

Abstract
Anti-tumor necrosis factor (TNF) antibodies are successfully used in the therapy of inflammatory bowel diseases (IBD). However, the molecular mechanism of action of these agents is still a matter of debate. Apart from neutralization of TNF, influence on the intestinal barrier function, induction of apoptosis in mucosal immune cells, formation of regulatory macrophages as well as other immune modulating properties have been discussed as central features. Nevertheless, clinically effective anti-TNF antibodies were shown to differ in their mode-of-action in vivo and in vitro. Furthermore, the anti-TNF agent etanercept is effective in the treatment of rheumatoid arthritis but failed to induce clinical response in Crohn’s disease patients, suggesting different contributions of TNF in the pathogenesis of these inflammatory diseases. In the following, we will review different aspects regarding the mechanism of action of anti-TNF agents in general and analyze comparatively different effects of each anti-TNF agent such as TNF neutralization, modulation of the immune system, reverse signaling and induction of apoptosis. We discuss the relevance of the membrane-bound form of TNF compared to the soluble form for the immunopathogenesis of IBD. Furthermore, we review reports that could lead to personalized medicine approaches regarding treatment with anti-TNF antibodies in chronic intestinal inflammation, by predicting response to therapy.
Keywords: Mucosal immunology, Lamina propria mononuclear cells, Crohn’s disease, Ulcerative colitis, Transmembrane tumor necrosis factor, Apoptosis
Core tip: To improve the response rates of patients with inflammatory bowel diseases to anti-tumor necrosis factor (TNF) therapy, it is mandatory to understand the pleiotropic effects of these agents. Herein, we overview and discuss several immunological targets of anti-TNF antibody application and highlight the most important findings.
INTRODUCTION
Inflammatory bowel diseases (IBD) are chronic inflammatory disorders mainly affecting the gut. The two main forms are Crohn’s disease (CD) and ulcerative colitis (UC) that differ in several aspects, e.g. regarding the location and distribution of inflammation and in the mucosal cell populations involved in the immune reaction. CD is a segmental, transmural disorder that can affect the whole gastrointestinal tract and type 1 T helper cells have been associated to be involved in the pathogenesis[1]. In contrast, UC is characterized by continuous inflammation of the colon, and a modified Th2 cytokine profile has been described[2]. Additionally, the recently described Th17 cells have been reported to be present in both CD and UC[3]. However, the precise etiology of IBD is still a matter of debate but there is general consensus that genetic predisposition, environmental factors and immunological dysfunction of tolerance against the intestinal microflora are involved in the immunopathogenesis[4]. In this regard, mucosal CD4+ T cells as key mediators in driving immune responses are critically involved in the pathogenesis of IBD and in accordance, a high number of infiltrating T lymphocytes can be found in both CD and UC in the gut[5]. Furthermore, recent studies strongly suggest that the proinflammatory cytokine tumor necrosis factor (TNF) is one of the major pathogenic cytokines involved in the pathogenesis of IBD as elevated levels of TNF are present in the serum of both UC and CD patients[6]. In addition, an elevated number of TNF-secreting cells in the inflamed mucosa of IBD patients has been repeatedly reported[7-9]. Herein, lamina propria mononuclear cells (LPMCs) isolated from colonic biopsies from IBD patients spontaneously produced increased amounts of TNF which correlated with the degree of tissue involvement and mucosal inflammation[10], strengthening the importance of TNF in the inflamed gut.
In accordance, application of antibodies targeting TNF in IBD patients have been shown to induce clinical response in up to 60% of CD patients, and inducing long-term maintenance of remission in a large amount of patients[11-13]. Comparable results have also been described for the therapy of UC patients[14,15]. Nevertheless, the molecular mechanism of action of TNF agents is still discussed and not all commercially available anti-TNF antagonists are effective in the therapy of IBD patients. Regarding the mechanism of action, TNF neutralization, diverse effects on the immune system, outside-to-inside signaling and importantly, induction of direct or indirect apoptosis have been suggested. Furthermore, first studies focusing on successful prediction of clinical response have been conducted, either via expression of various biomarkers[16,17] or by endoscopic in vivo molecular imaging[18]. In this review, TNF biology and different anti-TNF antibodies will be assessed, and the results of different aspects of the biological function of anti-TNF agents for the treatment of IBD will be discussed.
TNF BIOLOGY AND ITS SIGNALING PATHWAY
TNF is a crucial mediator in driving inflammatory processes in the gut. It is produced by a variety of mucosal cells, mainly macrophages and T cells, as a preform on the plasma membrane[19]. In addition, Paneth cells in CD affected segments of the terminal ileum were shown to strongly express TNF RNA in contrast to Paneth cells in normal tissue, indicating an induction under pathogenic conditions[20]. The transmembrane precursor form (mTNF), a homotrimer of 26 kDa subunits, is cleaved by the matrixmetalloproteinase TNF alpha converting enzyme (TACE/Adam17) into a soluble form (sTNF, a homotrimer of 17 kDa monomers)[21-23]. The expression of mTNF by CD14+ macrophages has been reported to be relevant in IBD[24] (Figure 1).
Figure 1.
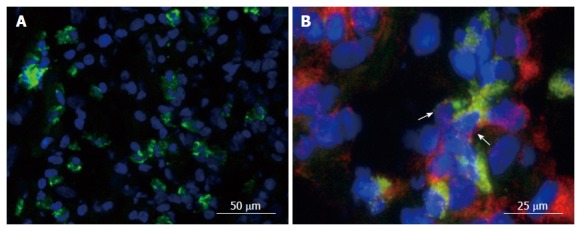
Expression of membrane-bound tumor necrosis factor in the gut. Gut tissue of patients with Crohn’s disease (CD) was cryo-frozen and stained for cell markers by immunofluorescence. Nuclei were counterstained with DAPI. A: Staining for mTNF (green). B: Staining for mTNF (green) and CD14 (red). Co-expressing cells are labeled with an arrow.
Both forms of TNF are biologically active and signal through two distinct receptors that differ in molecule mass: the 55-kDa TNFR1 (TNFRSF1A/CD120a) and the 75-kDa TNFR2 (TNFRSF1B/CD120b) glycoproteins[25]. TNFR2 is mainly expressed on lymphocytes and endothelial cells, whereas TNFR1 is ubiquitously expressed and possesses an intracellular death domain[26]. TNF and its receptors are crucially involved in the pathogenesis of IBD. For example, elevated levels of the soluble form of TNFR1 and TNFR2 have been detected in both CD and UC patients and their expression correlated with disease activity[27].
Signal transduction of the membrane-bound form of TNF can be transmitted through both TNFR1 and TNFR2, whereas sTNF mainly signals through TNFR1. Binding affinity studies revealed that sTNF preferentially binds to TNFR1 with higher affinity[28]. In contrast, TNFR2 is mainly activated by mTNF[29]. Activation of TNFR1 by TNF induces an intracellular signaling cascade with pleiotropic effects involving apoptosis, cell proliferation or cytokine secretion. Activation of the nuclear factor kappa B (NFκB) following stimulation of TNFR1 results in translocation to the nucleus and transcriptional upregulation of several genes such as IL-8, IL-1, IL-6, COX2 and TNF[21]. Alternatively, TNFR1 can activate a Caspase-8 dependent signaling pathway via FADD resulting in apoptosis. The TNFR2 pathway does not contain a death domain and its stimulation can result in proliferation, migration and cytokine production such as IL-1 and IL-6. Furthermore, binding of mTNF to TNFR2 not only activates an intracellular signaling pathway, but can also result in reverse signaling within the TNF-expressing cell[30] which will be later discussed in detail.
The role of the receptors of TNF in the pathogenesis of IBD remains only partly understood. A study using a colitis mouse model suggests that both TNFR1 and TNFR2 have protective functions in intestinal inflammation[31]. Herein, intestinal inflammation was provoked by oral application of dextran sulfate in mice deficient for TNFR1 or TNFR2 as well as wildtype controls. TNFR1 or TNFR2 ablation resulted in exacerbation of colitis, possibly due to increased apoptosis of colonic epithelial cells[31]. However, in other studies a central role of TNFR2 in mucosal inflammation has been proposed. For example, mutations in the gene of TNFR2 have been linked with IBD[32,33], suggesting that this polymorphism could increase the disease risk. In accordance, its overexpression drives inflammation in a transgenic mouse stain overexpressing human TNFR2, causing a severe multiorgan inflammatory syndrome mainly affecting liver, pancreas, kidney and lung[34]. Regarding IBD, TNFR2 expression was found to be upregulated on lamina propria and peripheral blood T cells in patients with CD[35]. In accordance, TNFR2 was found to promote experimental colitis. Herein, T cells overexpressing TNFR2 were transferred into SCID mice that do not express T and B cells. In comparison to SCID mice that received wildtype T cells, transfer of T cells overexpressing TNFR2 resulted in more severe colitis and enhanced expression of T helper cells type 1[35], underlining the importance of TNFR2 in intestinal inflammation. It could moreover be shown that activation of mucosal TNFR2 expressing CD4+ T cells by mTNF expressing CD14+ led to heightened resistance of the intestinal lymphocytes to apoptosis, resulting in perpetuation of chronic intestinal inflammation in IBD[24].
STRUCTURE AND APPLICATION OF ANTI-TNF ANTIBODIES
Infliximab
In 1998, infliximab (Remicade®) was the first antibody targeting TNF to be approved in the United States for the treatment of CD and later for the treatment of UC. It is a chimeric monoclonal antibody with 25% murine and 75% human sequences. Infliximab therapy is intravenously initiated at weeks 0, 2 and 6 and then applied every 8 wk for maintenance of remission. The starting concentration for both CD and UC is 5 mg/kg[6].
CT-P13
The infliximab biosimilar CT-P13 (Remsima®, Inflectra®) was approved in Europe for the treatment of both CD and UC based on a comprehensive non-clinical comparability exercise and extrapolation of clinical data from two studies with rheumatoid arthritis patients. The biosimilar product is highly similar to its originator biological drug infliximab and is therefore clinically used in the same way.
Adalimumab
In contrast to infliximab, adalimumab (Humira®) is a monoclonal human antibody produced by CHO cells. It is approved for the treatment of both CD and UC. Adalimumab is administered subcutaneously and can be injected by the patient himself. At therapy initiation, adalimumab is administered at 160 mg and then 80 mg two weeks later. For maintenance of remission adalimumab is administered every 2 wk at 40 mg.
Golimumab
In 2013 golimumab (Simponi®), a fully human monoclonal antibody was approved for the treatment of UC[36]. Like adalimumab, it is administered subcutaneously. The induction dose is 200 mg and then 100 mg two weeks later. Maintenance of remission is achieved by 50 mg (< 80 kg body weight) and 100 mg (≥ 80 kg bodyweight) every 4 wk.
Certolizumab pegol
Certolizumab pegol (Cimzia®), a PEGylated humanized Fab’ fragment is approved for the treatment of CD in the United States and Switzerland[37] but not by the European Medicines Agency. In contrast to the other anti-TNF antibodies, this agent does not possess a Fc fragment and correspondingly, is not recognized by Fc receptors. The subcutaneous administration is performed at week 0, 2 and 4 with 400 mg. For maintenance of remission, certolizumab pegol is given every 4 wk in a dose of 400 mg.
Etanercept
Etanercept (Enbrel®) is a chimeric fusion protein that consists of the p75 part of TNFR2 and a human IgG1 Fc domain. Importantly, in combination with methotrexate, etanercept has similar efficacy in the therapy of rheumatoid arthritis as infliximab and adalimumab[38]. It nevertheless failed to be effective in a clinical study with CD patients, when 25 mg were subcutaneously applied twice per week[39]. Etanercept is therefore not approved for treatment of CD.
MECHANISM OF ACTION OF ANTI-TNF ANTIBODIES
Although anti-TNF antibodies are approved for the therapy of IBD and other inflammatory diseases for many years, the mechanism of action is still a matter of debate. Initially, it was suggested that anti-TNF agents inactivate the proinflammatory cytokine TNF by direct neutralization, thus resulting in suppression of inflammation. Given the complexity of TNF signaling, a lot of studies indicate that anti-TNF antibodies exert more complex functions beyond simple blockade. Furthermore, functional characterization of etanercept and other anti-TNF agents can elucidate the signaling pathways that are important in IBD in contrast to other autoimmune diseases such as rheumatoid arthritis. In the following part we will list several described effects of anti-TNF antibody therapy that are suggested to be relevant in IBD (see also Figure 2).
Figure 2.
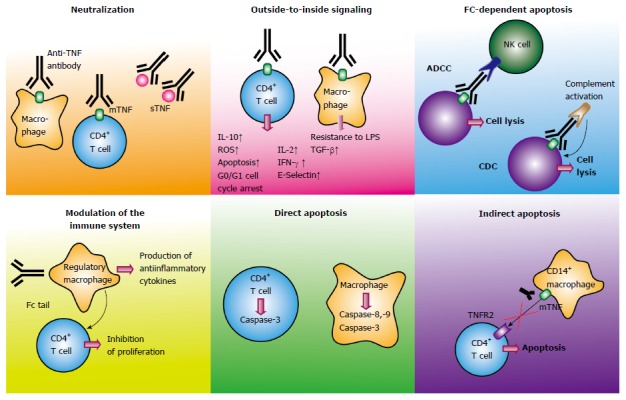
Mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel disease. Schematic illustration of different modes of action of anti-TNF antibodies in inflammatory bowel diseases. TNF: Tumor necrosis factor; mTNF: Membrane-bound TNF; sTNF: Soluble TNF; TNFR: TNF receptor; NK cell: Natural killer cell; ADCC: Antibody-dependent cellular cytotoxicity; CD: Crohn’s disease; CDC: Complement-dependent cytotoxicity; LPS: Lipopolysaccharide.
TNF NEUTRALIZATION
Although all anti-TNF agents have the same target, the affinity of the antibodies to TNF has been reported not to be equal. An overview of respective affinities and avidities of anti-TNF agents is given in Tables 1 and 2. In TNF bioassays, infliximab, etanercept, adalimumab, certolizumab pegol and golimumab all neutralize sTNF[23]. However, using the method of surface plasmon resonance, golimumab was shown to bind to soluble TNF with a similar affinity as etanercept, but with greater affinity than infliximab and adalimumab, the later showing the weakest affinity in the used setting[40]. However, affinity of adalimumab and infliximab were reported to be similar although less compared to etanercept in another study[41]. Regarding avidity, etanercept was shown to bind to sTNF with a 10- to 20-fold greater avidity as compared to adalimumab or infliximab[41].
Table 1.
Comparison of affinity to tumor necrosis factor by anti-tumor necrosis factor agents
| Target | Method /source | Affinity | Ref. |
| sTNF | SPR | GOL = ETA > IFX > ADA | [40] |
| sTNF | SPR | ETA > ADA = IFX | [41] |
| mTNF | mTNF-expressing cells (Sp2/0-11A5-1 myeloma cells) | IFX = ADA = ETA | [41] |
| mTNF | mTNF-expressing cells (K2 murine myeloma cells) | IFX = GOL > ADA > ETA | [40] |
| mTNF | PBMCs (T cells) | IFX = ADA > Certo >> No binding of ETA | [42] |
| Flow cytometry | |||
| mTNF | PBL | IFX >> No binding of ETA | [43] |
| Flow cytometry | |||
| sTNF | Immunoassay | IFX > ETA (rapid dissociation) | [44] |
ADA: Adalimumab; ETA: Etanercept; GOL: Golimumab; IFX: Infliximab; Certo: Certolizumab pegol; PBL: peripheral blood lymphocytes; TNF: Tumor necrosis factor; mTNF: Membrane-bound TNF; sTNF: Soluble TNF; SPR: Surface plasmon resonance.
Table 2.
Comparison of avidity to tumor necrosis factor by anti-tumor necrosis factor agents
| Target | Method/source | Avidity | Ref. |
| sTNF | KinExA® | ETA > ADA | [41] |
| (automated flow immunoassay system) | IFX | ||
| mTNF | [44] | IFX > ETA | [44] |
ADA: Adalimumab; ETA: Etanercept; IFX: Infliximab; TNF: Tumor necrosis factor; mTNF: Membrane-bound TNF; sTNF: Soluble TNF.
For mTNF, affinity of anti-TNF antibodies has been reported to be lower than to sTNF[40,41]. Using mTNF-transfected cells, binding affinities of infliximab, adalimumab and etanercept were shown to be similar[41]. In contrast, affinities for golimumab and infliximab to another mTNF-expressing cell line were comparable and greater than that of adalimumab, whereas all three anti-TNF agents had significantly greater affinity than etanercept[40]. Furthermore, in other studies binding of etanercept to mTNF could not be detected, perhaps due to the fact that peripheral blood mononuclear cells (PBMCs) and not cell lines stably expressing mTNF were examined[42,43], arising the question to what extent the binding of etanercept to mTNF expressing cell lines represents the situation in vivo. Furthermore and in discrepancy to the above mentioned study, etanercept was shown to form relatively unstable complexes with sTNF, resulting in fast release of dissociated TNF in contrast to infliximab that bound to both sTNF and mTNF by forming stable complexes with high stability[44]. However, interpretations of all presented data have to take into account the different cells, methods and techniques used for respective assessment of affinity, which could have a profound effect on the data obtained.
Importantly, another aspect is the variation of the different anti-TNF agents in regard to cross-linking towards mTNF. Herein, it has been reported that up to three infliximab molecules can bind to each TNF homotrimer. In contrast, etanercept was shown to bind to TNF trimer in a 1:1 ratio, suggesting that not all binding sides are blocked[44]. This aspect together with the fact that infliximab forms more stable complexes with mTNF[44] is a possible explanation why infliximab is effective in the therapy of IBD, whereas etanercept failed to show any clinical efficacy in a trial with Crohn’s disease patients and is therefore not approved for IBD treatment. Recent findings indicate that the neutralization of mTNF rather than sTNF is crucial in the therapy of IBD. For example, in a mouse model of IBD the neutralization of sTNF alone was ineffective, in contrast to anti-TNF antibodies that block both sTNF and mTNF, indicating that neutralization of mTNF is crucial in the therapy of IBD[45]. The pivotal role of mTNF was further demonstrated in an experimental colitis model by transferring CD4+ T cells into RAG2 deficient mice[46]. Herein, by transferring TNF mutant T cells, disease activity was compared in absence of TNF, in absence of sTNF and in presence of both sTNF and mTNF. Whereas the expression of TNF was essential for the induction of colitis, the absence of sTNF did not protect the mice from intestinal inflammation[46], clearly underlining the pathogenic role of mTNF in the pathogenesis of mucosal inflammation in the intestine.
MODULATION OF THE IMMUNE SYSTEM
Cytokines
Cytokines as mediators of inflammation are in imbalance in IBD. On the one hand proinflammatory cytokine levels such as TNF, IL-13, IL-17 or IFN-γ[1-3,6] are elevated and drive the massive tissue damage in the gut. On the other hand, anti-inflammatory cytokines that usually regulate overwhelming immune reactions are supposed to be also involved in the pathogenesis of IBD. For example, mice deficient of IL-10 spontaneously develop a chronic intestinal inflammation, which is dependent on the influence of the microflora[47]. Interestingly, the enterocolitis exacerbates in mice deficient for both IL-10 and TNF, indicating that TNF has some protective effects in this context[48]. Crucial mediators of inflammation in the gut are neutrophils and granulocytes that accumulate in inflamed areas and secrete proinflammatory cytokines, chemokines and recruit effector cells to the side of inflammation. In this setting, Agnholt et al[49] reported that GM-CSF, a growth factor for granulocytes and neutrophils, was decreased in CD patients after application of infliximab. In accordance, also reduced peripheral blood neutrophils and diminished infiltration of polymorphonuclear cells in the lamina propria were observed after anti-TNF application. Furthermore, in vitro experiments using an intestinal T cell culture system showed that GM-CSF secretion by T cells was inhibited by infliximab, suggesting direct effects of infliximab on T cells. Apart from GM-CSF, the release of the proinflammatory cytokine IL-1 beta after stimulation of human monocytes with lipopolysaccharide (LPS) was inhibited by anti-TNF agents. Herein, certolizumab pegol was the most potent inhibitor. Infliximab and adalimumab were also able to inhibit IL-1 beta release at higher concentrations whereas etanercept only partially prevented IL-1 beta release[50]. Furthermore, in vitro analysis using PBMCs from patients with UC showed that incubation of the cells with infliximab inhibited secretion of the proinflammatory cytokines IFN-γ, IL-13, IL-17A and TNF[51], cytokines reported to be critically involved in the pathogenesis of IBD. In addition, infliximab reduced the expression of TNF, IL-1, IL-6 and IL-8 by LPS-stimulated monocytes in vitro[21,52]. In addition, the key Th1 cytokine IL-12 and the anti-inflammatory cytokine IL-10 were described to be affected by anti-TNF antibodies. Monocytes that were isolated from blood of healthy controls or CD patients expressed less IL-10 or IL-12 when they were incubated in the presence of adalimumab or infliximab, whereas etanercept had no effect in this setting[53].
Interestingly, a recent study analysed the regulation of 14169 different genes by anti-TNF antibodies and compared the changes in anti-TNF responders versus non-responders. Intestinal biopsies from CD patients were used and compared to controls[54]. They identified some genes that were regulated by anti-TNF treatment, such as IL-6, CD69, GPR183 and MMP9. Interestingly, the proinflammatory cytokine IL-6 was downregulated by anti-TNF agents but independently of clinical response to therapy. Of note, IL-1 beta and IL-17A were found to be significantly upregulated in patients with active CD and patients refractory to anti-TNF, in accordance with an enhanced Th17 cell population in IBD[3], thereby representing possible targets in anti-TNF refractory patients. In contrast to this study, Katz el al[55] found IL-17 levels not to be affected by anti-TNF therapy in CD patients but showed IL-2 to be significantly decreased during anti-TNF therapy. These contradictory findings may reflect biological differences between gene expression and protein expression.
T helper cell subsets
T cells are critically involved in the pathogenesis of IBD and therefore, modulation of immune responses by influencing the presence of T helper cell subsets is suggested to be an important effect of anti-TNF agents. Response and clinical remission could be the result of either a reduction in proinflammatory T cell subsets such as Th1, Th2 or Th17 cells or an enhanced presence of anti-inflammatory T helper cells, or both. In this regard, several studies investigated the number of pro- and anti-inflammatory T helper cell subsets before and after anti-TNF therapy. The influence of infliximab in vitro on isolated T cells from the peripheral blood of UC patients was examined by Dahlén et al[51]. The T cell activation marker CD25 was reduced on both CD4+ and CD8+ blood T cells and their proliferation was inhibited after incubation with infliximab. In accordance, the levels of proinflammatory cytokines such as IFN-γ, IL-13, IL-17A, TNF and granzyme A were inhibited. The authors also investigated effects of infliximab on regulatory T cell populations; however, the results regarding CD4+ Foxp3+ T cells were ambiguous[51]. Importantly, effects of infliximab on blood or mucosal T cells were shown not to be identical. Herein, Li et al[56] described that infliximab therapy resulted in an increase in CD4+CD25+Foxp3+ and CD4+CD25-Foxp3+ regulatory T cell populations in the blood of both UC and CD patients; in contrast, Foxp3 mRNA and protein were downregulated in the mucosa, possibly reflecting increased apoptosis rates or downregulated T cell activation in the intestine during therapy. Other studies described general reduction of T cells in the mucosa after infliximab therapy for both CD[57] and UC patients[58]. A direct effect of adalimumab on T cell populations could be elegantly demonstrated by Maggi et al[59]. Local injection of adalimumab along perianal fistulas in CD patients resulted in decrease of infiltrating CD4+ CD161+ T cells whereas no systemic effects on circulating T cells could be detected[59].
In addition, infliximab has been shown not only to suppress CD4 T cell activation, but also to downregulate IL-21 expression by mucosal CD4+ T cells and consequently, inhibit Th17 differentiation. Using intestinal biopsies of CD patients before and after infliximab therapy, the authors could clearly show that intestinal mucosal healing during therapy was due to down-regulation of mucosal infiltration of the highly proinflammatory Th17 cells through diminished IL-21 expression, a cytokine that is pivotal to Th17 cell differentiation[60]. In summary, these studies clearly indicate that effects of anti-TNF antibodies on T cells are complex and differ between peripheral and mucosal T cells, as well as in vivo and in vitro.
Epithelial cells, angiogenesis and immunosuppressive macrophages
In addition to shifts in cell populations, effects of anti-TNF antibodies on the epithelial barrier function have been also examined. For example, adalimumab has been shown to prevent barrier dysfunction induced by TNF in vitro using the intestinal epithelial cell lines Caco-2 and T-84[61]. Furthermore, a role of infliximab in restoring epithelial barrier dysfunction in CD has been described. Herein, downregulation of epithelial apoptosis was noticed in 10 of 11 patients undergoing therapy with infliximab[62]. Additionally, effects of anti-TNF antibodies on the mucosal microcirculation have been reported. In this context, infliximab was shown to downregulate CD40 expression by intestinal microvessels in vivo in the mucosa of CD patients on the one hand and to reduce the levels of sCD40L in the circulation on the other hand, indicating an inhibition of vascular inflammation in the gut[63]. In addition, infliximab downregulated mucosal angiogenesis in CD patients[64]. A direct effect of infliximab on myofibroblasts has been described by Di Sabatino et al[65]. Myofibroblasts isolated from CD patients expressed more mTNF than control myofibroblasts and infliximab significantly augmented the migration ability from CD myofibroblasts in a tissue inhibitor of metalloproteinases (TIMP)-1-dependent manner, suggesting another mode-of-action of anti-TNF antibodies that results in enhanced mucosal healing. However, the TIMP-1 production was also increased by adalimumab and etanercept, so it is still unclear if this effect is relevant for induction of clinical remission in CD. Interestingly, the induction of immunosuppressive macrophages has been suggested as mechanism of action of anti-TNF antibodies[42]. Therefore, the influence of infliximab, adalimumab, certolizumab pegol and etanercept was investigated in PBMCs from healthy controls. The T cell proliferation was diminished by infliximab and adalimumab in a mixed lymphocyte reaction, whereas certolizumab pegol and etanercept had no effect. The inhibition of proliferation was driven by immunosuppressive CD14+ macrophages that secreted large amounts of the anti-inflammatory cytokine IL-10 and were activated by the Fc region of the antibodies[42]. The same group later showed that regulatory macrophages were induced in IBD patients with mucosal healing upon infliximab treatment and functionally, these macrophages had the ability to induce wound healing in vitro[66].
In summary, several effects of anti-TNF antibodies on the immune system have been described. The reduction of proinflammatory T cell subsets on the one hand and the induction of regulatory T cells on the other hand clearly contributes to resolution of inflammation. In addition, the induction of regulatory macrophages[42] and effects on mucosal healing[61,62,65] have been suggested.
OUTSIDE-TO-INSIDE SIGNALLING
As mentioned before, activation of the membrane-bound form of TNF can result in bidirectional signalling. The function of mTNF not only as a ligand but also as a receptor is called “outside-to-inside signal” or reverse signalling. Reverse signalling by mTNF on monocytes/macrophages confers resistance to bacterial LPS[67], suggesting that this pathway represents a silencing signal. A very recent paper demonstrated that the immune-suppressive effect of mTNF following LPS stimulation in macrophages was mediated by TGF-β via activation of the MAPK kinase, interestingly this effect was mediated by infliximab and golimumab whereas etanercept failed to induce TGF-β in human macrophages[68]. Functional studies showed that LPS resistance downstream of mTNF is mediated through protein kinase-C dependent and independent pathways in monocytic cells[67]. In addition, enhanced secretion of IL-2 and IFN-γ[69] and an upregulation of E-selectin in T cells have been reported[70]. Furthermore, apoptosis induction as consequence of reverse signalling has also been proposed in CD patients[43,71]. A study by Mitoma et al[72] shed light on the reverse signalling pathway induced by infliximab and etanercept. Herein, human Jurkat T cells were stably transfected with a wild-type and a mutant form of mTNF. E-selectin expression was induced by both agents, but only infliximab induced the expression of the anti-inflammatory cytokine IL-10, activation of ROS accumulation and apoptosis by upregulation of Bax and Bak[72]. Interestingly, their finding that infliximab further induced G0/G1 cell cycle arrest elucidated another mechanism of action of anti-TNF antibodies. Later the induction of cell cycle arrest was shown to be mediated by adalimumab as well[73]. In the context of reverse signalling, a downregulation of the growth and differentiation factor-1 (GDF-1) mediated by infliximab and certolizumab pegol has been described[74]. Using gene expression assays, GDF-1 was found to be upregulated in inflamed tissue from CD patients, and downregulated after infliximab treatment in clinical responders. Furthermore, GDF-1 was shown to act as proinflammatory mediator by IL-6 and STAT-3 induction[74], indicating that a down-regulation of proinflammatory effectors can also be a consequence of reverse signalling by anti-TNF agents in contrast to immune-suppressive effects.
APOPTOSIS
The programmed cell death of immune cells is a fundamental mechanism of resolution of inflammation. Apoptosis induction by anti-TNF antibodies has therefore been addressed by several groups. The induction of apoptosis can either be direct or indirect as side effect of TNF signaling. Furthermore, apoptosis can be induced by the Fc region of anti-TNF antibodies involving the complement or natural killer cells or can be a consequence of the activation of TNF itself by antibody binding. An overview of the apoptosis inducing capabilities of the different anti-TNF antibodies is given in Table 3.
Table 3.
Mechanism of action of anti-tumor necrosis factor agents
| Agent | Cell type | Mechanism of action | Ref. |
| ADA | CHO cell line stably expressing mTNF cocultured with human PBMCs | Induction of ADCC | [75] |
| ADA | CHO cell line stably expressing mTNF with human normal serum | Induction of CDC | [75] |
| IFX, ADA, ETA, Certo | TNF6.5 cells (murine NSO cell line stably expressing human TNF) with baby rabbit complement | CDC: IFX, ADA, ETA ++; Certo - | [50] |
| IFX, ADA, ETA, Certo | TNF6.5 cells in coculture with CD14-depleted PBMCs | ADCC: IFX, ADA ++, ETA+, Certo - | [50] |
| IFX, ADA, ETA, Certo | Peripheral blood lymphocytes | Apoptosis: IFX, ADA ++; ETA+, Certo - | [50] |
| IFX, ADA, ETA, Certo | Peripheral blood monocytes | Apoptosis: IFX, ADA ++; ETA+, Certo - | [50] |
| IFX, ADA | mTNF transfected cell line (SP2/0-11A5-1, murine) | Induction of CDC | [41] |
| IFX, ADA | Activated human PBMCs | No induction of CDC | [41] |
| GOL, ADA, IFX, Certo | mTNF expressing Jurkat cells with PBMCs/human serum | ADCC, CDC: GOL, ADA, IFX ++; ETA+, Certo- | [77] |
| ADA, IFX, GOL, Certo | mTNF expressing Jurkat cells | Apoptosis: ADA, IFX ++, | [77] |
| GOL+, Certo+ | |||
| ADA, IFX, ETA | cultured monocytes | Caspase-dependent apoptosis: ADA, IFX ++; ETA - | [53] |
| IFX, ETA | Peripheral blood lymphocytes and lamina propria T cells | Caspase-3 activation and apoptosis: IFX +; ETA - | [43] |
| ADA, IFX, ETA, Certo | Intestinal CD4/CD14 cells from IBD patients | Apoptosis by targeting mTNF/TNFR2: ADA, IFX, Certo +; ETA - | [24] |
Apoptosis induction of anti-TNF agents. ++: Strong effect; +: Effect visible; -: No effect; ADA: Adalimumab; ETA: Etanercept; IFX: Infliximab; Certo: Certolizumab pegol; GOL: Golimumab; TNF: Tumor necrosis factor; mTNF: Membrane-bound TNF; sTNF: Soluble TNF; PBMCs: Peripheral blood mononuclear cells; ADCC: Antibody-dependent cellular cytotoxicity.
ANTIBODY-DEPENDENT CELLULAR CYTOTOXICITY AND COMPLEMENT-DEPENDENT CYTOTOXICITY
Antibody-dependent cellular cytotoxicity (ADCC) is a mechanism of the adapted immune system in order to kill antibody-tagged target cells, for example infected cells. Therefore, after binding of an antibody to its target cell, the Fc domain is recognized by the Fc receptor of effector immune cells, typically natural killer cells. The natural killer cell then releases cytotoxic proteins such as perforins and granzymes that subsequently results in lysis of the target cell. ADCC is a mechanism of action of anti-TNF antibodies that possess a Fc domain. A recent study indicated that ADCC measured as cell lysis of co-incubation of mTNF-expressing CHO cells and PBMCs could be induced by adalimumab in up to 76% of cells in contrast to a control isotype that caused ADCC in less than 5% of the cells[75]. In a comparative study, infliximab and adalimumab were shown to induce ADCC in a similar way and more potent as etanercept whereas certolizumab pegol did not show any effect due the fact that the later agent does not possess the Fc domain[50].
Apart from ADCC, binding of antibodies to a target cells can result in complement activation. The complement consists of several different proteins that act as a cascade which finally leads to the formation of a membrane attacking complex, inducing a pore within the cell and subsequent cell death. In a CDC assay, 10 μg/mL adalimumab were shown to induce 90.6% cell death in the presence of heat-inactivated human serum[75]. Comparing different agents, infliximab and adalimumab had the same capacity in inducing CDC-dependent cell death and were more effective than etanercept and as expected, certolizumab pegol did not show any effect[50], which was in accordance to other studies[41,73]. Of note, CDC was induced in mTNF-transfected cells by adalimumab and infliximab, but not in activated human PBMCs[41], raising the question if cell lines over-expressing mTNF reflect the situation in vivo. In a functional study, affinity of infliximab and adalimumab to FcRγII and FcRγIII receptors was found to be low but significantly increased in the presence of exogenous TNF[76]. Similarly, both antibodies bound to the complement protein C1q in the presence of TNF. In both settings, etanercept binding was shown to be low. The more recently approved anti-TNF antibody golimumab induced both CDC and ADCC to a similar extent as infliximab and adalimumab[77].
Fc-mediated apoptosis has been shown to be most potently induced by infliximab and adalimumab. However, the fact that certolizumab pegol is effective in CD despite its incapacity to induce ADCC or CDC strongly suggests that Fc-mediated apoptosis by anti-TNF antibodies is not a central mechanism of action.
APOPTOSIS OF INFLAMMATORY IMMUNE CELLS
Apoptosis of inflammatory immune cells as consequence of anti-TNF treatment has been discussed as a major mechanism of action explaining the fast onset of therapeutic effects upon successful therapy. Thus, the induced cell death of monocytes/macrophages or T cells has been analyzed. Regarding monocytes, in a comparative study both adalimumab and infliximab induced apoptosis in cultured monocytes rapidly in a caspase-dependent way whereas etanercept did not[53]. In addition, apoptosis induction via transmembrane TNF in a human monocytic cell line by adalimumab and infliximab could be detected in a chimeric mouse model and furthermore, apoptosis could be abrogated by in vivo treatment with a pan-caspase inhibitor, indicating a caspase-dependent mechanism of action[78]. Infliximab-induced apoptosis of monocytes required the activation of members of the caspase-family, such as caspase-8, -9 and -3 in peripheral blood monocytes derived from CD patients[79]. Activation of caspase was induced independently from the CD95/CD95L signaling pathway by release of mitochondrial cytochrome C, apparently triggered by Bax and Bak[79].
The effect of anti-TNF agents on lymphocytes was studied by Van den Brande et al[43]. They could show that etanercept, which is ineffective in the treatment of CD, in contrast to infliximab was incapable of inducing apoptosis in peripheral blood lymphocytes from healthy controls and lamina propria T cells from CD patients. Here, infliximab rapidly activated caspase 3 which possibly can also be a consequence of reverse signaling. Later the same authors demonstrated that this was also relevant in vivo. A rapid increase of apoptosis was detected in experimental colitis and importantly, also in CD patients where infliximab mediated apoptosis occurred within 24 h after starting the therapy[80]. In this in vivo molecular imaging study, patients underwent single-photon emission computer tomography (SPECT) 24 h after infliximab infusion, and apoptosis was measured by radiolabeled annexinV uptake using scintigraphic imaging. A strong correlation of detected apoptosis signals and diseased regions was observed and furthermore, annexinV uptake was significantly higher in patients subsequently responding to infliximab therapy. In addition, in some patients LPMCs from biopsies were isolated and a marked increase in apoptosis of CD4 T cells after infliximab treatment was observed[80]. Another study analyzing apoptotic markers such as Bcl-2, Bax, Caspase3 and Fas in biopsies from CD patients before and after infliximab/adalimumab therapy showed that in LPMCs, a significant increase of active Caspase-3 and an increase in the pro-apoptotic Bax/Bcl-2 ratio could be detected upon anti-TNF therapy, but no change of TNFR1 or Fas expression could be measured[81].
In recent studies of our group, another mechanism of action apart from direct induction of apoptosis by clinically effective anti-TNF antibodies was identified. We showed that T cell apoptosis was only induced if mucosal T cells expressing TNFR2 were co-cultivated with mTNF-expressing CD14+ macrophages[24]. Binding of mTNF to TNFR2 on T cells resulted in activation of TNFR-2 dependent signaling pathways leading to TRAF2 and NF-κB induction followed by heightened IL-6 production, and subsequently resulting in T cell resistance to apoptosis, which leads to perpetuation of intestinal inflammation. This concept proposes an indirect apoptosis induction by neutralizing the mTNF binding side for TNFR2 by anti-TNF antibody treatment, resulting in apoptosis of TNFR2 expressing T cells[24].
PREDICTION OF CLINICAL RESPONSE
Given that approximately 30% of the patients fail to respond to anti-TNF therapy (primary non-responders), and up to 50% lose the response during therapy (secondary non-responders)[82], there is a clinical need for biomarkers allowing prediction of clinical response. Expression of biomarkers in blood or intestinal biopsies has been investigated in this regard. Table 4 presents a selection of so far collected data regarding possible predictors of response to anti-TNF therapy in IBD. A study using intestinal biopsies of patients with CD responding and non-responding to anti-TNF therapy revealed that modulation of TNF-dependent genes has also been observed in non-responsive patients, indicating biological activity of anti-TNF independently of clinical response[54]. On the other hand, in this study IL1B and IL17A remained altered in non-responders, suggesting these cytokines as relevant targets in this subgroup of patients[54]. A database search validated possible biomarkers in gene expression in patients with IBD under infliximab therapy. Of interest, datasets of PBMCs and biopsies were not comparable. Regarding intestinal biopsies, the genes IL13RA2, PTGS2 and WNT5A were shown to possibly predict the responsiveness to infliximab in IBD[16].
Table 4.
Prediction of response to anti-tumor necrosis factor therapy - possible biomarkers
| Material/method | Predictor for response | Disease | Ref. |
| Intestinal biopsies | IL1B (upregulated in non-responders) | CD | [54] |
| IL17A (upregulated in non-responders) | |||
| Intestinal biopsies | IL13RA2 | CD and UC | [16] |
| PTGS2 | |||
| WNT5A | |||
| Blood | high CAI | UC | [17] |
| negative ANCA | |||
| IL23R | |||
| Blood | High API | CD | [83] |
| Blood | C-reactive protein | CD and UC | [84] |
| Intestinal biopsies | TNFRSF11B (osteoprotegerin) | UC | [85] |
| STC1 (stanniocalcin-1) | |||
| PTGS2 (prostaglandin-endoperoxide synthase 2) | |||
| IL13Ralpha2 (IL13R alpha 2) | |||
| IL11 (IL-11) | |||
| SPECT | High apoptosis rate | CD | [80] |
| Endoscopic molecular imaging | High mucosal mTNF expression in vivo | CD | [18] |
CD: Crohn’s disease; UC: Ulcerative colitis; SPECT: Single-photon emission computer tomography.
Another study investigating UC patients before and after therapy with infliximab identified a high CAI, a negative antineutrophil cytoplasmatic autoantibody status and the IL23R genotype as predictors of response to therapy[17]. As apoptosis is a central mechanism of action of anti-TNF antibodies, a predictive model based on an apoptotic pharmacological index (API) has been proposed in another study. Herein, response rates of infliximab in CD patients have been investigated in regard to polymorphisms of apoptosis genes (FAS ligand -843C/T; Fas -670G/A; Caspase-9 93 C/T). Patients with an low API had the lowest response whereas patients with an high API displayed high response and remission rates[83]. In addition, high C-reactive protein levels have been described as predictors for response to anti-TNF therapy[84]. Gene array analyses of biopsies from UC patients taken before anti-TNF treatment identified mainly five differentially expressed genes involved in adaptive immunity (osteoprotegerin, stanniocalcin-1, prostaglandin-endoperoxide synthase 2, IL13R alpha 2 and IL-11) in patients later responding to infliximab as compared to patients that did not[85]. However, these promising biomarkers will have to be validated in larger cohorts of patients to verify if they serve as reliable markers for prediction of therapy in the clinic.
Another approach for prediction of response is based on biomarkers derived from the molecular mechanism of action of anti-TNF antibodies and transition into corresponding in vivo molecular imaging modalities. As described earlier, apoptosis induction by infliximab was determined by SPECT imaging and detected a significantly higher apoptosis rate in patients later responding to infliximab[80]. The visualization of mTNF expressing cells by molecular imaging was the fundament of another recently published clinical trial. Based on the previous finding that the interaction of mTNF expressing cells and TNFR2 expressing T cells is targeted by anti-TNF antibodies[24], it was possible to predict clinical response to subsequent anti-TNF therapy using GMP-conform fluorescent anti-TNF antibodies during endoscopy. Here, prior to initiation of anti-TNF therapy, fluorescent anti-TNF antibodies were topically applied in vivo to the inflamed mucosa in 25 CD patients and the number of mTNF positive mucosal cells were localized with molecular imaging using a confocal laser endomicroscope. It could be demonstrated that CD patients with high amounts of mTNF+ cells showed significantly higher response rates at week 12 (92%) upon subsequent anti-TNF therapy as compared to patients with low amounts of mTNF+ cells (15%). Clinical response was defined as the reduction of the Crohn’s Disease Activity Index > 100 points at week 12 after the initiation of the anti-TNF therapy. A high number of mTNF positive cells also predicted sustained clinical response at week 52, decreased steroid use and high mucosal healing rates[18]. These findings represent a first step towards individualized medicine, making it possible to determine the most suitable biological therapy based on molecular level analysis thereby circumventing ineffective antibody therapy in IBD patients.
DISCUSSION AND FUTURE RESEARCH DIRECTIONS
Given the complexity of TNF signaling, the identification of the mechanism of action of anti-TNF therapy remains challenging. In this review, several aspects of potential targets for anti-TNF antibodies have been discussed. However, as reviewed here, contradictory results concerning the influence of anti-TNF antibodies on cells in vivo and in vitro have been reported, which possibly reflect the different experimental conditions used in each study. Furthermore, although infliximab, adalimumab, golimumab, certolizumab pegol and etanercept target the same epitope, fundamental differences in their mode-of-action in vivo have been demonstrated, beginning with the fact that etanercept is effective in the treatment of RA, but did not show therapeutic efficacy in the treatment of CD. However, in summary some conclusions can be drawn: First, as many IBD patients benefit from long-term remission induced by anti-TNF therapy, additionally to TNF neutralization fundamental biological changes must be induced, especially in those patients that can stop the medication due to stable remission. Second, it is unclear if Fc-dependent apoptosis such as ADCC and CDC by anti-TNF agents is relevant in vivo in IBD. Certolizumab pegol is incapable of inducing ADCC and CDC due to its structure but is successfully used for the treatment of CD. Third, direct and indirect apoptosis induction of anti-TNF agents and the modulation of the immune system possibly might be connected. For example, apoptosis induction could possibly directly influence the mucosal cytokine production[53]. Fourth, the effects of anti-TNF agents apparently are dependent on the cell type; in some studies no detection of apoptosis induction in peripheral blood T cells could be observed, while induction of apoptosis in mucosal T cells were described in vitro and in vivo. Lastly, several findings suggest binding of anti-TNF antibodies to mTNF rather than binding to sTNF is critical in IBD. In order to improve the response rates of anti-TNF antibody treatment, beside pharmacokinetic reasons, it is mandatory to better understand the mechanism of action of these agents and unravel the key signaling pathways involved. First clinical studies using in vivo imaging are opening the field for individualized therapeutic approaches in the treatment of IBD.
Footnotes
Manuscript source: Invited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: Germany
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Conflict-of-interest statement: Raja Atreya has served as advisor for AbbVie and Markus F Neurath has served as advisor for MSD, AbbVie, Takeda, Boehringer, Giuliani.
Peer-review started: July 13, 2016
First decision: July 29, 2016
Article in press: October 10, 2016
P- Reviewer: Eder P, Ingle SB, Papamichael KX S- Editor: Gong ZM L- Editor: A E- Editor: Zhang FF
References
- 1.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA, Fiocchi C, Strober W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol. 1996;157:1261–1270. [PubMed] [Google Scholar]
- 2.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Bürgel N, Fromm M, et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology. 2005;129:550–564. doi: 10.1016/j.gastro.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Ueno A, Ghosh A, Hung D, Li J, Jijon H. Th17 plasticity and its changes associated with inflammatory bowel disease. World J Gastroenterol. 2015;21:12283–12295. doi: 10.3748/wjg.v21.i43.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang YZ, Li YY. Inflammatory bowel disease: pathogenesis. World J Gastroenterol. 2014;20:91–99. doi: 10.3748/wjg.v20.i1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monteleone G, Caprioli F. T-cell-directed therapies in inflammatory bowel diseases. Clin Sci (Lond) 2010;118:707–715. doi: 10.1042/CS20100027. [DOI] [PubMed] [Google Scholar]
- 6.Holtmann MH, Schütz M, Galle PR, Neurath MF. Functional relevance of soluble TNF-alpha, transmembrane TNF-alpha and TNF-signal transduction in gastrointestinal diseases with special reference to inflammatory bowel diseases. Z Gastroenterol. 2002;40:587–600. doi: 10.1055/s-2002-33418. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald TT, Hutchings P, Choy MY, Murch S, Cooke A. Tumour necrosis factor-alpha and interferon-gamma production measured at the single cell level in normal and inflamed human intestine. Clin Exp Immunol. 1990;81:301–305. doi: 10.1111/j.1365-2249.1990.tb03334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reimund JM, Wittersheim C, Dumont S, Muller CD, Baumann R, Poindron P, Duclos B. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. doi: 10.1007/BF01540912. [DOI] [PubMed] [Google Scholar]
- 9.Breese EJ, Michie CA, Nicholls SW, Murch SH, Williams CB, Domizio P, Walker-Smith JA, MacDonald TT. Tumor necrosis factor alpha-producing cells in the intestinal mucosa of children with inflammatory bowel disease. Gastroenterology. 1994;106:1455–1466. doi: 10.1016/0016-5085(94)90398-0. [DOI] [PubMed] [Google Scholar]
- 10.Reinecker HC, Steffen M, Witthoeft T, Pflueger I, Schreiber S, MacDermott RP, Raedler A. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- 12.Panaccione R, Colombel JF, Sandborn WJ, D’Haens G, Zhou Q, Pollack PF, Thakkar RB, Robinson AM. Adalimumab maintains remission of Crohn’s disease after up to 4 years of treatment: data from CHARM and ADHERE. Aliment Pharmacol Ther. 2013;38:1236–1247. doi: 10.1111/apt.12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schreiber S, Rutgeerts P, Fedorak RN, Khaliq-Kareemi M, Kamm MA, Boivin M, Bernstein CN, Staun M, Thomsen OØ, Innes A. A randomized, placebo-controlled trial of certolizumab pegol (CDP870) for treatment of Crohn’s disease. Gastroenterology. 2005;129:807–818. doi: 10.1053/j.gastro.2005.06.064. [DOI] [PubMed] [Google Scholar]
- 14.Armuzzi A, De Pascalis B, Lupascu A, Fedeli P, Leo D, Mentella MC, Vincenti F, Melina D, Gasbarrini G, Pola P, et al. Infliximab in the treatment of steroid-dependent ulcerative colitis. Eur Rev Med Pharmacol Sci. 2004;8:231–233. [PubMed] [Google Scholar]
- 15.Sandborn WJ, van Assche G, Reinisch W, Colombel JF, D’Haens G, Wolf DC, Kron M, Tighe MB, Lazar A, Thakkar RB. Adalimumab induces and maintains clinical remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2012;142:257–65.e1-3. doi: 10.1053/j.gastro.2011.10.032. [DOI] [PubMed] [Google Scholar]
- 16.Győrffy A, Kormos M, Bartha L, Szabó A, Győrffy B, Budczies J, Vásárhelyi B. Validation of Biomarkers in Gene Expression Datasets of Inflammatory Bowel Disease: IL13RA2, PTGS2 and WNT5A as Predictors of Responsiveness to Infliximab Therapy. J Proteomics Bioinform. 2014;7:272–277. [Google Scholar]
- 17.Jürgens M, Laubender RP, Hartl F, Weidinger M, Seiderer J, Wagner J, Wetzke M, Beigel F, Pfennig S, Stallhofer J, et al. Disease activity, ANCA, and IL23R genotype status determine early response to infliximab in patients with ulcerative colitis. Am J Gastroenterol. 2010;105:1811–1819. doi: 10.1038/ajg.2010.95. [DOI] [PubMed] [Google Scholar]
- 18.Atreya R, Neumann H, Neufert C, Waldner MJ, Billmeier U, Zopf Y, Willma M, App C, Münster T, Kessler H, et al. In vivo imaging using fluorescent antibodies to tumor necrosis factor predicts therapeutic response in Crohn’s disease. Nat Med. 2014;20:313–318. doi: 10.1038/nm.3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bradley JR. TNF-mediated inflammatory disease. J Pathol. 2008;214:149–160. doi: 10.1002/path.2287. [DOI] [PubMed] [Google Scholar]
- 20.Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nuñez G, Keshav S. Crohn’s disease and the NOD2 gene: a role for paneth cells. Gastroenterology. 2003;125:47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 21.Slevin SM, Egan LJ. New Insights into the Mechanisms of Action of Anti-Tumor Necrosis Factor-α Monoclonal Antibodies in Inflammatory Bowel Disease. Inflamm Bowel Dis. 2015;21:2909–2920. doi: 10.1097/MIB.0000000000000533. [DOI] [PubMed] [Google Scholar]
- 22.Taylor PC. Pharmacology of TNF blockade in rheumatoid arthritis and other chronic inflammatory diseases. Curr Opin Pharmacol. 2010;10:308–315. doi: 10.1016/j.coph.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP. Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther. 2008;117:244–279. doi: 10.1016/j.pharmthera.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 24.Atreya R, Zimmer M, Bartsch B, Waldner MJ, Atreya I, Neumann H, Hildner K, Hoffman A, Kiesslich R, Rink AD, et al. Antibodies against tumor necrosis factor (TNF) induce T-cell apoptosis in patients with inflammatory bowel diseases via TNF receptor 2 and intestinal CD14⁺ macrophages. Gastroenterology. 2011;141:2026–2038. doi: 10.1053/j.gastro.2011.08.032. [DOI] [PubMed] [Google Scholar]
- 25.Loetscher H, Schlaeger EJ, Lahm HW, Pan YC, Lesslauer W, Brockhaus M. Purification and partial amino acid sequence analysis of two distinct tumor necrosis factor receptors from HL60 cells. J Biol Chem. 1990;265:20131–20138. [PubMed] [Google Scholar]
- 26.Walczak H. TNF and ubiquitin at the crossroads of gene activation, cell death, inflammation, and cancer. Immunol Rev. 2011;244:9–28. doi: 10.1111/j.1600-065X.2011.01066.x. [DOI] [PubMed] [Google Scholar]
- 27.Hadziselimovic F, Emmons LR, Gallati H. Soluble tumour necrosis factor receptors p55 and p75 in the urine monitor disease activity and the efficacy of treatment of inflammatory bowel disease. Gut. 1995;37:260–263. doi: 10.1136/gut.37.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- 30.Eissner G, Kolch W, Scheurich P. Ligands working as receptors: reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 2004;15:353–366. doi: 10.1016/j.cytogfr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 31.Wang Y, Han G, Chen Y, Wang K, Liu G, Wang R, Xiao H, Li X, Hou C, Shen B, et al. Protective role of tumor necrosis factor (TNF) receptors in chronic intestinal inflammation: TNFR1 ablation boosts systemic inflammatory response. Lab Invest. 2013;93:1024–1035. doi: 10.1038/labinvest.2013.89. [DOI] [PubMed] [Google Scholar]
- 32.Sashio H, Tamura K, Ito R, Yamamoto Y, Bamba H, Kosaka T, Fukui S, Sawada K, Fukuda Y, Tamura K, et al. Polymorphisms of the TNF gene and the TNF receptor superfamily member 1B gene are associated with susceptibility to ulcerative colitis and Crohn’s disease, respectively. Immunogenetics. 2002;53:1020–1027. doi: 10.1007/s00251-001-0423-7. [DOI] [PubMed] [Google Scholar]
- 33.Pierik M, Vermeire S, Steen KV, Joossens S, Claessens G, Vlietinck R, Rutgeerts P. Tumour necrosis factor-alpha receptor 1 and 2 polymorphisms in inflammatory bowel disease and their association with response to infliximab. Aliment Pharmacol Ther. 2004;20:303–310. doi: 10.1111/j.1365-2036.2004.01946.x. [DOI] [PubMed] [Google Scholar]
- 34.Douni E, Kollias G. A critical role of the p75 tumor necrosis factor receptor (p75TNF-R) in organ inflammation independent of TNF, lymphotoxin alpha, or the p55TNF-R. J Exp Med. 1998;188:1343–1352. doi: 10.1084/jem.188.7.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holtmann MH, Douni E, Schütz M, Zeller G, Mudter J, Lehr HA, Gerspach J, Scheurich P, Galle PR, Kollias G, et al. Tumor necrosis factor-receptor 2 is up-regulated on lamina propria T cells in Crohn’s disease and promotes experimental colitis in vivo. Eur J Immunol. 2002;32:3142–3151. doi: 10.1002/1521-4141(200211)32:11<3142::AID-IMMU3142>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 36.Sandborn WJ, Feagan BG, Marano C, Zhang H, Strauss R, Johanns J, Adedokun OJ, Guzzo C, Colombel JF, Reinisch W, et al. Subcutaneous golimumab induces clinical response and remission in patients with moderate-to-severe ulcerative colitis. Gastroenterology. 2014;146:85–95; quiz e14-5. doi: 10.1053/j.gastro.2013.05.048. [DOI] [PubMed] [Google Scholar]
- 37.Vande Casteele N, Gils A. Pharmacokinetics of anti-TNF monoclonal antibodies in inflammatory bowel disease: Adding value to current practice. J Clin Pharmacol. 2015;55 Suppl 3:S39–S50. doi: 10.1002/jcph.374. [DOI] [PubMed] [Google Scholar]
- 38.Hochberg MC, Tracy JK, Hawkins-Holt M, Flores RH. Comparison of the efficacy of the tumour necrosis factor alpha blocking agents adalimumab, etanercept, and infliximab when added to methotrexate in patients with active rheumatoid arthritis. Ann Rheum Dis. 2003;62 Suppl 2:ii13–ii16. doi: 10.1136/ard.62.suppl_2.ii13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sandborn WJ, Hanauer SB, Katz S, Safdi M, Wolf DG, Baerg RD, Tremaine WJ, Johnson T, Diehl NN, Zinsmeister AR. Etanercept for active Crohn’s disease: a randomized, double-blind, placebo-controlled trial. Gastroenterology. 2001;121:1088–1094. doi: 10.1053/gast.2001.28674. [DOI] [PubMed] [Google Scholar]
- 40.Shealy DJ, Cai A, Staquet K, Baker A, Lacy ER, Johns L, Vafa O, Gunn G, Tam S, Sague S, et al. Characterization of golimumab, a human monoclonal antibody specific for human tumor necrosis factor α. MAbs. 2010;2:428–439. doi: 10.4161/mabs.2.4.12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK, Salfeld J, Sasso EH. Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol. 2009;131:308–316. doi: 10.1016/j.clim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Vos AC, Wildenberg ME, Duijvestein M, Verhaar AP, van den Brink GR, Hommes DW. Anti-tumor necrosis factor-α antibodies induce regulatory macrophages in an Fc region-dependent manner. Gastroenterology. 2011;140:221–230. doi: 10.1053/j.gastro.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 43.Van den Brande JM, Braat H, van den Brink GR, Versteeg HH, Bauer CA, Hoedemaeker I, van Montfrans C, Hommes DW, Peppelenbosch MP, van Deventer SJ. Infliximab but not etanercept induces apoptosis in lamina propria T-lymphocytes from patients with Crohn’s disease. Gastroenterology. 2003;124:1774–1785. doi: 10.1016/s0016-5085(03)00382-2. [DOI] [PubMed] [Google Scholar]
- 44.Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C. Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther. 2002;301:418–426. doi: 10.1124/jpet.301.2.418. [DOI] [PubMed] [Google Scholar]
- 45.Perrier C, de Hertogh G, Cremer J, Vermeire S, Rutgeerts P, Van Assche G, Szymkowski DE, Ceuppens JL. Neutralization of membrane TNF, but not soluble TNF, is crucial for the treatment of experimental colitis. Inflamm Bowel Dis. 2013;19:246–253. doi: 10.1002/ibd.23023. [DOI] [PubMed] [Google Scholar]
- 46.Corazza N, Brunner T, Buri C, Rihs S, Imboden MA, Seibold I, Mueller C. Transmembrane tumor necrosis factor is a potent inducer of colitis even in the absence of its secreted form. Gastroenterology. 2004;127:816–825. doi: 10.1053/j.gastro.2004.06.036. [DOI] [PubMed] [Google Scholar]
- 47.Kühn R, Löhler J, Rennick D, Rajewsky K, Müller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 48.Hale LP, Greer PK. A novel murine model of inflammatory bowel disease and inflammation-associated colon cancer with ulcerative colitis-like features. PLoS One. 2012;7:e41797. doi: 10.1371/journal.pone.0041797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agnholt J, Kelsen J, Brandsborg B, Jakobsen NO, Dahlerup JF. Increased production of granulocyte-macrophage colony-stimulating factor in Crohn’s disease--a possible target for infliximab treatment. Eur J Gastroenterol Hepatol. 2004;16:649–655. doi: 10.1097/01.meg.0000108344.41221.8b. [DOI] [PubMed] [Google Scholar]
- 50.Nesbitt A, Fossati G, Bergin M, Stephens P, Stephens S, Foulkes R, Brown D, Robinson M, Bourne T. Mechanism of action of certolizumab pegol (CDP870): in vitro comparison with other anti-tumor necrosis factor alpha agents. Inflamm Bowel Dis. 2007;13:1323–1332. doi: 10.1002/ibd.20225. [DOI] [PubMed] [Google Scholar]
- 51.Dahlén R, Strid H, Lundgren A, Isaksson S, Raghavan S, Magnusson MK, Simrén M, Sjövall H, Öhman L. Infliximab inhibits activation and effector functions of peripheral blood T cells in vitro from patients with clinically active ulcerative colitis. Scand J Immunol. 2013;78:275–284. doi: 10.1111/sji.12081. [DOI] [PubMed] [Google Scholar]
- 52.Ringheanu M, Daum F, Markowitz J, Levine J, Katz S, Lin X, Silver J. Effects of infliximab on apoptosis and reverse signaling of monocytes from healthy individuals and patients with Crohn’s disease. Inflamm Bowel Dis. 2004;10:801–810. doi: 10.1097/00054725-200411000-00015. [DOI] [PubMed] [Google Scholar]
- 53.Shen C, Assche GV, Colpaert S, Maerten P, Geboes K, Rutgeerts P, Ceuppens JL. Adalimumab induces apoptosis of human monocytes: a comparative study with infliximab and etanercept. Aliment Pharmacol Ther. 2005;21:251–258. doi: 10.1111/j.1365-2036.2005.02309.x. [DOI] [PubMed] [Google Scholar]
- 54.Leal RF, Planell N, Kajekar R, Lozano JJ, Ordás I, Dotti I, Esteller M, Masamunt MC, Parmar H, Ricart E, et al. Identification of inflammatory mediators in patients with Crohn’s disease unresponsive to anti-TNFα therapy. Gut. 2015;64:233–242. doi: 10.1136/gutjnl-2013-306518. [DOI] [PubMed] [Google Scholar]
- 55.Katz LH, Kopylov U, Fudim E, Yavzori M, Picard O, Ungar B, Eliakim R, Ben-Horin S, Chowers Y. Expression of IL-2, IL-17 and TNF-alpha in patients with Crohn’s disease treated with anti-TNF antibodies. Clin Res Hepatol Gastroenterol. 2014;38:491–498. doi: 10.1016/j.clinre.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 56.Li Z, Arijs I, De Hertogh G, Vermeire S, Noman M, Bullens D, Coorevits L, Sagaert X, Schuit F, Rutgeerts P, et al. Reciprocal changes of Foxp3 expression in blood and intestinal mucosa in IBD patients responding to infliximab. Inflamm Bowel Dis. 2010;16:1299–1310. doi: 10.1002/ibd.21229. [DOI] [PubMed] [Google Scholar]
- 57.Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 1999;116:22–28. doi: 10.1016/s0016-5085(99)70224-6. [DOI] [PubMed] [Google Scholar]
- 58.Olsen T, Cui G, Goll R, Husebekk A, Florholmen J. Infliximab therapy decreases the levels of TNF-alpha and IFN-gamma mRNA in colonic mucosa of ulcerative colitis. Scand J Gastroenterol. 2009;44:727–735. doi: 10.1080/00365520902803507. [DOI] [PubMed] [Google Scholar]
- 59.Maggi L, Capone M, Giudici F, Santarlasci V, Querci V, Liotta F, Ficari F, Maggi E, Tonelli F, Annunziato F, et al. CD4+CD161+ T lymphocytes infiltrate Crohn’s disease-associated perianal fistulas and are reduced by anti-TNF-α local therapy. Int Arch Allergy Immunol. 2013;161:81–86. doi: 10.1159/000343467. [DOI] [PubMed] [Google Scholar]
- 60.Liu C, Xia X, Wu W, Wu R, Tang M, Chen T, Xu F, Cong Y, Xu X, Liu Z. Anti-tumour necrosis factor therapy enhances mucosal healing through down-regulation of interleukin-21 expression and T helper type 17 cell infiltration in Crohn’s disease. Clin Exp Immunol. 2013;173:102–111. doi: 10.1111/cei.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fischer A, Gluth M, Pape UF, Wiedenmann B, Theuring F, Baumgart DC. Adalimumab prevents barrier dysfunction and antagonizes distinct effects of TNF-α on tight junction proteins and signaling pathways in intestinal epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2013;304:G970–G979. doi: 10.1152/ajpgi.00183.2012. [DOI] [PubMed] [Google Scholar]
- 62.Zeissig S, Bojarski C, Buergel N, Mankertz J, Zeitz M, Fromm M, Schulzke JD. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut. 2004;53:1295–1302. doi: 10.1136/gut.2003.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Danese S, Sans M, Scaldaferri F, Sgambato A, Rutella S, Cittadini A, Piqué JM, Panes J, Katz JA, Gasbarrini A, et al. TNF-alpha blockade down-regulates the CD40/CD40L pathway in the mucosal microcirculation: a novel anti-inflammatory mechanism of infliximab in Crohn’s disease. J Immunol. 2006;176:2617–2624. doi: 10.4049/jimmunol.176.4.2617. [DOI] [PubMed] [Google Scholar]
- 64.Rutella S, Fiorino G, Vetrano S, Correale C, Spinelli A, Pagano N, Arena V, Maggiano N, Repici A, Malesci A, et al. Infliximab therapy inhibits inflammation-induced angiogenesis in the mucosa of patients with Crohn’s disease. Am J Gastroenterol. 2011;106:762–770. doi: 10.1038/ajg.2011.48. [DOI] [PubMed] [Google Scholar]
- 65.Di Sabatino A, Pender SL, Jackson CL, Prothero JD, Gordon JN, Picariello L, Rovedatti L, Docena G, Monteleone G, Rampton DS, et al. Functional modulation of Crohn’s disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology. 2007;133:137–149. doi: 10.1053/j.gastro.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 66.Vos AC, Wildenberg ME, Arijs I, Duijvestein M, Verhaar AP, de Hertogh G, Vermeire S, Rutgeerts P, van den Brink GR, Hommes DW. Regulatory macrophages induced by infliximab are involved in healing in vivo and in vitro. Inflamm Bowel Dis. 2012;18:401–408. doi: 10.1002/ibd.21818. [DOI] [PubMed] [Google Scholar]
- 67.Kirchner S, Boldt S, Kolch W, Haffner S, Kazak S, Janosch P, Holler E, Andreesen R, Eissner G. LPS resistance in monocytic cells caused by reverse signaling through transmembrane TNF (mTNF) is mediated by the MAPK/ERK pathway. J Leukoc Biol. 2004;75:324–331. doi: 10.1189/jlb.0703343. [DOI] [PubMed] [Google Scholar]
- 68.Pallai A, Kiss B, Vereb G, Armaka M, Kollias G, Szekanecz Z, Szondy Z. Transmembrane TNF-α Reverse Signaling Inhibits Lipopolysaccharide-Induced Proinflammatory Cytokine Formation in Macrophages by Inducing TGF-β: Therapeutic Implications. J Immunol. 2016;196:1146–1157. doi: 10.4049/jimmunol.1501573. [DOI] [PubMed] [Google Scholar]
- 69.Higuchi M, Nagasawa K, Horiuchi T, Oike M, Ito Y, Yasukawa M, Niho Y. Membrane tumor necrosis factor-alpha (TNF-alpha) expressed on HTLV-I-infected T cells mediates a costimulatory signal for B cell activation--characterization of membrane TNF-alpha. Clin Immunol Immunopathol. 1997;82:133–140. doi: 10.1006/clin.1996.4291. [DOI] [PubMed] [Google Scholar]
- 70.Harashima S, Horiuchi T, Hatta N, Morita C, Higuchi M, Sawabe T, Tsukamoto H, Tahira T, Hayashi K, Fujita S, et al. Outside-to-inside signal through the membrane TNF-alpha induces E-selectin (CD62E) expression on activated human CD4+ T cells. J Immunol. 2001;166:130–136. doi: 10.4049/jimmunol.166.1.130. [DOI] [PubMed] [Google Scholar]
- 71.ten Hove T, van Montfrans C, Peppelenbosch MP, van Deventer SJ. Infliximab treatment induces apoptosis of lamina propria T lymphocytes in Crohn’s disease. Gut. 2002;50:206–211. doi: 10.1136/gut.50.2.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mitoma H, Horiuchi T, Hatta N, Tsukamoto H, Harashima S, Kikuchi Y, Otsuka J, Okamura S, Fujita S, Harada M. Infliximab induces potent anti-inflammatory responses by outside-to-inside signals through transmembrane TNF-alpha. Gastroenterology. 2005;128:376–392. doi: 10.1053/j.gastro.2004.11.060. [DOI] [PubMed] [Google Scholar]
- 73.Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, To K, Harashima S, Hatta N, Harada M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008;58:1248–1257. doi: 10.1002/art.23447. [DOI] [PubMed] [Google Scholar]
- 74.Derer S, Till A, Haesler R, Sina C, Grabe N, Jung S, Nikolaus S, Kuehbacher T, Groetzinger J, Rose-John S, et al. mTNF reverse signalling induced by TNFα antagonists involves a GDF-1 dependent pathway: implications for Crohn’s disease. Gut. 2013;62:376–386. doi: 10.1136/gutjnl-2011-300384. [DOI] [PubMed] [Google Scholar]
- 75.Yan L, Hu R, Tu S, Cheng WJ, Zheng Q, Wang JW, Kan WS, Ren YJ. Establishment of a cell model for screening antibody drugs against rheumatoid arthritis with ADCC and CDC. Int J Clin Exp Med. 2015;8:20065–20071. [PMC free article] [PubMed] [Google Scholar]
- 76.Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, Louie JS, Kohno T. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. 2009;45:124–131. doi: 10.1016/j.cyto.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 77.Ueda N, Tsukamoto H, Mitoma H, Ayano M, Tanaka A, Ohta S, Inoue Y, Arinobu Y, Niiro H, Akashi K, et al. The cytotoxic effects of certolizumab pegol and golimumab mediated by transmembrane tumor necrosis factor α. Inflamm Bowel Dis. 2013;19:1224–1231. doi: 10.1097/MIB.0b013e318280b169. [DOI] [PubMed] [Google Scholar]
- 78.Shen C, Van Assche G, Rutgeerts P, Ceuppens JL. Caspase activation and apoptosis induction by adalimumab: demonstration in vitro and in vivo in a chimeric mouse model. Inflamm Bowel Dis. 2006;12:22–28. doi: 10.1097/01.mib.0000194185.69800.07. [DOI] [PubMed] [Google Scholar]
- 79.Lügering A, Schmidt M, Lügering N, Pauels HG, Domschke W, Kucharzik T. Infliximab induces apoptosis in monocytes from patients with chronic active Crohn’s disease by using a caspase-dependent pathway. Gastroenterology. 2001;121:1145–1157. doi: 10.1053/gast.2001.28702. [DOI] [PubMed] [Google Scholar]
- 80.Van den Brande JM, Koehler TC, Zelinkova Z, Bennink RJ, te Velde AA, ten Cate FJ, van Deventer SJ, Peppelenbosch MP, Hommes DW. Prediction of antitumour necrosis factor clinical efficacy by real-time visualisation of apoptosis in patients with Crohn’s disease. Gut. 2007;56:509–517. doi: 10.1136/gut.2006.105379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eder P, Lykowska-Szuber L, Krela-Kazmierczak I, Stawczyk-Eder K, Zabel M, Linke K. The influence of infliximab and adalimumab on the expression of apoptosis-related proteins in lamina propria mononuclear cells and enterocytes in Crohn’s disease - an immunohistochemical study. J Crohns Colitis. 2013;7:706–716. doi: 10.1016/j.crohns.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 82.Olesen CM, Coskun M, Peyrin-Biroulet L, Nielsen OH. Mechanisms behind efficacy of tumor necrosis factor inhibitors in inflammatory bowel diseases. Pharmacol Ther. 2016;159:110–119. doi: 10.1016/j.pharmthera.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 83.Hlavaty T, Ferrante M, Henckaerts L, Pierik M, Rutgeerts P, Vermeire S. Predictive model for the outcome of infliximab therapy in Crohn’s disease based on apoptotic pharmacogenetic index and clinical predictors. Inflamm Bowel Dis. 2007;13:372–379. doi: 10.1002/ibd.20024. [DOI] [PubMed] [Google Scholar]
- 84.Vermeire S, Van Assche G, Rutgeerts P. C-reactive protein as a marker for inflammatory bowel disease. Inflamm Bowel Dis. 2004;10:661–665. doi: 10.1097/00054725-200409000-00026. [DOI] [PubMed] [Google Scholar]
- 85.Arijs I, Li K, Toedter G, Quintens R, Van Lommel L, Van Steen K, Leemans P, De Hertogh G, Lemaire K, Ferrante M, et al. Mucosal gene signatures to predict response to infliximab in patients with ulcerative colitis. Gut. 2009;58:1612–1619. doi: 10.1136/gut.2009.178665. [DOI] [PubMed] [Google Scholar]