

Abstract
Obesity is an excessive accumulation of body fat that may be harmful to health. Today, obesity is a major public health problem, affecting in greater or lesser proportion all demographic groups. Obesity is estimated by body mass index (BMI) in a clinical setting, but BMI reports neither body composition nor the location of excess body fat. Deaths from cardiovascular diseases, cancer and diabetes accounted for approximately 65% of all deaths, and adiposity and mainly abdominal adiposity are associated with all these disorders. Adipose tissue could expand to inflexibility levels. Then, adiposity is associated with a state of low-grade chronic inflammation, with increased tumor necrosis factor-α and interleukin-6 release, which interfere with adipose cell differentiation, and the action pattern of adiponectin and leptin until the adipose tissue begins to be dysfunctional. In this state the subject presents insulin resistance and hyperinsulinemia, probably the first step of a dysfunctional metabolic system. Subsequent to central obesity, insulin resistance, hyperglycemia, hypertriglyceridemia, hypoalphalipoproteinemia, hypertension and fatty liver are grouped in the so-called metabolic syndrome (MetS). In subjects with MetS an energy balance is critical to maintain a healthy body weight, mainly limiting the intake of high energy density foods (fat). However, high-carbohydrate rich (CHO) diets increase postprandial peaks of insulin and glucose. Triglyceride-rich lipoproteins are also increased, which interferes with reverse cholesterol transport lowering high-density lipoprotein cholesterol. In addition, CHO-rich diets could move fat from peripheral to central deposits and reduce adiponectin activity in peripheral adipose tissue. All these are improved with monounsaturated fatty acid-rich diets. Lastly, increased portions of ω-3 and ω-6 fatty acids also decrease triglyceride levels, and complement the healthy diet that is recommended in patients with MetS.
Keywords: Obesity, Metabolic syndrome, Metabolism, Adipokines, Insulin resistance, Lipotoxicity and nutrition
Core tip: Central obesity, the insulin resistance, hyperglycemia, hypertriglyceridemia, hypoalphalipoproteinemia, hypertension and fatty liver are grouped in the so-called metabolic syndrome (MetS). In subjects with MetS an energy balance is critical to maintain a healthy body weight, mainly limiting the intake of high energy density foods. However, high-carbohydrate rich (CHO) diets increase postprandial peaks of insulin and glucose. Triglyceride-rich lipoproteins are also increased, which interferes with reverse cholesterol transport lowering high-density lipoprotein cholesterol. In addition, CHO-rich diets could move fat from peripheral to central deposits and reduce adiponectin activity in peripheral adipose tissue. All these are improved with monounsaturated fatty acid-rich diets. Lastly, increased portions of ω-3 and ω-6 fatty acids also decrease triglyceride levels, and complement the healthy diet that is recommended in patients with MetS.
INTRODUCTION
Overweight and obesity are an excessive accumulation of body fat that may be harmful to health. Today, obesity is a major public health problem, affecting in greater or lesser proportion all demographic groups. Obesity is estimated by body mass index (BMI) in a clinical setting, but BMI reports neither body composition nor the location of excess body fat. People with normal weight but high body fat percentages could have a cardiovascular risk equal to that of people with obesity.
Deaths from cardiovascular diseases (CVD), cancer and diabetes accounted for approximately 65% of all deaths, and general adiposity and mainly abdominal adiposity are associated with increased risk of death for all these disorders. Adipose tissue could expand to levels of inflexibility. Then, adiposity is associated with a state of low-grade chronic inflammation, with increased tumor necrosis factor (TNF)-α and interleukin (IL)-6 release, which interfere with adipose cell differentiation, and the action pattern of adiponectin and leptin until the adipose tissue begins to be dysfunctional. In this state the subject presents insulin resistance (IR) and hyperinsulinemia, probably the first step of a dysfunctional metabolic system. Subsequent to central obesity, insulin resistance, hyperglycemia, hypertriglyceridemia, hypoalphalipoproteinemia, hypertension and fatty liver are grouped in the so-called metabolic syndrome (MetS).
In subjects with MetS an energy balance is critical to maintain a healthy body weight, mainly limiting high energy density foods. The first factor to be avoided in the prevention of MetS is obesity, and the percentage of fat in the diet has traditionally been associated with the development of obesity. However, it is well established that the type of fat consumed could be more decisive than the total amount of fat consumed when we only look at changes in body composition and distribution of adipose tissue. In addition, insulin resistance is a feature of MetS and is associated with other components of the syndrome. The beneficial impact of fat quality on insulin sensitivity (IS) was not seen in individuals with a high fat intake (> 37E%). Other dietary factors that can influence various components of MetS, like postprandial glycemic and insulin levels, triglycerides and high-density lipoprotein (HDL)-C levels, weight regulation and body composition, as well as fatty liver, are the glycemic load (GL) and the excess of fructose, and amount of dietary fiber content of food eaten. The increased levels of triglycerides associated with hypoalphalipoproteinemia are a feature of insulin resistance and MetS, and increase cardiovascular risk regardless of low-density lipoprotein (LDL) cholesterol levels.
High-carbohydrate rich (CHO) diets increase postprandial peaks of insulin and glucose. Triglyceride-rich lipoproteins are also increased, which interferes with reverse cholesterol transport lowering HDL cholesterol. In addition, CHO-rich diets could move fat from peripheral to central deposits and reduce adiponectin activity in peripheral adipose tissue. All these are improved with monounsaturated fatty acids (MUFA)-rich diets.
The American Diabetes Association (ADA) recommends an intake of dietary fiber of 20 to 35 g/d mainly because of the cholesterol-lowering and glucose-lowering effects of soluble fiber. However, more beneficial effects of a higher intake of dietary fiber, particularly of the soluble type, above the level recommended by the ADA, were reported to improve glycemic control, decreases hyperinsulinemia, and lower plasma lipid concentrations in patients with type 2 diabetes.
Lastly, the prevalence of enlarged waist circumference, hypertension and hypertriacylglycerolemia were reduced after the isoenergetic low fat high complex carbohydrates (LFHCC) supplemented with ω-3 diet. Thus, the prevalence of MetS fell by 20.5% after LFHCC ω-3 diet compared with the high saturated fatty acids (HSFA) (10.6%), high MUFA (HMUFA) (12%) or LFHCC (10.4%) diets. Therefore, increased fish intake instead of meat portions increases ω-3 fatty acids, and moderate portions of dried fruits (walnuts) increases ω-6, could complement the healthy diet that is recommended in patients with MetS.
In summary, an equilibrate calory diet, low in animal fat, sugar and fructose, high in MUFA and polyunsaturated fatty acids (PUFA), fresh vegetables high in fiber, and with moderate complex carbohydrates portions, could improve weight loss, lower postprandial glucose and insulin levels, and triglyceride levels could also decrease, and, eventually, increased HDL cholesterol levels are observed.
The maintenance of an ideal body weight, usually established between 18 and 25 years of age, requires achieving a life-long energy balance, where the amount of energy intake must equal the amount of energy expended. However, in the study of obesity in humans, if we look at only the imbalance between energy intake and energy expenditure, we have failed in its clinical application[1,2]. In humans, obesity depends on multiple factors apart from diet, like age and stage of development, genes and epigenetic factors, physical activity, environment, level of instruction and nutrition education, as well as several diseases that alter both physical and psychosocial interaction[3,4]. Therefore, the increase in overweight and obesity rates are classified as major public health issues, affecting in greater or lesser proportion all demographic groups, irrespective of age, sex, race, education or economic level[5]. World Health Organization (WHO) expects the 400 million obese adults worldwide registered in 2005[6] to double, and in the United States, obesity has been increasing in both adults and children in the last few years[7-9]. The age-standardized rate of death from any cause was generally lowest among subjects with an optimal BMI of 22.5 to 24.9 kg/m2[10-12].
Recently, it has been observed that death attributed to factors related to high BMI is in fourth place behind deaths from high blood pressure, smoking, and unhealthy diets; and is ahead of deaths attributable to diabetes, physical inactivity, high salt intake, alcoholism and high blood cholesterol levels[13]. In addition, epidemiology studies have established associations between food and nutrient intake with specific diseases such as cancer, diabetes and CVD[14,15] as well as with obesity, body fat distribution, hypertension, insulin resistance and hyperglycemia[16-18].
Deaths from CVD, cancer and diabetes accounted for up to approximately 65% of all deaths, and general adiposity and central adiposity are related with increased risk of death for all these disorders, shortened life expectancy and causes disability in addition to high economic costs. Where levels of BMI are higher than 25 kg/m2 a direct relationship with high mortality due to CVD is well established[3,19-23]. Cardiovascular disease accounts for approximately 38.5% of all deaths in EE.UU., although have declined substantially since the 1940s and 1960s[10]. This trend may be related with several primary prevention activities (for example, smoking cessation, sugar, trans fat and excess of saturated fat ingestion), improved treatment for ischemic acute phase and finally, improved secondary intervention (treatment of hypertension, hyperglycemia and hypercholesterolemia)[24,25]. The pattern of obesity may also influence this CVD risk and those with a waist-hip ratio higher to or equal than the average have in general an odds ratio of 3.0 (95%CI: 2.1-4.2) for ischemic cerebrovascular, even when BMI and other risk factors were adjusted[26]. Last, a weight loss of 10% maintained over time in obese subjects may decrease the expected events of coronary and stroke diseases[27].
On the other hand, concurrent with obesity rates during the 90s, there was an increase of diabetes to 61% in the United States (mainly approximately 90%-95% of type 2 diabetes, T2D)[28]. The mortality rate directly attributable to diabetes is about 3%, and diabetic patients have 2-4 times higher cardiovascular risk and many die of CVD[29]. Obesity and high body fat are related with diabetes in all ethnic groups. In the United States approximately the 70% of T2D prevalence could be attributed to overweight and obesity and, after 10 years, each kilogram gain from ideal body weight, raises the risk by 4.5%[10]. However, again “central obesity” is more strongly associated with metabolic complications linked to insulin resistance including diabetes[30,31]. For the prevention and treatment of T2D maintenance of a healthy body weight (BMI < 27-30 kg/m2) plus physical activity, limit the intake of sugar and saturated fat, and increase the consumption of mono and PUFA, as well as whole grains and fiber[32-34], is recommended.
Finally, all cancers combined accounted for approximately 23% of the total number of deaths[10]. The relationship between BMI and a high mortality due to cancer in most specific sites[12,35] is well established. Obesity may account for up to 14% of cancer in men and up to 20% of cancer in women, and the risk of death from cancer in people with BMI ≥ 40 kg/m2 increases up to 52% in men and 62% in women as compared with people with normal weight[36]. The underlying pathophysiological mechanisms that may be attributed to increase cancer rates are uncertain but can involve higher circulating levels of glucose, low-grade inflammatory state in many tissues, increased oxidative stress, as well as the bioavailability of hormones, mainly insulin, estrogens and androgens.
After obesity is developed most subjects present IR and hyperinsulinemia, probably the first step of a dysfunctional metabolic system. Subjects with more central obesity present a higher risk of IR, hyperglycemia, hypertriglyceridemia, hypoalphalipoproteinemia, hypertension and fatty liver, and different combination are grouped in so-called MetS. In subjects with MetS achieving an energy balance is critical to maintain a healthy body weight, limiting the consumption of food with high energy density (fat). However, high-carbohydrate rich (CHO) diets increase postprandial peaks of insulin and glucose, and triglyceride-rich lipoproteins are also increased, which interferes with reverse cholesterol transport lowering HDL cholesterol, and could deposit fat mainly in central deposits and reduce adiponectin activity in peripheral adipose tissue. However, all these were improved with MUFA-rich diets. In addition, food with high fiber content (vegetables and whole-grain) and food rich in ω-3 and ω-6 fatty acids could improve some components of this dysfunctional metabolic system.
The traditional Mediterranean diet is featured by a moderate to high ingestion of olive oil, a lower density of calories in the diet, legumes and vegetables, fruit, nuts, and whole cereals; a moderate to higher intake of poultry and fish; a moderate intake of dairy products, but more restrictive in higher caloric density foods such as red and processed meats, and sweets; finally, mainly red wine is drunk with meals[37]. Selected subjects at high cardiovascular risk, a Mediterranean style diet supplemented either with extra-virgin olive oil or nuts decrease the incidence of major cardiovascular events[38]. Finally, studies of healthy habits in the 50s[39] show that physical activity at work, walking and cycling as a means of transport all contributed to overall energy expenditure. However, these physical activities have decreased dramatically in societies today because of sedentary habits at work and in holiday life[40]. Thus, dietary habits, a major factor in controlling obesity, are made up of environmental, cultural, economic and technological aspects. These can be modified by agricultural policies that govern prices, extending the range and availability of food and regulating beneficial or harmful dietary components[41,42].
OBESITY ASSESSMENT
Obesity could be estimated only by measures of the body weight; however, relating body weight to height give us a more accurate measure of obesity[43]. The BMI or the Quetelet’s index is the measure that is currently used in clinical setting to graduate from the normal weight to obesity in adults, and is estimated by the weight/height ratio squared, and expressed as kg/m2. The approach taken by WHO is: (1) BMI between 18-25 kg/m2 is considered normal weight; (2) BMI between 25-29.9 kg/m2 is considered overweight; and (3) a BMI greater than or equal to 30 kg/m2 is defined as obesity[44,45]. However, BMI does not gives us information about body composition and body fat distribution, neither about individual variations in terms of amounts of lean body mass (fat-free muscle mass), or the pattern of depot on body fat distribution. Thus, the percentage of body fat (BF%) is a better measure as it relates the ratio of total weight of fatty body weight. However, it is more difficult to measure BF% than single BMI, but several methods of varying accuracy and complexity exist[46]. In a clinical setting the most commonly used anthropometric indicator of body composition analysis involving two components (body fat and free-fat mass) are estimated from measurements of skinfold thicknesses, that should be measured in several regions, in order to obtain a clearer picture of fat composition[47]. In research, the percentage of body fat determined by hydrostatic weighing (body weight by immersion), is the gold standard[48]. In addition, the bioelectrical impedance analysis technique is also used to measure body composition, and using a four-terminal bioimpedance analyzer has a prediction error less or equal to the standard anthropometry for estimating body fat[49]. Therefore, it is possible to estimate the amount of body water and the proportion of fat-free mass and by subtracting body fat from total body weight[50]. Furthermore, a relatively simple technique to evaluate the total and regional adiposity in an individual involves a study of the whole body with a scan densitometer (dual energy X-ray absorptiometry, DEXA)[51,52].
People with normal weight but high body fat percentages could have a cardiovascular risk equal to that of people with obesity. The range of normal body fat is 2%-5% in men and 10%-13% in women, while the obesity range of body fat percentage is above of 25% in men and 32% in women[53,54]. Experimentally, it was observed that BMI = 30 kg/m2 implies approximately 30% of BF% at 20 years of age but increase to 40% at 60 year in men, while in older women these values were to 40% and 50%, respectively. Therefore, body fat composition changes with age and sex. Body fat percentage for adults can be estimated from the BMI as follows: BF% = 1.2 × BMI + 0.23 × age - 5.4 - (10.8 × gender) (being 0 if gender is male and 1 if female; it differ for children). The correlation between BMI-BF% is r = 0.75 in male and r = 0.82 in females, for all ages[55].
On the other hand, BMI does not report on the location or distribution of excess body fat, it is to say about the distribution of body fat. Central obesity is characterized mainly by excess fat depot in the abdominal area and within the peritoneal cavity and lower expansion of peripheral adipose tissue. In a clinical setting, several parameters can be used to estimates central obesity; the most widely used being the perimeter of waist circumference (WC), hip ratio (HR) and waist-HR (WHR). Recently, the waist-to-height ratio, which relates waist circumference to height, has also been used to identify higher cardiometabolic risk in adults[56-58] and children[59,60]. This has advantages compared to the BMI, and even with WC and WHR, and a healthy individual should maintain a waist circumference to less than half their height[61]. All these parameters help to predict the risk of metabolic diseases such as T2D[62], and could be more effective in the case of CVD[63]. In addition, mortality due to any cause was increased with a BMI < 30 when the subjects have a large WC[64]. Thus, WC and WHR help to identify high-risk individuals regardless of their BMI[65]. The WC range that estimates mainly central adiposity varies with race and it is currently suggested that for individuals of the United States > 88 cm in women and > 102 cm in men; for the European Union ≥ 80 cm in women and ≥ 94 cm in men; for Chinese and South Asia > 90 cm and for Japanese > 85 cm for both women and men[66]. These assessments are used mainly in the clinic, but there are others more complex and more expensive techniques used in research, which are more accurate, such as DEXA, computed tomography (CT), and magnetic resonance imaging (MRI). Distribution of body fat is evaluated by DEXA by automatic scanning of default regions (arms, legs and trunk). The trunk is the area bounded by the horizontal line under the chin, side edges of the ribs and oblique lines through the femoral neck; and leg area includes the area under these oblique lines. This measure has a coefficient of variation of approximately 2%[51,52,67]. Central obesity is composed of abdominal subcutaneous fat and intraabdominal fat, as is seen by MRI and CT. In addition, intraabdominal adipose tissue is composed of visceral adipose tissue (VAT) as omental and mesenteric fat (intraperitoneal fat) and retroperitoneal fat mass[68]. Finally, single-voxel magnetic resonance spectroscopy is the gold-standard for ectopic fat quantification. Although very similar to MRI, it does not give anatomical information in image form, but gives information about the chemical composition as it is based on chemical shift. The water protons from (-OH) hydroxyl groups have a spectral peak at 4.7 ppm (parts-per-million). However, the triglycerides have the predominant protons from the (-CH2)n methylene groups[69,70]. Finally, ectopic fat is estimated with accurate methods that separate water and fat signals within each voxel (software such as jMRUI). Occasionally other techniques have been used in determining the ectopic fatty tissue including ultrasonography (US), with a highly significant correlation between CT and by US[71].
ADIPOCYTE AND ADIPOSITY DEVELOPMENT
Adipocyte differentiation
In humans there are two types of well-differentiated adipose tissue, which have different distribution and functions, and are referred to as white adipose tissue (WAT) and brown adipose tissue (BAT) (Figure 1A). The WAT is mainly related to the function of deposit of surplus energy as triacylglycerol (fat), which could be mobilized and offered through hormonal signaling and has a tremendous ability to expand; excess fat storage is associated with mechanical overload and slow to moderate increased risk of metabolic disorders. Mature WAT are characterized by the increased expression of transporters of glucose sensitive to insulin (GLUT4), and enzymes like fatty acid synthase (FAS) and glycerol-2-phosphate dehydrogenase[72,73]. By contrast, BAT is involved in thermogenesis functions and thus in energy expenditure and body weight regulation[74,75]. In mammals, BAT is the primary site of thermogenesis without accompanying muscle contraction. This function is stimulated by exposure to cold or after lipid-rich calorie food, and this process is called adaptive thermogenesis[76]. This thermogenic function of BAT is mediated by the activation of a specific mitochondrial uncoupling protein 1 (UCP1), which is ubiquitous in the inner mitochondrial membrane, uncoupling electron transport of mitochondrial respiration, where the saturation of the production of ATP is dissipated as heat (Figure 1B). The presence of functionally active BAT in rodents has been known for many years. In humans, the first evidence of BAT function was related to the control of body temperature after birth and in early childhood[77]. However, several data from adipose tissue samples together with evidence provided by positron emission tomography coupled with computed tomography have established the existence of functionally active brown adipose tissue in adult humans[78-81]. Furthermore, some of these studies have also related data between the size of activation of these sites with BAT and lower BMI, increased basal energy expenditure and decreased onset of diabetes[82]. Different amounts of BAT in adult humans can be found in the cervical and supraclavicular[83], and are known as canonical BAT. Although brown adipocytes are also observed infiltrating skeletal muscle and in different areas of WAT[84]. Therefore, a third fat cell or new functional adipose tissue is being defined[85,86].
Figure 1.
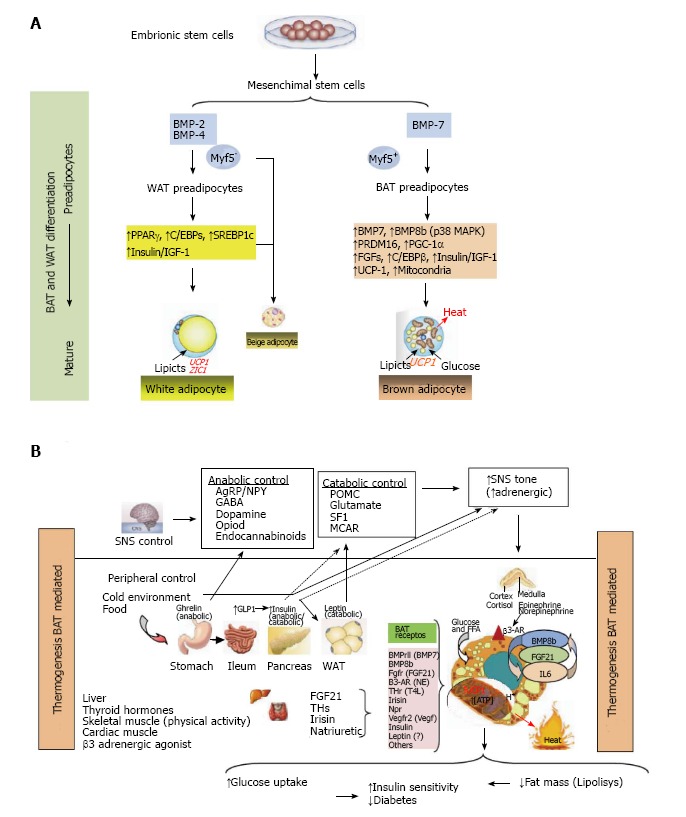
Thermogenesis brown adipose tissue (Bat) mediated. A: Adipocytes were developed because non adipocytes cells are unable to store calories as fat to meet fuel needs during long periods without eating. If the energy intake is more than energy expenditure, WAT is expanded and leads to obesity. However, a second type of adipose tissue, called BAT was developed especially for energy expenditure (thermogenesis). Today, research in identifying the main genes that control differentiation, development and activation of BAT is highly active, because, activation of BAT, in detriment of WAT, could have anti-obesity effects, which can be utilized to keep the system of fat deposit balanced. In this research, PRDM16, PPAR-γ and PGC-1α, have been identified as the key nodes in the regulation of inducible BAT; B: The thermogenic potential of BAT is controlled by the SNS, which densely innervates brown fat depots. In addition, BAT is activated in response to cold temperatures, hormones and possibly diet. BAT content and activation is highest in children and decreases with age. BAT activation is decreased in fatness, and BAT activity has been inversely correlated to BMI, body fat, and visceral obesity. In humans, BAT amount and activation is higher in women than in men. Of clinical relevance, BAT activation is very low in diabetic patients in comparison with non-diabetic subjects. Thyroid hormones play a main role in control of BAT activation, therefore the cold-induced enhancement of the enzyme 5’-deiodinase type II activity, which deiodinates thyroxine (T4) to T3. Catecholamines such as norepinephrine binds to β-ARs and induce PGC1α through p38 MAPK and finally triggers expression of UCP1. Whereas β1-AR is considered important for proliferation of classical brown adipocyte precursors in response to norepinephrine, β3-AR plays a major role in thermogenic function of mature brown adipocytes. Another signal, Irisin hormone which comes from muscle to fat tissue, is able to induce a robust browning programme, and mediates the beneficial effects of exercise and could reduce diet-induced obesity and insulin resistance. A more generalized program in the control of adipose tissue is conducted by FGF21 through regulating lipolysis in WAT as well as increasing substrate utilization by increasing fatty acid oxidation in the liver. Last, beige fat cell functions include either a like to “WAT” when energy balance is exceeded, or a like to “BAT” in response to many stimuli similar to BAT activation. WAT: White adipose tissue; BAT: Brown adipose tissue; PRDM16: PR domain containing 16; PPAR-γ: Peroxisome proliferator-activated receptor-γ; PGC-1α: Peroxisome proliferator-activated receptor γ coactivator 1α; SNS: Sympathetic nervous system; BMI: Body mass index; FGF21: Fibroblast growth factor 21.
Transcriptional signaling of adipocyte formation
Expansion of WAT in ideal weight or in obesity is not only the result of hypertrophy and/or hyperplasia of adipocytes, but supporting elements like vascular and mesenchymal stromal including immune cells, endothelial cells, and undifferentiated or adipocyte precursor cells (APs) must also be developed. Alterations in vascular tissue development and hypoxia is associated with adipocyte apoptosis and macrophage infiltration, and an appropriate induction of vascular endothelial growth factor A in adipose tissue is essential during expandability of adipose tissue (Figure 2)[87].
Figure 2.
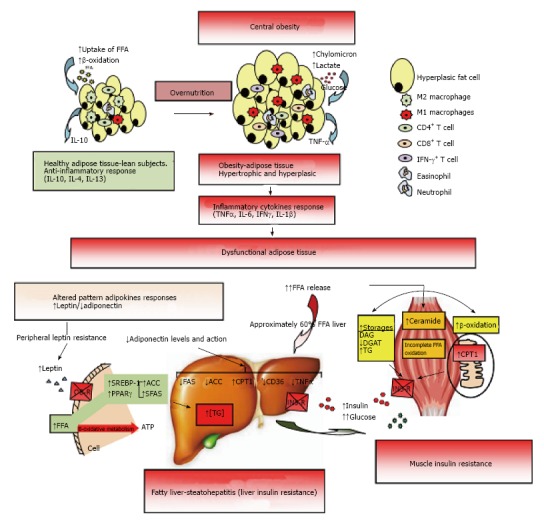
Dysfunctional adipose tissue. Early central obesity is associated with a low-grade chronic inflammatory state characterized by slow infiltration of macrophages which are an important source of inflammation of this adipose tissue[275,276]. Several macrophage subtypes can be found, and simply put, are divided in pro-inflammatory M1 or alternatively activated M2, although in vivo studies reveal a spectrum of macrophage phenotypes[277]. Adipocytes and immune cells such as T cells and macrophages participate in the activation and production of inflammatory cytokines[170,275,278,279]. The M1 macrophages mainly found in obesity, are induced from precursor M0 macrophages by stimulation of components of bacteria (lipopolysaccharide) and type 1 T-helper (Th1) inflammatory cytokines like IFN-γ and TNF-α. The M2 macrophages are activated by type 2 (Th2) cytokines such as IL-4 and IL-13. The M2 macrophages are abundant in adipose tissue of lean subjects and appear to be involved in remodeling, tissue repair, and maintenance of insulin sensitivity through the production and expression of IL-10, IL-1 receptor antagonist, and arginase-1. Whereas M1 macrophages use glucose for energy, M2 macrophages activate the β-oxidation of fatty acids[277,280]. Finally, M1 macrophages are the major source of inflammatory cytokines including TNF-α which inhibits adipose cell differentiation by activating Wnt signaling and suppressing expression of PPAR-γ transcription factor essential for the development and function of adipocyte, and reducing the effect on stored triglycerides[281,282]. The subcutaneous adipose tissue will continue to expand to an equilibrium point. When this capacity is exceeded, glucose and lipid uptake begins to decline and insulin levels are raised to maintain serum glucose in the normal range[215]. In addition, when WAT is unable to expand (inflexibility), associated with insulin resistance state, a continuous release of FFA to interstice begins, generating a systemic lipotoxic effect in muscle, liver, etc., (lipotoxicity). The adipose tissue itself begins a slow process of low-level chronic inflammation (macrophages, lymphocytes, etc.) which increases local release of TNF-α and IL-6[166]. TNF-α and IL-6 levels are inversely related with peripheral and hepatic glucose-uptake which is insulin-mediated[283]. The liver keeps excess uptake of FFA in serum to capacity by joining with glycerol (TAG) and slowly fatty liver is developed (NAFLD). It has been shown that peripheral fatty acids contribute approximately 60% of total TAG stored in the liver, whereas the novo lipogenesis in the liver is approximately 26% and approximately 15% is from the diet[284]. On the other hand, leptin levels respond directly to adipose expansion, while adiponectin levels tend to decrease when metabolic syndrome is developed. The elevated leptin levels should increase lipolysis in non-adipose tissues, decreasing excess fatty acids in these cells. However, this action of leptin may be partially blocked by the anabolic effect established by hyperinsulinemia, settling down leptin system dysfunction (peripheral leptin resistance)[115]. In addition, the decreased adiponectin levels are inversely related to peripheral glucose uptake and directly related with progressive development of chronic liver disease by fat infiltration. Adiponectin exerts a protective action on liver fat accumulation, favoring lipolysis by promoting the action of CPT-1, while interfering with the action of FAS, ACO and TNF-α, and decreasing the expression and action of CD-36 protein that promotes the transport of fatty acids[129]. Finally, both leptin and adiponectin seem to regulate the deposition of fat in insulin-sensitive tissues by increasing fat oxidation. IFN-γ: Interferon-γ; TNF-α: Tumor necrosis factor-α; IL: Interleukin; PPAR-γ: Peroxisome proliferator-activated receptor-γ; WAT: White adipose tissue; FFA: Free fatty acids; NAFLD: Non alcoholic fatty liver disease; CPT-1: Carnitine palmitoyltransferase-1; FAS: Fatty acid synthase; ACO: Acyl CoA carboxylase.
The hypertrophy of WAT only depends on its own renewal from APs which remain present during the entire life span and after suitable signaling can form different mature fat cells (Figure 1A)[88]. In WAT development several key transcription factors have been identified and among them the binding proteins CCAAT/enhancer (C/EBP) and peroxisome proliferator-activated receptor (PPAR) should be mentioned. Sterol regulatory element binding transcription factor 1 (SREBP1c) has been found as a pro-adipogenic basic helix-loop-helix transcription factor which activates peroxisome proliferator-activated receptor-γ (PPAR-γ) expression[89] and mediates the induction of lipid biosynthesis by insulin[90]. On the other hand, BAT derived from Myf5 + progenitors paraxial mesoderm layer shares a common origin with the development of skeletal myoblasts[91]. The development of BAT requires that PRDM16 interacts with either PPAR-γ coactivator (PGC-1α/β) or CtBPs to activate brown genes or the inhibition of several transcription factors that induce WAT, respectively[92,93]. In addition, it has been shown that bone morphogenetic protein 7 turn on a complete program of brown adipogenesis involving induction of early key regulatory transcription to brown cells as PRDM16 and PGC-1α, and increased expression of UCP-1 which is characteristic of brown cells[94]. Finally, Myf5 was found to drive the expression of classical BAT depots in retroperitoneal and anterior subcutaneous WATs, and the existence of Myf5 positive cells mixed in WAT has been confirmed[95]. The term “beige” has been used to describe those cells that are morphologically identical to white adipocytes, but may be inducible to cells expressing brown adipocytes definitive characteristics of UCP1 activity with β-adrenergic stimulation[96,97]. Adipose tissue located in the inguinal area is seen today as the largest and physiologically most relevant fat depot capable of inducing brought beige adipocytes[96]. In addition, it has been observed that in this fat depot the beige mature adipocytes can be interconvert in adipocytes with characteristics typical of white and brown adipocytes, without the need for “de novo” cell differentiation from precursors[97]. Thus, physiologically this could mean that the rate of lipid storage or lipid oxidation could be adapted and adjusted in response to external stimuli such as a decrease or increase in temperature, but it still requires further investigation.
EFFECT OF HORMONES AND ADIPOKINES ON ADIPOGENESIS
The adipose tissue can be expanded and developed by many factors such as hormones, growth factors, factors produced by adipose tissue itself (adipokines) and specific effects induced by nutritional factors and some pharmacological components (Figure 1B).
Hormones and growth factors
Insulin: In “in vitro” studies, a mixture of dexamethasone, isobutylmethylxanthine and insulin is regularly used to generate well-differentiated adipose tissue, insulin being the most potent of the three factors. Insulin within the physiological range induces lipogenesis and insulin receptor is required for adipocyte differentiation[98]. Insulin regulates brown preadipocyte determination through a necdin-E2F4 interaction that represses PPAR-γ transcription via a cyclic AMP response element binding protein-dependent pathway[99]. Hyperinsulinemia either undergone exogenously (treatment) or endogenously (secretion), is clearly related with weight gain, which is a feature of the MetS. However, several molecules such as TNF-α, leptin, resistin, interact and block multiple steps of insulin signaling and antagonize its effects on adipocytes.
Growth hormone and insulin like growth factor 1: Growth hormone (GH) is not only involved in postnatal somatic growth to adulthood, but also has a role in the regulation of metabolic substrates in the control of body composition and body fat distribution, through the combination of lipolytic and anabolic effects[100]. In fact, patients with GH deficiency have a smaller number of adipocytes which also has less volume, and these are partially normalized with GH replacement therapy[101]. GH is involved in the conversion of preadipocytes into mature adipocytes, and subsequently plays a role in the maturation of adipocytes which makes them sensitive to insulin and IGF-I[102]. The effect of GH on adipogenesis seems mainly mediated via stimulating Stat5A/5B inducing the transcriptional activity of PPAR in cooperation with C/EBPb/δ[103].
Thyroid hormones: Thyroid hormones are involved in the growth and maturation of several organs and tissues during fetal and neonatal development[104]. Finally, in adult life, thyroid hormones regulate energy metabolism and function of organs such as the adipose tissue, liver, heart, skin tissue, muscle or adipose tissue. It has been observed that thyroid function in BAT is mediated by the C/EBPs signaling which induces the expression of thyroid hormone receptor and PGC1α (PPAR-γ coactivator) and deiodinase (D2) activity determines grade of thyroid function “in situ”[105,106].
Glucocorticoids and sexual hormones: In humans, infusion of hydrocortisone for 6 h increased levels of circulating FFA, and several mechanisms for the lipolysis of glucocorticoids have been observed[107,108]. In addition, dexamethasone is involved in the expression of PPAR-γ transcription factors and C/EBPδ, and decreases the expression of pref-1 which is a negative regulator of adipogenesis[109]. Therefore, the central obesity phenotype is associated mainly with the consumption of peripheral adipose tissue (lipolysis), and it is observed in human hypercortisolism situations as in Cushing’s syndrome. The adrenal glands and gonads are the main primary source of serum levels of steroid hormones. However, adipose tissue has a full arsenal of enzymes that induce, interconvert, and inactivate peripheral steroid sex hormones[110]. The regulation of glucocorticoids levels is critical for the maintenance of homeostasis and the activity in some tissues of 11-β-hydroxysteroid dehydrogenase 1 and 2 (11 βHSD1 and 2) interconvert the active form of cortisol in other inactive product called cortisone and vice versa[111]. This enzyme is highly expressed in adipose tissue and an increase in its activity seems involved in an increased level of visceral adipose tissue[112,113]. Moreover, the distribution of body fat is characteristically different between men and women; while they are sexually active, resulting in so-called “android or apple” obesity with abdominal fat depot and “gynoid or pear” obesity where fat accumulates predominately in the buttock. However, the actions that sex steroids have on adipogenesis are poorly known. In addition, the main determinants of the action of sex steroids is given by free circulating levels of the hormone in question and the degree of expression in the target organ receptors. The prereceptor tissue-specific metabolism of steroid hormones is also involved in its function. Adipose tissue and preadipocytes have a great activity either cytochrome P450-dependent aromatase and 17βHSD enzymes. Aromatase regulate the rate of formation of androgens into estrogens: Androstenedione to estrone and testosterone to estradiol. Whereas, the 17βHSD is involved in the production of more active forms of testosterone and androstendiona from their weaker precursors, and the rate 17B-HSD/aromatasa in adipose tissue is correlated positively with central adiposity[110,114]. Finally, many men with insulin resistance, T2D or MetS present low testosterone concentrations with high or low gonadotropins (25% and 4%, respectively).
Adipokines: The developed and mature adipocyte acquires the ability to synthesize and release many proteins, known generally by Adipokines. These proteins and hormones are involved in energy homeostasis by regulating energy intake and basal metabolism. Therefore, adipose tissue is implicated in the metabolic control of energy substrates such as glucose and lipids, and interacts with several hormonal systems. The molecules produced by adipose tissue act remotely (endocrine) and locally as paracrine and autocrine on stroma and other components of the adipose tissue (blood vessels, inflammatory cells, etc.) and also other tissues such as muscle. All these actions will contribute in the regulation of the different adipose tissue depots, for expanding the size of peripheral adipose tissue or in fat redistribution to other depots.
In obese and insulin resistant patients increased levels of some adipokines (e.g., leptin, resistin) are often observed while others such as adiponectin levels are typically decreased[115] (Figure 2).
Major adipokines
Leptin: Leptin is specifically secreted by fat cells whose primary function assigned was to establish an adiposity signal between the amount of developed adipose tissue and satiety centers in the brain completing a negative feedback loop[116,117]. People who lose weight following a low calorie diet usually decrease circulating leptin levels. This decrease in leptin appears to mediate reversible decrease in thyroid activity, sympathetic tone, and a decrease in basal energy expenditure[118]. Treating leptin deficiency with recombinant leptin reduces food intake and body weight[119]. Therefore, in subjects with very low levels of serum leptin, the recombinant leptin treatment also improved several abnormalities including infertility, lipodystrophy and impaired glucose metabolism and impaired immunity[120-123]. The expression and release of leptin is controlled by several hormones and factors. Therefore, appears to be stimulated by insulin, glucocorticoids, TNF-α, estrogens, and C/EBPA; by contrast, is decreased by androgens, β3-adrenergic activity, GH, free fatty acids, and PPAR-γ agonist[124]. The action of leptin is essential for energy metabolism, but is also involved in the mobilization of lipids from different fat depots and may be related to the protection of some tissues on lipotoxicity syndrome[125,126]. Thus, lipid oxidation in cells that have this capacity (mitochondria) could be increased through the signal of leptin and could reduce excessive fatty acids and protect against lipotoxicity in the liver, pancreas, heart, kidney, and muscle tissue (Figures 2 and 3).
Figure 3.
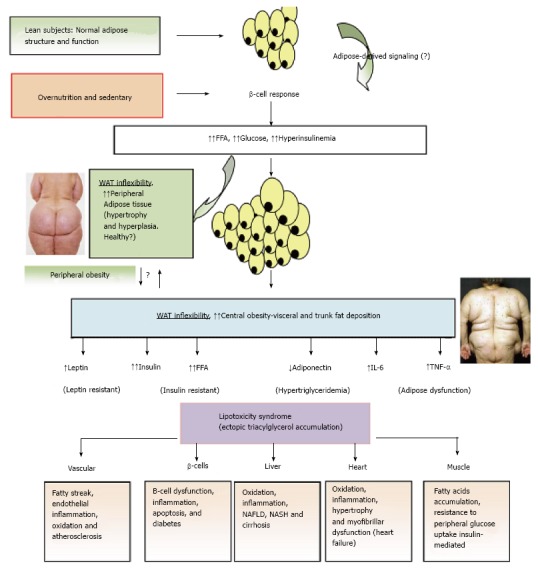
Adipose tissue expandability and metabolic syndrome. After a long period of overeating with positive energy balance, associated with increased hormones such as insulin, adipose tissue responds by increasing its storage capacity, which is determined by a number of factors. Individuals with a higher capacity for storing fat, mainly when peripheral WAT is expanded (WAT flexibility), most subjects will remain metabolically normal for a longer period, despite obesity developing. These subjects are observed to be metabolically healthy (MHO). Chronic inflammatory response leads to dysfunctional adipose tissue with increased local and endocrine secretion of acute phase reactants and inflammatory signaling pathways[285]. Abnormal cytokine and adipokines production is related to insulin resistance, hyperglycemia, altered lipid profile and cardiovascular diseases[115,286,287]. Insulin resistance slowly results from increased accumulation of lipids in other nonadipose tissues such as muscle (lipotoxicity) due to enhanced release of fatty acids from hypertrophic and hyperplasic adipocyte cells. In addition, when adipocytes achieve their maximal storage capacity, they begin to alter their adipokynes secretion profile. Therefore, a proinflammatory milieu with elevation in IL-6 and TNF-α and altered adipokines profile, with decreased adiponectin and increased leptin levels, with peripheral leptin resistance, in a dysfunctional adipose system is observed. This suggests that the limitation in storage capacity could be necessary and even precedes the development of metabolic factors. Ectopic lipid accumulation in non-adipocyte cells causes lipotoxicity in these organs and tissues, including inflammation and finally apoptosis. Thus, lipotoxicity in β-cell could decrease beta cell mass (dysfunction of β-cell secretion) and would cause diabetes. Increased fat in liver leads to hepatic steatosis (NAFLD) and steatohepatitis (NASH) and would cause hepatic dysfunction, in the heart would cause myocardiac dysfunction, in the endothelial fatty streak would be precursor of generalized arteriosclerosis, etc. At what point the adipose tissue begins to fail is likely to be determined by genetic and epigenetic factors. However, the question is: Can storage capacity in WAT be enhanced to meet an increased demand[288]? So far, in human trials, the PPAR-γ agonists (TZDs), that remove fat from central deposits toward more favorable peripheral deposits, have been shown to improve lipid profile, insulin-sensitivity, and reduce diabetes and NAFLD[269]. WAT: White adipose tissue; MHO: Metabolically healthy obese; IL: Interleukin; TNF-α: Tumor necrosis factor-α; NAFLD: Non alcoholic fatty liver disease; NASH: Nonalcoholic steatohepatitis; PPAR-γ: Peroxisome proliferator-activated receptor-γ; TZD: Thiazolidinedione.
Adiponectin: Adiponectin is produced specifically in mature adipocytes and RNA abundance is higher in peripheral adipose tissue compared with visceral adipose tissue[127]. Adiponectin receptors are G protein-coupled and have high expression in muscle and liver. Adiponectin is involved in lipid oxidation in skeletal muscle and in the liver, and moreover reduce hepatic production glucose load and postprandial hyperglycemic[128,129]. An inverse relationship has been found between plasma adiponectin levels and the development of obesity, insulin resistance and T2D[130]. However, conflicting data have been observed between adiponectin levels and the development of cardiovascular disease[131]. Adiponectin treatment decreases TNF-a plasma levels and its hepatic production. Adiponectin was able of improving hepatomegaly, steatosis, and alanine aminotransferase levels related with nonalcoholic obese subjects (Figure 2)[129]. Finally, adiponectin levels is early decreased in insulin resistance syndrome, even before the onset of obesity, and adiponectin administration improves IS[132].
TNF-α: TNF-α is a transmembrane protein released mostly by activated macrophages, and also by several other cell types including lymphoid cells, cardiac myocytes, endothelial cells, adipose tissue, etc.[133-135] (Figure 2). Therefore, TNF-α is regarded as an adipokine implicated in process of local and systemic inflammation and in proliferation and differentiation of the cells. TNF-α exerts its effects by binding two receptors, TNFR1 (TNF type 1 or CD120a) and TNFR2 (TNF type 2 or CD120b)[136]. Both TNF-α gene and its receptors are expressed and modulated in adipocytes and is expressed at higher levels in WAT[127]. Some metabolic effects induced by TNF-α implicates it in inhibiting differentiation to mature adipocyte. This in turn leads to insulin resistance, and finally an increase of free fatty acids could result[137,138]. In this way, TNF-α treatment decreased the expression of PPAR-γ and repressed genes involved in lipid and glucose uptake[138,139].
IL-6: IL-6 is secreted by T cells and macrophages involved in the immune response (Figure 2). Smooth muscle cells in blood vessels can also produce IL-6 as a pro-inflammatory cytokine. Finally, IL-6 is synthesized by adipocytes and appears to be associated with elevated levels of CRP and inflammatory states found in obese patients[140]. An important part of the total concentration of IL-6 (approximately 1/3) is produced in adipose tissue. However, the expression and release of IL-6 is two to three times higher in visceral adipose tissue compared to peripheral adipose tissue[127]. Finally, circulating levels of IL-6 have been found to be directly linked to both obesity and insulin resistance[141]. IL-6 inhibits the activity of lipoprotein lipase (LPL) and reduces the differentiation of human preadipocytes, both associated with adipogenesis[142].
Others main adipokines
Resistin: Resistin is a cytokine whose role is not well defined, although firstly was related to obesity, insulin resistance and development of T2D[143].
Visfatin: Visfatin is mainly synthesized in the abdominal adipose tissue of humans but not by peripheral adipose tissue, and the first role appeared to have insulin-mimetic actions[144,145]. However, the relevance of visfatin in the regulation of glucose metabolism is not clear[146].
Omentin 1: Plasma levels and omentin gene expression in visceral adipose are decreased in obesity[147]. Omentin 1 is decreased in obese women with polycystic ovary syndrome (PCOS), both glucose and insulin negatively regulate omentin-1 levels ex vivo and in vivo, and women with PCOS who were treated with metformin increased serum omentin levels[148,149].
Effect of fatty acid metabolism and enzymes on adipogenesis
Fatty acids (FFA) are energy-rich molecules that play a role in metabolism. The excess of calories ingested as fat, protein and carbohydrates, and unspent, are stored as triglycerides (TG; FFA plus glycerol) in mature white adipocytes. They are also an integral part of the cell membrane, conferring functions in fluidity and in the expression of receptors and transporters. In addition, FFA have hormone-like actions and can influence gene expression in preadipocytes, affecting adipogenesis through proliferation and differentiation[150]. In humans, food is an important source of FFAs, but biosynthesis could supply most of the fatty acids requirements[151]. However, humans are unable to synthesize certain PUFA. Therefore, some precursor in the diet are essentials for two series of PUFA, linoleic acid series (ω-6 series) and linolenic acid (ω-3 series), that are related with decreased CVD. Today, most diets in the world provide enough ω-6 and too little ω-3, with an increased ratio ω-6:ω-3. By contrast, diets with excess saturated fatty acids (and unsaturated trans) have been associated with a significantly increased risk of CVD.
In differentiation and maturation of adipocytes, insulin has a definitive influence increasing the expression and activity of LPL, which is needed for an effective FFA uptake and storage. Adipocytes release and express apo CII and apo CIII by regulating extracellular LPL activity[152]. In addition, fatty acid binding proteins (FABPs) are cytoplasmic proteins that carry out intracellular transport of FFA[153]. It appears that the expression of fatty acid binding protein-4 (FABP4) is involved in the balance between lipogenesis and lipolysis and in the process of differentiation of preadipocytes. Therefore, it is likely that FABPs serve as a critical link between lipid metabolism, hormone action and cellular function in adipocytes and other cells and thus contribute to systemic energy homeostasis involving glucose metabolism[154].
In humans, “de novo” synthesis of straight-chain fatty acids is formed predominantly in the liver where acetyl-CoA is formed from pyruvate, and to a lesser extent in adipose tissue. FFA can be endogenously synthesized from acetyl-CoA and malonyl-CoA precursors through two enzymatic steps, including acetyl-CoA carboxylase (ACC) and FAS. The ACC controls six recurring reactions until production of short fatty acids and then the fatty acids are elongated until 16-carbon palmitic acid is produced by the action of FAS (Citosol). Humans can synthesize nearly all fatty acids required from palmitic acid by combining several mechanisms of oxidation and elongation[155]. In mammals seven Elovl family enzymes (Elovl1-7) have been identified, and these enzymes are the limitations in control of production by fatty acid elongation[156]. The enzyme activity of Elovl3 is transcriptionally regulated by PPAR-γ, and in turn the levels of VLCFAs (C18: 1 and C20: 1) produced by the expression of Elov13 activate PPARγ. Therefore Elovl3-PPAR activity is implicated in the regulation of adipogenesis[157]. Saturated fatty acids are amply available from the food by humans, thus FAS enzyme has been shown to have less importance. However, the malonyl-CoA levels are determined by the rate of synthesis by ACC and FAS-mediated catabolic rate, and appear to be an important energy status sensor in the hypothalamus in the metabolic control of body weight[158]. Moreover, in the process of differentiation of preadipocytes to mature adipocytes a lower activity of FAS has effects reducing adipose tissue[159]. Finally, in the process of synthesis of triglycerides in adipose cells, several enzymes have been observed with an interest in adipogenesis[160]. The levels of mRNA and protein of diacylglycerol acyltransferase 1 (DGAT1) increase during the process of differentiation of preadipocytes. DGAT1-deficient mice are resistant to diet-induced obesity associated with a higher energy expenditure. While overexpression of DGAT1 resulting in increased adipose tissue without affecting IS, but increased the secretion of TNF, which interferes with insulin signaling[161].
OBESITY AND LIPOTOXICITY SYNDROME
After absorption in intestine and after synthesis in the liver triglycerides (TG) are packed in specialized lipoproteins [chylomicron and very like density lipoprotein (VLDL)]. They are transported in a network between different locations such as the digestive system, liver, adipose tissue and other tissues. The formation of TG can also be considered a cellular detoxification process by controlling the levels of diacylglycerol and the input and output flows of FFA and acyl-CoA[162]. In this regard, droplets containing TG were found in all investigated cells, and even brain tissue has this capacity to form TG. These fat droplets are surrounded by a monolayer of phospholipids hooked by a specific protein called Perilipin (ADRP) which appear to regulate, and are rate limiting factor in its formation, growth and dissolution[163].
Downloading and uptake of free fatty acids in non adipose tissues typically is coupled to its necessity. During periods of fasting and physical exercise should be increased the lipolysis, that is mediated by suppression of plasma insulin and elevation of contrainsulin hormones (glucagon, cortisol, etc.), generating a coupled fuel delivery. Thus, for an optimal mobilization and storage of lipid an efficient adipose tissue is required. By contrast, after a prolonged overfeeding state, fatty acid load offered may exceed the storage capacity of adipose tissue (inflexibility) (Figure 3). Nuclear receptor PPAR-γ is a key gene that regulates adipogenesis and lipid storage, but it appears that is also needed for the control of the lipolysis, dysregulation of which is a prominent characteristic of obesity-induced insulin resistance in humans[164]. In addition, the expression of leptin receptor is found in several tissues in the body involving leptin actions in many different sites, including as be a mediator of energy expenditure[124]. Leptin secretion rises in parallel with fat expansion in adipocytes and it has been proposed that this prevents lipotoxicity by minimizing ectopic accumulation of lipids into nonadipocytes because leptin induced β-oxidation increasing transcription of PPAR-α. Therefore, excess fatty acids will increase activation of PPAR-α which is a transcription factor of lipolytic enzymes such as carnitine palmitoyl transferase-1 and acyl CoA oxidase. Lipolysis is forced by increasing β-oxidation and uncoupling proteins activity, which corresponds with the observed increase in heat and finally would protect these tissues from the accumulation of fatty acids[165,166]. However, although insulin treatment acutely increases leptin levels, it has been observed that patients with insulin resistance syndrome have lower mRNA leptin abundance in adipocytes than IS patients[115,167]. In addition, a leptin resistance syndrome in humans for central hypothalamic action has also been found. Finally, this system of chronic increase of β-oxidation can already generate oxidative stress “per se” and an inflammatory condition, which can be harmful to these tissues. On the other hand, adiponectin have a key role like insulinsensitizing, anti-inflammatory, anti-apoptotic and pro-angiogenic properties increasing the metabolic flexibility of adipose tissue, i.e., to make adipose tissue more efficient at discharging FFAs when are required and upgrade the rate of FFA re-esterification during the postprandial state[168]. Finally, in insulin resistant patients early lower serum adiponectin levels that could not adequately prevent all these processes are observed[115]. When these mechanisms are exceeded, an accumulation of fatty acids occurs, and its derived metabolites, which generate lipotoxicity and increased cell death in those tissue not prepared to accumulate this excess of lipids such as muscle, β-cells pancreatic, liver, heart, kidneys, etc.[126].
FROM INSULIN RESISTANCE TO OTHER CARDIOVASCULAR RISK FACTORS
In conditions of overnutrition the adipose tissue (AT) expands to levels of inflexibility (adiposity), and in this state the subject presents a longer postprandial state which leads to hyperinsulinemia, probably the first step in this altered dysfunctional metabolic system (Figure 4). Thus, a lower capacity of disposal and storage of fatty acids associated with an increased lipolysis by AT, and dysfunctional pattern of adipocytokine release (e.g., decreased adiponectin, and increased leptin, TNF-α and IL-6), may result in inflexibility of AT and indirectly induce redistribution of fat towards undesired and toxic lipids ectopic accumulation. Therefore, when central obesity is slowly being developed, it is observed that hyperinsulinemia and hyperglycemia also progress slowly in postprandial state and later a global hyperglycemia (T2D), hypertriglyceridemia, hypoalphalipoproteinemia, hypertension and fatty liver (dysfunctional metabolism) are developed. When a combination of any of these factors cluster together in the same individual the concept of MetS is established[169].
Figure 4.
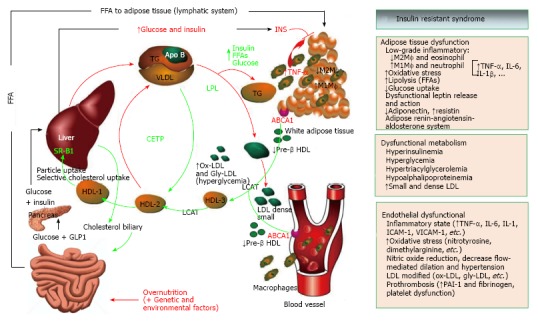
Insulin resistant syndrome and lipid metabolism. When obesity is developing, early abnormalities are observed at this time including hyperinsulinemia and low grade of proinflammatory state (↑ cytokines and PCR-hs), increase liberation of free fatty acids from adipose tissue (↑ lipolysis) and altered release of adipokines (↓ adiponectin, ↑leptin with leptin resistance). In some subjects, fatty liver develops later and consequently affects some functions of the liver. These include an early altered postprandial state (increasing glucose and triglyceride-rich VLDL particles), but finally these finding are observed in fasting state[289]. The VLDL particles undergo reduction by LPL and triglycerides are taken up by adipose tissue. The final result is the increase of cholesterol-rich small and dense LDL particles in serum. These LDL particles are highly susceptible to modifications like oxidation and glycation and the result is the increasing levels of ox-LDL, gly-LDL and the generation of antibodies to ox-LDL[190]. Finally, modified LDL are phagocytosed by macrophages in endothelial blood vessels and an inflammatory pattern that alters endothelial function initiating arteriosclerosis begins[177]. On the other hand, through ABC1 ligand the lipid efflux from peripheral cells to start the reverse transport of cholesterol is mediated. Mature HDL3 are generated from lipid-free apo A1 or lipid-poor pre-β1-HDL as the precursors, and LCAT-mediated sterification of cholesterol generates mature HDL3 and HDL2[189]. In T2D insulin-resistant patients, after adequate metabolic control the HDL3 cholesterol and APO A1 levels were increased. These findings were associated with a higher specific binding activity of HDL3 in those patients that showed improved insulin resistance[190]. Cholesterol efflux capacity has a strong inverse association with carotid intima-media thickness and was inversely associated with the incidence of cardiovascular events in a population-based cohort[188,290]. LCAT-mediated cholesterol esterification generates large spherical HDL2 particles, but large HDL2 can be converted in turn to small HDL3 upon CETP-mediated transfer of CE from HDL to apoB-containing lipoproteins, interfering with reverse cholesterol transport. Finally, SR-BI mediates the selective uptake of cholesteryl esters from HDL particles into mainly liver and steroidogenic organs[291]. VLDL: Very light density lipoprotein; LPL: Lipoprotein lipase; ox-LDL: Oxidized-LDL; gly-LDL: Glycated-LDL; ABC1: ATP-binding cassette transporter 1; LCAT: Lecithin cholesterol acyltransferase; CETP: Cholesteryl ester transfer protein; SR-BI: Scavenger receptor class-B, type I.
The elevated levels of TG are directed toward white adipose tissue and changes occur in adipocyte size, which leads to changes in its function, and an increase in secretion of TNF-α and Leptin, which stimulates the secretion of monocyte chemotactic protein (MCP-1)[170]. This attracts more macrophages to the adipose tissue. Increasing leptin secretion also stimulates macrophage transport to adipose tissue[171] and macrophage adhesion to endothelial cells[172]. Whatever the stimulus for attracting these macrophages, once present and the recruitment is active, the cytokine production of these macrophages interfere with the normal function of adipocytes (adipose tissue dysfunction)[173]. When an inflammatory environment is established in the adipose tissue, the lipid metabolism is altered, initiating postprandial hypertriglyceridemia, because the liver overproduction of VLDL is not removed in time and remains for longer in plasma (postprandial hyperlipidemia). Further, because lipolysis from peripheral adipose tissue is extended, the interstitial content of free fatty acids increases, which can be taken up by the adjacent muscle cells (↓ IS) or again transferred into lipoproteins to the plasma and could be taken up by the liver (↑ VLDL production) and other organs (lipotoxicity). However, not all obese individuals necessarily develop metabolic complications, as some remain insulin sensitive and do not develop fatty liver[115]. On top of all these factors, the link between obesity and associated metabolic abnormalities seems to be better related to the topography, anatomical distribution and/or the functional peculiarities of the adipose tissue, a phenomenon which seems to be more relevant in patients with relatively normal weight (Figures 2 and 3).
In obese people elevated triglyceride levels, that are independently associated with an increased risk of cardiovascular disease, are often observed. The liver frees VLDL which are carriers of triglycerides, cholesterol esters and phospholipids, and the hydrolysis of VLDL-TG macromolecule provides cholesterol to peripheral tissues and triglycerides mainly to adipose tissue. The metabolism of triglycerides in adipose tissue is affected by adipokines (leptin and adiponectin) and other factors such as LPL and cholesterol ester transferase protein (CETP)[174]. Moreover, the LDL molecules remain longer in plasma, and slowly lose some cholesterol and become small and dense particles, which make these particles more susceptible to changes in oxidation and glycosilation (ox-LDL, gly-LDL, etc). The removal and phagocytosis of oxidized and modified forms of LDL cholesterol (LDL-C) by macrophages located in blood vessel walls is a main event in the development of atherosclerosis[175]. Under these conditions, also possibly being affected by high insulin levels and increasing macrophage infiltration, which when activated produce proinflammatory cytokines and adhesion molecules (CRP, TNF-α, IL-6, VCAM, ICAM and MCP-1), blood vessels endothelial cells undergoes hypertrophy[176]. In early obese T2D patients, even serum ox-LDL levels are influenced by short-term serum glucose variations and flow-mediated endothelium-dependent dilation was decreased and inversely related with increments of circulating ox-LDL levels (endothelial dysfunction)[177]. Finally, HDL, which removes surplus cholesterol in peripheral tissues and moves it to the liver either to reuse or excretion, what is recognized as reverse cholesterol transport (RCT), are also lowered by effects at various points[178]. Therefore, elevated triglycerides and decreased HDL-C, also so-called atherosclerotic profile, are considered a risk factor for CVD, independent of LDL-C levels[174,179-183]. The RCT begins when small precursors of HDL (nascent Apo AI/HDL, pre-β HDL) accept the cholesterol and phospholipids through interaction with ATP-binding cassette (ABC) transporters ABCA1 and ABCG1[184]. ApoA-I is released mainly by the liver and small bowel as lipid-poor apoA-I and nascent phopholipid-rich cholesterol-poor HDL particles. In humans, various mutations in the ABCA1 gene outcome in lowered plasma HDL-C levels and great storage of cholesterol in macrophages located in lymph tissue, and they have an enhanced risk of atherosclerotic events. The liver X receptors LXRα (NR1H3) and LXRβ (NR1H2) have a key role in the control of cholesterol metabolism. Storage intracellular cholesterol levels results in increased cholesterol oxidized forms (oxysterol) which are endogenous ligands for LXRs; therefore, it is as sensors to keep cholesterol at suitable levels and to equilibrate it in all sites of body[185]. The LXR system could intervene in gene expression, controlling the efflux of cholesterol from peripheral cells (macrophages), the elimination of cholesterol from the liver, and the regulation of cholesterol absorption in the small bowel[186,187]. Although the efflux of cholesterol from macrophages is a small part of reverse cholesterol flux, it is the most significant component of atheroprotection. Thus, both plasma HDL cholesterol level and the ability to efflux are highly significant indicators of cardiovascular disease status[188]. In obesity HDL functions change dramatically during acute and chronic inflammation of adipose tissue, and changes in quality of HDL can contribute to the failure of atheroprotective capacity, and decreased efflux capacity in patients with MetS and diabetes have been shown[189]. In addition, after adequate metabolic control of diabetes in T2D insulin resistant patients, the HDL3 cholesterol and APO A1 levels, directly associated with higher specific binding activity of HDL3, were increased[190]. Moreover, LCAT (lecithin cholesterol acyl-transferase) enzymes bound to HDL particles play an important role in the change from nascent to mature HDL. LCAT converts free and unesterified cholesterol (form of efflux) in cholesteryl ester, a hydrophobic form of cholesterol (form of transport), that make particles of HDL more spherical and mature. The mature HDL2 and HDL3 particles in plasma are constantly remodeled by lipase and interact with other lipoproteins through lipid transfer. This can affect the normal reverse transport of HDL cholesterol to its routes of removal (mainly liver). Therefore, the CETP mediates exchange of HDL cholesteryl ester (CE) with VLDL-triglycerides lipoproteins, and this result in a CE reduction with higher amount of TG in HDL lipoproteins (Figure 4). Thus, in clinical situations of obesity, like insulin resistance and T2D, where VLDL particles are frequently increased (hypertriglyceridemia), HDL cholesterol levels are inversely lowered. In addition, HDL has a variety of anti-atherogenic properties apart from efflux of cholesterol and RCT. It improves endothelial function, inhibits thrombosis and has powerful antioxidant and anti-inflammatory effects.
Last, most patients with features of MetS have increased blood pressure. Several contributing factors such as hyperinsulinemia increases the reabsorption of Na+ and also activates the sympathetic nervous system. In addition, releasing factors from adipose tissue could stimulate aldosterone secretion independently of angiotensin II, K+ or ACTH[191]. Furthermore, local source of angiotensin II in adipose tissue may also be raised in obese hypertensive subjects establishing the participation of adipose-tissue renin-angiotensin system in insulin resistant syndrome[192].
FROM OBESITY AND INSULIN RESISTANCE TO METS
MetS was referred to as a group of related metabolic disorders for the first time in 1920 by Kylin. Decades before of the introduction of measurements with specific methods for insulin, Himsworth (1936) suggests that diabetes could be found two types, what he termed “insulin-sensitive” and “insulin-insensitive” types. Later, Reaven[193] (1988) observed that several risk factors (dyslipidemia, hypertension, hyperglycemia) commonly cluster together in insulin resistant subjects (Figure 4). He described it and underscored their clinical importance in their Banting lecture, and he used the name “Syndrome X” but obesity was not including in their definition. Today it is known as “MetS“ defined as a “set of metabolic disorders and cardiovascular risk factors, which foresee a high risk of developing diabetes and CVD”. The more clinical definition was advanced by Grundy[194] in 1999, who described MetS as “a set of metabolic disorders, many of which promoted the development of atherosclerosis and increase the risk of CVD”, and were established in the national cholesterol education program’s adult treatment panel III report (ATP III) and later updated in 2004[195]. It avoids the implication that insulin resistance is the primary or only cause of associated risk factors. In addition, because the presence of abdominal obesity is more highly correlated with the metabolic risk factors, measurement of waist circumference was included as a clinical method to identify patients susceptible to MetS[196]. When it is > 102 cm in men and > 88 cm in women it is called abdominal obesity, which is a high risk factor of MetS[194]. Other clinical criteria that Grundy established for the diagnosis of MetS were a blood pressure ≥ 135/85 mmHg[197], elevated fasting glucose levels ≥ 110[198], triglycerides ≥ 150 mg/dL[199] and HDL-C < 40 mg/dL for men and < 50 mg/dL for women (Atherogenic dislipemia). When any 3 of the 5 listed characteristics are present, a diagnosis of MetS must be made. A proinflammatory state, clinically observed by elevation of C-reactive protein (CRP-hs), and a prothrombotic state characterized by increased plasma levels of the inhibitor of plasminogen activator (PAI-1) and fibrinogen are also recognized in MetS.
At the same time (1999) the expert committee of the WHO described MetS as a cardiovascular disorder associated with insulin resistance. In order to diagnose MetS according to WHO criteria, insulin resistance should be identified, together with two or more risk factors, with minimal changes of the factors previously described, but including urinary albumin excretion rate ≥ 20 μg/min or albumin: Creatinine ratio ≥ 30 mg/g (microalbuminuria)[200,201].
Last, in order to unify both epidemiologic criteria as clinical, the International Diabetes Federation (IDF) established a set of criteria for diagnosing MetS[202]. While the pathogenesis of MetS and each of its components is complex, multifactorial and not well established, either central obesity and insulin resistance or both are recognized as the main causative requirements. Cardiometabolic risk is mainly associated with abdominal obesity because VAT triggers dyslipidemia, insulin resistance and hypertension[203,204]. This VAT could be assessed by CT, MRI and DEXA, costly measures and not for everyday use. However WC and WHR may be used as proxy measures of VAT, as they are correlated with it[205-207]. Waist circumference gives a closer approximation of abdominal obesity than BMI, the range being different between ethnic populations with respect to overall adiposity, abdominal obesity and visceral fat[208-210]. However, IDF dropped the WHO requirement for insulin resistance but made abdominal obesity necessary as 1 of 5 factors required in the diagnosis. IDF provides the following criteria to define MetS: Central (abdominal) obesity is readily measured using waist circumference and is particularly related with each of the other MetS components, singularly with insulin resistance, and “is a prerequisite risk factor”. Abnormality in the distribution of body fat, associated with central obesity and ethnic specific values for waist circumference (BMI ≥ 30 kg/m2; WC ≥ 94 and 80 cm and 102 and 84 cm, respectively for men and women in Europe and United States).
In addition, any two of the following four factors: The atherogenic dyslipidemia with: (1) high levels of triglycerides (≥ 150 mg/dL); (2) reduced cholesterol-HDL (< 40 mg/dL in men and < 50 mg/dL in women), and more precise analysis high level of apolipoprotein B (Apo B) and high number of small and thick LDL particles and small HDL particles[211]; (3) Treatment of previously diagnosed hypertension or high blood pressure (≥ 130 mmHg systolic and ≥ 85 mmHg diastolic); and (4) The hyperglycemia defined as impaired fasting glucose > 100 mg/dL or previously diagnosed T2D.
Other factors such as genetic profile, physical inactivity, aging, proinflammatory state and hormonal dysregulation could be considered[202].
Therefore, additional metabolic measurements are recommended. Lipodystrophic disorders, either genetic (e.g., Dunnigan familial partial lipodystrophy, Berardinelli-Seip congenital lipodystrophy, etc.) or acquired are almost associated with MetS, and occasionally a genetic study could be considered. Most components of MetS are correlated with a sedentary lifestyle. MetS prevalence and each of its components is directly related with age in most people on the world. Assessment of body fat distribution (DEXA) or central obesity (CT/MRI) or fatty liver content (spectroscopy) could be advised. Proinflammatory state presents an increased levels of CRP, and adipocytes and macrophages release inflammatory cytokines (TNF-α, IL-6), and decrease antiinflammatory adiponectin and increased leptin levels are associated with adipose dysfunction[212,213]. Prothrombotic state with increased PAI-1 and fibrinogen[214]. Vascular dysregulation (apart of hypertension) could be estimated with endothelial function and presence of microalbuminuria. Insulin resistance with measurements of fasting insulin/proinsulin levels, HOMA-IR[215], by Bergman Minimal Model[216], during oral glucose tolerance test[217], and gold standard from M value from euglycemic-hyperinsulinemic clamp[218,219].
Finally, several organizations have attempted to harmonize criteria for the definition of MetS [International Diabetes Federation Task Force on Epidemiology and Prevention, National Heart, Lung, and Blood Institute (NHLBI), American Heart Association (AHA), World Heart Federation; International Atherosclerosis Society, and International Association for the Study of Obesity]. They concluded that three abnormal findings out of five would be sufficient to diagnose a person as having MetS. The IDF and AHA/NHLBI agreed that central obesity may not be a prerequisite for diagnosing MetS but could be one of the 5 criteria[66].
EFFECTS OF NUTRITION ON METS COMPONENTS
The prevalence of MetS based on the ATP criteria rose from 28% in the Third National Health and Nutrition Examination and Survey (NHANES) 1988-1994, to 32% in NHANES 1999-2000. It is estimated that 11% of men and 18% of women between the age of 20-39 have MetS. But, rates increase to 40% in men and 46% in women older than 60 years of age, the frequencies being similar in many developed countries of the world[220]. However, at the moment epidemiological and clinical research has released complex and partial information to guide the development of finished nutrition prevention programs. The US Departments of Agriculture and Health and Human Services issued dietary recommendations in the Dietary Guidelines for Americans (DGA), to aid decrease the risk of CVD. This document was also recommended by the AHA (in 2005 and update in 2010) as a dietary proposal to decline the incidence of MetS[221,222]. The updated edition of the DGA accentuates about calory density of the nutrient, and recommends a reduced intake of saturated fat and a confined intake of trans fats, but a greater intake of whole grain, variety of fruit and vegetables, and its adherences have been related with a improve in incidence and prevalence of MetS[223,224].
Recently, Scientific Report of the 2015 Dietary Guidelines Advisory Committee (DGAC) also shows that the dietary standard of the majority of the United States people, as well as other developed countries, has a low intake of key food groups that are important sources of shortfall nutrients, including vegetables, fruits, whole grains, and dairy[225]. In addition, a higher intake of red and processed meats are shown as harmful compared with a lower intake, and higher ingestion of sugar-sweetened foods and beverages as well as derived of refined grains have been found damaging with moderate to strong evidence. Moreover, the DGAC also found that sodium and saturated fat are being over-consumed by Americans, and probably in many westernized countries as well. However, overweight and obesity rates have continued to increase despite actions to recommend decreasing the percentage of fat in food, suggesting that the actions on obesity are more complex. In addition, the healthy Mediterranean-style diet is one of three diets recommended by DGAC, because variations of this diet include many components associated with health benefits. Mediterranean diet is part of an ancient culture of nutrition and is being adopted by different peoples and countries. Previously, an elegant study identified the subjects with MetS as a target for dietary therapies to reduce several components of this syndrome. Patients with MetS, received elaborate advice on how to raise daily ingestion of whole grains, vegetables, fruits, nuts, and olive oil; whereas patients in the control group followed a prudent diet. After 2 years, patients that follow the Mediterranean diet had an intake higher in monounsaturated fat, as well as polyunsaturated fat, and fiber and had a decrease ratio of ω-6 to ω-3 fatty acids. At 2 years of follow-up, patients consuming the Mediterranean diet had significantly reduced serum concentrations of hs-CRP, interleukins, as well as IS, and endothelial function score were improved. Moreover, the Mediterranean diet prevented MetS compared with the control group[226]. Last, its beneficial effects have recently been reported among persons at high cardiovascular risk. A Mediterranean diet supplemented with extra-virgin olive oil or nuts reduced the incidence of major cardiovascular events and prevalence of MetS[38,227].
In the prevention and treatment of MetS it has been found that it is not one specific diet, but rather various changes of nutrients in the diet that should be recommended to treat or prevent the onset of each different component of the syndrome.
Effects of nutrition on obesity
The first factor to be avoided in the prevention of MetS is obesity, and the percentage of fat in the diet has traditionally been associated with the development of obesity. There is evidence to show that metabolic stressors including energy-dense high-fat diets develop obesity, and probably insulin resistance and MetS[228-230]. In overweight subjects, selected on the basis of impaired glucose tolerance, the prevalence of overweight and MetS decreased after two and four years of an extensive life-style intervention which mainly included a reduction of energy and SFA intake and an increase in physical activity[231,232]. However, other strong epidemiological evidence has reported contradictory results at this point. An important epidemiological analysis from the European Prospective Investigation into Cancer and Nutrition, which included 519978 participants, found no significant relationship between the amount and type of fat consumed and annual weight gain. Recently, in this cohort it has also been observed that higher SFA consumption was not related with higher ischemic heart disease risk[230,233]. But, residual confounding factors, such as cholesterol-lowering therapy and trans fat intake or limited variation in SFA and PUFA intake, may explain these findings. Moreover, in well-conducted intervention studies, in extremely obese subjects with a raised prevalence either diabetes or MetS, a higher weight loss was showed after six months on a carbohydrate-restricted diet than on a fat-restricted diet, with a relative upgrade in IS and triglyceride levels, even after control for the amount of weight lost[234]. Additionally, in a randomized controlled trial to observe weight loss in overweight premenopausal women, where four diets containing a gradual and inverse fat and carbohydrate content were compared, the diet with less carbohydrate content (Atkins) achieved greater weight loss and metabolic success[235]. It has also been found that high-protein and low glycemic index (GI) diets are better tolerated than low-protein with high GI. In addition, the low protein with high GI diet was associated with subsequent significant weight regain[236]. Further, higher weight loss with low-carbohydrate diets may be associated to the satiating effects of fat and protein content. We have previously found that following the intake of a standard breakfast, the glucagon like peptide-1 (GLP-1) postprandial release was significantly raised in those patients who had eaten an isocaloric olive oil-enriched meal compared to when they had a CHO-rich meal, further supporting the idea that monounsaturated (MUFA) fatty acids may act as secretagogues of GLP-1 (Figure 5)[237]. The biological effects of GLP-1 well know include stimulation of glucose-dependent insulin secretion which is lowered until a normal blood glucose level, delay gastric emptying and inhibition of food intake, increases the β-cell proliferation and inhibition of their cell death[237]. Finally, epidemiological studies have established an inverse relationship between the consumption of dietary fiber and body weight and waist perimeter[238,239]. Therefore, several controlled intervention studies demonstrated that dietary fiber content in the diet is negatively associated with weight gain, and may have a satiating effect and decreases the amount of calories ingested[240,241]. Thus, weight loss can be difficult to attain and maintain long-term with interventions of more or less experimental diets. Therefore, important data to reduce and maintain body weight should include the total amount of energy consumed, others characteristics and combinations of the nutrients ingested and the amount and type of physical exercise performed daily. The main interest of research today is to define the potential therapeutic effects of replacing SFA with MUFA or with a low-fat diet on regression of MetS or the effect on the different components of the syndrome.
Figure 5.
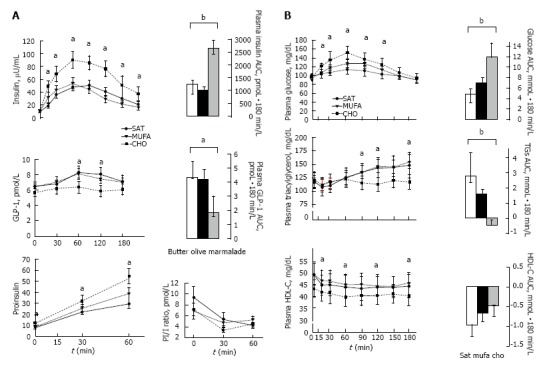
Mean (± SE) postprandial responses of insulin, proinsulin and glucagon-like peptide-1 levels (A), and glucose, triacylglycerol and high-density lipoprotein cholesterol levels (B), in 11 insulin-resistant subjects to three isocaloric (443 kcal) standard breakfasts. A breakfast rich in carbohydrates, a Mediterranean breakfast enriched with extra-virgin olive oil and standard breakfast high in saturated fat. The incremental AUC was calculated by the formula based on the trapezoid rule with adjustment for baseline concentrations. Repeated measures ANOVA and Tukey’s test. aP < 0.05; bP < 0.01[237]. CHO: High-carbohydrate; MUFA: Monounsaturated fatty acids; AUC: Area under the curve; ANOVA: Analysis of variance.
Effects of nutrition on central fat distribution
It is well established that the type of fat consumed could be more decisive than the total amount of fat consumed when we only look at changes in body composition and distribution of adipose tissue[242,243]. It has been proposed that high adiposity and central fat deposit is related to diets with a high ratio of saturated to unsaturated fatty acids[115]. In this regard, SFA refers mainly to Myristic (C14), Palmitic (C16) and Stearic (C18) acids; MUFA refers mainly to oleic acid (C18:1n-9) in Western and Mediterranean countries; PUFA refers mainly to linoleic acid (C18:2n-6), a less ratio of alpha-linolenic acid (C18:3n-3) and, in relation of seafood ingested, a changeable but lower rate of long chain PUFA such as Arachidonic, eicosapentaenoic (EPA), docosapentaenoic and docosahexaenoic (DHA) acids; and finally TFA remits to the main trans fatty acids which are isomers of 18:1 trans and are not found in nature and are the result of human processing (e.g., hydrogenation). In this issue, the Nurses’ Health Study showed just a weak direct association between whole fat intake and overweight. However, when the proportion of SFA and TFA was higher it showed a strong relationship to obesity, but the consumption of MUFA and PUFA were not associated[242]. In addition, MUFA and PUFA fat intake have been associated with healthy effects on body fat distribution and improved some other metabolic disorders, as compared with SFA and TFA intake, while maintaining stable body weight. Therefore, in subjects selected with central obesity, after a short intervention with a low-fat carbohydrate-rich diet, patients grouped according to insulin-resistant state (Matsuda < 4) showed a redistribution of their body fat from peripheral adipose tissue toward central body deposits as compared with isocaloric MUFA-rich diet (Table 1)[52]. Moreover, the substitution in the diet of saturated with unsaturated fat, mainly MUFA, resulted in little but consistent loss of body weight, decreased body fat content in limbs and trunk, while maintaining a high and isocaloric fat content (approximately 40%)[244]. Furthermore, the intake of n-3 PUFA, EPA and DHA have been linked to an effect on body weight and body composition. Therefore, higher plasma levels of total n-3 PUFA are related to a decreased BMI and waist and hip circumferences[245]. In addition, central fat distribution was negatively related with n-6 PUFA and MUFA in adipose tissue that correlated closely with fatty acids intake in obese patients from a Mediterranean area[246]. Recently, the long-term consumption of a LFHCC diet increased fasting FABP4 expression in adipose tissue, while it was reduced by the consumption of LFHCC supplemented with n-3 diet[247]. Finally, it was found that conjugated linoleic acids (CLA) produces a reduction on adiposity whereas the lean body mass was not altered or increased, and the waist-hip ratio decreased significantly compared with placebo in adults[248,249]. In another study it was found that the rate of body fat lowered in the CLA-treated group, whereas body weight, BMI, and central abdominal diameter were unmodified[250].
Table 1.
Composition and body fat distribution after three dietary interventions in insulin-resistant subjects
| Baseline | High-SAT | High-MUFA | High-CHO | P | |
| EE, (kJ/min) | 5.36 ± 0.40 | 5.49 ± 3.90 | 5.23 ± 0.37 | 5.02 ± 0.36 | 0.3 |
| Anthropometry | |||||
| Weight, kg | 84.4 ± 5.7 | 83.2 ± 5.7 | 83.6 ± 5.8 | 81.8 ± 6.03 | 0.3 |
| Total body fat, kg | 36.8 ± 4.1 | 35.0 ± 4.0 | 35.6 ± 4.0 | 34.9 ± 4.3 | 0.1 |
| Lean body mass, kg | 47.5 ± 2.5 | 48.1 ± 2.5 | 48.9 ± 2.6 | 46.8 ± 2.1 | 0.2 |
| Waist to hip ratio | 0.99 ± 0.01 | 0.99 ± 0.01 | 0.98 ± 0.01 | 0.98 ± 0.01 | 0.9 |
| DEXA analysis | |||||
| Total body trunk, g | - | 37101 ± 2026 | 38154 ± 1911 | 39134 ± 2104 | 0.3 |
| Fatty body trunk, g | - | 14313 ± 1362 | 14842 ± 1437 | 16459 ± 1653 | < 0.05 |
| Total body limb, g | - | 36420 ± 3886 | 36239 ± 3862 | 32887 ± 3825 | 0.7 |
| Fat in arm, g | - | 7097 ± 1528 | 7652 ± 1339 | 7225 ± 1830 | 0.4 |
| Fat in leg, g | - | 8517 ± 1588 | 8036 ± 1398 | 7358 ± 1253 | < 0.05 |
| Fat trunk:fat leg ratio | - | 1.9 ± 0.3 | 2.1 ± 0.2 | 2.50 ± 0.2 | < 0.05 |
Data are mean ± SE. P value is analysis of variance for repeated variables. Copyright 2007 American Diabetes Association. Diabetes Care 2007; 30: 1717-1723. Reprinted with permission from the American Diabetes Association. EE: Energy expenditure; SAT: Saturated fat; MUFA: Monounsaturated fat; CHO: Carbohydrates rich diets; DEXA: Dual energy X-ray absorptiometry.
Effects of nutrition on insulin resistance
Insulin resistance is a main characteristic of MetS and is related with other components of the syndrome. The KANWU study treated 162 healthy subjects selected at aleatory to eat a controlled, isoenergetic diet for 3 mo containing either a major rate of saturated (SAFA diet) or monounsaturated (MUFA diet) fatty acids. After 3 mo, subjects lowering saturated fatty acid and increasing monounsaturated fatty acid, enhanced IS but had no action on insulin secretion. This favorable effect of different proportion and fat quality on IS was not found in subjects with a fat proportion ingested higher than > 37% of energy eaten[251]. In addition, in healthy subjects, it has been shown that isoenergetic substitution of SFA for MUFA or complex carbohydrates (CCHO) improved IS, and other components of MetS such as blood pressure[252,253]. In selected subjects with central obesity and insulin-resistance on weight maintenance, a MUFA-rich diet improved IS (HOMA-IR) and fasting proinsulin levels as compared to the CHO-rich diet[237]. Finally, in subjects with early diagnosed non alcoholic fatty liver disease (NAFLD), those with more adiposity, higher trunk fat:leg fat ratio (by DEXA) and lower IS, had a higher ratio SAT:MUFA fat intake than insulin sensitive (IS) subjects[115]. By contrast, the LIPGENE was the largest human intervention study, pan-European and multicentre, developed to observe the effects and efficacy of changing the type and proportion of dietary fat eaten on IS and other metabolic components that integrate the MetS. This intervention was isoenergetic to avoid the effects of weight modification. At the time, it is partially known the metabolic consequence of adhering to low-SFA diets enriched in MUFA or to LFHCC diets, and whether LC n-3 PUFA can improve the negative effects of a low-fat high-carbohydrate diet in MetS. In conclusion, LC n-3 PUFA supplementation significantly lowered TG and FFA levels in men with MetS. The reduction of dietary SFA had no action on IS, blood pressure, LDL cholesterol levels and factors of inflammation. The LIPGENE study observed that the previous dietary consumed and environment may determine responsiveness to dietary fat modification with respect to IS. More specific dietary fat modifications may be necessary to significantly improve IS and other components of MetS; perhaps in combination with dietary restriction and weight loss[254]. There is evidence that a proportion of fat in the diet in excess of 40% worsens IS, especially when ingested fat is saturated[251]. However, recently in this same study those MetS subjects when were selected from the upper HOMA-IR were improved IR, with lowered insulin and HOMA-IR levels after ingestion of the HMUFA and LFHCC n-3 diets. Therefore, specifically insulin-resistant MetS subjects with more metabolic components make a response differently to dietary fat change, being more sensitive to a healthy effect from the exchange of the high SFAs diet by the HMUFA and LFHCC n–3 diets[255].
Effects of nutrition on glucose metabolism
Other dietary factors that can influence various components of MetS, like postprandial glycemic and insulin levels, triglycerides and HDL-C levels, weight regulation and body composition, as well as fatty liver, are the glycemic load (GL) and the excess of fructose and dietary fiber content of food eaten. On the glycemic index (GI) of a food we identify the area under the curve of blood glucose levels two hours after ingestion of a set amount of CHO where glucose is set to equal 100%. So a low GI food will cause a small rise (≤ 55), while a high GI food will trigger a dramatic spike (≥ 70)[256]. Diets higher in fat and a lower content of CHO necessarily have a lower GL and lower GI. Therefore, the beneficial effect of an olive oil enriched diet avoiding simple carbohydrates, e.g., a typical Mediterranean breakfast with wheat bread and olive oil instead of white bread and marmalade, is also found during the postprandial state where lower glucose and insulin AUCs are observed, as compared with CHO-rich diets (Figure 5)[237]. By contrast, during an isocaloric low carbohydrate high fat (better MUFA) diet, after absorption the free fatty acids are transported via the lymphatic system without stimulating the secretion of insulin, so the fatty acids are carried directly to the peripheral adipose tissue; thus, postprandial insulin peak and hyperglycemia are reduced[237]. These higher postprandial levels of glucose and insulin after eating foods with a high GI or GL may mediate changes on adiposity and central fat redistribution observed in selected insulin-resistant subjects (Table 1)[52]. Following intestinal absorption of excess carbohydrates these are transported via portal and, after signaling insulin secretion in the pancreas, are deposited in the liver. However, in obese subjects, when the storage limit is exceeded, and through several metabolic pathways, that mainly include the transcription protein carbohydrate response element binding protein, which is activated by a high-carbohydrate diet, the glucose can be used to synthesize fatty acids which are released into plasma as VLDL rich in triglyceride[257]. Thus, triglycerides can be captured more widely and again can reach the central depot (Figure 4). Once the function of liver buffer is lost, a state of concomitant hyperglycemia, hyperinsulinemia and hypertriglyceridemia and fatty liver results, due to the consumption of diets high in carbohydrates and high GI. However, conflicting data have been published addressing this concept. It is possible that the type of CHO eaten as well as other macronutrients accompanying these diets could modify and partially explain these discrepant results. Therefore, intervention studies looking at the effects of GI and GL have not had clarifying results. The comparisons of four diets of varying GL on weight loss and cardiovascular risk reduction in a randomized controlled trial was made in 129 overweight and obese young adults[258]. The authors concluded that either high-protein or low-GI regimes could have effect on body fat loss, but effects on cardiovascular risk factors are improved by a high-carbohydrate but low-GI diet.
Effects of nutrition on atherogenic dyslipidemia
The increased levels of triglycerides associated with hypoalphalipoproteinemia, are a feature of insulin resistance and MetS, and increase cardiovascular risk regardless of LDL cholesterol levels. The high insulin levels in MetS constantly target the peripheral adipose tissue and stimulates its hypertrophy, which initiates an aberrant inflammatory condition (↑M1 Ø) with elevated levels of TNF-α and IL-6 resulting in adipose dysfunction. Therefore the activity of lipoprotein lipase is reduced in AT and the triglyceride clearance is decreased. Adiponectin levels are reduced and the β-oxidation can be lowered by muscles and liver as well as lowering the sensitivity to insulin (Figure 2)[52,259,260]. Furthermore, the increase of VLDL (↑TG) can interact with reverse cholesterol transport by exchanging triglycerides for cholesterol of HDL-C molecules, which eventually can be reduced in plasma. In fact, low HDL-C levels can be considered as one of the earliest signs of a state of insulin resistance. The consumption of a extra-virgin olive-oil-based breakfast by central-obese insulin-resistant subjects lowered postprandial glucose and insulin postprandial excursions, and increased GLP-1 levels as compared with a isocaloric standard CHO-rich breakfast (Figure 5)[237]. In addition, the effects of these dietary interventions on the plasma lipid profile in these insulin-resistant subjects independently of weight loss were also investigated. Serum total cholesterol and Apo B levels tended to decrease after the CHO diets, but a potentially harmful result lowering HDL-C concentrations (approximately 11%) was also observed. By contrast, the consumption of a hight MUFA diet was associated with significantly higher HDL-C levels. However, fasting serum triacylglycerol concentrations were not altered by any of the three diets (SAT, MUFA and CHO). These effects could be associated to the fact that body weight was maintained unchanged during the three dietary periods, suggesting that triglycerides levels are mainly related with total body fat[237]. By contrast, in the LIPGENE human dietary intervention study, MetS subjects (n = 472) from 8 European countries were randomly assigned 4 diets: A HSFA; a HMUFA diet; a LFHCC diet supplemented with long-chain n–3 polyunsaturated fatty acids (1.2 g/d); or a LFHCC diet supplemented with placebo for 12 wk (control). The LFHCC n-3 PUFA diet reduced plasma TG and FFA concentrations, particularly in men[254]. Finally, in this study, was made a post hoc analysis, selecting only those patients who had been diagnosed of MetS syndrome (according to NCEP MetS criteria updated by the joint scientific statement harmonizing the MetS criteria) to observe the effect after 12 wk of an isoenergetic dietary fat exchange on final incidence of each component of MetS. In addition, final regression of MetS and each component of MetS post-intervention were also investigated. This study concluded that an isoenergetic LFHCC diet supplemented with LC n-3 PUFA reduced some features of MetS compared with high-fat (HSFA and HMUFA) diets and low-fat diet without LC n-3 PUFA. The prevalence of enlarged waist circumference, hypertension and hypertriacylglycerolemia were reduced after the isoenergetic LFHCC n-3 diet. Thus, the prevalence of MetS fell by 20.5% after LFHCC n-3 diet compared with the HSFA (10.6%), HMUFA (12%) or LFHCC (10.4%) diets (Figure 6)[261]. Interestingly, the prevalence of hypertension was reduced after consumption of LFHCC diet supplemented with VLC n-3 PUFA. In a population-based study on food n-3 PUFA intake, an independent inverse relation of total n-3 PUFA intake to systolic and diastolic pressure has previously been shown[262]. In addition, the capacity of PUFAs to target the signaling on gene expression of SREBP-dependent, which controls genes implicated in cholesterol metabolism, gives an evidence of the potential effects of fatty acids on gene expression, beyond of purely nutritional[263]. Further, it has been observed that n-3 fatty acids but not SAT fatty acids are important activators of PPAR-α implicated in triglycerides reduction. Therefore, because of their capacity to repress inflammatory pathways and control the expression of a great quantity of genes associated to lipid metabolism and adipose tissue, n-3 fatty acids are being using as therapeutic agents in lipids, T2D, steatohepatitis and MetS.
Figure 6.
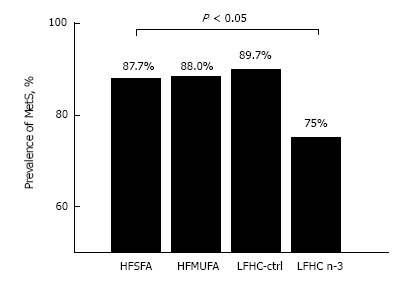
Prevalence of metabolic syndrome after 12-wk of diet assignment. HFSFA is a high fat diet rich in saturated fat and HFMUFA is a high fat diet rich in monounsaturated fat. LFHC-control is a low-fat, high complex carbohydrate diet and LFHC n-3 is a low-fat, high complex carbohydrate diet supplemented with 1.24 g/d of very long chain n-3 polyunsaturated fatty acid (VLC n-3 PUFA). χ2 test, P < 0.05)[261]. MetS: Metabolic syndrome; HFSFA: High fat diets rich in saturated fat; HFMUFA: High fat diets rich in monounsaturated fat; PUFA: Polyunsaturated fatty acids.
Effect of fiber on glucose and lipid metabolism
Different fiber content of the diet can influence several components of MetS. The ADA recommends an consumption of dietary fiber of 20 to 35 g per day mainly since of the cholesterol-lowering and glucose-lowering results of soluble fiber. However, more beneficial actions of a higher ingestion of dietary fiber, specially of the soluble form, over the amount advised by the ADA, were reported to get better glycemic control, lowers hyperinsulinemia, and decreases plasma lipid levels in type 2 diabetic patients[264]. This should warn us that the intake of complex carbohydrates with high fiber content (e.g., whole bread) have healthier effects compared with refined CHO food popular in modern nutrition. Therefore, maintaining a diet that includes a high intake of fruits, vegetables, and whole grains, a rich sources of dietary fiber, such as a Mediterranean diet, should be strongly emphasized.
Effect of nutrition on adipokines
We have recently analyzed the repertoire of adipokines in patients diagnosed with fatty liver, a human model of central obesity, much of them with MetS. We confirmed that IR patients had lower serum adiponectin level than IS patients, and a positive correlation between IS index (ISi) and serum adiponectin levels was observed[115]. It has been shown that hypoadiponectinemia may play a pathophysiological role in the progression from NAFLD to NASH. Adiponectin exerts a endocrine protective action on liver fat accumulation favoring lipolysis (Figure 2)[129]. In addition, we have previously documented a differential postprandial regulation of adiponectin gene expression on peripheral adipose tissue in response to differences in the isocaloric macronutrient composition of diets. Therefore, after a CHO-rich breakfast a lowered adiponectin mRNA expression levels were found as compared when a MUFA-rich breakfast were eaten[52]. The paracrine effects of adiponectin can increase insulin sensitivity by increasing fat β-oxidation and energy expenditure on skeletal muscle[265]. Therefore, these actions and a direct adiponectin effect on the ability of adipose tissue to expand it seems play a key role for the regulation in differences in insulin sensitivity and the prevention of central-obesity in responses to different macronutrient composition of diets, in the context of isoenergetic diets and energy balance[52,115]. Finally, a recent review on the effects of diet on adiponectin levels summarizes that daily consumption of sea foods or omega-3 supplementation could increase adiponectin concentrations by 14%-60%. In addition, weight loss performed with a low-calorie diet more physical activity raised adiponectin concentrations by 18%-48%. Last, with fiber supplementation were improved adiponectin levels until a 60%-115%[266].
Adiponectin and leptin seem to regulate the deposition of fat in insulin-sensitive tissues by increasing fat oxidation. However, whereas leptin acts on peripheral target and through CNS, adiponectin seems to act mainly on peripheral tissue and liver. Therefore, deposition of fat in the trunk but not in the legs was directly related with increased liver enzyme levels. Fatty liver patients with IR show lower leptin (LEP) mRNA expression in peripheral adipose tissue in comparison with IS patients[115]. In these patients we failed to show differences in LEP serum levels between IR and IS patients. Nevertheless, since IR patients were more obese and had higher energy intake in comparison with IS subjects, we speculate that IR patients should have exhibited relatively higher plasma leptin concentrations. This may indicate a dysfunction of adipose tissue in maintaining appropriate levels of leptin to overcome the state of leptin resistance observed in obese subjects particularly where insulin resistance is developed[115]. With respect to the response to diets with different macronutrient composition, there is evidence that long-term change in diet (approximately 1 year), including decreased intake of SATs and increased PUFA reduced plasma LEP concentration regardless of changes in fat mass[267]. The results of a study conducted in our laboratory, are interesting. The study was on the acute effects of different isocaloric diets during postprandial state and after an insulin treatment on molecular markers characteristically involved in the process of WAT expansion. All patients were previously diagnosed with fatty liver and IR (n = 15) and were stabilized for 2 wk with an isocaloric standard diet (National Cholesterol Education Program step 1) for fasting peripheral adipose biopsy. They then randomly eat each one of three isocaloric-specific diets for 4 wk, finally undergoing a postprandial biopsy of adipose tissue after three specific meal test meals; a high saturated fat (SAS), high monounsaturated (MUFA) and low-FAT high-carbohydrates (CHO) (Table 2). The gene-expression array profiles in IR patients showed that acute response after an isocaloric-specific diet (180 min) (MUFA, SAT and CHO) presented similar postprandial transcripts. Our gene-expression arrays further confirmed that an anabolic stimulus induced by insulin treatment can acutely increase LEP and PPARG gene expression in WAT per se. It has been observed that PPARG and insulin are involved in the nutritional regulation of the fsp27 gene in WAT, which is needed for the most favorable energy storage and performs a key role regulating whole-body energy equilibrium[268]. Additionally, this is important because PPARG pharmacologic ligands, such as the thiazolidinediones, increase peripheral AT capacity and decreases liver fat deposition, resulting in a healthier insulin action and liver enzymatic profile[269]. However, as with glitazone treatment, insulin therapy is often associated with weight gain due to its lipogenic effects. Therefore, this approach has the potential of initiating a vicious cycle ultimately leading to further obesity and metabolic stress, and eventually to more IR.
Table 2.
Gene expression ARRAYS of peripheral-white adipose tissue in fasting state and its responses to postprandial specific diets and insulin stimulus
| Baseline (n = 8) | P1 | High-MUFA (n = 9) | High-SAT (n = 9) | High-CHO (n = 9) | P2 | Postinsulin (n = 9) | P3 | P4 | |
| INSR | 6.74 ± 0.11 | 0.69 | 6.94 ± 0.06 | 6.95 ± 0.01 | 6.98 ± 0.04 | 0.95 | 6.99 ± 0.04 | 0.93 | 0.10 |
| GCGR | 6.25 ± 0.13 | 0.03 | 5.92 ± 0.06 | 5.90 ± 0.07 | 5.89 ± 0.06 | 0.95 | 5.75 ± 0.07 | 0.93 | 0.04 |
| BMP7 | 4.80 ± 0.08 | 0.69 | 4.49 ± 0.07 | 4.45 ± 0.09 | 4.53 ± 0.11 | 0.95 | 4.55 ± 0.09 | 0.93 | 0.33 |
| BMP2 | 6.21 ± 0.10 | 0.62 | 5.89 ± 0.09 | 5.96 ± 0.06 | 5.92 ± 0.09 | 0.95 | 5.89 ± 0.07 | 0.93 | 0.12 |
| PPARG | 9.06 ± 0.28 | 0.11 | 9.64 ± 0.08 | 9.44 ± 0.10 | 9.57 ± 0.09 | 0.95 | 9.97 ± 0.13 | 0.14 | 0.04 |
| ADIPOQ | 10.89 ± 0.4 | 0.69 | 11.65 ± 0.06 | 11.33 ± 0.22 | 11.46 ± 0.11 | 0.95 | 11.53 ± 0.16 | 0.93 | 0.10 |
| LEP | 9.37 ± 0.34 | 0.60 | 9.87 ± 0.16 | 9.64 ± 0.22 | 9.91 ± 0.18 | 0.95 | 10.11 ± 0.14 | 0.93 | 0.04 |
| ADRβ3 | 5.23 ± 0.12 | 0.69 | 5.06 ± 0.12 | 5.01 ± 0.10 | 5.05 ± 0.17 | 0.95 | 5.22 ± 0.11 | 0.93 | 0.82 |
| RETN | 6.19 ± 0.05 | 0.02 | 5.59 ± 0.10 | 5.63 ± 0.07 | 5.72 ± 0.09 | 0.95 | 5.68 ± 0.08 | 0.93 | 0.10 |
| IL-6 | 4.33 ± 0.12 | 0.69 | 4.24 ± 0.10 | 4.27 ± 0.08 | 4.43 ± 0.16 | 0.95 | 4.26 ± 0.11 | 0.93 | 0.60 |
| IL6R | 6.50 ± 0.08 | 0.11 | 6.83 ± 0.06 | 6.86 ± 0.06 | 6.86 ± 0.07 | 0.95 | 6.85 ± 0.05 | 0.93 | 0.10 |
| TNF-α | 5.25 ± 0.05 | 0.69 | 5.11 ± 0.07 | 5.13 ± 0.06 | 5.26 ± 0.08 | 0.95 | 5.08 ± 0.07 | 0.93 | 0.10 |
| TNFRSF1A | 7.49 ± 0.11 | 0.69 | 7.68 ± 0.05 | 7.79 ± 0.07 | 7.74 ± 0.06 | 0.95 | 7.70 ± 0.06 | 0.93 | 0.38 |
| TNFRSF1B | 8.19 ± 0.09 | 0.20 | 8.51 ± 0.04 | 8.52 ± 0.04 | 8.56 ± 0.03 | 0.95 | 8.52 ± 0.04 | 0.93 | 0.10 |
Gene-expression profiles of the same patient with NAFLD (n = 9) from human peripheral (white) adipose tissue were generated with the use of the Affymetrix U133 Plus 2.0 platform. Gene-chip normalization avoids the use of unstable single housekeeping genes and thus can be technically superior for clinical biomarker studies. Data are mean ± SE. P1 value compare baseline in fasting state and postprandial MUFA, SAT and CHO dietary periods with ANOVA repeated measured (n = 8). P2 value compares postprandial MUFA, SAT and CHO dietary periods with ANOVA repeated measured (n = 9). P3 value compares post-insulin infusion state (180’) and postprandial MUFA, SAT and CHO dietary periods with ANOVA repeated measured (n = 9). P4 value compares baseline in fasting state and post-insulin infusion state (180’) with paired t-test analysis (n = 8). P-values for multiple comparisons were adjusted by Hommel’s test. P < 0.05 was considered significant. INSR: Insulin receptor; GCGR: Glucagon receptor; BMP: Bone morphogenetic protein; PPARG: Peroxisome proliferator-activated receptor gamma; ADIPOQ: Adiponectin; LEP: Leptin; ADRβ3: Adrenergic β-3 receptor; RETN: Resistin; IL-6: Interleukin-6; IL-6R: Interleukin-6 receptor; TNF-α: Tumor necrosis factor-α; TNFRSF1A: Tumor necrosis factor receptor superfamily, member 1A; TNFRSF1B: Tumor necrosis factor receptor superfamily, member 1B; MUFA: High monounsaurated fatty acid diet; SAT: High saturated fatty acid diet; CHO: Low fat-high carbohydrate diet.
Effect of nutrition on oxidative stress
Finally, we have gathered information about the energy balance of these patients. Our results indicate that in early stages of the disease, changes in REE, RQ, and CHO, fat and protein oxidation could not be differentiated between IR and IS patients. However, the higher waist-to-hip ratio early correlated negatively with CHO oxidation and directly with fat-oxidation, suggesting that central adipose fat distribution could decrease glucose utilization as fuel. In addition, the increase in energy intake in IR patients seemed to be primarily related to their apparent preference for higher saturated fat and refined cereal, sugar and soft drink intake. Several mechanisms have implicated high SAT and sugar diets with the development of fatty liver. This includes its association with higher insulin resistance, as well as an increase in markers associated with endoplasmic reticulum stress, excessive production of reactive oxygen species, leading to inflammatory and proapoptotic responses.
The ability of the adipose tissue to keep in reserve fats in obesity is associated with different cellular actions. Thus, a key function in this issue is improving performance of the endoplasmic reticulum (ER) in the adipocytes. ER is a main organelle that regulates nutrient storage, and the surplus of nutrients increases the amount of altered proteins synthesis witch accumulate in the ER. ER stress has been related to many effects of disability cellular that include activation of inflammation and stress networks, closely linked to turn on with by oxidative stress and insulin resistance. The actions of different dietary fat compositions in functions of ER stress on adipose tissue has been investigated in patients with obesity and MetS. In a substudy accomplished within the LIPGENE study, 39 MetS patients were assigned to one of four isocaloric diets: High-SFA (38% E from fat, 16% as SFA), high MUFA (38% E from fat, 20% MUFA), and two low-fat, high-complex carbohydrate (28% E from fat) diets supplemented with 1.24 g/d of long-chain n-3 PUFA or placebo for 12 wk each. This study observed that during the postprandial state, several genes linked to ER stress, such as sXBP-1 and BiP, independent of the fat consumed, in peripheral adipose tissue of patients with MetS, are activated. In addition, after the 12 wk of HSFA diet the expression of PDIA3 gene was twice higher than after 12 wk of LFHCC n-3 diet. Overall, these data indicate that increase of ER stress in adipose tissue, by amount and different types of fat intake, could play a key role for regulating the capacity of glucose and TG clearance. Thus, ER capacity of AT may modulate metabolic flexibility, initially during postprandial state, accelerating remove of glucose and lipid[270].
Moreover, a high oxidative stress is found in MetS patients, which is showed by a raised activity of NADPH-oxidase and a reduced expression of antioxidant enzymes in the adipose tissue. In patients from the LIPGENE study it was observed that MUFA fat intake decreases oxidative stress as compared with high SAT fat diet by increasing postprandial antioxidant reaction in adipose tissue. Therefore, changing a proportion of SFA by MUFA in the diets could have any beneficial effect to decrease the oxidative stress in MetS patients[271]. Last, MetS patients normally present higher inflammatory state in AT, which is increased during postprandial response, which was seen with independence of the fat eaten. We have found that p65, IκBα, MCP-1 and IL-1β gene transcripts were induced during the postprandial response, also with independence of fat intake. Of note, IL-6 expression was only identify after the postprandial responses[272].
In summary, in patients at risk, achieving and maintaining an ideal body weight, adjusting energy balance between calorie intake and daily regular exercise is essential in preventing the development of MetS, regardless of the distribution of macronutrient energy. However, the composition of macronutrients can have beneficial or harmful effects on several factors of the metabolic profile, and this can be very important in the dietary counseling of patients with MetS.
Therefore, in subjects with early central obesity associated with other components of MetS, the first recommendation would be to reduce calorie intake and ensure daily physical exercise in order to achieve an ideal weight. Secondly, avoid the intake of trans fat, mainly cakes, biscuits, pastries, etc., and moderate the intake of saturated fat, mainly red meat, processed meats and meat sauces. Thirdly, avoid eating simple carbohydrates, such as sugar, soft drinks, and fruit and juices in excess. This will prevent insulin spike, an increment in triglycerides levels, and also improve the reverse transport cholesterol, and probably fatty liver and central obesity. It is preferable to increase the intake of complex carbohydrates with a lower glycemic index such as wholemeal bread and legumes. Moderate intake of white pasta, potatoes, white rice, etc., is permitted, but these should be eaten with plenty of vegetables, thus increasing fiber content will decrease its GI. The fourth recommendation, is to moderate protein intake of high biological value associated with polyunsaturated fatty acids ω-3, which can be achieved by replacing portions of meat with seafood. Lastly, take abundant and varied vegetables daily in the two main dishes, fresh and steamed, seasoned with moderate portions of extra virgin olive oil and small portions of dried fruits. This will not only ensure vitamin and mineral requirements are met, but will also give the meal a high fiber volume, flatten postprandial blood glucose of carbohydrates eaten, and the dried fruits will ensure that ω-6 polyunsaturated needs are met. In addition, olive oil should be used in moderate amounts, not more than 20 cc (approximately 180 kcal) per 1000 calories consumed, thus avoiding their overuse, which can lead to obesity. Olive oil is a healthy fat with obvious improvements in atherogenic lipid profile, and contains polyphenols as well as some fat-soluble vitamins like vitamin E which are natural antioxidants[273]. A modest reduction in salt consumption causes significant decreases in blood pressure either hypertensive or normotensive individuals. Thus, the current guidance to decrease salt ingestion to 5-6 g/d should be advised, but a further reduction lower 3 g/d could be required in MetS[16]. In addition, moderate ingestion of red wine is related with a inferior prevalence of MetS, as well as with beneficial effects on central adiposity, lipid profile and fasting insulin levels[274]. Finally, until more conclusive data, it is essential that 2-3 servings per day of semi-skimmed milk and derivatives, and at least 2-3 eggs per week should be included in the diet. Both nutrients provide proteins of high biological value, provide some needs in essential minerals, and are reasonably low in fat.
Footnotes
Conflict-of-interest statement: The author has no potential conflict of interest.
Manuscript source: Invited manuscript
Specialty type: Endocrinology and metabolism
Country of origin: Spain
Peer-review report classification
Grade A (Excellent): A
Grade B (Very good): B
Grade C (Good): 0
Grade D (Fair): 0
Grade E (Poor): 0
Peer-review started: March 25, 2016
First decision: May 13, 2016
Article in press: September 9, 2016
P- Reviewer: Parvizi N, Szkudelska K S- Editor: Gong XM L- Editor: A E- Editor: Lu YJ
References
- 1.Bray GA. Medical consequences of obesity. J Clin Endocrinol Metab. 2004;89:2583–2589. doi: 10.1210/jc.2004-0535. [DOI] [PubMed] [Google Scholar]
- 2.Hill JO, Pagliassotti MJ, Peters JC. Nongenetic determinants of obesity and fat topography. Genetic determinants of obesity. In: Bouchard C, editor. Boca Raton, FL: CRC Press; 1994. pp. 35–48. [Google Scholar]
- 3.Kopelman PG. Obesity as a medical problem. Nature. 2000;404:635–643. doi: 10.1038/35007508. [DOI] [PubMed] [Google Scholar]
- 4.Christakis NA, Fowler JH. The spread of obesity in a large social network over 32 years. N Engl J Med. 2007;357:370–379. doi: 10.1056/NEJMsa066082. [DOI] [PubMed] [Google Scholar]
- 5.Berghöfer A, Pischon T, Reinhold T, Apovian CM, Sharma AM, Willich SN. Obesity prevalence from a European perspective: a systematic review. BMC Public Health. 2008;8:200. doi: 10.1186/1471-2458-8-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization. Obesity and overweight. Available from: http://www.who.int/dietphysicalactivity/publications/facts/obesity/en/print.
- 7.Engeland A, Bjørge T, Søgaard AJ, Tverdal A. Body mass index in adolescence in relation to total mortality: 32-year follow-up of 227,000 Norwegian boys and girls. Am J Epidemiol. 2003;157:517–523. doi: 10.1093/aje/kwf219. [DOI] [PubMed] [Google Scholar]
- 8.Flegal KM, Carroll MD, Ogden CL, Johnson CL. Prevalence and trends in obesity among US adults, 1999-2000. JAMA. 2002;288:1723–1727. doi: 10.1001/jama.288.14.1723. [DOI] [PubMed] [Google Scholar]
- 9.Flegal KM, Carroll MD, Kuczmarski RJ, Johnson CL. Overweight and obesity in the United States: prevalence and trends, 1960-1994. Int J Obes Relat Metab Disord. 1998;22:39–47. doi: 10.1038/sj.ijo.0800541. [DOI] [PubMed] [Google Scholar]
- 10.Eyre H, Kahn R, Robertson RM. Preventing cancer, cardiovascular disease, and diabetes: a common agenda for theAmerican Cancer Society, the American Diabetes Association, and the American Heart Association. CA Cancer J Clin. 2004;54:190–207. doi: 10.3322/canjclin.54.4.190. [DOI] [PubMed] [Google Scholar]
- 11.Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, van der Schouw YT, Spencer E, Moons KG, Tjønneland A, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. 2008;359:2105–2120. doi: 10.1056/NEJMoa0801891. [DOI] [PubMed] [Google Scholar]
- 12.Berrington de Gonzalez A, Hartge P, Cerhan JR, Flint AJ, Hannan L, MacInnis RJ, Moore SC, Tobias GS, Anton-Culver H, Freeman LB, et al. Body-mass index and mortality among 1.46 million white adults. N Engl J Med. 2010;363:2211–2219. doi: 10.1056/NEJMoa1000367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ezzati M, Riboli E. Behavioral and dietary risk factors for noncommunicable diseases. N Engl J Med. 2013;369:954–964. doi: 10.1056/NEJMra1203528. [DOI] [PubMed] [Google Scholar]
- 14.No Author. Washington, DC: American Institute for Cancer Research; 2007. Food, nutrition, physical activity, and the prevention of cancer: A global perspective. [Google Scholar]
- 15.Mozaffarian D, Appel LJ, Van Horn L. Components of a cardioprotective diet: new insights. Circulation. 2011;123:2870–2891. doi: 10.1161/CIRCULATIONAHA.110.968735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He FJ, Li J, Macgregor GA. Effect of longer term modest salt reduction on blood pressure: Cochrane systematic review and meta-analysis of randomised trials. BMJ. 2013;346:f1325. doi: 10.1136/bmj.f1325. [DOI] [PubMed] [Google Scholar]
- 17.Sacks FM, Bray GA, Carey VJ, Smith SR, Ryan DH, Anton SD, McManus K, Champagne CM, Bishop LM, Laranjo N, et al. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. N Engl J Med. 2009;360:859–873. doi: 10.1056/NEJMoa0804748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mozaffarian D, Hao T, Rimm EB, Willett WC, Hu FB. Changes in diet and lifestyle and long-term weight gain in women and men. N Engl J Med. 2011;364:2392–2404. doi: 10.1056/NEJMoa1014296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manson JE, Willett WC, Stampfer MJ, Colditz GA, Hunter DJ, Hankinson SE, Hennekens CH, Speizer FE. Body weight and mortality among women. N Engl J Med. 1995;333:677–685. doi: 10.1056/NEJM199509143331101. [DOI] [PubMed] [Google Scholar]
- 20.Willett WC, Manson JE, Stampfer MJ, Colditz GA, Rosner B, Speizer FE, Hennekens CH. Weight, weight change, and coronary heart disease in women. Risk within the ‘normal’ weight range. JAMA. 1995;273:461–465. doi: 10.1001/jama.1995.03520300035033. [DOI] [PubMed] [Google Scholar]
- 21.Stevens J, Plankey MW, Williamson DF, Thun MJ, Rust PF, Palesch Y, O’Neil PM. The body mass index-mortality relationship in white and African American women. Obes Res. 1998;6:268–277. doi: 10.1002/j.1550-8528.1998.tb00349.x. [DOI] [PubMed] [Google Scholar]
- 22.Lindsted KD, Singh PN. Body mass and 26 y risk of mortality among men who never smoked: a re-analysis among men from the Adventist Mortality Study. Int J Obes Relat Metab Disord. 1998;22:544–548. doi: 10.1038/sj.ijo.0800623. [DOI] [PubMed] [Google Scholar]
- 23.Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW. Body-mass index and mortality in a prospective cohort of U.S. adults. N Engl J Med. 1999;341:1097–1105. doi: 10.1056/NEJM199910073411501. [DOI] [PubMed] [Google Scholar]
- 24.American Heart Association. Dallas, TX: American Heart Association; 2004. Heart disease and stroke statistics. [Google Scholar]
- 25.American Heart Association. Dallas, TX: American Heart Association; 2003. Heart disease and stroke statistics. [Google Scholar]
- 26.Suk SH, Sacco RL, Boden-Albala B, Cheun JF, Pittman JG, Elkind MS, Paik MC; Northern Manhattan Stroke Study. Abdominal obesity and risk of ischemic stroke: the Northern Manhattan Stroke Study. Stroke. 2003;34:1586–1592. doi: 10.1161/01.STR.0000075294.98582.2F. [DOI] [PubMed] [Google Scholar]
- 27.Oster G, Thompson D, Edelsberg J, Bird AP, Colditz GA. Lifetime health and economic benefits of weight loss among obese persons. Am J Public Health. 1999;89:1536–1542. doi: 10.2105/ajph.89.10.1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mokdad AH, Ford ES, Bowman BA, Nelson DE, Engelgau MM, Vinicor F, Marks JS. Diabetes trends in the U.S.: 1990-1998. Diabetes Care. 2000;23:1278–1283. doi: 10.2337/diacare.23.9.1278. [DOI] [PubMed] [Google Scholar]
- 29.Laakso M. Hyperglycemia and cardiovascular disease in type 2 diabetes. Diabetes. 1999;48:937–942. doi: 10.2337/diabetes.48.5.937. [DOI] [PubMed] [Google Scholar]
- 30.Landin K, Krotkiewski M, Smith U. Importance of obesity for the metabolic abnormalities associated with an abdominal fat distribution. Metabolism. 1989;38:572–576. doi: 10.1016/0026-0495(89)90219-9. [DOI] [PubMed] [Google Scholar]
- 31.Peiris AN, Mueller RA, Smith GA, Struve MF, Kissebah AH. Splanchnic insulin metabolism in obesity. Influence of body fat distribution. J Clin Invest. 1986;78:1648–1657. doi: 10.1172/JCI112758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, Willett WC. Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med. 2001;345:790–797. doi: 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- 33.American Diabetes Association. (4) Foundations of care: education, nutrition, physical activity, smoking cessation, psychosocial care, and immunization. Diabetes Care. 2015;38 Suppl:S20–S30. doi: 10.2337/dc15-S007. [DOI] [PubMed] [Google Scholar]
- 34.Kayıkçıoğlu M, Özdoğan Ö. [Nutrition and cardiovascular health: 2015 American Dietary Guidelines Advisory Report] Turk Kardiyol Dern Ars. 2015;43:667–672. doi: 10.5543/tkda.2015.80963. [DOI] [PubMed] [Google Scholar]
- 35.Lew EA, Garfinkel L. Variations in mortality by weight among 750,000 men and women. J Chronic Dis. 1979;32:563–576. doi: 10.1016/0021-9681(79)90119-x. [DOI] [PubMed] [Google Scholar]
- 36.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 37.Willett WC, Sacks F, Trichopoulou A, Drescher G, Ferro-Luzzi A, Helsing E, Trichopoulos D. Mediterranean diet pyramid: a cultural model for healthy eating. Am J Clin Nutr. 1995;61:1402S–1406S. doi: 10.1093/ajcn/61.6.1402S. [DOI] [PubMed] [Google Scholar]
- 38.Estruch R, Ros E, Salas-Salvadó J, Covas MI, Corella D, Arós F, Gómez-Gracia E, Ruiz-Gutiérrez V, Fiol M, Lapetra J, et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med. 2013;368:1279–1290. doi: 10.1056/NEJMc1806491. [DOI] [PubMed] [Google Scholar]
- 39.Morris JN, Heady JA, Raffle PA, Roberts CG, Parks JW. Coronary heart-disease and physical activity of work. Lancet. 1953;265:1053–1057; contd. doi: 10.1016/s0140-6736(53)90665-5. [DOI] [PubMed] [Google Scholar]
- 40.Levine JA, Weisell R, Chevassus S, Martinez CD, Burlingame B, Coward WA. The work burden of women. Science. 2001;294:812. doi: 10.1126/science.1064627. [DOI] [PubMed] [Google Scholar]
- 41.Ezzati M, Riboli E. Can noncommunicable diseases be prevented? Lessons from studies of populations and individuals. Science. 2012;337:1482–1487. doi: 10.1126/science.1227001. [DOI] [PubMed] [Google Scholar]
- 42.Mozaffarian D, Afshin A, Benowitz NL, Bittner V, Daniels SR, Franch HA, Jacobs DR, Kraus WE, Kris-Etherton PM, Krummel DA, et al. Population approaches to improve diet, physical activity, and smoking habits: a scientific statement from the American Heart Association. Circulation. 2012;126:1514–1563. doi: 10.1161/CIR.0b013e318260a20b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Magnusson I, Rothman DL, Katz LD, Shulman RG, Shulman GI. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults--The Evidence Report. National Institutes of Health. Obes Res. 1998;6 Suppl 2:51S–209S. [PubMed] [Google Scholar]
- 45.Physical status: the use and interpretation of anthropometry. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser. 1995;854:1–452. [PubMed] [Google Scholar]
- 46.Seidell JC, Flegal KM. Assessing obesity: classification and epidemiology. Br Med Bull. 1997;53:238–252. doi: 10.1093/oxfordjournals.bmb.a011611. [DOI] [PubMed] [Google Scholar]
- 47.Wang J, Thornton JC, Kolesnik S, Pierson RN. Anthropometry in body composition. An overview. Ann N Y Acad Sci. 2000;904:317–326. doi: 10.1111/j.1749-6632.2000.tb06474.x. [DOI] [PubMed] [Google Scholar]
- 48.Ferland M, Després JP, Tremblay A, Pinault S, Nadeau A, Moorjani S, Lupien PJ, Thériault G, Bouchard C. Assessment of adipose tissue distribution by computed axial tomography in obese women: association with body density and anthropometric measurements. Br J Nutr. 1989;61:139–148. doi: 10.1079/bjn19890104. [DOI] [PubMed] [Google Scholar]
- 49.Lukaski HC, Bolonchuk WW, Hall CB, Siders WA. Validation of tetrapolar bioelectrical impedance method to assess human body composition. J Appl Physiol (1985) 1986;60:1327–1332. doi: 10.1152/jappl.1986.60.4.1327. [DOI] [PubMed] [Google Scholar]
- 50.Kyle UG, Bosaeus I, De Lorenzo AD, Deurenberg P, Elia M, Gómez JM, Heitmann BL, Kent-Smith L, Melchior JC, Pirlich M, et al. Bioelectrical impedance analysis--part I: review of principles and methods. Clin Nutr. 2004;23:1226–1243. doi: 10.1016/j.clnu.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 51.Taylor RW, Jones IE, Williams SM, Goulding A. Evaluation of waist circumference, waist-to-hip ratio, and the conicity index as screening tools for high trunk fat mass, as measured by dual-energy X-ray absorptiometry, in children aged 3-19 y. Am J Clin Nutr. 2000;72:490–495. doi: 10.1093/ajcn/72.2.490. [DOI] [PubMed] [Google Scholar]
- 52.Paniagua JA, Gallego de la Sacristana A, Romero I, Vidal-Puig A, Latre JM, Sanchez E, Perez-Martinez P, Lopez-Miranda J, Perez-Jimenez F. Monounsaturated fat-rich diet prevents central body fat distribution and decreases postprandial adiponectin expression induced by a carbohydrate-rich diet in insulin-resistant subjects. Diabetes Care. 2007;30:1717–1723. doi: 10.2337/dc06-2220. [DOI] [PubMed] [Google Scholar]
- 53.Giusti M, Mortara L, Degrandi R, Cecoli F, Mussap M, Rodriguez G, Ferone D, Minuto F. Metabolic and cardiovascular risk in patients with a history of differentiated thyroid carcinoma: A case-controlled cohort study. Thyroid Res. 2008;1:2. doi: 10.1186/1756-6614-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Katherine Zeratsky . Mayo Clinic: Obesity Expert Answers; 2009. Normal weight obesity: A hidden health risk? Can you be considered obese if you have a normal body weight? [Google Scholar]
- 55.Deurenberg P, Weststrate JA, Seidell JC. Body mass index as a measure of body fatness: age- and sex-specific prediction formulas. Br J Nutr. 1991;65:105–114. doi: 10.1079/bjn19910073. [DOI] [PubMed] [Google Scholar]
- 56.Ashwell M, Cole TJ, Dixon AK. Ratio of waist circumference to height is strong predictor of intra-abdominal fat. BMJ. 1996;313:559–560. doi: 10.1136/bmj.313.7056.559d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cox BD, Whichelow M. Ratio of waist circumference to height is better predictor of death than body mass index. BMJ. 1996;313:1487. doi: 10.1136/bmj.313.7070.1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hsieh SD, Yoshinaga H. Waist/height ratio as a simple and useful predictor of coronary heart disease risk factors in women. Intern Med. 1995;34:1147–1152. doi: 10.2169/internalmedicine.34.1147. [DOI] [PubMed] [Google Scholar]
- 59.Savva SC, Tornaritis M, Savva ME, Kourides Y, Panagi A, Silikiotou N, Georgiou C, Kafatos A. Waist circumference and waist-to-height ratio are better predictors of cardiovascular disease risk factors in children than body mass index. Int J Obes Relat Metab Disord. 2000;24:1453–1458. doi: 10.1038/sj.ijo.0801401. [DOI] [PubMed] [Google Scholar]
- 60.Hara M, Saitou E, Iwata F, Okada T, Harada K. Waist-to-height ratio is the best predictor of cardiovascular disease risk factors in Japanese schoolchildren. J Atheroscler Thromb. 2002;9:127–132. doi: 10.5551/jat.9.127. [DOI] [PubMed] [Google Scholar]
- 61.Savva SC, Lamnisos D, Kafatos AG. Predicting cardiometabolic risk: waist-to-height ratio or BMI. A meta-analysis. Diabetes Metab Syndr Obes. 2013;6:403–419. doi: 10.2147/DMSO.S34220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vazquez G, Duval S, Jacobs DR, Silventoinen K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: a meta-analysis. Epidemiol Rev. 2007;29:115–128. doi: 10.1093/epirev/mxm008. [DOI] [PubMed] [Google Scholar]
- 63.Huxley R, Mendis S, Zheleznyakov E, Reddy S, Chan J. Body mass index, waist circumference and waist: hip ratio as predictors of cardiovascular risk--a review of the literature. Eur J Clin Nutr. 2010;64:16–22. doi: 10.1038/ejcn.2009.68. [DOI] [PubMed] [Google Scholar]
- 64.Boggs DA, Rosenberg L, Cozier YC, Wise LA, Coogan PF, Ruiz-Narvaez EA, Palmer JR. General and abdominal obesity and risk of death among black women. N Engl J Med. 2011;365:901–908. doi: 10.1056/NEJMoa1104119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lean ME, Han TS, Morrison CE. Waist circumference as a measure for indicating need for weight management. BMJ. 1995;311:158–161. doi: 10.1136/bmj.311.6998.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 67.Snijder MB, Dekker JM, Visser M, Bouter LM, Stehouwer CD, Yudkin JS, Heine RJ, Nijpels G, Seidell JC. Trunk fat and leg fat have independent and opposite associations with fasting and postload glucose levels: the Hoorn study. Diabetes Care. 2004;27:372–377. doi: 10.2337/diacare.27.2.372. [DOI] [PubMed] [Google Scholar]
- 68.Wajchenberg BL. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev. 2000;21:697–738. doi: 10.1210/edrv.21.6.0415. [DOI] [PubMed] [Google Scholar]
- 69.Hamilton G, Middleton MS, Bydder M, Yokoo T, Schwimmer JB, Kono Y, Patton HM, Lavine JE, Sirlin CB. Effect of PRESS and STEAM sequences on magnetic resonance spectroscopic liver fat quantification. J Magn Reson Imaging. 2009;30:145–152. doi: 10.1002/jmri.21809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim H, Taksali SE, Dufour S, Befroy D, Goodman TR, Petersen KF, Shulman GI, Caprio S, Constable RT. Comparative MR study of hepatic fat quantification using single-voxel proton spectroscopy, two-point dixon and three-point IDEAL. Magn Reson Med. 2008;59:521–527. doi: 10.1002/mrm.21561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armellini F, Zamboni M, Robbi R, Todesco T, Rigo L, Bergamo-Andreis IA, Bosello O. Total and intra-abdominal fat measurements by ultrasound and computerized tomography. Int J Obes Relat Metab Disord. 1993;17:209–214. [PubMed] [Google Scholar]
- 72.Spiegelman BM, Frank M, Green H. Molecular cloning of mRNA from 3T3 adipocytes. Regulation of mRNA content for glycerophosphate dehydrogenase and other differentiation-dependent proteins during adipocyte development. J Biol Chem. 1983;258:10083–10089. [PubMed] [Google Scholar]
- 73.Mandrup S, Lane MD. Regulating adipogenesis. J Biol Chem. 1997;272:5367–5370. doi: 10.1074/jbc.272.9.5367. [DOI] [PubMed] [Google Scholar]
- 74.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 75.Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerbäck S, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 76.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 77.Lean ME. Brown adipose tissue in humans. Proc Nutr Soc. 1989;48:243–256. doi: 10.1079/pns19890036. [DOI] [PubMed] [Google Scholar]
- 78.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, Tseng YH. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495:379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ouellet V, Routhier-Labadie A, Bellemare W, Lakhal-Chaieb L, Turcotte E, Carpentier AC, Richard D. Outdoor temperature, age, sex, body mass index, and diabetic status determine the prevalence, mass, and glucose-uptake activity of 18F-FDG-detected BAT in humans. J Clin Endocrinol Metab. 2011;96:192–199. doi: 10.1210/jc.2010-0989. [DOI] [PubMed] [Google Scholar]
- 81.Zingaretti MC, Crosta F, Vitali A, Guerrieri M, Frontini A, Cannon B, Nedergaard J, Cinti S. The presence of UCP1 demonstrates that metabolically active adipose tissue in the neck of adult humans truly represents brown adipose tissue. FASEB J. 2009;23:3113–3120. doi: 10.1096/fj.09-133546. [DOI] [PubMed] [Google Scholar]
- 82.Cypess AM, Kahn CR. The role and importance of brown adipose tissue in energy homeostasis. Curr Opin Pediatr. 2010;22:478–484. doi: 10.1097/MOP.0b013e32833a8d6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cypess AM, White AP, Vernochet C, Schulz TJ, Xue R, Sass CA, Huang TL, Roberts-Toler C, Weiner LS, Sze C, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19:635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cinti S. The adipose organ at a glance. Dis Model Mech. 2012;5:588–594. doi: 10.1242/dmm.009662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Young P, Arch JR, Ashwell M. Brown adipose tissue in the parametrial fat pad of the mouse. FEBS Lett. 1984;167:10–14. doi: 10.1016/0014-5793(84)80822-4. [DOI] [PubMed] [Google Scholar]
- 86.Loncar D. Convertible adipose tissue in mice. Cell Tissue Res. 1991;266:149–161. doi: 10.1007/BF00678721. [DOI] [PubMed] [Google Scholar]
- 87.Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, Nelson SM, Cowling R, Nagy K, Michael IP, et al. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17:61–72. doi: 10.1016/j.cmet.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 88.Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Näslund E, Britton T, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453:783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 89.Kim JB, Sarraf P, Wright M, Yao KM, Mueller E, Solanes G, Lowell BB, Spiegelman BM. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J Clin Invest. 1998;101:1–9. doi: 10.1172/JCI1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim JB, Wright HM, Wright M, Spiegelman BM. ADD1/SREBP1 activates PPARgamma through the production of endogenous ligand. Proc Natl Acad Sci USA. 1998;95:4333–4337. doi: 10.1073/pnas.95.8.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maroto M, Bone RA, Dale JK. Somitogenesis. Development. 2012;139:2453–2456. doi: 10.1242/dev.069310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tseng YH, Kokkotou E, Schulz TJ, Huang TL, Winnay JN, Taniguchi CM, Tran TT, Suzuki R, Espinoza DO, Yamamoto Y, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454:1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shan T, Liang X, Bi P, Zhang P, Liu W, Kuang S. Distinct populations of adipogenic and myogenic Myf5-lineage progenitors in white adipose tissues. J Lipid Res. 2013;54:2214–2224. doi: 10.1194/jlr.M038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Waldén TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am J Physiol Endocrinol Metab. 2012;302:E19–E31. doi: 10.1152/ajpendo.00249.2011. [DOI] [PubMed] [Google Scholar]
- 97.Rosenwald M, Perdikari A, Rülicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol. 2013;15:659–667. doi: 10.1038/ncb2740. [DOI] [PubMed] [Google Scholar]
- 98.Accili D, Taylor SI. Targeted inactivation of the insulin receptor gene in mouse 3T3-L1 fibroblasts via homologous recombination. Proc Natl Acad Sci USA. 1991;88:4708–4712. doi: 10.1073/pnas.88.11.4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tseng YH, Butte AJ, Kokkotou E, Yechoor VK, Taniguchi CM, Kriauciunas KM, Cypess AM, Niinobe M, Yoshikawa K, Patti ME, et al. Prediction of preadipocyte differentiation by gene expression reveals role of insulin receptor substrates and necdin. Nat Cell Biol. 2005;7:601–611. doi: 10.1038/ncb1259. [DOI] [PubMed] [Google Scholar]
- 100.Nam SY, Lobie PE. The mechanism of effect of growth hormone on preadipocyte and adipocyte function. Obes Rev. 2000;1:73–86. doi: 10.1046/j.1467-789x.2000.00015.x. [DOI] [PubMed] [Google Scholar]
- 101.Salomon F, Cuneo RC, Hesp R, Sönksen PH. The effects of treatment with recombinant human growth hormone on body composition and metabolism in adults with growth hormone deficiency. N Engl J Med. 1989;321:1797–1803. doi: 10.1056/NEJM198912283212605. [DOI] [PubMed] [Google Scholar]
- 102.Wabitsch M, Hauner H, Heinze E, Teller WM. The role of growth hormone/insulin-like growth factors in adipocyte differentiation. Metabolism. 1995;44:45–49. doi: 10.1016/0026-0495(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 103.Kawai M, Namba N, Mushiake S, Etani Y, Nishimura R, Makishima M, Ozono K. Growth hormone stimulates adipogenesis of 3T3-L1 cells through activation of the Stat5A/5B-PPARgamma pathway. J Mol Endocrinol. 2007;38:19–34. doi: 10.1677/jme.1.02154. [DOI] [PubMed] [Google Scholar]
- 104.Morreale de Escobar G, Obregon MJ, Escobar del Rey F. Role of thyroid hormone during early brain development. Eur J Endocrinol. 2004;151 Suppl 3:U25–U37. doi: 10.1530/eje.0.151u025. [DOI] [PubMed] [Google Scholar]
- 105.Park EA, Song S, Olive M, Roesler WJ. CCAAT-enhancer-binding protein alpha (C/EBP alpha) is required for the thyroid hormone but not the retinoic acid induction of phosphoenolpyruvate carboxykinase (PEPCK) gene transcription. Biochem J. 1997;322(Pt 1):343–349. doi: 10.1042/bj3220343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Carmona MC, Iglesias R, Obregón MJ, Darlington GJ, Villarroya F, Giralt M. Mitochondrial biogenesis and thyroid status maturation in brown fat require CCAAT/enhancer-binding protein alpha. J Biol Chem. 2002;277:21489–21498. doi: 10.1074/jbc.M201710200. [DOI] [PubMed] [Google Scholar]
- 107.Divertie GD, Jensen MD, Miles JM. Stimulation of lipolysis in humans by physiological hypercortisolemia. Diabetes. 1991;40:1228–1232. doi: 10.2337/diab.40.10.1228. [DOI] [PubMed] [Google Scholar]
- 108.Xu C, He J, Jiang H, Zu L, Zhai W, Pu S, Xu G. Direct effect of glucocorticoids on lipolysis in adipocytes. Mol Endocrinol. 2009;23:1161–1170. doi: 10.1210/me.2008-0464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smas CM, Chen L, Zhao L, Latasa MJ, Sul HS. Transcriptional repression of pref-1 by glucocorticoids promotes 3T3-L1 adipocyte differentiation. J Biol Chem. 1999;274:12632–12641. doi: 10.1074/jbc.274.18.12632. [DOI] [PubMed] [Google Scholar]
- 110.Bélanger C, Luu-The V, Dupont P, Tchernof A. Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res. 2002;34:737–745. doi: 10.1055/s-2002-38265. [DOI] [PubMed] [Google Scholar]
- 111.Pereira CD, Azevedo I, Monteiro R, Martins MJ. 11β-Hydroxysteroid dehydrogenase type 1: relevance of its modulation in the pathophysiology of obesity, the metabolic syndrome and type 2 diabetes mellitus. Diabetes Obes Metab. 2012;14:869–881. doi: 10.1111/j.1463-1326.2012.01582.x. [DOI] [PubMed] [Google Scholar]
- 112.Bujalska IJ, Walker EA, Tomlinson JW, Hewison M, Stewart PM. 11Beta-hydroxysteroid dehydrogenase type 1 in differentiating omental human preadipocytes: from de-activation to generation of cortisol. Endocr Res. 2002;28:449–461. doi: 10.1081/erc-120016822. [DOI] [PubMed] [Google Scholar]
- 113.Stewart PM, Tomlinson JW. Cortisol, 11 beta-hydroxysteroid dehydrogenase type 1 and central obesity. Trends Endocrinol Metab. 2002;13:94–96. doi: 10.1016/s1043-2760(02)00566-0. [DOI] [PubMed] [Google Scholar]
- 114.Meseguer A, Puche C, Cabero A. Sex steroid biosynthesis in white adipose tissue. Horm Metab Res. 2002;34:731–736. doi: 10.1055/s-2002-38249. [DOI] [PubMed] [Google Scholar]
- 115.Paniagua JA, Escandell-Morales JM, Gil-Contreras D, Berral de la Rosa FJ, Romero-Jimenez M, Gómez-Urbano A, Sanchez-Lopez A, Bellido E, Poyato A, Calatayud B, et al. Central obesity and altered peripheral adipose tissue gene expression characterize the NAFLD patient with insulin resistance: Role of nutrition and insulin challenge. Nutrition. 2014;30:177–185. doi: 10.1016/j.nut.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 116.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 117.Friedman JM. Leptin, leptin receptors, and the control of body weight. Nutr Rev. 1998;56:s38–46; discussion s54-75. doi: 10.1111/j.1753-4887.1998.tb01685.x. [DOI] [PubMed] [Google Scholar]
- 118.Chan JL, Heist K, DePaoli AM, Veldhuis JD, Mantzoros CS. The role of falling leptin levels in the neuroendocrine and metabolic adaptation to short-term starvation in healthy men. J Clin Invest. 2003;111:1409–1421. doi: 10.1172/JCI17490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S. Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med. 1999;341:879–884. doi: 10.1056/NEJM199909163411204. [DOI] [PubMed] [Google Scholar]
- 120.Welt CK, Chan JL, Bullen J, Murphy R, Smith P, DePaoli AM, Karalis A, Mantzoros CS. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997. doi: 10.1056/NEJMoa040388. [DOI] [PubMed] [Google Scholar]
- 121.Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, Yu C, Cline GW, DePaoli AM, Taylor SI, Gorden P, et al. Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest. 2002;109:1345–1350. doi: 10.1172/JCI15001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Oral EA, Simha V, Ruiz E, Andewelt A, Premkumar A, Snell P, Wagner AJ, DePaoli AM, Reitman ML, Taylor SI, et al. Leptin-replacement therapy for lipodystrophy. N Engl J Med. 2002;346:570–578. doi: 10.1056/NEJMoa012437. [DOI] [PubMed] [Google Scholar]
- 123.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394:897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 124.Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–1433. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- 125.Unger RH, Zhou YT. Lipotoxicity of beta-cells in obesity and in other causes of fatty acid spillover. Diabetes. 2001;50 Suppl 1:S118–S121. doi: 10.2337/diabetes.50.2007.s118. [DOI] [PubMed] [Google Scholar]
- 126.Unger RH, Orci L. Diseases of liporegulation: new perspective on obesity and related disorders. FASEB J. 2001;15:312–321. doi: 10.1096/fj.00-0590. [DOI] [PubMed] [Google Scholar]
- 127.Fain JN, Madan AK, Hiler ML, Cheema P, Bahouth SW. Comparison of the release of adipokines by adipose tissue, adipose tissue matrix, and adipocytes from visceral and subcutaneous abdominal adipose tissues of obese humans. Endocrinology. 2004;145:2273–2282. doi: 10.1210/en.2003-1336. [DOI] [PubMed] [Google Scholar]
- 128.Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci USA. 2001;98:2005–2010. doi: 10.1073/pnas.041591798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu A, Wang Y, Keshaw H, Xu LY, Lam KS, Cooper GJ. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest. 2003;112:91–100. doi: 10.1172/JCI17797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288:H2031–H2041. doi: 10.1152/ajpheart.01058.2004. [DOI] [PubMed] [Google Scholar]
- 131.Kanhai DA, Kranendonk ME, Uiterwaal CS, van der Graaf Y, Kappelle LJ, Visseren FL. Adiponectin and incident coronary heart disease and stroke. A systematic review and meta-analysis of prospective studies. Obes Rev. 2013;14:555–567. doi: 10.1111/obr.12027. [DOI] [PubMed] [Google Scholar]
- 132.Hotta K, Funahashi T, Bodkin NL, Ortmeyer HK, Arita Y, Hansen BC, Matsuzawa Y. Circulating concentrations of the adipocyte protein adiponectin are decreased in parallel with reduced insulin sensitivity during the progression to type 2 diabetes in rhesus monkeys. Diabetes. 2001;50:1126–1133. doi: 10.2337/diabetes.50.5.1126. [DOI] [PubMed] [Google Scholar]
- 133.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. 1984;312:724–729. doi: 10.1038/312724a0. [DOI] [PubMed] [Google Scholar]
- 135.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 136.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 137.Ruan H, Lodish HF. Insulin resistance in adipose tissue: direct and indirect effects of tumor necrosis factor-alpha. Cytokine Growth Factor Rev. 2003;14:447–455. doi: 10.1016/s1359-6101(03)00052-2. [DOI] [PubMed] [Google Scholar]
- 138.Xing H, Northrop JP, Grove JR, Kilpatrick KE, Su JL, Ringold GM. TNF alpha-mediated inhibition and reversal of adipocyte differentiation is accompanied by suppressed expression of PPARgamma without effects on Pref-1 expression. Endocrinology. 1997;138:2776–2783. doi: 10.1210/endo.138.7.5242. [DOI] [PubMed] [Google Scholar]
- 139.Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes. 2002;51:1319–1336. doi: 10.2337/diabetes.51.5.1319. [DOI] [PubMed] [Google Scholar]
- 140.Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99:2221–2222. [PubMed] [Google Scholar]
- 141.Fernández-Real JM, Ricart W. Insulin resistance and chronic cardiovascular inflammatory syndrome. Endocr Rev. 2003;24:278–301. doi: 10.1210/er.2002-0010. [DOI] [PubMed] [Google Scholar]
- 142.Wang B, Jenkins JR, Trayhurn P. Expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture: integrated response to TNF-alpha. Am J Physiol Endocrinol Metab. 2005;288:E731–E740. doi: 10.1152/ajpendo.00475.2004. [DOI] [PubMed] [Google Scholar]
- 143.Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA. The hormone resistin links obesity to diabetes. Nature. 2001;409:307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- 144.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, et al. Visfatin: a protein secreted by visceral fat that mimics the effects of insulin. Science. 2005;307:426–430. doi: 10.1126/science.1097243. [DOI] [PubMed] [Google Scholar]
- 145.Fukuhara A, Matsuda M, Nishizawa M, Segawa K, Tanaka M, Kishimoto K, Matsuki Y, Murakami M, Ichisaka T, Murakami H, et al. Retraction. Science. 2007;318:565. doi: 10.1126/science.318.5850.565b. [DOI] [PubMed] [Google Scholar]
- 146.Arner P. Visfatin--a true or false trail to type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:28–30. doi: 10.1210/jc.2005-2391. [DOI] [PubMed] [Google Scholar]
- 147.de Souza Batista CM, Yang RZ, Lee MJ, Glynn NM, Yu DZ, Pray J, Ndubuizu K, Patil S, Schwartz A, Kligman M, et al. Omentin plasma levels and gene expression are decreased in obesity. Diabetes. 2007;56:1655–1661. doi: 10.2337/db06-1506. [DOI] [PubMed] [Google Scholar]
- 148.Tan BK, Adya R, Farhatullah S, Lewandowski KC, O’Hare P, Lehnert H, Randeva HS. Omentin-1, a novel adipokine, is decreased in overweight insulin-resistant women with polycystic ovary syndrome: ex vivo and in vivo regulation of omentin-1 by insulin and glucose. Diabetes. 2008;57:801–808. doi: 10.2337/db07-0990. [DOI] [PubMed] [Google Scholar]
- 149.Tan BK, Adya R, Farhatullah S, Chen J, Lehnert H, Randeva HS. Metformin treatment may increase omentin-1 levels in women with polycystic ovary syndrome. Diabetes. 2010;59:3023–3031. doi: 10.2337/db10-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Duplus E, Glorian M, Forest C. Fatty acid regulation of gene transcription. J Biol Chem. 2000;275:30749–30752. doi: 10.1074/jbc.R000015200. [DOI] [PubMed] [Google Scholar]
- 151.Food and Agriculture Organization of the United Nations. Geneva, Rome: Food and Agriculture Organization of the United Nations; 2010. Fats and fatty acids in human nutrition: Report of an expert consultation. [Google Scholar]
- 152.Gonzales AM, Orlando RA. Role of adipocyte-derived lipoprotein lipase in adipocyte hypertrophy. Nutr Metab (Lond) 2007;4:22. doi: 10.1186/1743-7075-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Storch J, Thumser AE. The fatty acid transport function of fatty acid-binding proteins. Biochim Biophys Acta. 2000;1486:28–44. doi: 10.1016/s1388-1981(00)00046-9. [DOI] [PubMed] [Google Scholar]
- 154.Hotamisligil GS, Johnson RS, Distel RJ, Ellis R, Papaioannou VE, Spiegelman BM. Uncoupling of obesity from insulin resistance through a targeted mutation in aP2, the adipocyte fatty acid binding protein. Science. 1996;274:1377–1379. doi: 10.1126/science.274.5291.1377. [DOI] [PubMed] [Google Scholar]
- 155.Jakobsson A, Westerberg R, Jacobsson A. Fatty acid elongases in mammals: their regulation and roles in metabolism. Prog Lipid Res. 2006;45:237–249. doi: 10.1016/j.plipres.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 156.Wang Y, Botolin D, Christian B, Busik J, Xu J, Jump DB. Tissue-specific, nutritional, and developmental regulation of rat fatty acid elongases. J Lipid Res. 2005;46:706–715. doi: 10.1194/jlr.M400335-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kobayashi T, Fujimori K. Very long-chain-fatty acids enhance adipogenesis through coregulation of Elovl3 and PPARγ in 3T3-L1 cells. Am J Physiol Endocrinol Metab. 2012;302:E1461–E1471. doi: 10.1152/ajpendo.00623.2011. [DOI] [PubMed] [Google Scholar]
- 158.Blouet C, Schwartz GJ. Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res. 2010;209:1–12. doi: 10.1016/j.bbr.2009.12.024. [DOI] [PubMed] [Google Scholar]
- 159.Schmid B, Rippmann JF, Tadayyon M, Hamilton BS. Inhibition of fatty acid synthase prevents preadipocyte differentiation. Biochem Biophys Res Commun. 2005;328:1073–1082. doi: 10.1016/j.bbrc.2005.01.067. [DOI] [PubMed] [Google Scholar]
- 160.Coleman RA, Lee DP. Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res. 2004;43:134–176. doi: 10.1016/s0163-7827(03)00051-1. [DOI] [PubMed] [Google Scholar]
- 161.Yu YH, Zhang Y, Oelkers P, Sturley SL, Rader DJ, Ginsberg HN. Posttranscriptional control of the expression and function of diacylglycerol acyltransferase-1 in mouse adipocytes. J Biol Chem. 2002;277:50876–50884. doi: 10.1074/jbc.M207353200. [DOI] [PubMed] [Google Scholar]
- 162.Coleman R, Bell RM. Triacylglycerol synthesis in isolated fat cells. Studies on the microsomal diacylglycerol acyltransferase activity using ethanol-dispersed diacylglycerols. J Biol Chem. 1976;251:4537–4543. [PubMed] [Google Scholar]
- 163.Greenberg AS, Egan JJ, Wek SA, Garty NB, Blanchette-Mackie EJ, Londos C. Perilipin, a major hormonally regulated adipocyte-specific phosphoprotein associated with the periphery of lipid storage droplets. J Biol Chem. 1991;266:11341–11346. [PubMed] [Google Scholar]
- 164.Rodriguez-Cuenca S, Carobbio S, Velagapudi VR, Barbarroja N, Moreno-Navarrete JM, Tinahones FJ, Fernandez-Real JM, Orešic M, Vidal-Puig A. Peroxisome proliferator-activated receptor γ-dependent regulation of lipolytic nodes and metabolic flexibility. Mol Cell Biol. 2012;32:1555–1565. doi: 10.1128/MCB.06154-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 2010;21:345–352. doi: 10.1016/j.tem.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Vidal-Puig A, Unger RH. Special issue on lipotoxicity. Biochim Biophys Acta. 2010;1801:207–208. doi: 10.1016/j.bbalip.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 167.Kolaczynski JW, Nyce MR, Considine RV, Boden G, Nolan JJ, Henry R, Mudaliar SR, Olefsky J, Caro JF. Acute and chronic effects of insulin on leptin production in humans: Studies in vivo and in vitro. Diabetes. 1996;45:699–701. doi: 10.2337/diab.45.5.699. [DOI] [PubMed] [Google Scholar]
- 168.Asterholm IW, Scherer PE. Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol. 2010;176:1364–1376. doi: 10.2353/ajpath.2010.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Marchesini G, Brizi M, Bianchi G, Tomassetti S, Bugianesi E, Lenzi M, McCullough AJ, Natale S, Forlani G, Melchionda N. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 170.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Sierra-Honigmann MR, Nath AK, Murakami C, García-Cardeña G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, et al. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–1686. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- 172.Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med. 2002;8:731–737. doi: 10.1038/nm724. [DOI] [PubMed] [Google Scholar]
- 173.Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003;112:1785–1788. doi: 10.1172/JCI20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.van de Woestijne AP, Monajemi H, Kalkhoven E, Visseren FL. Adipose tissue dysfunction and hypertriglyceridemia: mechanisms and management. Obes Rev. 2011;12:829–840. doi: 10.1111/j.1467-789X.2011.00900.x. [DOI] [PubMed] [Google Scholar]
- 175.Glass CK, Witztum JL. Atherosclerosis. the road ahead. Cell. 2001;104:503–516. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 176.Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW, DeFuria J, Jick Z, Greenberg AS, Obin MS. Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes. 2007;56:2910–2918. doi: 10.2337/db07-0767. [DOI] [PubMed] [Google Scholar]
- 177.Paniagua JA, López-Miranda J, Pérez-Martínez P, Marín C, Vida JM, Fuentes F, Fernández de la Puebla RA, Pérez-Jiménez F. Oxidized-LDL levels are changed during short-term serum glucose variations and lowered with statin treatment in early Type 2 diabetes: a study of endothelial function and microalbuminuria. Diabet Med. 2005;22:1647–1656. doi: 10.1111/j.1464-5491.2005.01703.x. [DOI] [PubMed] [Google Scholar]
- 178.Brown MS, Goldstein JL. Receptor-mediated control of cholesterol metabolism. Science. 1976;191:150–154. doi: 10.1126/science.174194. [DOI] [PubMed] [Google Scholar]
- 179.Steinberg D. The cholesterol controversy is over. Why did it take so long? Circulation. 1989;80:1070–1078. doi: 10.1161/01.cir.80.4.1070. [DOI] [PubMed] [Google Scholar]
- 180.Moreton JR. Atherosclerosis and Alimentary Hyperlipemia. Science. 1947;106:190–191. doi: 10.1126/science.106.2748.190. [DOI] [PubMed] [Google Scholar]
- 181.Hokanson JE, Austin MA. Plasma triglyceride level is a risk factor for cardiovascular disease independent of high-density lipoprotein cholesterol level: a meta-analysis of population-based prospective studies. J Cardiovasc Risk. 1996;3:213–219. [PubMed] [Google Scholar]
- 182.Patel A, Barzi F, Jamrozik K, Lam TH, Ueshima H, Whitlock G, Woodward M; Asia Pacific Cohort Studies Collaboration. Serum triglycerides as a risk factor for cardiovascular diseases in the Asia-Pacific region. Circulation. 2004;110:2678–2686. doi: 10.1161/01.CIR.0000145615.33955.83. [DOI] [PubMed] [Google Scholar]
- 183.Sarwar N, Danesh J, Eiriksdottir G, Sigurdsson G, Wareham N, Bingham S, Boekholdt SM, Khaw KT, Gudnason V. Triglycerides and the risk of coronary heart disease: 10,158 incident cases among 262,525 participants in 29 Western prospective studies. Circulation. 2007;115:450–458. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- 184.Oram JF, Lawn RM. ABCA1. The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42:1173–1179. [PubMed] [Google Scholar]
- 185.Chen W, Chen G, Head DL, Mangelsdorf DJ, Russell DW. Enzymatic reduction of oxysterols impairs LXR signaling in cultured cells and the livers of mice. Cell Metab. 2007;5:73–79. doi: 10.1016/j.cmet.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Calkin AC, Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhang Y, Breevoort SR, Angdisen J, Fu M, Schmidt DR, Holmstrom SR, Kliewer SA, Mangelsdorf DJ, Schulman IG. Liver LXRα expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J Clin Invest. 2012;122:1688–1699. doi: 10.1172/JCI59817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Oram JF, Vaughan AM. ATP-Binding cassette cholesterol transporters and cardiovascular disease. Circ Res. 2006;99:1031–1043. doi: 10.1161/01.RES.0000250171.54048.5c. [DOI] [PubMed] [Google Scholar]
- 190.Paniagua JA, López-Miranda J, Jansen S, Zambrana JL, López Segura F, Jiménez Perepérez JA, Pérez-Jiménez F. Increased high-density lipoprotein-3 binding to leukocytes following weight loss and improved glycemic control in type 2 diabetic patients. Metabolism. 2000;49:692–697. doi: 10.1053/meta.2000.6248. [DOI] [PubMed] [Google Scholar]
- 191.Jeon JH, Kim KY, Kim JH, Baek A, Cho H, Lee YH, Kim JW, Kim D, Han SH, Lim JS, et al. A novel adipokine CTRP1 stimulates aldosterone production. FASEB J. 2008;22:1502–1511. doi: 10.1096/fj.07-9412com. [DOI] [PubMed] [Google Scholar]
- 192.Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, Teboul M, Massiéra F, Sharma AM. The adipose-tissue renin-angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35:807–825. doi: 10.1016/s1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 193.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 194.Grundy SM. Hypertriglyceridemia, insulin resistance, and the metabolic syndrome. Am J Cardiol. 1999;83:25F–29F. doi: 10.1016/s0002-9149(99)00211-8. [DOI] [PubMed] [Google Scholar]
- 195.Grundy SM, Brewer HB, Cleeman JI, Smith SC, Lenfant C; American Heart Association; National Heart, Lung, and Blood Institute. Definition of metabolic syndrome: Report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- 196.National Heart, Lung, and Blood Institute. Bethesda, Md: National Institutes of Health; 1968. Clinical Guidelines on the Identification, Evaluation, and Treatment of Overweight and Obesity in Adults. The Evidence Report. [Google Scholar]
- 197.Williams RR, Hopkins PN, Hunt SC, Schumacher MC, Elbein SC, Wilson DE, Stults BM, Wu LL, Hasstedt SJ, Lalouel JM. Familial dyslipidaemic hypertension and other multiple metabolic syndromes. Ann Med. 1992;24:469–475. doi: 10.3109/07853899209166998. [DOI] [PubMed] [Google Scholar]
- 198.Stern MP. Impaired glucose tolerance: Risk factor or diagnostic category. Diabetes Mellitus. A Fundamental and Clinical Text. In: LeRoith D, Taylor SI, Olefsky JM, editors. Philadelphia: Lippincott-Raven Publishers; 1996. pp. 467–474. [Google Scholar]
- 199.Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827–832. doi: 10.1016/0735-1097(90)90282-t. [DOI] [PubMed] [Google Scholar]
- 200.Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15:539–553. doi: 10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 201.World Health Organization. Geneva, Switzerland: World Health Organization, 1999. [accessed 2003 Dec 12]; Definition, diagnosis and classification of diabetes mellitus and its complications: Report of a WHO Consultation. Part 1: Diagnosis and classification of diabetes mellitus. Available from: http://whqlibdoc.who.int/hq/1999/WHO_NCD_NCS_99.2.pdf. [Google Scholar]
- 202.Alberti KG, Zimmet P, Shaw J. Metabolic syndrome--a new world-wide definition. A Consensus Statement from the International Diabetes Federation. Diabet Med. 2006;23:469–480. doi: 10.1111/j.1464-5491.2006.01858.x. [DOI] [PubMed] [Google Scholar]
- 203.Tchernof A, Lamarche B, Prud’Homme D, Nadeau A, Moorjani S, Labrie F, Lupien PJ, Després JP. The dense LDL phenotype. Association with plasma lipoprotein levels, visceral obesity, and hyperinsulinemia in men. Diabetes Care. 1996;19:629–637. doi: 10.2337/diacare.19.6.629. [DOI] [PubMed] [Google Scholar]
- 204.Després JP, Moorjani S, Lupien PJ, Tremblay A, Nadeau A, Bouchard C. Regional distribution of body fat, plasma lipoproteins, and cardiovascular disease. Arteriosclerosis. 1990;10:497–511. doi: 10.1161/01.atv.10.4.497. [DOI] [PubMed] [Google Scholar]
- 205.Kamel EG, McNeill G, Han TS, Smith FW, Avenell A, Davidson L, Tothill P. Measurement of abdominal fat by magnetic resonance imaging, dual-energy X-ray absorptiometry and anthropometry in non-obese men and women. Int J Obes Relat Metab Disord. 1999;23:686–692. doi: 10.1038/sj.ijo.0800904. [DOI] [PubMed] [Google Scholar]
- 206.Onat A, Avci GS, Barlan MM, Uyarel H, Uzunlar B, Sansoy V. Measures of abdominal obesity assessed for visceral adiposity and relation to coronary risk. Int J Obes Relat Metab Disord. 2004;28:1018–1025. doi: 10.1038/sj.ijo.0802695. [DOI] [PubMed] [Google Scholar]
- 207.Pouliot MC, Després JP, Lemieux S, Moorjani S, Bouchard C, Tremblay A, Nadeau A, Lupien PJ. Waist circumference and abdominal sagittal diameter: best simple anthropometric indexes of abdominal visceral adipose tissue accumulation and related cardiovascular risk in men and women. Am J Cardiol. 1994;73:460–468. doi: 10.1016/0002-9149(94)90676-9. [DOI] [PubMed] [Google Scholar]
- 208.Després JP, Couillard C, Gagnon J, Bergeron J, Leon AS, Rao DC, Skinner JS, Wilmore JH, Bouchard C. Race, visceral adipose tissue, plasma lipids, and lipoprotein lipase activity in men and women: the Health, Risk Factors, Exercise Training, and Genetics (HERITAGE) family study. Arterioscler Thromb Vasc Biol. 2000;20:1932–1938. doi: 10.1161/01.atv.20.8.1932. [DOI] [PubMed] [Google Scholar]
- 209.Tan CE, Ma S, Wai D, Chew SK, Tai ES. Can we apply the National Cholesterol Education Program Adult Treatment Panel definition of the metabolic syndrome to Asians? Diabetes Care. 2004;27:1182–1186. doi: 10.2337/diacare.27.5.1182. [DOI] [PubMed] [Google Scholar]
- 210.Lear SA, Toma M, Birmingham CL, Frohlich JJ. Modification of the relationship between simple anthropometric indices and risk factors by ethnic background. Metabolism. 2003;52:1295–1301. doi: 10.1016/s0026-0495(03)00196-3. [DOI] [PubMed] [Google Scholar]
- 211.Carr MC, Brunzell JD. Abdominal obesity and dyslipidemia in the metabolic syndrome: importance of type 2 diabetes and familial combined hyperlipidemia in coronary artery disease risk. J Clin Endocrinol Metab. 2004;89:2601–2607. doi: 10.1210/jc.2004-0432. [DOI] [PubMed] [Google Scholar]
- 212.Lemieux I, Pascot A, Prud’homme D, Alméras N, Bogaty P, Nadeau A, Bergeron J, Després JP. Elevated C-reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol. 2001;21:961–967. doi: 10.1161/01.atv.21.6.961. [DOI] [PubMed] [Google Scholar]
- 213.Yudkin JS. Adipose tissue, insulin action and vascular disease: inflammatory signals. Int J Obes Relat Metab Disord. 2003;27 Suppl 3:S25–S28. doi: 10.1038/sj.ijo.0802496. [DOI] [PubMed] [Google Scholar]
- 214.Devaraj S, Rosenson RS, Jialal I. Metabolic syndrome: an appraisal of the pro-inflammatory and procoagulant status. Endocrinol Metab Clin North Am. 2004;33:431–453, table of contents. doi: 10.1016/j.ecl.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 215.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883. [DOI] [PubMed] [Google Scholar]
- 216.Bergman RN, Ider YZ, Bowden CR, Cobelli C. Quantitative estimation of insulin sensitivity. Am J Physiol. 1979;236:E667–E677. doi: 10.1152/ajpendo.1979.236.6.E667. [DOI] [PubMed] [Google Scholar]
- 217.Nakajima M, Sawada H, Yamada Y, Watanabe A, Tatsumi M, Yamashita J, Matsuda M, Sakaguchi T, Hirao T, Nakano H. The prognostic significance of amplification and overexpression of c-met and c-erb B-2 in human gastric carcinomas. Cancer. 1999;85:1894–1902. doi: 10.1002/(sici)1097-0142(19990501)85:9<1894::aid-cncr3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 218.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237:E214–E223. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 219.Tam CS, Xie W, Johnson WD, Cefalu WT, Redman LM, Ravussin E. Defining insulin resistance from hyperinsulinemic-euglycemic clamps. Diabetes Care. 2012;35:1605–1610. doi: 10.2337/dc11-2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Ford ES, Giles WH, Mokdad AH. Increasing prevalence of the metabolic syndrome among u.s. Adults. Diabetes Care. 2004;27:2444–2449. doi: 10.2337/diacare.27.10.2444. [DOI] [PubMed] [Google Scholar]
- 221.Grundy SM, Hansen B, Smith SC, Cleeman JI, Kahn RA; American Heart Association; National Heart, Lung, and Blood Institute; American Diabetes Association. Clinical management of metabolic syndrome: report of the American Heart Association/National Heart, Lung, and Blood Institute/American Diabetes Association conference on scientific issues related to management. Circulation. 2004;109:551–556. doi: 10.1161/01.CIR.0000112379.88385.67. [DOI] [PubMed] [Google Scholar]
- 222.Report of the Dietary Guidelines Advisory Committee on the dietary guidelines for Americans, 2010: To the Secretary of Agriculture and the Secretary of Health and Human Services. Washington, D.C: United States Department of Agriculture, United States Depatment of Health and Human Services; 2010. [Google Scholar]
- 223.Fogli-Cawley JJ, Dwyer JT, Saltzman E, McCullough ML, Troy LM, Meigs JB, Jacques PF. The 2005 Dietary Guidelines for Americans and risk of the metabolic syndrome. Am J Clin Nutr. 2007;86:1193–1201. doi: 10.1093/ajcn/86.4.1193. [DOI] [PubMed] [Google Scholar]
- 224.Fogli-Cawley JJ, Dwyer JT, Saltzman E, McCullough ML, Troy LM, Meigs JB, Jacques PF. The 2005 Dietary Guidelines for Americans and insulin resistance in the Framingham Offspring Cohort. Diabetes Care. 2007;30:817–822. doi: 10.2337/dc06-1927. [DOI] [PubMed] [Google Scholar]
- 225.Office of Disease Prevention and Health Promotion. Scientific report of the 2015 dietary guidelines advisory committee. [accessed 2015 Feb 20] Available from: http://www.health.gov/dietaryguidelines/2015-scientific-report/PDFs/Scientific-Report-of-the-2015-Dietary-Guidelines-Advisory-Committee.pdf. [Google Scholar]
- 226.Esposito K, Marfella R, Ciotola M, Di Palo C, Giugliano F, Giugliano G, D’Armiento M, D’Andrea F, Giugliano D. Effect of a mediterranean-style diet on endothelial dysfunction and markers of vascular inflammation in the metabolic syndrome: a randomized trial. JAMA. 2004;292:1440–1446. doi: 10.1001/jama.292.12.1440. [DOI] [PubMed] [Google Scholar]
- 227.Babio N, Toledo E, Estruch R, Ros E, Martínez-González MA, Castañer O, Bulló M, Corella D, Arós F, Gómez-Gracia E, et al. Mediterranean diets and metabolic syndrome status in the PREDIMED randomized trial. CMAJ. 2014;186:E649–E657. doi: 10.1503/cmaj.140764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Storlien LH, Baur LA, Kriketos AD, Pan DA, Cooney GJ, Jenkins AB, Calvert GD, Campbell LV. Dietary fats and insulin action. Diabetologia. 1996;39:621–631. doi: 10.1007/BF00418533. [DOI] [PubMed] [Google Scholar]
- 229.Feskens EJ, Virtanen SM, Räsänen L, Tuomilehto J, Stengård J, Pekkanen J, Nissinen A, Kromhout D. Dietary factors determining diabetes and impaired glucose tolerance. A 20-year follow-up of the Finnish and Dutch cohorts of the Seven Countries Study. Diabetes Care. 1995;18:1104–1112. doi: 10.2337/diacare.18.8.1104. [DOI] [PubMed] [Google Scholar]
- 230.Melanson EL, Astrup A, Donahoo WT. The relationship between dietary fat and fatty acid intake and body weight, diabetes, and the metabolic syndrome. Ann Nutr Metab. 2009;55:229–243. doi: 10.1159/000229004. [DOI] [PubMed] [Google Scholar]
- 231.Orchard TJ, Temprosa M, Goldberg R, Haffner S, Ratner R, Marcovina S, Fowler S; Diabetes Prevention Program Research Group. The effect of metformin and intensive lifestyle intervention on the metabolic syndrome: the Diabetes Prevention Program randomized trial. Ann Intern Med. 2005;142:611–619. doi: 10.7326/0003-4819-142-8-200504190-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tuomilehto J, Lindström J, Eriksson JG, Valle TT, Hämäläinen H, Ilanne-Parikka P, Keinänen-Kiukaanniemi S, Laakso M, Louheranta A, Rastas M, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344:1343–1350. doi: 10.1056/NEJM200105033441801. [DOI] [PubMed] [Google Scholar]
- 233.Praagman J, Beulens JW, Alssema M, Zock PL, Wanders AJ, Sluijs I, van der Schouw YT. The association between dietary saturated fatty acids and ischemic heart disease depends on the type and source of fatty acid in the European Prospective Investigation into Cancer and Nutrition-Netherlands cohort. Am J Clin Nutr. 2016;103:356–365. doi: 10.3945/ajcn.115.122671. [DOI] [PubMed] [Google Scholar]
- 234.Samaha FF, Iqbal N, Seshadri P, Chicano KL, Daily DA, McGrory J, Williams T, Williams M, Gracely EJ, Stern L. A low-carbohydrate as compared with a low-fat diet in severe obesity. N Engl J Med. 2003;348:2074–2081. doi: 10.1056/NEJMoa022637. [DOI] [PubMed] [Google Scholar]
- 235.Gardner CD, Kiazand A, Alhassan S, Kim S, Stafford RS, Balise RR, Kraemer HC, King AC. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA. 2007;297:969–977. doi: 10.1001/jama.297.9.969. [DOI] [PubMed] [Google Scholar]
- 236.Larsen TM, Dalskov SM, van Baak M, Jebb SA, Papadaki A, Pfeiffer AF, Martinez JA, Handjieva-Darlenska T, Kunešová M, Pihlsgård M, et al. Diets with high or low protein content and glycemic index for weight-loss maintenance. N Engl J Med. 2010;363:2102–2113. doi: 10.1056/NEJMoa1007137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Paniagua JA, de la Sacristana AG, Sánchez E, Romero I, Vidal-Puig A, Berral FJ, Escribano A, Moyano MJ, Peréz-Martinez P, López-Miranda J, et al. A MUFA-rich diet improves posprandial glucose, lipid and GLP-1 responses in insulin-resistant subjects. J Am Coll Nutr. 2007;26:434–444. doi: 10.1080/07315724.2007.10719633. [DOI] [PubMed] [Google Scholar]
- 238.Du H, van der A DL, Boshuizen HC, Forouhi NG, Wareham NJ, Halkjaer J, Tjønneland A, Overvad K, Jakobsen MU, Boeing H, et al. Dietary fiber and subsequent changes in body weight and waist circumference in European men and women. Am J Clin Nutr. 2010;91:329–336. doi: 10.3945/ajcn.2009.28191. [DOI] [PubMed] [Google Scholar]
- 239.Romaguera D, Angquist L, Du H, Jakobsen MU, Forouhi NG, Halkjaer J, Feskens EJ, van der A DL, Masala G, Steffen A, et al. Dietary determinants of changes in waist circumference adjusted for body mass index - a proxy measure of visceral adiposity. PLoS One. 2010;5:e11588. doi: 10.1371/journal.pone.0011588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Parnell JA, Reimer RA. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am J Clin Nutr. 2009;89:1751–1759. doi: 10.3945/ajcn.2009.27465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Cani PD, Joly E, Horsmans Y, Delzenne NM. Oligofructose promotes satiety in healthy human: a pilot study. Eur J Clin Nutr. 2006;60:567–572. doi: 10.1038/sj.ejcn.1602350. [DOI] [PubMed] [Google Scholar]
- 242.Field AE, Willett WC, Lissner L, Colditz GA. Dietary fat and weight gain among women in the Nurses’ Health Study. Obesity (Silver Spring) 2007;15:967–976. doi: 10.1038/oby.2007.616. [DOI] [PubMed] [Google Scholar]
- 243.Soriguer F, Almaraz MC, García-Almeida JM, Cardona I, Linares F, Morcillo S, García-Escobar E, Dobarganes MC, Olveira G, Hernando V, et al. Intake and home use of olive oil or mixed oils in relation to healthy lifestyles in a Mediterranean population. Findings from the prospective Pizarra study. Br J Nutr. 2010;103:114–122. doi: 10.1017/S0007114509991498. [DOI] [PubMed] [Google Scholar]
- 244.Piers LS, Walker KZ, Stoney RM, Soares MJ, O’Dea K. Substitution of saturated with monounsaturated fat in a 4-week diet affects body weight and composition of overweight and obese men. Br J Nutr. 2003;90:717–727. doi: 10.1079/bjn2003948. [DOI] [PubMed] [Google Scholar]
- 245.Micallef M, Munro I, Phang M, Garg M. Plasma n-3 Polyunsaturated Fatty Acids are negatively associated with obesity. Br J Nutr. 2009;102:1370–1374. doi: 10.1017/S0007114509382173. [DOI] [PubMed] [Google Scholar]
- 246.Garaulet M, Pérez-Llamas F, Pérez-Ayala M, Martínez P, de Medina FS, Tebar FJ, Zamora S. Site-specific differences in the fatty acid composition of abdominal adipose tissue in an obese population from a Mediterranean area: relation with dietary fatty acids, plasma lipid profile, serum insulin, and central obesity. Am J Clin Nutr. 2001;74:585–591. doi: 10.1093/ajcn/74.5.585. [DOI] [PubMed] [Google Scholar]
- 247.Camargo A, Meneses ME, Perez-Martinez P, Delgado-Lista J, Jimenez-Gomez Y, Cruz-Teno C, Tinahones FJ, Paniagua JA, Perez-Jimenez F, Roche HM, et al. Dietary fat differentially influences the lipids storage on the adipose tissue in metabolic syndrome patients. Eur J Nutr. 2014;53:617–626. doi: 10.1007/s00394-013-0570-2. [DOI] [PubMed] [Google Scholar]
- 248.Gaullier JM, Halse J, Høye K, Kristiansen K, Fagertun H, Vik H, Gudmundsen O. Conjugated linoleic acid supplementation for 1 y reduces body fat mass in healthy overweight humans. Am J Clin Nutr. 2004;79:1118–1125. doi: 10.1093/ajcn/79.6.1118. [DOI] [PubMed] [Google Scholar]
- 249.Gaullier JM, Halse J, Høivik HO, Høye K, Syvertsen C, Nurminiemi M, Hassfeld C, Einerhand A, O’Shea M, Gudmundsen O. Six months supplementation with conjugated linoleic acid induces regional-specific fat mass decreases in overweight and obese. Br J Nutr. 2007;97:550–560. doi: 10.1017/S0007114507381324. [DOI] [PubMed] [Google Scholar]
- 250.Smedman A, Vessby B. Conjugated linoleic acid supplementation in humans--metabolic effects. Lipids. 2001;36:773–781. doi: 10.1007/s11745-001-0784-7. [DOI] [PubMed] [Google Scholar]
- 251.Vessby B, Uusitupa M, Hermansen K, Riccardi G, Rivellese AA, Tapsell LC, Nälsén C, Berglund L, Louheranta A, Rasmussen BM, et al. Substituting dietary saturated for monounsaturated fat impairs insulin sensitivity in healthy men and women: The KANWU Study. Diabetologia. 2001;44:312–319. doi: 10.1007/s001250051620. [DOI] [PubMed] [Google Scholar]
- 252.Salas J, López Miranda J, Jansen S, Zambrana JL, Castro P, Paniagua JA, Blanco A, López Segura F, Jiménez Perepérez JA, Pérez Jiménez F. [The diet rich in monounsaturated fat modifies in a beneficial way carbohydrate metabolism and arterial pressure] Med Clin (Barc) 1999;113:765–769. [PubMed] [Google Scholar]
- 253.Pérez-Jiménez F, López-Miranda J, Pinillos MD, Gómez P, Paz-Rojas E, Montilla P, Marín C, Velasco MJ, Blanco-Molina A, Jiménez Perepérez JA, et al. A Mediterranean and a high-carbohydrate diet improve glucose metabolism in healthy young persons. Diabetologia. 2001;44:2038–2043. doi: 10.1007/s001250100009. [DOI] [PubMed] [Google Scholar]
- 254.Tierney AC, McMonagle J, Shaw DI, Gulseth HL, Helal O, Saris WH, Paniagua JA, Gołąbek-Leszczyñska I, Defoort C, Williams CM, et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome--LIPGENE: a European randomized dietary intervention study. Int J Obes (Lond) 2011;35:800–809. doi: 10.1038/ijo.2010.209. [DOI] [PubMed] [Google Scholar]
- 255.Yubero-Serrano EM, Delgado-Lista J, Tierney AC, Perez-Martinez P, Garcia-Rios A, Alcala-Diaz JF, Castaño JP, Tinahones FJ, Drevon CA, Defoort C, et al. Insulin resistance determines a differential response to changes in dietary fat modification on metabolic syndrome risk factors: the LIPGENE study. Am J Clin Nutr. 2015;102:1509–1517. doi: 10.3945/ajcn.115.111286. [DOI] [PubMed] [Google Scholar]
- 256.Atkinson FS, Foster-Powell K, Brand-Miller JC. International tables of glycemic index and glycemic load values: 2008. Diabetes Care. 2008;31:2281–2283. doi: 10.2337/dc08-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Uyeda K, Repa JJ. Carbohydrate response element binding protein, ChREBP, a transcription factor coupling hepatic glucose utilization and lipid synthesis. Cell Metab. 2006;4:107–110. doi: 10.1016/j.cmet.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 258.McMillan-Price J, Petocz P, Atkinson F, O’neill K, Samman S, Steinbeck K, Caterson I, Brand-Miller J. Comparison of 4 diets of varying glycemic load on weight loss and cardiovascular risk reduction in overweight and obese young adults: a randomized controlled trial. Arch Intern Med. 2006;166:1466–1475. doi: 10.1001/archinte.166.14.1466. [DOI] [PubMed] [Google Scholar]
- 259.Jeppesen J, Hollenbeck CB, Zhou MY, Coulston AM, Jones C, Chen YD, Reaven GM. Relation between insulin resistance, hyperinsulinemia, postheparin plasma lipoprotein lipase activity, and postprandial lipemia. Arterioscler Thromb Vasc Biol. 1995;15:320–324. doi: 10.1161/01.atv.15.3.320. [DOI] [PubMed] [Google Scholar]
- 260.Panarotto D, Rémillard P, Bouffard L, Maheux P. Insulin resistance affects the regulation of lipoprotein lipase in the postprandial period and in an adipose tissue-specific manner. Eur J Clin Invest. 2002;32:84–92. doi: 10.1046/j.1365-2362.2002.00945.x. [DOI] [PubMed] [Google Scholar]
- 261.Paniagua JA, Pérez-Martinez P, Gjelstad IM, Tierney AC, Delgado-Lista J, Defoort C, Blaak EE, Risérus U, Drevon CA, Kiec-Wilk B, et al. A low-fat high-carbohydrate diet supplemented with long-chain n-3 PUFA reduces the risk of the metabolic syndrome. Atherosclerosis. 2011;218:443–450. doi: 10.1016/j.atherosclerosis.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 262.Ueshima H, Stamler J, Elliott P, Chan Q, Brown IJ, Carnethon MR, Daviglus ML, He K, Moag-Stahlberg A, Rodriguez BL, et al. Food omega-3 fatty acid intake of individuals (total, linolenic acid, long-chain) and their blood pressure: INTERMAP study. Hypertension. 2007;50:313–319. doi: 10.1161/HYPERTENSIONAHA.107.090720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Deckelbaum RJ, Worgall TS, Seo T. n-3 fatty acids and gene expression. Am J Clin Nutr. 2006;83:1520S–1525S. doi: 10.1093/ajcn/83.6.1520S. [DOI] [PubMed] [Google Scholar]
- 264.Chandalia M, Garg A, Lutjohann D, von Bergmann K, Grundy SM, Brinkley LJ. Beneficial effects of high dietary fiber intake in patients with type 2 diabetes mellitus. N Engl J Med. 2000;342:1392–1398. doi: 10.1056/NEJM200005113421903. [DOI] [PubMed] [Google Scholar]
- 265.Berg AH, Combs TP, Du X, Brownlee M, Scherer PE. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med. 2001;7:947–953. doi: 10.1038/90992. [DOI] [PubMed] [Google Scholar]
- 266.Silva FM, de Almeida JC, Feoli AM. Effect of diet on adiponectin levels in blood. Nutr Rev. 2011;69:599–612. doi: 10.1111/j.1753-4887.2011.00414.x. [DOI] [PubMed] [Google Scholar]
- 267.Reseland JE, Anderssen SA, Solvoll K, Hjermann I, Urdal P, Holme I, Drevon CA. Effect of long-term changes in diet and exercise on plasma leptin concentrations. Am J Clin Nutr. 2001;73:240–245. doi: 10.1093/ajcn/73.2.240. [DOI] [PubMed] [Google Scholar]
- 268.Karbowska J, Kochan Z. Intermittent fasting up-regulates Fsp27/Cidec gene expression in white adipose tissue. Nutrition. 2012;28:294–299. doi: 10.1016/j.nut.2011.06.009. [DOI] [PubMed] [Google Scholar]
- 269.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, Balas B, Gastaldelli A, Tio F, Pulcini J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–2307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 270.Camargo A, Meneses ME, Rangel-Zuñiga OA, Perez-Martinez P, Marin C, Delgado-Lista J, Paniagua JA, Tinahones FJ, Roche H, Malagon MM, et al. Endoplasmic reticulum stress in adipose tissue determines postprandial lipoprotein metabolism in metabolic syndrome patients. Mol Nutr Food Res. 2013;57:2166–2176. doi: 10.1002/mnfr.201300036. [DOI] [PubMed] [Google Scholar]
- 271.Peña-Orihuela P, Camargo A, Rangel-Zuñiga OA, Perez-Martinez P, Cruz-Teno C, Delgado-Lista J, Yubero-Serrano EM, Paniagua JA, Tinahones FJ, Malagon MM, et al. Antioxidant system response is modified by dietary fat in adipose tissue of metabolic syndrome patients. J Nutr Biochem. 2013;24:1717–1723. doi: 10.1016/j.jnutbio.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 272.Meneses ME, Camargo A, Perez-Martinez P, Delgado-Lista J, Cruz-Teno C, Jimenez-Gomez Y, Paniagua JA, Gutierrez-Mariscal FM, Tinahones FJ, Vidal-Puig A, et al. Postprandial inflammatory response in adipose tissue of patients with metabolic syndrome after the intake of different dietary models. Mol Nutr Food Res. 2011;55:1759–1770. doi: 10.1002/mnfr.201100200. [DOI] [PubMed] [Google Scholar]
- 273.López-Miranda J, Pérez-Jiménez F, Ros E, De Caterina R, Badimón L, Covas MI, Escrich E, Ordovás JM, Soriguer F, Abiá R, et al. Olive oil and health: summary of the II international conference on olive oil and health consensus report, Jaén and Córdoba (Spain) 2008. Nutr Metab Cardiovasc Dis. 2010;20:284–294. doi: 10.1016/j.numecd.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 274.Freiberg MS, Cabral HJ, Heeren TC, Vasan RS, Curtis Ellison R; Third National Health and Nutrition Examination Survey. Alcohol consumption and the prevalence of the Metabolic Syndrome in the US.: a cross-sectional analysis of data from the Third National Health and Nutrition Examination Survey. Diabetes Care. 2004;27:2954–2959. doi: 10.2337/diacare.27.12.2954. [DOI] [PubMed] [Google Scholar]
- 275.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Bulló M, García-Lorda P, Megias I, Salas-Salvadó J. Systemic inflammation, adipose tissue tumor necrosis factor, and leptin expression. Obes Res. 2003;11:525–531. doi: 10.1038/oby.2003.74. [DOI] [PubMed] [Google Scholar]
- 277.Prieur X, Mok CY, Velagapudi VR, Núñez V, Fuentes L, Montaner D, Ishikawa K, Camacho A, Barbarroja N, O’Rahilly S, et al. Differential lipid partitioning between adipocytes and tissue macrophages modulates macrophage lipotoxicity and M2/M1 polarization in obese mice. Diabetes. 2011;60:797–809. doi: 10.2337/db10-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 279.Rosen BS, Cook KS, Yaglom J, Groves DL, Volanakis JE, Damm D, White T, Spiegelman BM. Adipsin and complement factor D activity: an immune-related defect in obesity. Science. 1989;244:1483–1487. doi: 10.1126/science.2734615. [DOI] [PubMed] [Google Scholar]
- 280.Kasbi Chadli F, Andre A, Prieur X, Loirand G, Meynier A, Krempf M, Nguyen P, Ouguerram K. n-3 PUFA prevent metabolic disturbances associated with obesity and improve endothelial function in golden Syrian hamsters fed with a high-fat diet. Br J Nutr. 2012;107:1305–1315. doi: 10.1017/S0007114511004387. [DOI] [PubMed] [Google Scholar]
- 281.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Sun K, Kusminski CM, Scherer PE. Adipose tissue remodeling and obesity. J Clin Invest. 2011;121:2094–2101. doi: 10.1172/JCI45887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95:2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Hotamisligil GS. Philadelphia, PA, United States: Lippincott-Raven Publishers; 2003. Inflammation, TNF-alpha and insulin resistance. [Google Scholar]
- 286.Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 287.Després JP, Lemieux I, Bergeron J, Pibarot P, Mathieu P, Larose E, Rodés-Cabau J, Bertrand OF, Poirier P. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–1049. doi: 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
- 288.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117:2621–2637. doi: 10.1172/JCI31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Adiels M, Borén J, Caslake MJ, Stewart P, Soro A, Westerbacka J, Wennberg B, Olofsson SO, Packard C, Taskinen MR. Overproduction of VLDL1 driven by hyperglycemia is a dominant feature of diabetic dyslipidemia. Arterioscler Thromb Vasc Biol. 2005;25:1697–1703. doi: 10.1161/01.ATV.0000172689.53992.25. [DOI] [PubMed] [Google Scholar]
- 290.Rohatgi A, Khera A, Berry JD, Givens EG, Ayers CR, Wedin KE, Neeland IJ, Yuhanna IS, Rader DR, de Lemos JA, et al. HDL cholesterol efflux capacity and incident cardiovascular events. N Engl J Med. 2014;371:2383–2393. doi: 10.1056/NEJMoa1409065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev. 2006;58:342–374. doi: 10.1124/pr.58.3.1. [DOI] [PubMed] [Google Scholar]