelling findings, the functional role of TERT promoter mutations in HCC remains unclear and must be further explored.
To date, recurrent somatic mutations in the TERT promoter have been identified as the most frequent non-coding mutations in multiple cancer types, suggesting that TERT promoter mutations are driver mutations in these cancers[154,159,160]. The frequency of TERT promoter mutations in HCC varies substantially across the different geographical regions studied. For example, cases of HCC with TERT promoter mutations have been reported from the United States[161], Europe[104,158,162], Africa[163], and East Asia (except for Japan)[103,104,164-166], with mutation frequencies of 44%, 47%-59%, 53%, and 20.7%-38.8%, respectively. These data indicate that TERT promoter mutations are less frequent among Asian patients with HBV-related HCC than among those with HCV-related HCC. The lower rate of TERT promoter mutations in patients with HBV-related HCC might be partially explained by the frequent insertion of HBV DNA in the TERT promoter, which is known to induce telomerase transcription[103,155]. These findings suggest that various etiological factors could be involved in different mechanisms that preserve telomeres during the carcinogenesis of HCC[164]. Despite these differences, TERT promoter mutations are currently considered the most frequent somatic genetic alterations in HCC regardless of patients’ geographical origin[163,167]. In the past few years, many investigators have explored the role of TERT mutations in HCC. In a recent study, TERT promoter mutations were found in 6% of low-grade dysplastic nodules (LGDNs), 19% of HGDNs, 61% of early HCCs and 42% of small and progressed HCCs. However, mutations in other classic HCC driver genes (i.e., CTNNB1, TP53, ARID1A, or ARID2) were not identified in LGDNs, HGDNs, or early HCC[105]. In another recent study, TERT mutations were found to occur at an early stage of tumorigenesis. Specifically, they were observed in 57% of preneoplastic lesions and in 30% of stage I HCCs[165], indicating that TERT promoter mutations occur early during malignant transformation and persist throughout tumor progression. These findings have been further confirmed by two recent studies using exome or DNA sequencing of liver tumor samples in which TERT promoter mutations occurred early during hepatocarcinogenesis[106,164]. In addition, when hTERT mRNA was measured via real-time quantitative RT-PCR, the hTERT mRNA levels were found to be increased in association with the progression of hepatocarcinogenesis, and most HGDNs strongly expressed hTERT mRNA at levels similar to those in HCC samples[168]. In a recent study, the authors found that the activation and expression of hTERT played extremely critical roles in the incidence and progression of HCC[169]. Previous studies also showed that telomere shortening and telomerase reactivation occurred in DNs during the early stages of hepatocarcinogenesis[163]. Indeed, alterations in telomerase restriction fragment (TRF) length, telomerase activity (TA), and hTERT and hTR expression were identified in both the early and late stages of hepatocarcinogenesis[170]. These findings demonstrate that telomere status is a factor in hepatocarcinogenesis.
hTERT mRNA has been reported to be detectable in the serum of patients with HCC, and it has been reported that the sensitivity and specificity for serum hTERT mRNA in detecting HCC were 77.14% and 100%, respectively, which are higher than the sensitivity and specificity for AFP in the early detection of HCC[171]. In another report, the sensitivity/specificity for serum hTERT mRNA in diagnosing HCC was found to be 90.2%/85.4%, which is superior to using alpha-fetoprotein (AFP), AFP-L3, and des-gamma-carboxy prothrombin (DCP) in the diagnosis of HCC at an early stage[172]. Therefore, measuring serum hTERT mRNA levels might serve as a potential diagnostic tool for HCC.
Taken together, these findings suggest that TERT promoter mutations are among the earliest genetic alterations in hepatocarcinogenesis, occurring at preneoplastic stages and behaving as a “gatekeeper” during the malignant transformation sequence[173,174].
Considering that TERT promoter mutations are among the earliest recurrent genetic events in tumorigenesis and are also the most frequent somatic genetic alterations in HCC, telomerase inhibition shows potential as an ideal therapeutic target in treating HCC. Currently, different strategies for telomerase inhibition, such as the use of nucleoside analogs, oligonucleotides, small molecule inhibitors, G-quadruplex stabilizers, immunotherapy, and gene therapy in different cancers, are currently in development, preclinical studies or clinical trials[175].
ARID1A and ARID2
Increasing evidence has demonstrated that the misregulation of ATP-dependent chromatin remodeling complexes (chromatin remodelers) contributes to tumorigenesis[176], tumor heterogeneity[177], and the cellular response to anticancer drugs[178-182]. Among the different ATP-dependent chromatin remodelers, genes encoding SWitch/sucrose nonfermentable (SWI/SNF) complex subunits are now recognized as among the most commonly mutated targets affecting chromatin remodeling, as they are present in 20% of human cancers[183-185]. SWI/SNF chromatin remodeling has been linked to a variety of epigenetic processes, including roles in maintaining nucleosome positioning and interacting with other chromatin modifiers[186]. The SWI/SNF complexes can be divided into two broad categories based on the presence of the AT-rich interactive domain containing protein 1A-B (ARID1A/B) subunits (BAF complex) or ARID2 and polybromo 1 (PBMR1) subunits (PBAF complex)[187].
Recent exome and WGS studies of HCC have shown that recurrent inactivating mutations in SWI/SNF subunits are involved in the molecular basis of hepatocarcinogenesis[101,102,188-190]. However, the functional role and molecular mechanisms underlying these mutations in the initiation and progression of HCC are not yet completely understood. Genes involved in coding for chromatin-modifying proteins are commonly mutated in HCC. In particular, two inactivating mutations in genes encoding subunits of the SWI/SNF complex, and ARID1A and ARID2 have been identified in approximately 10% of HCC cases[101,106,107,189,191]. Therefore, it is not surprising that chromatin remodeling complex alterations might play important roles in the initiation and progression of HCC. Interestingly, the frequency of ARID1A and ARID2 mutations occurring in HCC varies considerably across HCC cases, depending on the different etiologies of the disease. For example, ARID1A mutations are significantly more frequent in HCC related to alcohol intake than in tumors of other etiologies[102], and ARID2 mutations commonly occur in HCV-associated HCC[188,192]. However, several studies did not observe an association between ARID1A and ARID2 mutations and the etiology of HCC. ARID1A was mutated in 13% of HBV-associated cases of HCC[189], and ARID2 mutations were not significantly associated with HCV infections[102]. A recent study also demonstrated that ARID1A alterations were not correlated with HBV infection, HCV infection or the heavy use of alcohol[193]. These findings suggest that ARID1A and ARID2 mutations are universally present in association with HCC related to hepatitis virus infection and alcohol intake.
The mechanisms by which mutations in SWI/SNF subunits drive tumorigenesis are unclear. Most ARID1A and ARID2 mutations detected in cancer cells to date are inactivating mutations, suggesting that both proteins function as tumor suppressors[194]. Several possible mechanisms for this effect have been suggested. ARID1A has been indicated in preventing DNA entanglements during mitosis. Hence, its mutational inactivation could lead to genomic instability and alter gene expression, which could contribute to tumorigenesis[195]. In addition, it has been found that ARID1A mutations tend to interact with the activation of the PI3K/AKT pathway in promoting tumorigenesis in many human cancers of diverse origins[196-203]. Furthermore, a recent study found that ARID1A mutations alone did not cause the development or progression of cancer but that a combination of ARID1A inactivation and a PI3K/AKT pathway aberration was sufficient to initiate tumorigenesis[204]. Theoretically, the two mechanisms mentioned above in other solid tumors might also apply to HCC. The functional significance of ARID1A and ARID2 mutations remains to be elucidated in relation to the initiation and progression of HCC.
In a recent study, HCC cases with altered ARID1A expression showed inverse correlations with the nuclear localization of P53 and beta-catenin, suggesting that the ARID1A pathway might represent an alternative pathway to the p53 and beta-catenin pathways in HCC. Thus, ARID1A might constitute a promising therapeutic target for treating a subset of HCCs[193].
NFE2L2/NRF2 and KEAP1
Oxidative stress involves elevated intracellular levels of reactive oxygen species (ROS) that cause damage to lipids, proteins and DNA[205]. Recent studies have shown that persistent oxidative stress due to elevated ROS levels is associated with carcinogenesis and the progression of cancer[206-210]. The NRF2-KEAP1 pathway is the major regulator of cytoprotective responses to endogenous and exogenous stresses caused by ROS and electrophiles[211,212]. The key proteins within the NRF2-KEAP1 pathway are the transcription factor NRF2, which mediates oxidative stress responses, and KEAP1, which is a negative regulator of NRF2 activity.
NRF2 has been traditionally considered a tumor suppressor because of its cytoprotective functions[213]. In fact, accumulating evidence from genetic analyses of human tumors suggests that the deregulation of NRF2 is a critical determinant in oncogenesis, and somatic mutations of either NRF2 or KEAP1 have frequently been detected in a variety of cancer types[107,214-216]. These findings indicate that mutations in NRF2 and KEAP1 frequently play important roles in carcinogenesis.
Recent exome sequencing of HCC samples has revealed that the oxidative stress pathway is activated in 12% of HCC patients, primarily as a result of mutations of NRF2 or KEAP1[106]. Numerous genomic studies on cancer have reported somatic mutations of NRF2 and inactivating mutations of KEAP1(6%-10% and 3%-8% of HCC patients, respectively)[102,106,107,138,217,218]. A recent functional experiment found that NRF2/KEAP1 mutations were present in 71% of early preneoplastic lesions and in 78.6% and 59.3% of early and advanced HCCs[219], respectively, suggesting that the onset of NRF2/KEAP1 mutations is a very early event in rat hepatocarcinogenesis. In contrast, mutations of NRF2 and KEAP1 in humans were observed only in advanced HCC and not in premalignant nodules or early HCC, suggesting that these mutations are late events in hepatocarcinogenesis in humans[105,106]. Despite some differences in the role of mutations of NRF2 and KEAP1 between rats and humans, it is evident that the dysregulation of the NRF2/KEAP1 pathway and mutations of these genes play important roles in hepatocarcinogenesis in both species. The NRF2/KEAP1 pathway might contribute to hepatocarcinogenesis through the following mechanisms. First, the NRF2/KEAP1 pathway might cause epigenetic instability, leading to HCC[220]. Second, either NRF2 acts by itself as a proto-oncogene or NRF2 or KEAP1 mutations support the accumulation of additional mutations of proto-oncogenes[215,221]. Third, the NRF2/KEAP1 pathway could alter the chromatin status, leading to abnormal methylation of TSGs, which might contribute to hepatocarcinogenesis[222]. Interestingly, recent analyses of somatic mutations in HCC have revealed that mutations in NRF2 or KEAP1 are significantly correlated with the deregulation of the Wnt/β-catenin pathway via CTNNB1 or AXIN1 mutations[102,217]. These results suggest that the NRF2/KEAP1 pathway might interact with Wnt/β-catenin signaling to promote hepatocarcinogenesis. Nevertheless, the exact molecular mechanism underlying the role of NRF2 in the pathogenesis of HCC must still be investigated.
The finding of recurrent mutations in HCC revealed that NRF2 activation was a driver event in the progression of tumors[102,138]. Collectively, NRF2/KEAP1 mutations might be involved in the pathogenesis and progression of HCC. The genetic or pharmacologic inhibition of NRF2 expression/activity in HCC cells increased the anticancer activity of erastin and sorafenib in vitro and in tumor xenograft models[223]. Intriguingly, the accumulation of phosphorylated P62, a selective autophagy substrate, was found to cause the persistent activation of NRF2, contributing to the development of HCC[223-225]. In addition, in Japanese HCC patients, NRF2 activation was associated with the phosphorylation of P62 but not with the KEAP1 status[226].These results suggest that there might be crosstalk between the NRF2/KEAP1 pathway and P62-mediated selective autophagy, and selective NRF2 inhibitors or inhibitors of the interaction between phosphorylated P62 and KEAP1 should be developed as potential therapeutic agents against human HCC.
Janus kinase 1
The JAK/STAT signaling pathways have been identified as promoters of carcinogenesis in a subset of HCCs via cytokine-induced JAK/STAT pathway activation[28,227,228]. A previous study using single-strand conformational polymorphisms (SSCPs) and direct sequencing reported a low frequency (1/84, 1.2%) of Janus kinase 1 (JAK1) mutations in HCC[229]. Recently, a comprehensive whole genome analysis revealed that JAK1 mutations appeared in 9.1% of HCCs, and the JAK/STAT pathway was altered in 45.5% of HCCs[28].These findings indicate that the JAK/STAT pathway might act as one of the major oncogenic drivers in HCC and suggest the possibility of its use as a promising therapeutic approach for HCC treatment.
Ribosomal protein S6 kinase polypeptide 3
Ribosomal protein S6 kinase polypeptide 3 (RPS6KA3) encodes a component of the RAS/MAPK signaling pathway, i.e., a gene located on chromosome X that encodes ribosomal S6 protein kinase 2 (RSK2). Recurrent mutations in RPS6KA3 have been found in 2%-9% of HCCs[102,106,189], suggesting that RPS6KA3 could act as a newly identified potential driver of the pathogenesis of HCC. Specifically, RPS6KA3 tended to be mutated in poorly differentiated HCCs[230] and was found in HCCs that developed without cirrhosis[102]. In addition, RPS6KA3 mutations were frequently associated with AXIN1 mutations[102], suggesting that RPS6KA3 inactivation might cooperate with Wnt/β-catenin signaling to promote hepatocarcinogenesis.
SIGNALING PATHWAYS IMPLICATED IN HCC
The recurrent mutated genes reviewed above were found to be highly enriched in multiple key driver signaling processes, including telomere maintenance, TP53, cell cycle regulation, the Wnt/β-catenin pathway (CTNNB1 and AXIN1), chromatin remodeling (ARID1A and ARID2), the phosphatidylinositol-3 kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway, and oxidative/endoplasmic reticulum stress (NFE2L2 and KEAP1). In the following section, we briefly summarize two of the most common molecular cellular pathways, Wnt/β-catenin and PI3K/AKT/ mTOR, in human HCC[28,35,107,231]. Other pathways are summarized in the section above.
Wnt/β-catenin signaling pathway
The WNT/β-catenin pathway can be classified into canonical (β-catenin dependent) and noncanonical (β-catenin independent) pathways[232]. In the absence of Wnt proteins, β-catenin is phosphorylated at amino-terminal serine and threonine residues by casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK-3β)[233]. β-catenin phosphorylation is facilitated by the axis inhibition protein (AXIN) and adenomatous polyposis coli (APC). Wnt signaling is activated upon Wnt-ligand binding to frizzled receptors (FZD), followed by the cytosolic accumulation of β-catenin through the prevention of GSK-3β-mediated phosphorylation of the β-catenin Ser/Thr domain[234]. The absence of β-catenin phosphorylation releases it from the degradation complex composed of APC, AXIN, GSK-3β and CK1, resulting in an accumulation of β-catenin in the cytoplasm[234]. Subsequently, cytosolic β-catenin can translocate to the nucleus to initiate the transcription of target genes through interactions with T-cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors[234]. Hepatocytes with the nuclear translocation of β-catenin displayed abnormal cellular proliferation and expressed membrane proteins associated with HCC, metastatic behavior, and cancer stem cells[235].
The deregulation of WNT/β-catenin signaling has been found in 40%-70% of HCC patients[236]. Increasing evidence suggests that the Wnt/β-catenin signaling cascade plays a major role in the pathogenesis of HCC[234,237]. Some studies have suggested possible mechanisms for this role. For example, research has found that the occurrence of HCC may be closely related to allelic loss, chromosomal changes and mutations in Wnt/β-catenin signaling pathway genes[238]. In addition, the Wnt/β-catenin signaling pathway contributes to angiogenesis, infiltration and metastasis in HCC by regulating the expression of angiogenic factors, such as matrix metalloproteinase2 (MMP-2), matrix metalloproteinase9 (MMP-9), vascular endothelial growth factorA (VEGF-A), vascular endothelial growth factor-C (VEGF-C) and basic fibroblast growth factor (bFGF)[239]. However, the precise molecular mechanism remains uncertain.
Mutations in exon 3 of the CTNNB1 gene, which encodes β-catenin, constitute a crucial molecular mechanism leading to the aberrant activation of the Wnt/β-catenin pathway, which is strongly associated with hepatocarcinogenesis[240]. In addition to gain-of-function mutations in positive modulators of Wnt signaling, such as β-catenin, the Wnt pathway can be activated by loss-of-function mutations in negative modulators, such as AXIN and APC[241]. It has been suggested that AXIN might play an important role in the pathogenesis and progression of HCC via the Wnt signaling pathway[242]. Moreover, the overexpression of the Frizzled-7 (FZD-7) receptor and glycogen synthase kinase-3 (GSK-3) inactivation may also lead to aberrant β-catenin pathway activation[243] as the FZD-7 receptor has been found to be up-regulated in 90% of human HCCs[244,245], suggesting that the consequent activation of Wnt/Frizzled-mediated signaling plays a key role in hepatic carcinogenesis. Specifically, one study analyzed the spectrum of mutations in a series of 125 cases of HCC, and the authors identified significant associations between mutations in ARID1A, RPSK6KA3 or NFE2L2 and mutations in CTNNB1 or AXIN1, suggesting that Wnt/β-catenin signaling might interact with oxidative stress responses, chromatin remodeling or the RAS/MAPK pathway to promote hepatocarcinogenesis[217].
Mutations in the Wnt/β-catenin pathway have been described in 20%-40% of HCCs[246]. In HBV-related HCC, β-catenin mutations have been found at a lower frequency[103,246,247], whereas higher incidences of β-catenin mutations have been shown to occur mainly in alcohol- and HCV-related HCCs[101,102,188,248]. These findings suggest that β-catenin mutations are associated with the etiology of the HCC, which might be explained in part by actions of the HCV core protein synergizing Wnt-induced stabilization and the accumulation of β-catenin, perhaps playing an important role in the pathogenesis of HCV[249]. In HCC occurring in association with HBV, patients display β-catenin activation, which is induced in a mutation-dependent manner by the expression of the HBx protein[250]. Furthermore, one explanation for why β-catenin mutations tend to occur in non-HBV-associated casesis that AXIN mutations (and rarely β-catenin mutations) are mainly found in chromosome-unstable tumors associated with HBV infections, and β-catenin mutations are mainly found in non-HBV, well-differentiated, chromosome-stable tumors[251]. Thus, these two components of the Wnt pathway, β-catenin and AXIN1, could operate in distinct ways in human HCC[252].
The verdict on the role of β-catenin mutations in the initiation and progression of HCC is currently uncertain. A few studies have demonstrated that β-catenin mutations are found only in association with HCC and not in DNs[104,253,254].These results suggest that β-catenin mutations might be a late event in malignant progression rather than β-catenin being an early event gene or a gatekeeper gene in the multistep process of hepatocarcinogenesis. Nevertheless, another study concluded that β-catenin accumulates in the cytoplasm and the nuclei in precancerous lesions of the liver and might contribute, at least in part, to hepatic carcinogenesis[255]. Moreover, a clonality analysis predicted that the CTNNB1 mutation was clonal and occurred earlier during hepatocarcinogenesis[256]. To date, numerous studies have investigated the possible mechanisms underlying the role of β-catenin mutations in the initiation and progression of HCC. For example, CTNNB1 mutations are likely to occur as late events in the context of aflatoxin exposure and chronic HBV infection, whereas CTNNB1 mutations might represent early events in carcinogenesis without a typical mutational pattern in areas with low AFB1 intake[135]. Transcription complexes, formed by a combination of intranuclear β-catenin and transcription factors, activate downstream target genes and regulate the expression of corresponding genes, leading to HCC tumorigenesis[257]. Although a β-catenin mutation might represent an important event leading to tumorigenic changes in hepatocytes, several studies using transgenic animal models have shown that the overexpression of mutant or stable forms of β-catenin on its own is not sufficient to induce HCC[258,259]. A recent study found that the up-regulated genes v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog G (MAFG) and synovial sarcoma, X breakpoint 1 (SSX1) significantly synergized with the transcriptional activity of β-catenin, and the overexpression of the downregulated genes one cut homeobox 1 (Onecut1) and forkhead box protein A3 (FOXA3) potently inhibited the growth of a CTNNB1-mutation-positive (HepG2) cell line and negative (Huh-7 and Hep3B) cell lines[260]. In another study, the over-expression of cysteine-rich protein 61 (Cyr61/CCN1) was positively correlated with increased levels of β-catenin in human HCC samples, indicating that Cyr61 is a direct target of β-catenin signaling in HCC[261]. Therefore, the findings of these studies indicate that β-catenin mutations can interact with other oncogenic alterations or pathways to result in hepatocarcinogenesis more often than previously recognized.
Similarly, there have been conflicting data in the literature on the question of whether β-catenin mutations in HCC are associated with favorable or unfavorable prognoses[262]. Some studies have found associations of β-catenin mutation or activation with worse outcomes, such as moderately/poorly differentiated HCV-related HCC, larger tumor sizes, multiple nodules and increased vascular invasion[263,264]. In contrast, other studies have reported that HCCs harboring β-catenin mutations had better outcomes, such as less invasive and less frequent portal vein involvement[138,260,265-268]. A recent meta-analysis also revealed that βcatenin mutations could predict a favorable prognosis in patients with HCC[269]. In addition, one study reported that β-catenin mutations were not associated with prognoses in patients with advanced HCC[238]. Interestingly, the expression of the noncanonical Wnt5a, which is known to inhibit canonical Wnt signaling, was increased in poorly differentiated HCC cell lines[270]. Based on this result, the authors proposed that canonical and noncanonical Wnt pathways play complementary roles in HCC, with canonical signaling contributing to tumor initiation and noncanonical signaling contributing to tumor progression[270]. Accordingly, the noncanonical activation of Wnt in HCC deserves further research. Furthermore, a possible mechanism underlying β-catenin mutations with favorable outcomes was proposed in another study. In this study, the presence of cytokeratin 19 (CK19) expression or the absence of β-catenin mutations was found to be predictive of early tumor recurrence (ETR), and CK19 expression abolished the suppressive effects of β-catenin mutations on the progression of HCC. CK19 expression and β-catenin mutations were found to play dramatically opposite roles in vascular invasion, ETR and the prognosis of HCC patients[271].
Considering these findings, future prospective studies to determine the initiation, progression and outcome of HCC as a function of the WNT/β-catenin pathway will be essential. Specifically, such studies should consider the geographical origin, etiology and heterogeneity of the patients as well as the modes of WNT/β-catenin pathway activation[272].
PI3K-AKT-mTOR pathway
The phosphoinositide 3-kinase-AKT-mammalian target of rapamycin (PI3K-AKT-mTOR) pathway is one of the most frequently deregulated pathways in human cancers, and it is a master regulator of processes that contribute to tumorigenesis and tumor maintenance[273]. The membrane lipid phosphatidylinositol 4, 5-bisphosphate (PIP2) is phosphorylated by PI3K into phosphatidylinositol 3, 4, 5-triphosphate (PIP3), which binds to and activates the serine/threonine kinase AKT[274]. The tumor suppressor gene product PTEN deleted on the chromosome is antagonistic to PI3K activity; the inactivation of PTEN through gene deletion increases PIP3 levels and activates AKT, which inhibits apoptosis, leading to the development of tumors[275]. Activated AKT initiates a cascade of downstream signaling events, including the mTOR pathway. Once activated by AKT, mTOR promotes cell growth and proliferation by stimulating protein synthesis through the phosphorylation of 4E-BPs and the S6 kinases[275].
The PI3K/AKT/mTOR pathway is frequently deregulated in human hepatocarcinogenesis[276]. Furthermore, the deregulation of key genes of the PI3K/AKT/mTOR pathway has clinical importance in HCC[277,278]. As a negative regulator of the PI3K/AKT/mTOR pathway, PTEN is considered a tumor suppressor gene. PTEN mutations rarely occur in HCC, whereas PTEN heterozygosity, resulting in reduced PTEN expression, has been observed in 32%-44% of HCC patients[279]. Recent studies have demonstrated that the underexpression of PTEN is associated with poorly differentiated HCC, advanced TNM (tumor-node-metastasis) stage and intrahepatic metastasis, and poor patient survival[278,280-282]. PI3KCA is an upstream regulator of AKT, although there is some controversy regarding the role of PI3KCA mutations in HCC. A recent study identified PIK3CA mutations in 14% of patients.These mutations were strongly correlated with tumor size, suggesting that PIK3CA mutations could be used as prognostic markers in HCC[283]. However, other more recent studies have shown that hot spot mutations in PIK3CA are completely absent or rare in HCC[263,284-287]; PI3K mutations were not associated with either hepatic carcinogenesis or the postoperative prognosis of HCC patients[284,285,288].
AKT, also known as protein kinase B, is a central effector in the PI3K pathway. Many HCCs have demonstrated the activation of AKT, and it has been reported that both hepatitis B and hepatitis C could activate PI3K/AKT signaling[289]. It is well established that AKT plays a key role in tumorigenesis by stimulating cell proliferation and inhibiting apoptosis. The phosphorylation of AKT at S473 was detected in up to 71% of HCC samples and was associated with the invasion, metastasis, and vascularization of HCC[278]. As an AKT effector, S6 ribosomal protein (pS6) could be used as a prognostic indicator of HCC[290]. In addition, phospho-AKT (pAKT) expression showed a significant correlation with decreased OS[291], suggesting a worse prognosis for HCC patients with activated AKT[292].
mTOR is a key component of the PI3K and AKT pathways that activate downstream kinases required for G1 to S phase transition[293]. mTOR deregulation has been reported to play a significant role in the pathogenesis and progression of HCC. A recent study showed that high mTOR expression levels were correlated with Edmondson tumor grades and cirrhosis[294]. Additionally, data from preclinical studies have indicated that the deregulated expression of mTOR pathway effectors occurred in 40%-50% of HCCs, and the activation of the mTOR pathway was associated with less differentiated tumors, earlier tumor recurrence, and lower survival rates[290,295]. mTOR acts by directly activating p70S6 kinase (p70S6K/S6K1) and inhibiting 4E binding protein 1 (4E-BP1)[296]. mTOR forms two multiprotein complexes, called mTORC1 (mTOR complexed with raptor) and mTORC2 (mTOR complexed with rictor)[297]. Both mTORC1 and mTORC2 participate in regulating the migration and invasion of HCC cells[298]. A recent study showed that a high ratio of the levels of rictor and raptor mRNAs in tumors was an independent prognostic indicator of DFS[297]. This finding suggests that an analysis of mTOR expression in cancer tissues could serve as a predictive marker of HCC recurrence after curative treatment.
Currently, many inhibitors targeting the PI3K/AKT/mTOR pathway are being evaluated for treating HCC in preclinical and clinical studies[299,300]. It is hoped that the efficacy of inhibitors of the PI3K/AKT/mTOR pathway, in combination with other anticancer agents, might represent a promising new strategy for treating HCC patients.
PROBLEMS AND PERSPECTIVES
Although numerous genes are altered in association with HCC, only a small number of them are considered alterations that drive clonal expansion and invasion.Most of the somatic alterations appear to be passengers that are neutral for tumor cell selection[301]. So far, most of the genetic events that initiate HCC remain unknown. Therefore, the identification of key driver genes in HCC is crucial to elucidating the genetic mechanism of hepatocarcinogenesis and providing new molecularly targeted therapies for HCC patients.
Recent advancements in NGS technology have allowed for the identification of recurrently mutated genes in the pathogenesis of HCC. For example, a recent study of NGS analyses was performed to identify mutations in the TERT promoter, TP53, and CTNNB1 genes that are major drivers of the development of HCC[103]. To date, however, no potential drivers of specific oncogenes (oncogene addiction, which is a term used when a cancer cell is found to be dependent on a single gene to survive) corresponding to targeted therapies have emerged, likely due to the genomic heterogeneity of HCC. In addition, the most prevalent of the critical driver mutations that have been identified in HCC are not yet drug-accessible targets[302]. Although several molecularly targeted agents have been evaluated in clinical trials in advanced HCC, no novel, fully effective molecularly targeted agents for the treatment of patients with advanced HCC have been produced, except for sorafenib. There are two factors, i.e., the lack of a clearly identified driver oncogene and the presence of underlying cirrhosis, that are primarily responsible for the frequently unsuccessful results in studies on the use of novel drugs in treating HCC[303].
It is anticipated that studies including large sample sizes combined with the integration of multiple levels of data, such as data on genomic instability, SNPs, and somatic mutations, in conjunction with integrative functional genomic approaches, will contribute to identifying driver genes in the pathogenesis of HCC. The identification of these driver genes will lead to the development of effective molecularly targeted therapies and personalized medicine.
Currently, it has been widely realized that signaling pathways, rather than individual genes, govern the course of carcinogenesis[304]. In fact, HCC is considered a multigenic disease with a multifactorial etiology, and hepatocarcinogenesis is an extremely complex multistep process, in which multiple signaling pathways are altered to some extent. In brief, due to the high complexity and heterogeneity of HCC genomes, it is important to emphasize that identifying the altered signaling pathways implicated in HCC, rather than individual mutated genes, may be the key in elucidating the genetic mechanisms underlying hepatocarcinogenesis. Furthermore, insights into the key signaling pathways will likely aid in defining previously unrecognized oncogenic addiction loops in HCC and in developing more effective targeted therapies[305]. Recent extensive research has identified multiple signaling pathways implicated in the pathogenesis of HCC; however, unfortunately, no single dominant signaling pathway is specifically altered in HCC. Future investigations into associated signaling pathways should elucidate the crosstalk between different signaling pathways, i.e., how different signaling pathways interact and how they are coordinately regulated in HCC. It is hoped that targeting these crosstalk pathways will result in superior clinical efficacy in treating HCC patients.
Taken together, current evidence suggests that there are no major mutated genes and signaling pathways corresponding to the development of tumors in the majority of cases of HCC, which might primarily be due to the heterogeneity in their geographic and etiologic backgrounds. Due to the intertumor and intratumor heterogeneity of HCCs, future studies must evaluate in detail genetic alterations in relation to the geographic origin of the disease, both across and within individual patients, and chronologically during tumor progression[306]. At the same time, the geographic and etiologic backgrounds of cases of HCC should also be considered in the design of future clinical trials testing molecularly targeted therapies. Such consideration will aid in identifying personalized therapies for treating HCC patients.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: The authors declare no conflict of interests.
Peer-review started: August 19, 2016
First decision: September 6, 2016
Article in press: October 19, 2016
P- Reviewer: Herrera B, Kasprzak A S- Editor: Yu J L- Editor: Wang TQ E- Editor: Wang CH
References
- 1.Lamarca A, Mendiola M, Barriuso J. Hepatocellular carcinoma: Exploring the impact of ethnicity on molecular biology. Crit Rev Oncol Hematol. 2016;105:65–72. doi: 10.1016/j.critrevonc.2016.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Bosetti C, Turati F, La Vecchia C. Hepatocellular carcinoma epidemiology. Best Pract Res Clin Gastroenterol. 2014;28:753–770. doi: 10.1016/j.bpg.2014.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Kulik LM, Chokechanachaisakul A. Evaluation and management of hepatocellular carcinoma. Clin Liver Dis. 2015;19:23–43. doi: 10.1016/j.cld.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 4.Ahn SM, Jang SJ, Shim JH, Kim D, Hong SM, Sung CO, Baek D, Haq F, Ansari AA, Lee SY, et al. Genomic portrait of resectable hepatocellular carcinomas: implications of RB1 and FGF19 aberrations for patient stratification. Hepatology. 2014;60:1972–1982. doi: 10.1002/hep.27198. [DOI] [PubMed] [Google Scholar]
- 5.Hai H, Tamori A, Kawada N. Role of hepatitis B virus DNA integration in human hepatocarcinogenesis. World J Gastroenterol. 2014;20:6236–6243. doi: 10.3748/wjg.v20.i20.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fantini M, Benvenuto M, Masuelli L, Frajese GV, Tresoldi I, Modesti A, Bei R. In vitro and in vivo antitumoral effects of combinations of polyphenols, or polyphenols and anticancer drugs: perspectives on cancer treatment. Int J Mol Sci. 2015;16:9236–9282. doi: 10.3390/ijms16059236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takai A, Dang HT, Wang XW. Identification of drivers from cancer genome diversity in hepatocellular carcinoma. Int J Mol Sci. 2014;15:11142–11160. doi: 10.3390/ijms150611142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nishida N, Goel A. Genetic and epigenetic signatures in human hepatocellular carcinoma: a systematic review. Curr Genomics. 2011;12:130–137. doi: 10.2174/138920211795564359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budczies J, Pfarr N, Stenzinger A, Treue D, Endris V, Ismaeel F, Bangemann N, Blohmer JU, Dietel M, Loibl S, et al. Ioncopy: a novel method for calling copy number alterations in amplicon sequencing data including significance assessment. Oncotarget. 2016;7:13236–13247. doi: 10.18632/oncotarget.7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Facciorusso A, Villani R, Bellanti F, Mitarotonda D, Vendemiale G, Serviddio G. Mitochondrial Signaling and Hepatocellular Carcinoma: Molecular Mechanisms and Therapeutic Implications. Curr Pharm Des. 2016;22:2689–2696. doi: 10.2174/1381612822666160209153624. [DOI] [PubMed] [Google Scholar]
- 11.Vincent K, Pichler M, Lee GW, Ling H. MicroRNAs, genomic instability and cancer. Int J Mol Sci. 2014;15:14475–14491. doi: 10.3390/ijms150814475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denisenko TV, Sorokina IV, Gogvadze V, Zhivotovsky B. Mitotic catastrophe and cancer drug resistance: A link that must to be broken. Drug Resist Updat. 2016;24:1–12. doi: 10.1016/j.drup.2015.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Giam M, Rancati G. Aneuploidy and chromosomal instability in cancer: a jackpot to chaos. Cell Div. 2015;10:3. doi: 10.1186/s13008-015-0009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Langie SA, Koppen G, Desaulniers D, Al-Mulla F, Al-Temaimi R, Amedei A, Azqueta A, Bisson WH, Brown DG, Brunborg G, et al. Causes of genome instability: the effect of low dose chemical exposures in modern society. Carcinogenesis. 2015;36 Suppl 1:S61–S88. doi: 10.1093/carcin/bgv031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011;3:1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 16.Lee JK, Choi YL, Kwon M, Park PJ. Mechanisms and Consequences of Cancer Genome Instability: Lessons from Genome Sequencing Studies. Annu Rev Pathol. 2016;11:283–312. doi: 10.1146/annurev-pathol-012615-044446. [DOI] [PubMed] [Google Scholar]
- 17.Kantidakis T, Saponaro M, Mitter R, Horswell S, Kranz A, Boeing S, Aygün O, Kelly GP, Matthews N, Stewart A, et al. Mutation of cancer driver MLL2 results in transcription stress and genome instability. Genes Dev. 2016;30:408–420. doi: 10.1101/gad.275453.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pikor L, Thu K, Vucic E, Lam W. The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 2013;32:341–352. doi: 10.1007/s10555-013-9429-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bastians H. Causes of Chromosomal Instability. Recent Results Cancer Res. 2015;200:95–113. doi: 10.1007/978-3-319-20291-4_5. [DOI] [PubMed] [Google Scholar]
- 21.McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. doi: 10.1038/embor.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin SA, Hewish M, Lord CJ, Ashworth A. Genomic instability and the selection of treatments for cancer. J Pathol. 2010;220:281–289. doi: 10.1002/path.2631. [DOI] [PubMed] [Google Scholar]
- 23.Pinkel D, Albertson DG. Array comparative genomic hybridization and its applications in cancer. Nat Genet. 2005;37 Suppl:S11–S17. doi: 10.1038/ng1569. [DOI] [PubMed] [Google Scholar]
- 24.Yeh YT, Dai HY, Chien CY. Amplification of MPZL1/PZR gene in hepatocellular carcinoma. Hepatobiliary Surg Nutr. 2014;3:87–90. doi: 10.3978/j.issn.2304-3881.2014.02.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lonigro RJ, Grasso CS, Robinson DR, Jing X, Wu YM, Cao X, Quist MJ, Tomlins SA, Pienta KJ, Chinnaiyan AM. Detection of somatic copy number alterations in cancer using targeted exome capture sequencing. Neoplasia. 2011;13:1019–1025. doi: 10.1593/neo.111252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Li X, Cheng Y, Sun X, Sun X, Self S, Kooperberg C, Dai JY. Copy number alterations detected by whole-exome and whole-genome sequencing of esophageal adenocarcinoma. Hum Genomics. 2015;9:22. doi: 10.1186/s40246-015-0044-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vosberg S, Herold T, Hartmann L, Neumann M, Opatz S, Metzeler KH, Schneider S, Graf A, Krebs S, Blum H, et al. Close correlation of copy number aberrations detected by next-generation sequencing with results from routine cytogenetics in acute myeloid leukemia. Genes Chromosomes Cancer. 2016;55:553–567. doi: 10.1002/gcc.22359. [DOI] [PubMed] [Google Scholar]
- 28.Kan Z, Zheng H, Liu X, Li S, Barber TD, Gong Z, Gao H, Hao K, Willard MD, Xu J, et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013;23:1422–1433. doi: 10.1101/gr.154492.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishida N, Kudo M, Nishimura T, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, Minami Y, et al. Unique association between global DNA hypomethylation and chromosomal alterations in human hepatocellular carcinoma. PLoS One. 2013;8:e72312. doi: 10.1371/journal.pone.0072312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roessler S, Long EL, Budhu A, Chen Y, Zhao X, Ji J, Walker R, Jia HL, Ye QH, Qin LX, et al. Integrative genomic identification of genes on 8p associated with hepatocellular carcinoma progression and patient survival. Gastroenterology. 2012;142:957–966.e12. doi: 10.1053/j.gastro.2011.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, Zhu Z, Wang Y, Pocalyko D, Yang WJ, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58:706–717. doi: 10.1002/hep.26402. [DOI] [PubMed] [Google Scholar]
- 32.Homayounfar K, Schwarz A, Enders C, Cameron S, Baumhoer D, Ramadori G, Lorf T, Gunawan B, Sander B. Etiologic influence on chromosomal aberrations in European hepatocellular carcinoma identified by CGH. Pathol Res Pract. 2013;209:380–387. doi: 10.1016/j.prp.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Fernandez-Banet J, Lee NP, Chan KT, Gao H, Liu X, Sung WK, Tan W, Fan ST, Poon RT, Li S, et al. Decoding complex patterns of genomic rearrangement in hepatocellular carcinoma. Genomics. 2014;103:189–203. doi: 10.1016/j.ygeno.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Wilkens L, Flemming P, Gebel M, Bleck J, Terkamp C, Wingen L, Kreipe H, Schlegelberger B. Induction of aneuploidy by increasing chromosomal instability during dedifferentiation of hepatocellular carcinoma. Proc Natl Acad Sci USA. 2004;101:1309–1314. doi: 10.1073/pnas.0305817101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bertino G, Demma S, Ardiri A, Proiti M, Gruttadauria S, Toro A, Malaguarnera G, Bertino N, Malaguarnera M, Malaguarnera M, et al. Hepatocellular carcinoma: novel molecular targets in carcinogenesis for future therapies. Biomed Res Int. 2014;2014:203693. doi: 10.1155/2014/203693. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Mínguez B, Tovar V, Chiang D, Villanueva A, Llovet JM. Pathogenesis of hepatocellular carcinoma and molecular therapies. Curr Opin Gastroenterol. 2009;25:186–194. doi: 10.1097/MOG.0b013e32832962a1. [DOI] [PubMed] [Google Scholar]
- 37.Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z, Zhao Y, Ge C, Zhao F, Chen T, et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology. 2011;54:1227–1236. doi: 10.1002/hep.24495. [DOI] [PubMed] [Google Scholar]
- 38.Chen L, Chan TH, Guan XY. Chromosome 1q21 amplification and oncogenes in hepatocellular carcinoma. Acta Pharmacol Sin. 2010;31:1165–1171. doi: 10.1038/aps.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma NF, Hu L, Fung JM, Xie D, Zheng BJ, Chen L, Tang DJ, Fu L, Wu Z, Chen M, et al. Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology. 2008;47:503–510. doi: 10.1002/hep.22072. [DOI] [PubMed] [Google Scholar]
- 40.Yim SH, Chung YJ. An overview of biomarkers and molecular signatures in HCC. Cancers (Basel) 2010;2:809–823. doi: 10.3390/cancers2020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Wu MC, Sham JS, Zhang W, Wu WQ, Guan XY. Prognostic significance of c-myc and AIB1 amplification in hepatocellular carcinoma. A broad survey using high-throughput tissue microarray. Cancer. 2002;95:2346–2352. doi: 10.1002/cncr.10963. [DOI] [PubMed] [Google Scholar]
- 42.Liu YJ, Zhou Y, Yeh MM. Recurrent genetic alterations in hepatitis C-associated hepatocellular carcinoma detected by genomic microarray: a genetic, clinical and pathological correlation study. Mol Cytogenet. 2014;7:81. doi: 10.1186/s13039-014-0081-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hyeon J, Ahn S, Park CK. CHD1L Is a Marker for Poor Prognosis of Hepatocellular Carcinoma after Surgical Resection. Korean J Pathol. 2013;47:9–15. doi: 10.4132/KoreanJPathol.2013.47.1.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang SG, Song WQ, Gao YT, Yang B, Du Z. CD1d gene is a target for a novel amplicon at 1q22-23.1 in human hepatocellular carcinoma. Mol Biol Rep. 2010;37:381–387. doi: 10.1007/s11033-009-9817-7. [DOI] [PubMed] [Google Scholar]
- 45.Jia D, Jing Y, Zhang Z, Liu L, Ding J, Zhao F, Ge C, Wang Q, Chen T, Yao M, et al. Amplification of MPZL1/PZR promotes tumor cell migration through Src-mediated phosphorylation of cortactin in hepatocellular carcinoma. Cell Res. 2014;24:204–217. doi: 10.1038/cr.2013.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaposi-Novak P, Libbrecht L, Woo HG, Lee YH, Sears NC, Coulouarn C, Conner EA, Factor VM, Roskams T, Thorgeirsson SS. Central role of c-Myc during malignant conversion in human hepatocarcinogenesis. Cancer Res. 2009;69:2775–2782. doi: 10.1158/0008-5472.CAN-08-3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pedica F, Ruzzenente A, Bagante F, Capelli P, Cataldo I, Pedron S, Iacono C, Chilosi M, Scarpa A, Brunelli M, et al. A re-emerging marker for prognosis in hepatocellular carcinoma: the add-value of fishing c-myc gene for early relapse. PLoS One. 2013;8:e68203. doi: 10.1371/journal.pone.0068203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tameda M, Sugimoto K, Shiraki K, Yamamoto N, Okamoto R, Usui M, Ito M, Takei Y, Nobori T, Kojima T, et al. Collagen triple helix repeat containing 1 is overexpressed in hepatocellular carcinoma and promotes cell proliferation and motility. Int J Oncol. 2014;45:541–548. doi: 10.3892/ijo.2014.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tanaka Y, Kanai F, Tada M, Tateishi R, Sanada M, Nannya Y, Ohta M, Asaoka Y, Seto M, Shiina S, et al. Gain of GRHL2 is associated with early recurrence of hepatocellular carcinoma. J Hepatol. 2008;49:746–757. doi: 10.1016/j.jhep.2008.06.019. [DOI] [PubMed] [Google Scholar]
- 50.Chung KY, Cheng IK, Ching AK, Chu JH, Lai PB, Wong N. Block of proliferation 1 (BOP1) plays an oncogenic role in hepatocellular carcinoma by promoting epithelial-to-mesenchymal transition. Hepatology. 2011;54:307–318. doi: 10.1002/hep.24372. [DOI] [PubMed] [Google Scholar]
- 51.Tsuji K, Yasui K, Gen Y, Endo M, Dohi O, Zen K, Mitsuyoshi H, Minami M, Itoh Y, Taniwaki M, et al. PEG10 is a probable target for the amplification at 7q21 detected in hepatocellular carcinoma. Cancer Genet Cytogenet. 2010;198:118–125. doi: 10.1016/j.cancergencyto.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 52.Dong H, Zhang H, Liang J, Yan H, Chen Y, Shen Y, Kong Y, Wang S, Zhao G, Jin W. Digital karyotyping reveals probable target genes at 7q21.3 locus in hepatocellular carcinoma. BMC Med Genomics. 2011;4:60. doi: 10.1186/1755-8794-4-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou L, Zhou W, Wu L, Yu X, Xing C, Zheng S. The association of frequent allelic loss on 17p13.1 with early metastatic recurrence of hepatocellular carcinoma after liver transplantation. J Surg Oncol. 2010;102:802–808. doi: 10.1002/jso.21743. [DOI] [PubMed] [Google Scholar]
- 54.Okuno T, Ueda M, Tsuruyama T, Haga H, Takada Y, Maetani Y, Tamaki K, Manabe T, Tanaka K, Uemoto S. Loss of heterozygosity on 10q23 is involved in metastatic recurrence of hepatocellular carcinoma. Cancer Sci. 2009;100:520–528. doi: 10.1111/j.1349-7006.2008.01056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Midorikawa Y, Yamamoto S, Tsuji S, Kamimura N, Ishikawa S, Igarashi H, Makuuchi M, Kokudo N, Sugimura H, Aburatani H. Allelic imbalances and homozygous deletion on 8p23.2 for stepwise progression of hepatocarcinogenesis. Hepatology. 2009;49:513–522. doi: 10.1002/hep.22698. [DOI] [PubMed] [Google Scholar]
- 56.Zhang H, Ma H, Wang Q, Chen M, Weng D, Wang H, Zhou J, Li Y, Sun J, Chen Y, et al. Analysis of loss of heterozygosity on chromosome 4q in hepatocellular carcinoma using high-throughput SNP array. Oncol Rep. 2010;23:445–455. [PubMed] [Google Scholar]
- 57.Moinzadeh P, Breuhahn K, Stützer H, Schirmacher P. Chromosome alterations in human hepatocellular carcinomas correlate with aetiology and histological grade--results of an explorative CGH meta-analysis. Br J Cancer. 2005;92:935–941. doi: 10.1038/sj.bjc.6602448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qi LN, Li LQ, Chen YY, Chen ZH, Bai T, Xiang BD, Qin X, Xiao KY, Peng MH, Liu ZM, et al. Genome-wide and differential proteomic analysis of hepatitis B virus and aflatoxin B1 related hepatocellular carcinoma in Guangxi, China. PLoS One. 2013;8:e83465. doi: 10.1371/journal.pone.0083465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tornillo L, Carafa V, Sauter G, Moch H, Minola E, Gambacorta M, Vecchione R, Bianchi L, Terracciano LM. Chromosomal alterations in hepatocellular nodules by comparative genomic hybridization: high-grade dysplastic nodules represent early stages of hepatocellular carcinoma. Lab Invest. 2002;82:547–553. doi: 10.1038/labinvest.3780449. [DOI] [PubMed] [Google Scholar]
- 60.Lu T, Hano H. Identification of minimal regions of deletion at 8p23.1-22 associated with metastasis of hepatocellular carcinoma. Liver Int. 2007;27:782–790. doi: 10.1111/j.1478-3231.2007.01504.x. [DOI] [PubMed] [Google Scholar]
- 61.Pang JZ, Qin LX, Ren N, Hei ZY, Ye QH, Jia WD, Sun BS, Lin GL, Liu DY, Liu YK, et al. Loss of heterozygosity at D8S298 is a predictor for long-term survival of patients with tumor-node-metastasis stage I of hepatocellular carcinoma. Clin Cancer Res. 2007;13:7363–7369. doi: 10.1158/1078-0432.CCR-07-0593. [DOI] [PubMed] [Google Scholar]
- 62.Peng C, Zhang Z, Wu J, Lv Z, Tang J, Xie H, Zhou L, Zheng S. A critical role for ZDHHC2 in metastasis and recurrence in human hepatocellular carcinoma. Biomed Res Int. 2014;2014:832712. doi: 10.1155/2014/832712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang GL, Li BK, Zhang MY, Zhang HZ, Wei RR, Yuan YF, Shi M, Chen XQ, Huang L, Li AH, et al. LOH analysis of genes around D4S2964 identifies ARD1B as a prognostic predictor of hepatocellular carcinoma. World J Gastroenterol. 2010;16:2046–2054. doi: 10.3748/wjg.v16.i16.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jang HS, Kang KM, Choi BO, Chai GY, Hong SC, Ha WS, Jirtle RL. Clinical significance of loss of heterozygosity for M6P/IGF2R in patients with primary hepatocellular carcinoma. World J Gastroenterol. 2008;14:1394–1398. doi: 10.3748/wjg.14.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Luzhna L, Kathiria P, Kovalchuk O. Micronuclei in genotoxicity assessment: from genetics to epigenetics and beyond. Front Genet. 2013;4:131. doi: 10.3389/fgene.2013.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Terradas M, Martín M, Tusell L, Genescà A. Genetic activities in micronuclei: is the DNA entrapped in micronuclei lost for the cell? Mutat Res. 2010;705:60–67. doi: 10.1016/j.mrrev.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 67.Samanta S, Dey P, Nijhawan R. Micronucleus in cervical intraepithelial lesions and carcinoma. Acta Cytol. 2011;55:42–47. doi: 10.1159/000320792. [DOI] [PubMed] [Google Scholar]
- 68.Lee YH, Oh BK, Yoo JE, Yoon SM, Choi J, Kim KS, Park YN. Chromosomal instability, telomere shortening, and inactivation of p21(WAF1/CIP1) in dysplastic nodules of hepatitis B virus-associated multistep hepatocarcinogenesis. Mod Pathol. 2009;22:1121–1131. doi: 10.1038/modpathol.2009.76. [DOI] [PubMed] [Google Scholar]
- 69.Guido M, Fassan M, Giacomelli L, Cillo U, Farinati F, Burra P, Fagiuoli S, Rugge M. Micronuclei and broken eggs in human liver carcinogenesis. Anticancer Res. 2008;28:2507–2511. [PubMed] [Google Scholar]
- 70.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, Pleasance ED, Lau KW, Beare D, Stebbings LA, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144:27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Crasta K, Ganem NJ, Dagher R, Lantermann AB, Ivanova EV, Pan Y, Nezi L, Protopopov A, Chowdhury D, Pellman D. DNA breaks and chromosome pulverization from errors in mitosis. Nature. 2012;482:53–58. doi: 10.1038/nature10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kloosterman WP, Koster J, Molenaar JJ. Prevalence and clinical implications of chromothripsis in cancer genomes. Curr Opin Oncol. 2014;26:64–72. doi: 10.1097/CCO.0000000000000038. [DOI] [PubMed] [Google Scholar]
- 73.Kim TM, Park PJ. A genome-wide view of microsatellite instability: old stories of cancer mutations revisited with new sequencing technologies. Cancer Res. 2014;74:6377–6382. doi: 10.1158/0008-5472.CAN-14-1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dore MP, Realdi G, Mura D, Onida A, Massarelli G, Dettori G, Graham DY, Sepulveda AR. Genomic instability in chronic viral hepatitis and hepatocellular carcinoma. Hum Pathol. 2001;32:698–703. doi: 10.1053/hupa.2001.25593. [DOI] [PubMed] [Google Scholar]
- 75.Kawai H, Suda T, Aoyagi Y, Isokawa O, Mita Y, Waguri N, Kuroiwa T, Igarashi M, Tsukada K, Mori S, et al. Quantitative evaluation of genomic instability as a possible predictor for development of hepatocellular carcinoma: comparison of loss of heterozygosity and replication error. Hepatology. 2000;31:1246–1250. doi: 10.1053/jhep.2000.7298. [DOI] [PubMed] [Google Scholar]
- 76.Zhang SH, Cong WM, Xian ZH, Wu MC. Clinicopathological significance of loss of heterozygosity and microsatellite instability in hepatocellular carcinoma in China. World J Gastroenterol. 2005;11:3034–3039. doi: 10.3748/wjg.v11.i20.3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pang JZ, Qin LX, Ren N, Ye QH, Ying WD, Liu YK, Tang ZY. Microsatellite alterations of circulating DNA in the plasma of patients with hepatocellular carcinoma. Zhonghua Yi Xue Za Zhi. 2006;86:1662–1665. [PubMed] [Google Scholar]
- 78.Chiappini F, Gross-Goupil M, Saffroy R, Azoulay D, Emile JF, Veillhan LA, Delvart V, Chevalier S, Bismuth H, Debuire B, et al. Microsatellite instability mutator phenotype in hepatocellular carcinoma in non-alcoholic and non-virally infected normal livers. Carcinogenesis. 2004;25:541–547. doi: 10.1093/carcin/bgh035. [DOI] [PubMed] [Google Scholar]
- 79.Togni R, Bagla N, Muiesan P, Miquel R, O’Grady J, Heaton N, Knisely AS, Portmann B, Quaglia A. Microsatellite instability in hepatocellular carcinoma in non-cirrhotic liver in patients older than 60 years. Hepatol Res. 2009;39:266–273. doi: 10.1111/j.1872-034X.2008.00455.x. [DOI] [PubMed] [Google Scholar]
- 80.Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis--A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000;32:970–979. doi: 10.1053/jhep.2000.19797. [DOI] [PubMed] [Google Scholar]
- 81.Wang B, Yeh CB, Lein MY, Su CM, Yang SF, Liu YF, Tang CH. Effects of HMGB1 Polymorphisms on the Susceptibility and Progression of Hepatocellular Carcinoma. Int J Med Sci. 2016;13:304–309. doi: 10.7150/ijms.14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang H, Zhai Y, Hu Z, Wu C, Qian J, Jia W, Ma F, Huang W, Yu L, Yue W, et al. Genome-wide association study identifies 1p36.22 as a new susceptibility locus for hepatocellular carcinoma in chronic hepatitis B virus carriers. Nat Genet. 2010;42:755–758. doi: 10.1038/ng.638. [DOI] [PubMed] [Google Scholar]
- 83.Qu LS, Jin F, Guo YM, Liu TT, Xue RY, Huang XW, Xu M, Chen TY, Ni ZP, Shen XZ. Nine susceptibility loci for hepatitis B virus-related hepatocellular carcinoma identified by a pilot two-stage genome-wide association study. Oncol Lett. 2016;11:624–632. doi: 10.3892/ol.2015.3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen JH, Wang YY, Lv WB, Gan Y, Chang W, Tian NN, Huang XH, Liu L, Yu XF, Chen SD. Effects of interactions between environmental factors and KIF1B genetic variants on the risk of hepatocellular carcinoma in a Chinese cohort. World J Gastroenterol. 2016;22:4183–4190. doi: 10.3748/wjg.v22.i16.4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sopipong W, Tangkijvanich P, Payungporn S, Posuwan N, Poovorawan Y. The KIF1B (rs17401966) single nucleotide polymorphism is not associated with the development of HBV-related hepatocellular carcinoma in Thai patients. Asian Pac J Cancer Prev. 2013;14:2865–2869. doi: 10.7314/apjcp.2013.14.5.2865. [DOI] [PubMed] [Google Scholar]
- 86.Sawai H, Nishida N, Mbarek H, Matsuda K, Mawatari Y, Yamaoka M, Hige S, Kang JH, Abe K, Mochida S, et al. No association for Chinese HBV-related hepatocellular carcinoma susceptibility SNP in other East Asian populations. BMC Med Genet. 2012;13:47. doi: 10.1186/1471-2350-13-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Al-Qahtani A, Al-Anazi M, Viswan NA, Khalaf N, Abdo AA, Sanai FM, Al-Ashgar H, Al-Ahdal M. Role of single nucleotide polymorphisms of KIF1B gene in HBV-associated viral hepatitis. PLoS One. 2012;7:e45128. doi: 10.1371/journal.pone.0045128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Huang M, Pan Y, Liu J, Qi F, Wen J, Xie K, Ma H, Shen H, Liu Y, Dai J. A genetic variant at KIF1B predicts clinical outcome of HBV-related hepatocellular carcinoma in Chinese. Cancer Epidemiol. 2014;38:608–612. doi: 10.1016/j.canep.2014.07.012. [DOI] [PubMed] [Google Scholar]
- 89.Pan H, Su C, Lin Y, Niu J. [The relationship between the KIF1B (rs17401966) single nucleotide polymorphism and the genetic susceptibility to Hepatocellular carcinoma] Zhonghua Yu Fang Yi Xue Za Zhi. 2015;49:419–423. [PubMed] [Google Scholar]
- 90.Miki D, Ochi H, Hayes CN, Abe H, Yoshima T, Aikata H, Ikeda K, Kumada H, Toyota J, Morizono T, et al. Variation in the DEPDC5 locus is associated with progression to hepatocellular carcinoma in chronic hepatitis C virus carriers. Nat Genet. 2011;43:797–800. doi: 10.1038/ng.876. [DOI] [PubMed] [Google Scholar]
- 91.Kumar V, Kato N, Urabe Y, Takahashi A, Muroyama R, Hosono N, Otsuka M, Tateishi R, Omata M, Nakagawa H, et al. Genome-wide association study identifies a susceptibility locus for HCV-induced hepatocellular carcinoma. Nat Genet. 2011;43:455–458. doi: 10.1038/ng.809. [DOI] [PubMed] [Google Scholar]
- 92.Kato N, Muroyama R, Goto K. [Hepatitis C virus induced hepatocellular carcinoma associated genes] Nihon Rinsho. 2015;73:333–338. [PubMed] [Google Scholar]
- 93.Burza MA, Motta BM, Mancina RM, Pingitore P, Pirazzi C, Lepore SM, Spagnuolo R, Doldo P, Russo C, Lazzaro V, et al. DEPDC5 variants increase fibrosis progression in Europeans with chronic hepatitis C virus infection. Hepatology. 2016;63:418–427. doi: 10.1002/hep.28322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li S, Qian J, Yang Y, Zhao W, Dai J, Bei JX, Foo JN, McLaren PJ, Li Z, Yang J, et al. GWAS identifies novel susceptibility loci on 6p21.32 and 21q21.3 for hepatocellular carcinoma in chronic hepatitis B virus carriers. PLoS Genet. 2012;8:e1002791. doi: 10.1371/journal.pgen.1002791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Moonesinghe R, Ioannidis JP, Flanders WD, Yang Q, Truman BI, Khoury MJ. Estimating the contribution of genetic variants to difference in incidence of disease between population groups. Eur J Hum Genet. 2012;20:831–836. doi: 10.1038/ejhg.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Clifford RJ, Zhang J, Meerzaman DM, Lyu MS, Hu Y, Cultraro CM, Finney RP, Kelley JM, Efroni S, Greenblum SI, et al. Genetic variations at loci involved in the immune response are risk factors for hepatocellular carcinoma. Hepatology. 2010;52:2034–2043. doi: 10.1002/hep.23943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, et al. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet. 2013;45:72–75. doi: 10.1038/ng.2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Khalifa RH, Bahgat DM, Darwish HA, Shahin RM. Significant association between FasL gene -844T/C polymorphism and risk to hepatocellular carcinoma in Egyptian patients. Immunol Lett. 2016;172:84–88. doi: 10.1016/j.imlet.2016.02.007. [DOI] [PubMed] [Google Scholar]
- 99.Xie CR, Sun HG, Sun Y, Zhao WX, Zhang S, Wang XM, Yin ZY. Significance of genetic variants in DLC1 and their association with hepatocellular carcinoma. Mol Med Rep. 2015;12:4203–4209. doi: 10.3892/mmr.2015.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chen Y, Zhang H, Liao W, Zhou J, He G, Xie X, Fei R, Qin L, Wei L, Chen H. FOXP3 gene polymorphism is associated with hepatitis B-related hepatocellular carcinoma in China. J Exp Clin Cancer Res. 2013;32:39. doi: 10.1186/1756-9966-32-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fujimoto A, Totoki Y, Abe T, Boroevich KA, Hosoda F, Nguyen HH, Aoki M, Hosono N, Kubo M, Miya F, et al. Whole-genome sequencing of liver cancers identifies etiological influences on mutation patterns and recurrent mutations in chromatin regulators. Nat Genet. 2012;44:760–764. doi: 10.1038/ng.2291. [DOI] [PubMed] [Google Scholar]
- 102.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, Calderaro J, Bioulac-Sage P, Letexier M, Degos F, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–698. doi: 10.1038/ng.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kawai-Kitahata F, Asahina Y, Tanaka S, Kakinuma S, Murakawa M, Nitta S, Watanabe T, Otani S, Taniguchi M, Goto F, et al. Comprehensive analyses of mutations and hepatitis B virus integration in hepatocellular carcinoma with clinicopathological features. J Gastroenterol. 2016;51:473–486. doi: 10.1007/s00535-015-1126-4. [DOI] [PubMed] [Google Scholar]
- 104.Nault JC, Mallet M, Pilati C, Calderaro J, Bioulac-Sage P, Laurent C, Laurent A, Cherqui D, Balabaud C, Zucman-Rossi J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat Commun. 2013;4:2218. doi: 10.1038/ncomms3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nault JC, Calderaro J, Di Tommaso L, Balabaud C, Zafrani ES, Bioulac-Sage P, Roncalli M, Zucman-Rossi J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology. 2014;60:1983–1992. doi: 10.1002/hep.27372. [DOI] [PubMed] [Google Scholar]
- 106.Schulze K, Imbeaud S, Letouzé E, Alexandrov LB, Calderaro J, Rebouissou S, Couchy G, Meiller C, Shinde J, Soysouvanh F, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–511. doi: 10.1038/ng.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Totoki Y, Tatsuno K, Covington KR, Ueda H, Creighton CJ, Kato M, Tsuji S, Donehower LA, Slagle BL, Nakamura H, et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat Genet. 2014;46:1267–1273. doi: 10.1038/ng.3126. [DOI] [PubMed] [Google Scholar]
- 108.Li S, Mao M. Next generation sequencing reveals genetic landscape of hepatocellular carcinomas. Cancer Lett. 2013;340:247–253. doi: 10.1016/j.canlet.2012.09.027. [DOI] [PubMed] [Google Scholar]
- 109.Buendia MA, Neuveut C. Hepatocellular carcinoma. Cold Spring Harb Perspect Med. 2015;5:a021444. doi: 10.1101/cshperspect.a021444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bruix J, Han KH, Gores G, Llovet JM, Mazzaferro V. Liver cancer: Approaching a personalized care. J Hepatol. 2015;62:S144–S156. doi: 10.1016/j.jhep.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ang C, Miura JT, Gamblin TC, He R, Xiu J, Millis SZ, Gatalica Z, Reddy SK, Yee NS, Abou-Alfa GK. Comprehensive multiplatform biomarker analysis of 350 hepatocellular carcinomas identifies potential novel therapeutic options. J Surg Oncol. 2016;113:55–61. doi: 10.1002/jso.24086. [DOI] [PubMed] [Google Scholar]
- 112.Villanueva A, Llovet JM. Liver cancer in 2013: Mutational landscape of HCC--the end of the beginning. Nat Rev Clin Oncol. 2014;11:73–74. doi: 10.1038/nrclinonc.2013.243. [DOI] [PubMed] [Google Scholar]
- 113.Hirotsu Y, Zheng TH, Amemiya K, Mochizuki H, Guleng B, Omata M. Targeted and exome sequencing identified somatic mutations in hepatocellular carcinoma. Hepatol Res. 2016;46:1145–1151. doi: 10.1111/hepr.12663. [DOI] [PubMed] [Google Scholar]
- 114.Liao W, Yang H, Xu H, Wang Y, Ge P, Ren J, Xu W, Lu X, Sang X, Zhong S, et al. Noninvasive detection of tumor-associated mutations from circulating cell-free DNA in hepatocellular carcinoma patients by targeted deep sequencing. Oncotarget. 2016;7:40481–40490. doi: 10.18632/oncotarget.9629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–1239.e4. doi: 10.1053/j.gastro.2015.05.061. [DOI] [PubMed] [Google Scholar]
- 116.Wang Z, Jiang Y, Guan D, Li J, Yin H, Pan Y, Xie D, Chen Y. Critical roles of p53 in epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma cells. PLoS One. 2013;8:e72846. doi: 10.1371/journal.pone.0072846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.El-Din HG, Ghafar NA, Saad NE, Aziz M, Rasheed D, Hassan EM. Relationship between codon 249 mutation in exon 7 of p53 gene and diagnosis of hepatocellular carcinoma. Arch Med Sci. 2010;6:348–355. doi: 10.5114/aoms.2010.14254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ierardi E, Rosania R, Zotti M, Giorgio F, Prencipe S, Valle ND, Francesco VD, Panella C. From chronic liver disorders to hepatocellular carcinoma: Molecular and genetic pathways. World J Gastrointest Oncol. 2010;2:259–264. doi: 10.4251/wjgo.v2.i6.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gouas D, Shi H, Hainaut P. The aflatoxin-induced TP53 mutation at codon 249 (R249S): biomarker of exposure, early detection and target for therapy. Cancer Lett. 2009;286:29–37. doi: 10.1016/j.canlet.2009.02.057. [DOI] [PubMed] [Google Scholar]
- 120.Kirk GD, Lesi OA, Mendy M, Szymañska K, Whittle H, Goedert JJ, Hainaut P, Montesano R. 249(ser) TP53 mutation in plasma DNA, hepatitis B viral infection, and risk of hepatocellular carcinoma. Oncogene. 2005;24:5858–5867. doi: 10.1038/sj.onc.1208732. [DOI] [PubMed] [Google Scholar]
- 121.Tanase AM, Marchio A, Dumitrascu T, Dima S, Herlea V, Oprisan G, Dejean A, Popescu I, Pineau P. Mutation spectrum of hepatocellular carcinoma from eastern-European patients betrays the impact of a complex exposome. J Expo Sci Environ Epidemiol. 2015;25:256–263. doi: 10.1038/jes.2014.16. [DOI] [PubMed] [Google Scholar]
- 122.Zhang XD, Wang Y, Ye LH. Hepatitis B virus X protein accelerates the development of hepatoma. Cancer Biol Med. 2014;11:182–190. doi: 10.7497/j.issn.2095-3941.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gouas DA, Villar S, Ortiz-Cuaran S, Legros P, Ferro G, Kirk GD, Lesi OA, Mendy M, Bah E, Friesen MD, et al. TP53 R249S mutation, genetic variations in HBX and risk of hepatocellular carcinoma in The Gambia. Carcinogenesis. 2012;33:1219–1224. doi: 10.1093/carcin/bgs135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ortiz-Cuaran S, Villar S, Gouas D, Ferro G, Plymoth A, Khuhaprema T, Kalalak A, Sangrajrang S, Friesen MD, Groopman JD, et al. Association between HBX status, aflatoxin-induced R249S TP53 mutation and risk of hepatocellular carcinoma in a case-control study from Thailand. Cancer Lett. 2013;331:46–51. doi: 10.1016/j.canlet.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 125.Jiang W, Wang XW, Unger T, Forgues M, Kim JW, Hussain SP, Bowman E, Spillare EA, Lipsky MM, Meck JM, et al. Cooperation of tumor-derived HBx mutants and p53-249(ser) mutant in regulating cell proliferation, anchorage-independent growth and aneuploidy in a telomerase-immortalized normal human hepatocyte-derived cell line. Int J Cancer. 2010;127:1011–1020. doi: 10.1002/ijc.25118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zender L, Villanueva A, Tovar V, Sia D, Chiang DY, Llovet JM. Cancer gene discovery in hepatocellular carcinoma. J Hepatol. 2010;52:921–929. doi: 10.1016/j.jhep.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lu JW, Yang WY, Tsai SM, Lin YM, Chang PH, Chen JR, Wang HD, Wu JL, Jin SL, Yuh CH. Liver-specific expressions of HBx and src in the p53 mutant trigger hepatocarcinogenesis in zebrafish. PLoS One. 2013;8:e76951. doi: 10.1371/journal.pone.0076951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Selimovic D, El-Khattouti A, Ghozlan H, Haikel Y, Abdelkader O, Hassan M. Hepatitis C virus-related hepatocellular carcinoma: An insight into molecular mechanisms and therapeutic strategies. World J Hepatol. 2012;4:342–355. doi: 10.4254/wjh.v4.i12.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Shen Y, Zhang S, Huang X, Chen K, Shen J, Wang Z. Involvement of p53 mutation and mismatch repair proteins dysregulation in NNK-induced malignant transformation of human bronchial epithelial cells. Biomed Res Int. 2014;2014:920275. doi: 10.1155/2014/920275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Nishimura T, Kohara M, Izumi K, Kasama Y, Hirata Y, Huang Y, Shuda M, Mukaidani C, Takano T, Tokunaga Y, et al. Hepatitis C virus impairs p53 via persistent overexpression of 3beta-hydroxysterol Delta24-reductase. J Biol Chem. 2009;284:36442–36452. doi: 10.1074/jbc.M109.043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Long J, Wang Y, Li M, Tong WM, Jia JD, Huang J. Correlation of TP53 mutations with HCV positivity in hepatocarcinogenesis: identification of a novel TP53 microindel in hepatocellular carcinoma with HCV infection. Oncol Rep. 2013;30:119–124. doi: 10.3892/or.2013.2430. [DOI] [PubMed] [Google Scholar]
- 132.Kasprzak A, Adamek A, Przybyszewska W, Czajka A, Olejniczak K, Juszczyk J, Biczysko W, Zabel M. p53 immunocytochemistry and TP53 gene mutations in patients with chronic hepatitis C virus (HCV) infection. Folia Histochem Cytobiol. 2009;47:35–42. doi: 10.2478/v10042-009-0003-5. [DOI] [PubMed] [Google Scholar]
- 133.Chittmittrapap S, Chieochansin T, Chaiteerakij R, Treeprasertsuk S, Klaikaew N, Tangkijvanich P, Komolmit P, Poovorawan Y. Prevalence of aflatoxin induced p53 mutation at codon 249 (R249s) in hepatocellular carcinoma patients with and without hepatitis B surface antigen (HBsAg) Asian Pac J Cancer Prev. 2013;14:7675–7679. doi: 10.7314/apjcp.2013.14.12.7675. [DOI] [PubMed] [Google Scholar]
- 134.Teufel A, Staib F, Kanzler S, Weinmann A, Schulze-Bergkamen H, Galle PR. Genetics of hepatocellular carcinoma. World J Gastroenterol. 2007;13:2271–2282. doi: 10.3748/wjg.v13.i16.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Galy O, Chemin I, Le Roux E, Villar S, Le Calvez-Kelm F, Lereau M, Gouas D, Vieco B, Suarez I, Navas MC, et al. Mutations in TP53 and CTNNB1 in Relation to Hepatitis B and C Infections in Hepatocellular Carcinomas from Thailand. Hepat Res Treat. 2011;2011:697162. doi: 10.1155/2011/697162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qi LN, Bai T, Chen ZS, Wu FX, Chen YY, De Xiang B, Peng T, Han ZG, Li LQ. The p53 mutation spectrum in hepatocellular carcinoma from Guangxi, China: role of chronic hepatitis B virus infection and aflatoxin B1 exposure. Liver Int. 2015;35:999–1009. doi: 10.1111/liv.12460. [DOI] [PubMed] [Google Scholar]
- 137.Nogueira JA, Ono-Nita SK, Nita ME, de Souza MM, do Carmo EP, Mello ES, Scapulatempo C, Paranaguá-Vezozzo DC, Carrilho FJ, Alves VA. 249 TP53 mutation has high prevalence and is correlated with larger and poorly differentiated HCC in Brazilian patients. BMC Cancer. 2009;9:204. doi: 10.1186/1471-2407-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cleary SP, Jeck WR, Zhao X, Chen K, Selitsky SR, Savich GL, Tan TX, Wu MC, Getz G, Lawrence MS, et al. Identification of driver genes in hepatocellular carcinoma by exome sequencing. Hepatology. 2013;58:1693–1702. doi: 10.1002/hep.26540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Liu J, Ma Q, Zhang M, Wang X, Zhang D, Li W, Wang F, Wu E. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and meta-analysis. Eur J Cancer. 2012;48:2328–2338. doi: 10.1016/j.ejca.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zhan P, Ji YN, Yu LK. TP53 mutation is associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a meta-analysis. Hepatobiliary Surg Nutr. 2013;2:260–265. doi: 10.3978/j.issn.2304-3881.2013.07.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Amaddeo G, Cao Q, Ladeiro Y, Imbeaud S, Nault JC, Jaoui D, Gaston Mathe Y, Laurent C, Laurent A, Bioulac-Sage P, et al. Integration of tumour and viral genomic characterizations in HBV-related hepatocellular carcinomas. Gut. 2015;64:820–829. doi: 10.1136/gutjnl-2013-306228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Villanueva A, Hoshida Y. Depicting the role of TP53 in hepatocellular carcinoma progression. J Hepatol. 2011;55:724–725. doi: 10.1016/j.jhep.2011.03.018. [DOI] [PubMed] [Google Scholar]
- 143.Subbiah IM, Falchook GS, Kaseb AO, Hess KR, Tsimberidou AM, Fu S, Subbiah V, Hong DS, Naing A, Piha-Paul SA, et al. Exploring response signals and targets in aggressive unresectable hepatocellular carcinoma: an analysis of targeted therapy phase 1 trials. Oncotarget. 2015;6:28453–28462. doi: 10.18632/oncotarget.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Parrales A, Iwakuma T. Targeting Oncogenic Mutant p53 for Cancer Therapy. Front Oncol. 2015;5:288. doi: 10.3389/fonc.2015.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sarek G, Marzec P, Margalef P, Boulton SJ. Molecular basis of telomere dysfunction in human genetic diseases. Nat Struct Mol Biol. 2015;22:867–874. doi: 10.1038/nsmb.3093. [DOI] [PubMed] [Google Scholar]
- 146.Bell RJ, Rube HT, Kreig A, Mancini A, Fouse SD, Nagarajan RP, Choi S, Hong C, He D, Pekmezci M, et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science. 2015;348:1036–1039. doi: 10.1126/science.aab0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Makowski MM, Willems E, Fang J, Choi J, Zhang T, Jansen PW, Brown KM, Vermeulen M. An interaction proteomics survey of transcription factor binding at recurrent TERT promoter mutations. Proteomics. 2016;16:417–426. doi: 10.1002/pmic.201500327. [DOI] [PubMed] [Google Scholar]
- 148.Akincilar SC, Unal B, Tergaonkar V. Reactivation of telomerase in cancer. Cell Mol Life Sci. 2016;73:1659–1670. doi: 10.1007/s00018-016-2146-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 151.Huang FW, Bielski CM, Rinne ML, Hahn WC, Sellers WR, Stegmeier F, Garraway LA, Kryukov GV. TERT promoter mutations and monoallelic activation of TERT in cancer. Oncogenesis. 2015;4:e176. doi: 10.1038/oncsis.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Li Y, Tergaonkar V. Telomerase reactivation in cancers: Mechanisms that govern transcriptional activation of the wild-type vs. mutant TERT promoters. Transcription. 2016;7:44–49. doi: 10.1080/21541264.2016.1160173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Borah S, Xi L, Zaug AJ, Powell NM, Dancik GM, Cohen SB, Costello JC, Theodorescu D, Cech TR. TERT promoter mutations and telomerase reactivation in urothelial cancer. Science. 2015;347:1006–1010. doi: 10.1126/science.1260200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Vinagre J, Almeida A, Pópulo H, Batista R, Lyra J, Pinto V, Coelho R, Celestino R, Prazeres H, Lima L, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185. doi: 10.1038/ncomms3185. [DOI] [PubMed] [Google Scholar]
- 155.Sung WK, Zheng H, Li S, Chen R, Liu X, Li Y, Lee NP, Lee WH, Ariyaratne PN, Tennakoon C, et al. Genome-wide survey of recurrent HBV integration in hepatocellular carcinoma. Nat Genet. 2012;44:765–769. doi: 10.1038/ng.2295. [DOI] [PubMed] [Google Scholar]
- 156.Paterlini-Bréchot P, Saigo K, Murakami Y, Chami M, Gozuacik D, Mugnier C, Lagorce D, Bréchot C. Hepatitis B virus-related insertional mutagenesis occurs frequently in human liver cancers and recurrently targets human telomerase gene. Oncogene. 2003;22:3911–3916. doi: 10.1038/sj.onc.1206492. [DOI] [PubMed] [Google Scholar]
- 157.Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, Ronaghi M, Toh HC, Chow PK, Chung AY, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. 2013;34:787–798. doi: 10.1093/carcin/bgs406. [DOI] [PubMed] [Google Scholar]
- 158.Pezzuto F, Izzo F, Buonaguro L, Annunziata C, Tatangelo F, Botti G, Buonaguro FM, Tornesello ML. Tumor specific mutations in TERT promoter and CTNNB1 gene in hepatitis B and hepatitis C related hepatocellular carcinoma. Oncotarget. 2016 doi: 10.18632/oncotarget.9801. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet. 2014;46:1160–1165. doi: 10.1038/ng.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet. 2014;46:1258–1263. doi: 10.1038/ng.3141. [DOI] [PubMed] [Google Scholar]
- 161.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA, Friedman AH, Friedman H, Gallia GL, Giovanella BC, et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci USA. 2013;110:6021–6026. doi: 10.1073/pnas.1303607110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Quaas A, Oldopp T, Tharun L, Klingenfeld C, Krech T, Sauter G, Grob TJ. Frequency of TERT promoter mutations in primary tumors of the liver. Virchows Arch. 2014;465:673–677. doi: 10.1007/s00428-014-1658-7. [DOI] [PubMed] [Google Scholar]
- 163.Cevik D, Yildiz G, Ozturk M. Common telomerase reverse transcriptase promoter mutations in hepatocellular carcinomas from different geographical locations. World J Gastroenterol. 2015;21:311–317. doi: 10.3748/wjg.v21.i1.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen YL, Jeng YM, Chang CN, Lee HJ, Hsu HC, Lai PL, Yuan RH. TERT promoter mutation in resectable hepatocellular carcinomas: a strong association with hepatitis C infection and absence of hepatitis B infection. Int J Surg. 2014;12:659–665. doi: 10.1016/j.ijsu.2014.05.066. [DOI] [PubMed] [Google Scholar]
- 165.Yang X, Guo X, Chen Y, Chen G, Ma Y, Huang K, Zhang Y, Zhao Q, Winkler CA, An P, et al. Telomerase reverse transcriptase promoter mutations in hepatitis B virus-associated hepatocellular carcinoma. Oncotarget. 2016;7:27838–27847. doi: 10.18632/oncotarget.8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Huang DS, Wang Z, He XJ, Diplas BH, Yang R, Killela PJ, Meng Q, Ye ZY, Wang W, Jiang XT, et al. Recurrent TERT promoter mutations identified in a large-scale study of multiple tumour types are associated with increased TERT expression and telomerase activation. Eur J Cancer. 2015;51:969–976. doi: 10.1016/j.ejca.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Donati B, Valenti L. Telomeres, NAFLD and Chronic Liver Disease. Int J Mol Sci. 2016;17:383. doi: 10.3390/ijms17030383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Oh BK, Kim YJ, Park YN, Choi J, Kim KS, Park C. Quantitative assessment of hTERT mRNA expression in dysplastic nodules of HBV-related hepatocarcinogenesis. Am J Gastroenterol. 2006;101:831–838. doi: 10.1111/j.1572-0241.2006.00532.x. [DOI] [PubMed] [Google Scholar]
- 169.Zhou XU, Lu J, Zhu H. Correlation between the expression of hTERT gene and the clinicopathological characteristics of hepatocellular carcinoma. Oncol Lett. 2016;11:111–115. doi: 10.3892/ol.2015.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.El Idrissi M, Hervieu V, Merle P, Mortreux F, Wattel E. Cause-specific telomere factors deregulation in hepatocellular carcinoma. J Exp Clin Cancer Res. 2013;32:64. doi: 10.1186/1756-9966-32-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.El-Mazny A, Sayed M, Sharaf S. Human telomerase reverse transcriptase messenger RNA (TERT mRNA) as a tumour marker for early detection of hepatocellular carcinoma. Arab J Gastroenterol. 2014;15:68–71. doi: 10.1016/j.ajg.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 172.Miura N, Osaki Y, Nagashima M, Kohno M, Yorozu K, Shomori K, Kanbe T, Oyama K, Kishimoto Y, Maruyama S, et al. A novel biomarker TERTmRNA is applicable for early detection of hepatoma. BMC Gastroenterol. 2010;10:46. doi: 10.1186/1471-230X-10-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nault JC, Zucman-Rossi J. TERT promoter mutations in primary liver tumors. Clin Res Hepatol Gastroenterol. 2016;40:9–14. doi: 10.1016/j.clinre.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 174.Pinyol R, Tovar V, Llovet JM. TERT promoter mutations: gatekeeper and driver of hepatocellular carcinoma. J Hepatol. 2014;61:685–687. doi: 10.1016/j.jhep.2014.05.028. [DOI] [PubMed] [Google Scholar]
- 175.Gomez DL, Armando RG, Cerrudo CS, Ghiringhelli PD, Gomez DE. Telomerase as a Cancer Target. Development of New Molecules. Curr Top Med Chem. 2016;16:2432–2440. doi: 10.2174/1568026616666160212122425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M, Elsner M, et al. Toward understanding and exploiting tumor heterogeneity. Nat Med. 2015;21:846–853. doi: 10.1038/nm.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lacoste N, Woolfe A, Tachiwana H, Garea AV, Barth T, Cantaloube S, Kurumizaka H, Imhof A, Almouzni G. Mislocalization of the centromeric histone variant CenH3/CENP-A in human cells depends on the chaperone DAXX. Mol Cell. 2014;53:631–644. doi: 10.1016/j.molcel.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 179.Banelli B, Carra E, Barbieri F, Würth R, Parodi F, Pattarozzi A, Carosio R, Forlani A, Allemanni G, Marubbi D, et al. The histone demethylase KDM5A is a key factor for the resistance to temozolomide in glioblastoma. Cell Cycle. 2015;14:3418–3429. doi: 10.1080/15384101.2015.1090063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Husain A, Begum NA, Taniguchi T, Taniguchi H, Kobayashi M, Honjo T. Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat Commun. 2016;7:10549. doi: 10.1038/ncomms10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wijdeven RH, Pang B, van der Zanden SY, Qiao X, Blomen V, Hoogstraat M, Lips EH, Janssen L, Wessels L, Brummelkamp TR, et al. Genome-Wide Identification and Characterization of Novel Factors Conferring Resistance to Topoisomerase II Poisons in Cancer. Cancer Res. 2015;75:4176–4187. doi: 10.1158/0008-5472.CAN-15-0380. [DOI] [PubMed] [Google Scholar]
- 183.Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, Crabtree GR. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601. doi: 10.1038/ng.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Shain AH, Pollack JR. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS One. 2013;8:e55119. doi: 10.1371/journal.pone.0055119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 186.Skulte KA, Phan L, Clark SJ, Taberlay PC. Chromatin remodeler mutations in human cancers: epigenetic implications. Epigenomics. 2014;6:397–414. doi: 10.2217/epi.14.37. [DOI] [PubMed] [Google Scholar]
- 187.Luchini C, Veronese N, Solmi M, Cho H, Kim JH, Chou A, Gill AJ, Faraj SF, Chaux A, Netto GJ, et al. Prognostic role and implications of mutation status of tumor suppressor gene ARID1A in cancer: a systematic review and meta-analysis. Oncotarget. 2015;6:39088–39097. doi: 10.18632/oncotarget.5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, Pawlik TM, Daniel HD, Kannangai R, Offerhaus GJ, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–829. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Huang J, Deng Q, Wang Q, Li KY, Dai JH, Li N, Zhu ZD, Zhou B, Liu XY, Liu RF, et al. Exome sequencing of hepatitis B virus-associated hepatocellular carcinoma. Nat Genet. 2012;44:1117–1121. doi: 10.1038/ng.2391. [DOI] [PubMed] [Google Scholar]
- 190.Zhong R, Liu L, Tian Y, Wang Y, Tian J, Zhu BB, Chen W, Qian JM, Zou L, Xiao M, et al. Genetic variant in SWI/SNF complexes influences hepatocellular carcinoma risk: a new clue for the contribution of chromatin remodeling in carcinogenesis. Sci Rep. 2014;4:4147. doi: 10.1038/srep04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Zhu AX, Chen D, He W, Kanai M, Voi M, Chen LT, Daniele B, Furuse J, Kang YK, Poon RT, et al. Integrative biomarker analyses indicate etiological variations in hepatocellular carcinoma. J Hepatol. 2016;65:296–304. doi: 10.1016/j.jhep.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 192.Zhao H, Wang J, Han Y, Huang Z, Ying J, Bi X, Zhao J, Fang Y, Zhou H, Zhou J, et al. ARID2: a new tumor suppressor gene in hepatocellular carcinoma. Oncotarget. 2011;2:886–891. doi: 10.18632/oncotarget.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Abe H, Hayashi A, Kunita A, Sakamoto Y, Hasegawa K, Shibahara J, Kokudo N, Fukayama M. Altered expression of AT-rich interactive domain 1A in hepatocellular carcinoma. Int J Clin Exp Pathol. 2015;8:2763–2770. [PMC free article] [PubMed] [Google Scholar]
- 194.Dhanasekaran R, Bandoh S, Roberts LR. Molecular pathogenesis of hepatocellular carcinoma and impact of therapeutic advances. F1000Res. 2016;5 doi: 10.12688/f1000research.6946.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, Pfister S, Cho YJ, Zhao K, Crabtree GR. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature. 2013;497:624–627. doi: 10.1038/nature12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Bosse T, ter Haar NT, Seeber LM, v Diest PJ, Hes FJ, Vasen HF, Nout RA, Creutzberg CL, Morreau H, Smit VT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations, TP53 and microsatellite instability in endometrial cancer. Mod Pathol. 2013;26:1525–1535. doi: 10.1038/modpathol.2013.96. [DOI] [PubMed] [Google Scholar]
- 197.Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43. doi: 10.1158/2159-8290.CD-12-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Wu RC, Wang TL, Shih IeM. The emerging roles of ARID1A in tumor suppression. Cancer Biol Ther. 2014;15:655–664. doi: 10.4161/cbt.28411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Chandler RL, Damrauer JS, Raab JR, Schisler JC, Wilkerson MD, Didion JP, Starmer J, Serber D, Yee D, Xiong J, et al. Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat Commun. 2015;6:6118. doi: 10.1038/ncomms7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Anglesio MS, Bashashati A, Wang YK, Senz J, Ha G, Yang W, Aniba MR, Prentice LM, Farahani H, Li Chang H, et al. Multifocal endometriotic lesions associated with cancer are clonal and carry a high mutation burden. J Pathol. 2015;236:201–209. doi: 10.1002/path.4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Huang HN, Lin MC, Huang WC, Chiang YC, Kuo KT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod Pathol. 2014;27:983–990. doi: 10.1038/modpathol.2013.216. [DOI] [PubMed] [Google Scholar]
- 202.Yamamoto S, Tsuda H, Takano M, Tamai S, Matsubara O. Loss of ARID1A protein expression occurs as an early event in ovarian clear-cell carcinoma development and frequently coexists with PIK3CA mutations. Mod Pathol. 2012;25:615–624. doi: 10.1038/modpathol.2011.189. [DOI] [PubMed] [Google Scholar]
- 203.Zhang Q, Yan HB, Wang J, Cui SJ, Wang XQ, Jiang YH, Feng L, Yang PY, Liu F. Chromatin remodeling gene AT-rich interactive domain-containing protein 1A suppresses gastric cancer cell proliferation by targeting PIK3CA and PDK1. Oncotarget. 2016 doi: 10.18632/oncotarget.10060. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Guan B, Rahmanto YS, Wu RC, Wang Y, Wang Z, Wang TL, Shih IeM. Roles of deletion of Arid1a, a tumor suppressor, in mouse ovarian tumorigenesis. J Natl Cancer Inst. 2014;106 doi: 10.1093/jnci/dju146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.He J, Jiang BH. Interplay between Reactive oxygen Species and MicroRNAs in Cancer. Curr Pharmacol Rep. 2016;2:82–90. doi: 10.1007/s40495-016-0051-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Shiota M, Yokomizo A. [Prostate cancer and oxidative stress] Nihon Rinsho. 2016;74 Suppl 3:71–74. [PubMed] [Google Scholar]
- 208.Toyokuni S. Oxidative stress as an iceberg in carcinogenesis and cancer biology. Arch Biochem Biophys. 2016;595:46–49. doi: 10.1016/j.abb.2015.11.025. [DOI] [PubMed] [Google Scholar]
- 209.Khurana RK, Kaur R, Lohan S, Singh KK, Singh B. Mangiferin: a promising anticancer bioactive. Pharm Pat Anal. 2016;5:169–181. doi: 10.4155/ppa-2016-0003. [DOI] [PubMed] [Google Scholar]
- 210.Srivastava KC, Austin RD, Shrivastava D. Evaluation of oxidant-antioxidant status in tissue samples in oral cancer: A case control study. Dent Res J (Isfahan) 2016;13:181–187. doi: 10.4103/1735-3327.178210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Kansanen E, Jyrkkänen HK, Levonen AL. Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic Biol Med. 2012;52:973–982. doi: 10.1016/j.freeradbiomed.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 212.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Menegon S, Columbano A, Giordano S. The Dual Roles of NRF2 in Cancer. Trends Mol Med. 2016;22:578–593. doi: 10.1016/j.molmed.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 214.Gañán-Gómez I, Wei Y, Yang H, Boyano-Adánez MC, García-Manero G. Oncogenic functions of the transcription factor Nrf2. Free Radic Biol Med. 2013;65:750–764. doi: 10.1016/j.freeradbiomed.2013.06.041. [DOI] [PubMed] [Google Scholar]
- 215.Geismann C, Arlt A, Sebens S, Schäfer H. Cytoprotection “gone astray”: Nrf2 and its role in cancer. Onco Targets Ther. 2014;7:1497–1518. doi: 10.2147/OTT.S36624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Araujo LH, Timmers C, Bell EH, Shilo K, Lammers PE, Zhao W, Natarajan TG, Miller CJ, Zhang J, Yilmaz AS, et al. Genomic Characterization of Non-Small-Cell Lung Cancer in African Americans by Targeted Massively Parallel Sequencing. J Clin Oncol. 2015;33:1966–1973. doi: 10.1200/JCO.2014.59.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Amaddeo G, Guichard C, Imbeaud S, Zucman-Rossi J. Next-generation sequencing identified new oncogenes and tumor suppressor genes in human hepatic tumors. Oncoimmunology. 2012;1:1612–1613. doi: 10.4161/onci.21480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Shibata T, Aburatani H. Exploration of liver cancer genomes. Nat Rev Gastroenterol Hepatol. 2014;11:340–349. doi: 10.1038/nrgastro.2014.6. [DOI] [PubMed] [Google Scholar]
- 219.Zavattari P, Perra A, Menegon S, Kowalik MA, Petrelli A, Angioni MM, Follenzi A, Quagliata L, Ledda-Columbano GM, Terracciano L, et al. Nrf2, but not β-catenin, mutation represents an early event in rat hepatocarcinogenesis. Hepatology. 2015;62:851–862. doi: 10.1002/hep.27790. [DOI] [PubMed] [Google Scholar]
- 220.Nishida N, Kudo M. Oxidative stress and epigenetic instability in human hepatocarcinogenesis. Dig Dis. 2013;31:447–453. doi: 10.1159/000355243. [DOI] [PubMed] [Google Scholar]
- 221.Karin M, Dhar D. Liver carcinogenesis: from naughty chemicals to soothing fat and the surprising role of NRF2. Carcinogenesis. 2016;37:541–546. doi: 10.1093/carcin/bgw060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nishida N, Arizumi T, Takita M, Kitai S, Yada N, Hagiwara S, Inoue T, Minami Y, Ueshima K, Sakurai T, et al. Reactive oxygen species induce epigenetic instability through the formation of 8-hydroxydeoxyguanosine in human hepatocarcinogenesis. Dig Dis. 2013;31:459–466. doi: 10.1159/000355245. [DOI] [PubMed] [Google Scholar]
- 223.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–184. doi: 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol. 2011;193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013;51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 226.Shimizu T, Inoue K, Hachiya H, Shibuya N, Aoki T, Kubota K. Accumulation of phosphorylated p62 is associated with NF-E2-related factor 2 activation in hepatocellular carcinoma. J Hepatobiliary Pancreat Sci. 2016;23:467–471. doi: 10.1002/jhbp.364. [DOI] [PubMed] [Google Scholar]
- 227.He G, Karin M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011;21:159–168. doi: 10.1038/cr.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Wilson GS, Tian A, Hebbard L, Duan W, George J, Li X, Qiao L. Tumoricidal effects of the JAK inhibitor Ruxolitinib (INC424) on hepatocellular carcinoma in vitro. Cancer Lett. 2013;341:224–230. doi: 10.1016/j.canlet.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 229.Xie HJ, Bae HJ, Noh JH, Eun JW, Kim JK, Jung KH, Ryu JC, Ahn YM, Kim SY, Lee SH, et al. Mutational analysis of JAK1 gene in human hepatocellular carcinoma. Neoplasma. 2009;56:136–140. doi: 10.4149/neo_2009_02_136. [DOI] [PubMed] [Google Scholar]
- 230.Zhang Y, Qiu Z, Wei L, Tang R, Lian B, Zhao Y, He X, Xie L. Integrated analysis of mutation data from various sources identifies key genes and signaling pathways in hepatocellular carcinoma. PLoS One. 2014;9:e100854. doi: 10.1371/journal.pone.0100854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Kumar M, Zhao X, Wang XW. Molecular carcinogenesis of hepatocellular carcinoma and intrahepatic cholangiocarcinoma: one step closer to personalized medicine? Cell Biosci. 2011;1:5. doi: 10.1186/2045-3701-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Gao C, Xiao G, Hu J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014;4:13. doi: 10.1186/2045-3701-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cervello M, McCubrey JA, Cusimano A, Lampiasi N, Azzolina A, Montalto G. Targeted therapy for hepatocellular carcinoma: novel agents on the horizon. Oncotarget. 2012;3:236–260. doi: 10.18632/oncotarget.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Lachenmayer A, Alsinet C, Savic R, Cabellos L, Toffanin S, Hoshida Y, Villanueva A, Minguez B, Newell P, Tsai HW, et al. Wnt-pathway activation in two molecular classes of hepatocellular carcinoma and experimental modulation by sorafenib. Clin Cancer Res. 2012;18:4997–5007. doi: 10.1158/1078-0432.CCR-11-2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Herencia C, Martínez-Moreno JM, Herrera C, Corrales F, Santiago-Mora R, Espejo I, Barco M, Almadén Y, de la Mata M, Rodríguez-Ariza A, et al. Nuclear translocation of β-catenin during mesenchymal stem cells differentiation into hepatocytes is associated with a tumoral phenotype. PLoS One. 2012;7:e34656. doi: 10.1371/journal.pone.0034656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Yang S, Luo C, Gu Q, Xu Q, Wang G, Sun H, Qian Z, Tan Y, Qin Y, Shen Y, et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget. 2016;7:5461–5469. doi: 10.18632/oncotarget.6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wands JR, Kim M. WNT/β-catenin signaling and hepatocellular carcinoma. Hepatology. 2014;60:452–454. doi: 10.1002/hep.27081. [DOI] [PubMed] [Google Scholar]
- 238.Yam JW, Wong CM, Ng IO. Molecular and functional genetics of hepatocellular carcinoma. Front Biosci (Schol Ed) 2010;2:117–134. doi: 10.2741/s51. [DOI] [PubMed] [Google Scholar]
- 239.Qu B, Liu BR, DU YJ, Chen J, Cheng YQ, Xu W, Wang XH. Wnt/β-catenin signaling pathway may regulate the expression of angiogenic growth factors in hepatocellular carcinoma. Oncol Lett. 2014;7:1175–1178. doi: 10.3892/ol.2014.1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Lu LC, Shao YY, Lee YH, Hsieh MS, Hsiao CH, Lin HH, Kao HF, Ma YY, Yen FC, Cheng AL, et al. β-catenin (CTNNB1) mutations are not associated with prognosis in advanced hepatocellular carcinoma. Oncology. 2014;87:159–166. doi: 10.1159/000362821. [DOI] [PubMed] [Google Scholar]
- 241.Takigawa Y, Brown AM. Wnt signaling in liver cancer. Curr Drug Targets. 2008;9:1013–1024. doi: 10.2174/138945008786786127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Guan CN, Chen XM, Lou HQ, Liao XH, Chen BY, Zhang PW. Clinical significance of axin and β-catenin protein expression in primary hepatocellular carcinomas. Asian Pac J Cancer Prev. 2012;13:677–681. doi: 10.7314/apjcp.2012.13.2.677. [DOI] [PubMed] [Google Scholar]
- 243.Oishi N, Wang XW. Novel therapeutic strategies for targeting liver cancer stem cells. Int J Biol Sci. 2011;7:517–535. doi: 10.7150/ijbs.7.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Merle P, Kim M, Herrmann M, Gupte A, Lefrançois L, Califano S, Trépo C, Tanaka S, Vitvitski L, de la Monte S, et al. Oncogenic role of the frizzled-7/beta-catenin pathway in hepatocellular carcinoma. J Hepatol. 2005;43:854–862. doi: 10.1016/j.jhep.2005.05.018. [DOI] [PubMed] [Google Scholar]
- 245.Kim M, Lee HC, Tsedensodnom O, Hartley R, Lim YS, Yu E, Merle P, Wands JR. Functional interaction between Wnt3 and Frizzled-7 leads to activation of the Wnt/beta-catenin signaling pathway in hepatocellular carcinoma cells. J Hepatol. 2008;48:780–791. doi: 10.1016/j.jhep.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Waly Raphael S, Yangde Z, Yuxiang C. Hepatocellular carcinoma: focus on different aspects of management. ISRN Oncol. 2012;2012:421673. doi: 10.5402/2012/421673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 2016;64:S84–101. doi: 10.1016/j.jhep.2016.02.021. [DOI] [PubMed] [Google Scholar]
- 248.Shin JW, Chung YH. Molecular targeted therapy for hepatocellular carcinoma: current and future. World J Gastroenterol. 2013;19:6144–6155. doi: 10.3748/wjg.v19.i37.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Liu J, Ding X, Tang J, Cao Y, Hu P, Zhou F, Shan X, Cai X, Chen Q, Ling N, et al. Enhancement of canonical Wnt/β-catenin signaling activity by HCV core protein promotes cell growth of hepatocellular carcinoma cells. PLoS One. 2011;6:e27496. doi: 10.1371/journal.pone.0027496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Srisuttee R, Koh SS, Kim SJ, Malilas W, Boonying W, Cho IR, Jhun BH, Ito M, Horio Y, Seto E, et al. Hepatitis B virus X (HBX) protein upregulates β-catenin in a human hepatic cell line by sequestering SIRT1 deacetylase. Oncol Rep. 2012;28:276–282. doi: 10.3892/or.2012.1798. [DOI] [PubMed] [Google Scholar]
- 251.Laurent-Puig P, Zucman-Rossi J. Genetics of hepatocellular tumors. Oncogene. 2006;25:3778–3786. doi: 10.1038/sj.onc.1209547. [DOI] [PubMed] [Google Scholar]
- 252.Zucman-Rossi J, Benhamouche S, Godard C, Boyault S, Grimber G, Balabaud C, Cunha AS, Bioulac-Sage P, Perret C. Differential effects of inactivated Axin1 and activated beta-catenin mutations in human hepatocellular carcinomas. Oncogene. 2007;26:774–780. doi: 10.1038/sj.onc.1209824. [DOI] [PubMed] [Google Scholar]
- 253.Park JY, Park WS, Nam SW, Kim SY, Lee SH, Yoo NJ, Lee JY, Park CK. Mutations of beta-catenin and AXIN I genes are a late event in human hepatocellular carcinogenesis. Liver Int. 2005;25:70–76. doi: 10.1111/j.1478-3231.2004.0995.x. [DOI] [PubMed] [Google Scholar]
- 254.Prange W, Breuhahn K, Fischer F, Zilkens C, Pietsch T, Petmecky K, Eilers R, Dienes HP, Schirmacher P. Beta-catenin accumulation in the progression of human hepatocarcinogenesis correlates with loss of E-cadherin and accumulation of p53, but not with expression of conventional WNT-1 target genes. J Pathol. 2003;201:250–259. doi: 10.1002/path.1448. [DOI] [PubMed] [Google Scholar]
- 255.Murata M, Miyoshi Y, Ohsawa M, Shibata K, Ohta T, Imai Y, Nishikawa M, Iwao K, Tateishi H, Shimano T, et al. Accumulation of beta-catenin in the cytoplasm and the nuclei during the early hepatic tumorigenesis. Hepatol Res. 2001;21:126–135. doi: 10.1016/s1386-6346(01)00116-4. [DOI] [PubMed] [Google Scholar]
- 256.Vilarinho S, Erson-Omay EZ, Harmanci AS, Morotti R, Carrion-Grant G, Baranoski J, Knisely AS, Ekong U, Emre S, Yasuno K, et al. Paediatric hepatocellular carcinoma due to somatic CTNNB1 and NFE2L2 mutations in the setting of inherited bi-allelic ABCB11 mutations. J Hepatol. 2014;61:1178–1183. doi: 10.1016/j.jhep.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 257.Li P, Cao Y, Li Y, Zhou L, Liu X, Geng M. Expression of Wnt-5a and β-catenin in primary hepatocellular carcinoma. Int J Clin Exp Pathol. 2014;7:3190–3195. [PMC free article] [PubMed] [Google Scholar]
- 258.Nejak-Bowen KN, Thompson MD, Singh S, Bowen WC, Dar MJ, Khillan J, Dai C, Monga SP. Accelerated liver regeneration and hepatocarcinogenesis in mice overexpressing serine-45 mutant beta-catenin. Hepatology. 2010;51:1603–1613. doi: 10.1002/hep.23538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Colnot S, Decaens T, Niwa-Kawakita M, Godard C, Hamard G, Kahn A, Giovannini M, Perret C. Liver-targeted disruption of Apc in mice activates beta-catenin signaling and leads to hepatocellular carcinomas. Proc Natl Acad Sci USA. 2004;101:17216–17221. doi: 10.1073/pnas.0404761101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Ding X, Yang Y, Han B, Du C, Xu N, Huang H, Cai T, Zhang A, Han ZG, Zhou W, et al. Transcriptomic characterization of hepatocellular carcinoma with CTNNB1 mutation. PLoS One. 2014;9:e95307. doi: 10.1371/journal.pone.0095307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Li ZQ, Ding W, Sun SJ, Li J, Pan J, Zhao C, Wu WR, Si WK. Cyr61/CCN1 is regulated by Wnt/β-catenin signaling and plays an important role in the progression of hepatocellular carcinoma. PLoS One. 2012;7:e35754. doi: 10.1371/journal.pone.0035754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cieply B, Zeng G, Proverbs-Singh T, Geller DA, Monga SP. Unique phenotype of hepatocellular cancers with exon-3 mutations in beta-catenin gene. Hepatology. 2009;49:821–831. doi: 10.1002/hep.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tornesello ML, Buonaguro L, Tatangelo F, Botti G, Izzo F, Buonaguro FM. Mutations in TP53, CTNNB1 and PIK3CA genes in hepatocellular carcinoma associated with hepatitis B and hepatitis C virus infections. Genomics. 2013;102:74–83. doi: 10.1016/j.ygeno.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 264.Behari J. The Wnt/β-catenin signaling pathway in liver biology and disease. Expert Rev Gastroenterol Hepatol. 2010;4:745–756. doi: 10.1586/egh.10.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Audard V, Grimber G, Elie C, Radenen B, Audebourg A, Letourneur F, Soubrane O, Vacher-Lavenu MC, Perret C, Cavard C, et al. Cholestasis is a marker for hepatocellular carcinomas displaying beta-catenin mutations. J Pathol. 2007;212:345–352. doi: 10.1002/path.2169. [DOI] [PubMed] [Google Scholar]
- 266.Boyault S, Rickman DS, de Reyniès A, Balabaud C, Rebouissou S, Jeannot E, Hérault A, Saric J, Belghiti J, Franco D, et al. Transcriptome classification of HCC is related to gene alterations and to new therapeutic targets. Hepatology. 2007;45:42–52. doi: 10.1002/hep.21467. [DOI] [PubMed] [Google Scholar]
- 267.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Solé M, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–6788. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Yuan RH, Chang KT, Chen YL, Hsu HC, Lee PH, Lai PL, Jeng YM. S100P expression is a novel prognostic factor in hepatocellular carcinoma and predicts survival in patients with high tumor stage or early recurrent tumors. PLoS One. 2013;8:e65501. doi: 10.1371/journal.pone.0065501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Wang Z, Sheng YY, Gao XM, Wang CQ, Wang XY, Lu XU, Wei JW, Zhang KL, Dong QZ, Qin LX. β-catenin mutation is correlated with a favorable prognosis in patients with hepatocellular carcinoma. Mol Clin Oncol. 2015;3:936–940. doi: 10.3892/mco.2015.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yuzugullu H, Benhaj K, Ozturk N, Senturk S, Celik E, Toylu A, Tasdemir N, Yilmaz M, Erdal E, Akcali KC, et al. Canonical Wnt signaling is antagonized by noncanonical Wnt5a in hepatocellular carcinoma cells. Mol Cancer. 2009;8:90. doi: 10.1186/1476-4598-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yuan RH, Jeng YM, Hu RH, Lai PL, Lee PH, Cheng CC, Hsu HC. Role of p53 and β-catenin mutations in conjunction with CK19 expression on early tumor recurrence and prognosis of hepatocellular carcinoma. J Gastrointest Surg. 2011;15:321–329. doi: 10.1007/s11605-010-1373-x. [DOI] [PubMed] [Google Scholar]
- 272.Monga SP. Role of Wnt/β-catenin signaling in liver metabolism and cancer. Int J Biochem Cell Biol. 2011;43:1021–1029. doi: 10.1016/j.biocel.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Brown KK, Toker A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015;7:13. doi: 10.12703/P7-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kudo M. Signaling pathway/molecular targets and new targeted agents under development in hepatocellular carcinoma. World J Gastroenterol. 2012;18:6005–6017. doi: 10.3748/wjg.v18.i42.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, Shokat KM. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang C, Cigliano A, Delogu S, Armbruster J, Dombrowski F, Evert M, Chen X, Calvisi DF. Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis: implications for the treatment of human liver cancer. Cell Cycle. 2013;12:1999–2010. doi: 10.4161/cc.25099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Zhou Q, Lui VW, Yeo W. Targeting the PI3K/Akt/mTOR pathway in hepatocellular carcinoma. Future Oncol. 2011;7:1149–1167. doi: 10.2217/fon.11.95. [DOI] [PubMed] [Google Scholar]
- 278.Chen JS, Wang Q, Fu XH, Huang XH, Chen XL, Cao LQ, Chen LZ, Tan HX, Li W, Bi J, et al. Involvement of PI3K/PTEN/AKT/mTOR pathway in invasion and metastasis in hepatocellular carcinoma: Association with MMP-9. Hepatol Res. 2009;39:177–186. doi: 10.1111/j.1872-034X.2008.00449.x. [DOI] [PubMed] [Google Scholar]
- 279.Augello G, Puleio R, Emma MR, Cusimano A, Loria GR, McCubrey JA, Montalto G, Cervello M. A PTEN inhibitor displays preclinical activity against hepatocarcinoma cells. Cell Cycle. 2016;15:573–583. doi: 10.1080/15384101.2016.1138183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Zhu X, Qin X, Fei M, Hou W, Greshock J, Bachman KE, Wooster R, Kang J, Qin CY. Combined phosphatase and tensin homolog (PTEN) loss and fatty acid synthase (FAS) overexpression worsens the prognosis of Chinese patients with hepatocellular carcinoma. Int J Mol Sci. 2012;13:9980–9991. doi: 10.3390/ijms13089980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Sze KM, Wong KL, Chu GK, Lee JM, Yau TO, Ng IO. Loss of phosphatase and tensin homolog enhances cell invasion and migration through AKT/Sp-1 transcription factor/matrix metalloproteinase 2 activation in hepatocellular carcinoma and has clinicopathologic significance. Hepatology. 2011;53:1558–1569. doi: 10.1002/hep.24232. [DOI] [PubMed] [Google Scholar]
- 282.Su R, Nan H, Guo H, Ruan Z, Jiang L, Song Y, Nan K. Associations of components of PTEN/AKT/mTOR pathway with cancer stem cell markers and prognostic value of these biomarkers in hepatocellular carcinoma. Hepatol Res. 2016 doi: 10.1111/hepr.12687. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 283.Kim DC, Chung WJ, Lee JH, Jang BK, Hwang JS, Kang KJ, Kwon SY. Clinicopathological characteristics of PIK3CA and HBx mutations in Korean patients with hepatocellular carcinomas. APMIS. 2014;122:1001–1006. doi: 10.1111/apm.12245. [DOI] [PubMed] [Google Scholar]
- 284.Li X, Zhang Q, He W, Meng W, Yan J, Zhang L, Zhu X, Liu T, Li Y, Bai Z. Low frequency of PIK3CA gene mutations in hepatocellular carcinoma in Chinese population. Pathol Oncol Res. 2012;18:57–60. doi: 10.1007/s12253-011-9416-5. [DOI] [PubMed] [Google Scholar]
- 285.Zuo Q, Huang H, Shi M, Zhang F, Sun J, Bin J, Liao Y, Liao W. Multivariate analysis of several molecular markers and clinicopathological features in postoperative prognosis of hepatocellular carcinoma. Anat Rec (Hoboken) 2012;295:423–431. doi: 10.1002/ar.21531. [DOI] [PubMed] [Google Scholar]
- 286.Hou W, Liu J, Chen P, Wang H, Ye BC, Qiang F. Mutation analysis of key genes in RAS/RAF and PI3K/PTEN pathways in Chinese patients with hepatocellular carcinoma. Oncol Lett. 2014;8:1249–1254. doi: 10.3892/ol.2014.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Kim H, Park CK, Lee SJ, Rha SY, Park KH, Lim HY. PIK3CA mutations in hepatocellular carcinoma in Korea. Yonsei Med J. 2013;54:883–887. doi: 10.3349/ymj.2013.54.4.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Bassullu N, Turkmen I, Dayangac M, Yagiz Korkmaz P, Yasar R, Akyildiz M, Yaprak O, Tokat Y, Yuzer Y, Bulbul Dogusoy G. The Predictive and Prognostic Significance of c-erb-B2, EGFR, PTEN, mTOR, PI3K, p27, and ERCC1 Expression in Hepatocellular Carcinoma. Hepat Mon. 2012;12:e7492. doi: 10.5812/hepatmon.7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Vinciguerra M, Foti M. PTEN at the crossroad of metabolic diseases and cancer in the liver. Ann Hepatol. 2008;7:192–199. [PubMed] [Google Scholar]
- 290.Zhou L, Huang Y, Li J, Wang Z. The mTOR pathway is associated with the poor prognosis of human hepatocellular carcinoma. Med Oncol. 2010;27:255–261. doi: 10.1007/s12032-009-9201-4. [DOI] [PubMed] [Google Scholar]
- 291.Schmitz KJ, Wohlschlaeger J, Lang H, Sotiropoulos GC, Malago M, Steveling K, Reis H, Cicinnati VR, Schmid KW, Baba HA. Activation of the ERK and AKT signalling pathway predicts poor prognosis in hepatocellular carcinoma and ERK activation in cancer tissue is associated with hepatitis C virus infection. J Hepatol. 2008;48:83–90. doi: 10.1016/j.jhep.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 292.Yu L, Zhang J, Guo X, Li Z, Zhang P. MicroRNA-224 upregulation and AKT activation synergistically predict poor prognosis in patients with hepatocellular carcinoma. Cancer Epidemiol. 2014;38:408–413. doi: 10.1016/j.canep.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 293.Rowinsky EK. Targeting the molecular target of rapamycin (mTOR) Curr Opin Oncol. 2004;16:564–575. doi: 10.1097/01.cco.0000143964.74936.d1. [DOI] [PubMed] [Google Scholar]
- 294.Kang GH, Lee BS, Lee ES, Kim SH, Lee HY, Kang DY. Prognostic significance of p53, mTOR, c-Met, IGF-1R, and HSP70 overexpression after the resection of hepatocellular carcinoma. Gut Liver. 2014;8:79–87. doi: 10.5009/gnl.2014.8.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855–865. doi: 10.1016/j.jhep.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–537. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 297.Kaibori M, Shikata N, Sakaguchi T, Ishizaki M, Matsui K, Iida H, Tanaka Y, Miki H, Nakatake R, Okumura T, et al. Influence of Rictor and Raptor Expression of mTOR Signaling on Long-Term Outcomes of Patients with Hepatocellular Carcinoma. Dig Dis Sci. 2015;60:919–928. doi: 10.1007/s10620-014-3417-7. [DOI] [PubMed] [Google Scholar]
- 298.Liao H, Huang Y, Guo B, Liang B, Liu X, Ou H, Jiang C, Li X, Yang D. Dramatic antitumor effects of the dual mTORC1 and mTORC2 inhibitor AZD2014 in hepatocellular carcinoma. Am J Cancer Res. 2015;5:125–139. [PMC free article] [PubMed] [Google Scholar]
- 299.Janku F, Kaseb AO, Tsimberidou AM, Wolff RA, Kurzrock R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 2014;5:3012–3022. doi: 10.18632/oncotarget.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gao JJ, Shi ZY, Xia JF, Inagaki Y, Tang W. Sorafenib-based combined molecule targeting in treatment of hepatocellular carcinoma. World J Gastroenterol. 2015;21:12059–12070. doi: 10.3748/wjg.v21.i42.12059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Llovet JM, Zucman-Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, Gores G. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2:16018. doi: 10.1038/nrdp.2016.18. [DOI] [PubMed] [Google Scholar]
- 303.Montella L, Palmieri G, Addeo R, Del Prete S. Hepatocellular carcinoma: Will novel targeted drugs really impact the next future? World J Gastroenterol. 2016;22:6114–6126. doi: 10.3748/wjg.v22.i27.6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Zhang J, Wu LY, Zhang XS, Zhang S. Discovery of co-occurring driver pathways in cancer. BMC Bioinformatics. 2014;15:271. doi: 10.1186/1471-2105-15-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Sia D, Villanueva A. Signaling pathways in hepatocellular carcinoma. Oncology. 2011;81 Suppl 1:18–23. doi: 10.1159/000333254. [DOI] [PubMed] [Google Scholar]
- 306.Lu LC, Hsu CH, Hsu C, Cheng AL. Tumor Heterogeneity in Hepatocellular Carcinoma: Facing the Challenges. Liver Cancer. 2016;5:128–138. doi: 10.1159/000367754. [DOI] [PMC free article] [PubMed] [Google Scholar]