

Abstract
The incidence of obesity in the population is increasing at an alarming rate, with this comes an increased risk of insulin resistance (IR). Obesity and IR increase an individual’s risk of having a stroke and they have been linked to several forms of dementia. Stroke and dementia are associated with, or exacerbated by, reduced cerebral blood flow, which has recently been described in obese patients. In this review we will discuss the effects of obesity on cerebral artery function and structure. Regarding their function, we will focus on the endothelium and nitric oxide (NO) dependent dilation. NO dependent dilation is impaired in cerebral arteries from obese rats, and the majority of evidence suggests this is a result of increased oxidative stress. We will also describe the limited studies showing that inward cerebral artery remodeling occurs in models of obesity, and that the remodeling is associated with an increase in the damage caused by cerebral ischemia. We will also discuss some of the more paradoxical findings associated with stroke and obesity, including the evidence that obesity is a positive factor for stroke survival. Finally we will discuss the evidence that links these changes in vascular structure and function to cognitive decline and dementia.
Keywords: Obesity, insulin resistance, cerebral ischemia, dementia
1. INTRODUCTION
The brain is one of the most highly perfused organs in the body, and proper brain function requires that cerebral blood flow be held at a constant level irrespective of the metabolic demands of other organs. The detrimental effects of obesity on the cerebral vasculature are striking, with several studies showing that obese patients, or patients with an elevated body mass index (BMI), have reduced cerebral blood flow [1–3]. Here we will describe some potential mechanisms and possible ramifications of reduced cerebral perfusion in obesity. Several differences in the cerebral vasculature have been identified between control and obese subjects, but very few studies have attempted to define if the obesity-associated pathology is the result of IR. Therefore, we will focus primarily on obesity studies but will also discuss the effect of IR without concomitant obesity. First, we must consider some features of the cerebral circulation that make it unique.
1.1. Cerebral Vessel Anatomy
The anatomy of the cerebral circulation has recently been described in detail [4]. Research on large cerebral arteries has primarily been conducted in the basilar artery and the middle cerebral artery (MCA). The basilar artery runs along the midline of the brain stem before connecting with the circle of Willis on the base of the brain. Three pairs of large arteries branch off of the circle of Willis; these are the posterior and anterior cerebral arteries and the MCA (Fig. 1A). The pial or leptomeningial arteries branch from these large arteries and ramify over the brain’s surface. The pial arteries form a collateral network that serves to maintain perfusion in the event of an arterial occlusion. These arteries receive extrinsic innervation from the peripheral nervous system [4].
Fig. 1.
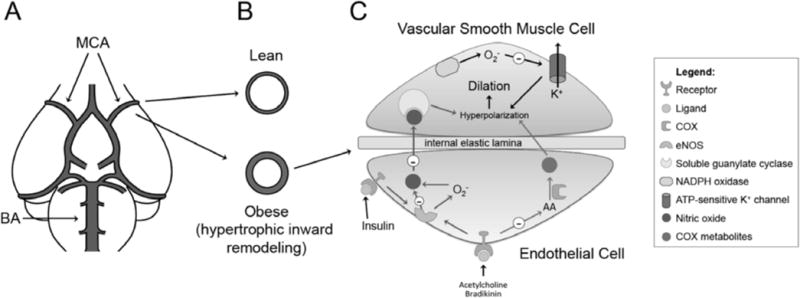
Overview of obesity associated changes in the cerebral vasculature. A) Graphic depiction of the large cerebral arteries and the Circle of Willis; BA: basilar artery, MCA: middle cerebral artery. B) Diet-induced obesity leads to hypertrophic inward remodeling of the MCA in rats [53]. The MCAs from high fat fed rats have a smaller lumen diameter and increased wall thickness (bottom image) when compared to the MCAs from lean control rats (upper image). C) Obesity and IR-induced endothelium dysfunction. The evidence in the literature suggests that obesity mainly impairs nitric oxide bioavailability and production by eNOS (blue arrows). Excessive superoxide generation seems to play a major role in the reduction of NO-induced dilation, because treatment with antioxidants improves NO-mediated dilation. Superoxide is generated by either uncoupling of eNOS (as shown in insulin-mediated dilation) or by increased NADPH-oxidase activity. On the other hand, IR-induced endothelial dysfunction appears to be a consequence of impaired production of COX metabolites with vasodilator properties (red arrows). This is supported by the fact that inhibition of COX with indomethacin has no effect on the already reduced dilation of cerebral arteries from IR rats, suggesting that these vessels no longer depend on COX metabolites to dilate. Independently of the mechanism (NO or COX metabolites), dilation is ultimately a consequence of hyperpolarization of smooth muscle cells by opening of K+ channels (black arrows). The function of these channels, particularly KATP, is impaired during both obesity and IR, through a mechanism that is dependent on superoxide, since antioxidant treatment increases dilation to KATP activators.
The pial arteries are connected to the parenchymal arterioles by penetrating arterioles, and the penetrating and parenchymal arterioles perfuse the parenchyma. The penetrating arterioles are located in the Virchow-Robin space and have very few anastomoses [5]; therefore they control blood flow to discrete regions of the cortex [6]. The parenchymal arterioles are surrounded by astrocytic end feet [7], and receive intrinsic innervation from within the neuropil [8]. The parenchymal arterioles give rise to capillaries. The cerebral capillaries contain endothelial cells and pericytes encased in the basal lamina. Astrocytic end feet wrap around the capillaries. The endothelial cells, pericytes, astrocytes, and associated neurons, make up the neurovascular unit, which may be a therapeutic target for ischemic strokes [9]. The blood brain barrier (BBB) is a selectively permeable physical and biochemical barrier that maintains central nervous system homeostasis. Most studies of BBB function take place using capillary endothelial cells, however, the large arteries and arterioles also contribute to the BBB properties [4, 10].
1. 2. What Makes the Cerebral Vasculature Unique?
There are several features of cerebral arteries that make them different from other vascular beds, and preclude us from extrapolating the effects of obesity on peripheral vessels to the brain [4, 9, 11]. The large cerebral arteries including the MCA contribute to the regulation of cerebral blood flow because they carry a significant portion of the cerebral vascular resistance [4, 12]. The cells in the cerebral arteries also interact with several other cell types including neurons, astrocytes, pericytes and glial cells. This feature is important in the microvessels where the neurons, astrocytes and pericytes are involved in blood flow control. The BBB also confers unique properties on the cerebral circulation [13]. Another difference that is particularly important in obesity is the absence of perivascular adipose tissue around the cerebral arteries. There is a growing literature describing the effects of perivascular adipose tissue on vascular function (for reviews see [14–16]). Because cerebral vessels are not surrounded by adipose tissue the effects of obesity are most likely a response to changes in blood pressure, innervation, and circulating factors.
The cerebral circulation has a unique ability to maintain constant parenchymal perfusion when blood pressure is fluctuating; this occurs through the mechanism of autoregulation. In humans, autoregulation occurs between mean arterial pressures of 60 mmHg and 150 mmHg. Outside this range blood flow is directly related to blood pressure [17], and perfusion pressures above and below this range have detrimental effects on the brain. Pressures above the autoregulatory range cause increased flow and vasogenic edema, and pressures below this range cause ischemic injury [4]. Several mechanisms have been proposed to contribute to the control of autoregulation in the cerebral circulation. These include neuronal nitric oxide (NO) production [17–19], metabolic byproducts [17], sympathetic [20] and cholinergic [21] nerve activity and flow itself [22].
Although not peculiar to the brain, myogenic activity in cerebral arteries plays a key role in the regulation of blood flow and cerebrovascular resistance [23]. The myogenic response is the ability of an artery to constrict in response to increasing intraluminal pressures, and this maintains blood flow at a constant rate. This is important at higher pressures to prevent overperfusion and damage of the downstream arterioles and capillaries [24]. The myogenic response is an intrinsic property of the smooth muscle cells, but the response is modulated by the endothelium [25, 26]. This modulation occurs through many mechanisms including NO, cyclooxygenase (COX) metabolites and endothelium derived hyperpolarizing factor (EDHF).
Despite its importance, little is known about how obesity affects cerebral artery myogenic reactivity and tone. One study investigated myogenic responses in isolated MCAs from obese Zucker rats (OZR) [27]. The OZR has a mutation in the leptin receptor that renders it non-functional. OZR are hyperphagic, hypercholesterolemic, hyperinsulinemic and mildly hypertensive. Lean Zucker rats (LZR), the control strain for OZR, have a functional leptin receptor and are metabolically normal. Adult OZR (14–16 weeks of age) show increased myogenic tone generation ex vivo at intraluminal pressures ranging from 60 to 120mmHg when compared to LZR. At this age the OZR are mildly hypertensive and this could account for the increased myogenic tone. However, tone generation appears to change as the OZR age. Young OZR (6–8 week) are obese but normotensive, and they show a reduction in MCA tone generation at high intraluminal pressures when compared to age matched LZR [27]. There is no difference in myogenic tone generation between OZR and LZR at 12 weeks of age [28]. The enhanced pressure mediated contraction observed in older OZR [27] could increase cerebrovascular resistance and reduce blood flow, which can increase the risk of developing dementia and exacerbate damage caused by ischemia. However, from this study it is not possible to truly define the effects of prolonged obesity versus hypertension.
2. OBESITY AND CEREBRAL ARTERY ENDOTHELIAL FUNCTION
One striking characteristic of the cerebral circulation is its ability to match perfusion to the regional metabolic demands of active neurons. This process, known as functional hyperemia, is dependent on the dilator capacity of penetrating and parenchymal arterioles. Functional hyperemia is impaired in obese rats fed a high fat (HF) and high carbohydrate cafeteria-style diet [29]. In the cerebral circulation, the endothelium plays a prominent role in regulating blood flow by inhibiting excessive constriction and causing dilation. Three signaling mechanisms contribute to the control of endothelium-dependent dilation. These are, 1) NO produced by endothelial NO synthase (eNOS), 2) arachidonic acid metabolites produced by COX, and 3) EDHF. We will focus on NO because this pathway has been widely studied in obesity. At present there are no published studies reporting alterations in EDHF dependent dilation with obesity. EDHF is an important cerebral artery dilator [30], and therefore we propose that efforts should be made to fill this knowledge gap.
2.1. Nitric Oxide Dependent Dilation in the Obese Zucker Rat
Impaired NO-dependent dilation has been reported in cerebral arteries from adult (12–17 weeks) and aged (36 weeks) OZR, and both sexes are affected. This has been observed ex vivo in pressurized MCAs from OZR [31]. In vivo cranial window studies suggest the same is true for pial arterioles [32]. The mechanism for the impaired NO-mediated dilation has been studied in 12 week old OZR using acetylcholine as a dilator [33]. Inhibition of eNOS with L-NG-nitroarginine methyl ester (L-NAME) blunted the acetylcholine-induced dilator response in LZR, but had little effect in OZR; this suggests the NO dilatory pathway is impaired in the OZR. NO donor administration restored dilation, suggesting a reduction in NO production or bioavailability rather than reduced smooth muscle cell sensitivity to NO in the OZR [33]. The impaired NO mediated dilation in OZR appears to be the result of increased reactive oxygen species generation, and the superoxide radical seems particularly important. Superoxide reacts with NO to generate peroxynitrite; this reduces NO bioavailability and impairs NO mediated dilation. Treatment of basilar arteries with superoxide dismutase (SOD), the endogenous enzyme that converts superoxide to hydrogen peroxide, improved dilation in OZR [33]. The authors concluded that SOD reduced superoxide levels, and therefore increased NO bioavailability. However, it is possible that these effects were mediated by hydrogen peroxide, which is an EDHF that induces cerebral artery dilation [34]. It is also interesting to note that eNOS protein expression is elevated in the cerebral arteries from OZR [28], even though NO production is reduced. This adds weight to the argument that eNOS is dysfunctional and likely uncoupled in the cerebral arteries from OZR [28]. Impaired acetylcholine-induced dilation was also observed in the basilar artery of aged female OZR. In this case administration of an NO-donor did not improve dilation [35], suggesting that at an advanced age the smooth muscle cells in the OZR might lose their sensitivity to NO. It is however not clear if this effect is sex- or age-dependent. The male rats used in the studies described above [33] were considerably younger than the female rats that exhibited insensitivity to NO [35].
Other studies have suggested a role for superoxide in impaired cerebral artery dilation in obese rats. In control rats, including LZR, hypoxia induces cerebral artery dilation. This is not the case in MCAs from 17-week-old OZR where hypoxia causes constriction. These effects appear to be dependent on superoxide, which is increased in MCAs from OZR. Pre-incubation of the MCAs with tempol, a SOD mimetic, quenched the superoxide levels and improved the hypoxia-mediated dilation [31]. Impaired hypoxia-mediated dilation may be particularly important in situations where cerebral blood flow is reduced such as cerebral ischemia.
2.2. Potassium Channels and Dilation in the Obese Zucker Rat
Dilation is dependent on endothelial and smooth muscle cell hyperpolarization after K+ channel opening. Thus, K+ channel dysfunction is a possible mechanism for the impaired dilation in obesity. In fact, young OZR show reduced dilation after incubation with cromakalim, an ATP-sensitive K+ channel (KATP) activator. This appears to be the result of oxidative stress because SOD [33] and rosuvastatin [36] restored cromakalim-induced dilation by reducing superoxide levels. Part of this dysfunction seems to occur in mitochondrial KATP channels. Activation of these channels causes dilation, and this is impaired in OZR [28]. Interestingly, mitochondrial KATP channel activation causes both smooth muscle and endothelial cell dependent cerebral artery dilation. In endothelial cells this dilation is dependent on eNOS activation by ROS-dependent and -independent mechanisms [37]. This could have important implications for the outcome of cerebral ischemia. The activity of mitochondrial KATP channels has been linked to infarct size and ischemic preconditioning in the heart following transient ischemia [38]. The effects of this channel on cerebral ischemic preconditioning have not been investigated.
2.3 Insulin Mediated Dilation in the Obese Zucker Rat
Recently, a novel role for insulin as a cerebral artery dilator has emerged. The insulin receptor is expressed in cerebral endothelial cells, and insulin increases blood flow in the cerebral cortex [39]. Despite the presence of the insulin receptor in cerebral arteries, it appears that endothelin receptor type B activation is responsible for the insulin-mediated dilation [39]. Pressurized isolated MCAs and posterior cerebral arteries dilate in response to insulin, this effect is lost after endothelium removal, eNOS blockade and K+ channel inhibition [39]. The dilator response to insulin is impaired in OZR. This is not a consequence of reduced insulin receptor levels; instead the insulin mediated NO production is reduced as a result of eNOS uncoupling. eNOS becomes uncoupled when availability of co-factors such as tetrahydrobiopterin is reduced. Uncoupled eNOS produces superoxide instead of NO [40]. Incubation of cerebral arteries from OZR with sepiapterin, a tetrahydrobiopterin donor, improved insulin mediated dilation in OZR, but not in LZR, suggesting eNOS uncoupling in OZR [41]. In keeping with this, basal eNOS levels were not different between the OZR and LZR in this study. Insulin caused eNOS activation but this was reduced in the OZR compared to the LZR. Similarly, the expression of GTP cyclohydrolase, the enzyme that produces tetrahydrobiopterin, was reduced in the OZR at basal levels and after insulin stimulation [41]. There is some variability in the reports of eNOS levels in OZR with some studies suggesting eNOS levels are increased in the obese rats [28]. It is unclear if insulin-mediated dilation of cerebral arteries has a NO-independent component.
While insulin appears to be primarily a dilator it the cerebral vasculature, it has been shown to cause constriction at low concentrations [39]. This appears to be mediated by constrictor prostanoid production by COX, and cerebral arteries from OZR appear to produce more of these than arteries from LZR [41]. Therefore, in the OZR the impaired dilation and enhanced constriction in response to insulin is likely to reduce cerebral blood flow and this may be an important determinant of the cognitive decline observed with obesity.
2.4 Effects of Insulin Resistance Without Obesity on Endothelial Function
From the studies using OZR it is tempting to assume that the deleterious effects of hyperinsulinemia and IR on endothelial function are all mediated by impairments in the NO pathway. However, studies using fructose-fed rats, a model of IR without obesity, suggest that COX metabolites are more important in such cases. MCAs from these rats showed reduced dilation to bradykinin that was completely abolished with L-NAME, but unaffected by COX inhibition with indomethacin. Bradykinin-induced dilation of cerebral arteries occurs through release of NO [42] and COX metabolites [43] by the endothelium. This suggests that arteries from the fructose-fed rats are not dependent on COX metabolites to dilate [44]. It is unclear if the impaired COX-dependent dilation is due to reduced production of COX metabolites or alterations in the signaling pathways they activate that lead to hyperpolarization. Blockade of the K+ channels involved in bradykinin-induced hyperpolarization did not further reduce MCA dilation in fructose-fed rats, this suggests COX production may be impaired. Interestingly, activation of KATP caused dilation in both control and fructose-fed rats, although to a lesser extent in the later [45].
3. EFFECTS OF OBESITY AND IR ON ARTERY STRUCTURE
Remodeling is the process by which arteries alter their structure in response to changes in blood pressure, flow and circulating factors. Remodeling has been widely studied and reviewed in the hypertension field [46–50]. We know less about the effects of obesity on artery structure. It should be recognized that artery remodeling is not always detrimental and often occurs to improve perfusion of a specific organ or region. Remodeling occurs in a variety of ways. Inward remodeling reflects a vessel with a smaller lumen and an increased wall-to-lumen ratio, the converse situation is known as outward remodeling [51]. Hypertrophic remodeling occurs when the wall area is increased, whereas the wall area and wall-to-lumen ratio are reduced in hypotrophic remodeling [50]. Eutrophic remodeling occurs when the amount of wall material is unchanged, but is rearranged in a manner that increases the wall-to-lumen ratio. Studies of artery remodeling are conducted on fully relaxed vessels; this is achieved by placing the arteries in calcium-free solutions with the possible addition of calcium chelators and vasodilators. The effects of artery remodeling are most important in situations where the ability of the vessel to contract and dilate is lost, such as occurs during cerebral ischemia. In this situation, small changes in passive lumen diameter will dramatically affect blood flow.
3.1. The Effects of Diet Induced Obesity on Artery Structure
Studies from our laboratory have shown that MCA remodeling occurs in a model of diet-induced obesity. Male Sprague Dawley rats fed a HF diet for 10 weeks from 3 weeks of age develop abdominal obesity, mild hypertension and increased fasting blood glucose and insulin [52]. MCAs from these rats and age matched regular chow fed rats were studied. The lumen diameter and outer diameter of the MCA was markedly reduced in the HF fed rats, whereas, the wall-to-lumen ratio and wall thickness were increased. This caused an increase in the MCA wall stiffness that was associated with an increased expression of collagen type I and an increase in the activity of matrix metalloproteinase (MMP)-2 [53].
3.2. The Effects of Genetic Obesity on Artery Structure
Other studies of MCA structure have been conducted in OZR. Young (6–7 week old) and adult (14–16 week old) OZR were compared by Osmond et al [27]. Both groups were obese, but only the adult rats where hypertensive, and this difference appears to contribute to MCA remodeling. In the adult rats the MCAs exhibited an inward remodeling that included a reduction in the lumen and outer diameter and a small increase in wall thickness. These changes were not apparent in the young OZR, suggesting that the artery remodeling may be the result of hypertension. In a follow up study, Osmond et al investigated the effects of hydrochlorothiazide, an anti-hypertensive, on MCA structure. Hydrochlorothiazide prevented the hypertension and the MCA remodeling observed in the OZR. These effects were observed without a change in body weight, insulin, triglyceride or cholesterol levels. This strongly suggests that blood pressure may be a key determinant of vascular remodeling in the OZR [54]. These studies are at odds with an earlier report from Phillips et al that failed to show differences in the lumen diameter, distensibility or stiffness of the MCA from 17 week-old OZR compared to LZR [31]. Body weight and blood pressure cannot account for the disparity in the results as the OZR used by Phillips et al were about 100 grams heavier than those used by Osmond et al [27], and their blood pressure was 10 mmHg higher. The same group also reported no difference in MCA structure in 13–15 week old OZR [55]. Other studies have failed to show a difference in the passive lumen diameter of the MCA in obese rats fed a cafeteria-style diet [29].
The studies from Osmond et al [27, 54] suggest hypertension plays a role in artery remodeling; there are no studies assessing the molecular mechanisms of artery remodeling. In models of hypertension, angiotensin II [56–59] and aldosterone [60–62] have been linked to cerebral artery remodeling. Aldosterone is elevated in obese patients [63], and some studies suggest that adipose tissue produces aldosterone [64], although this has been disputed [65]. We observed increased aldosterone production in rats fed a HF diet [66], we have also observed vascular remodeling in this model [53]. Therefore it is possible that aldosterone is the driving force behind obesity induced cerebrovascular remodeling.
We still have much to learn about the effects of obesity on artery structure. In some situations rarefaction or loss of arteries occurs, and this reduces cerebral perfusion and increases cerebrovascular resistance. To the best of our knowledge there are no studies documenting cerebral artery rarefaction in obesity. That is not to say that this does not occur, and in fact the studies of hypertensive rats very much suggest that this is a possibility [67]. Similarly there appear to be no studies of the penetrating or parenchymal arteries in obese rats. Given the mounting evidence that obesity contributes to dementia as result of cerebral hypoperfusion these studies need to be completed. BBB dysfunction has been identified in OZR [68]. The same is true in patients with metabolic syndrome, which is associated with obesity and IR [69]. The mechanisms responsible for the breakdown of the BBB have not been investigated.
4. EFFECTS OF OBESITY ON STROKE RISK AND STROKE OUTCOMES
4.1. Human Studies
Stroke is a leading cause of disability and the second most common cause of death worldwide [70, 71]. Strokes can be divided into three subtypes. Ischemic strokes, resulting from a blocked vessel, account for approximately 80% of strokes. The other two subtypes of strokes result from hemorrhages that can be intracerebral or subarachnoid [72]. For reviews of the etiology of stroke and the choice of stroke models see references [73, 74].
There are several studies that describe obesity and IR as a risk factor for stroke. We have elected to direct readers to two recent meta-analyses of the literature. The first assessed the relationship between stroke and obesity. The authors generated a pooled estimate of risk that showed a 22 and 64% increase in ischemic stroke risk for overweight and obese patients respectively. There was no association between obesity and hemorrhagic stroke. The association between increased body weight and stroke risk was greatest for studies of European and North American populations, and was the same for men and women [75]. Importantly, increased BMI is an independent risk factor for stroke, and not just a subsequence of the hypertension, hypercholesterolemia and diabetes that is often found in obese patients [76]. IR and obesity are key components of metabolic syndrome, and a meta-analysis has been conducted for studies of stroke risk and metabolic syndrome [77]. It showed that metabolic syndrome causes a 2-fold increase in the risk of stroke. This study did not separate ischemic from hemorrhagic strokes, so it is unclear if the increased risk is only associated with ischemic strokes as is the case with obesity. However, the analysis did show that the risk of stroke with metabolic syndrome is independent of type-II diabetes.
Not all studies have shown as strong a link between obesity and stroke risk. In a study of Danish women the patients with cerebrovascular events had a slightly increased BMI, but when BMI was considered as a continuous variable it had no effect on stroke risk [78]. Another study showed that having an increased waist-to-hip ratio increased the risk of stroke, but BMI was not associated with stroke risk. However, the number of stroke patients in this study was very small (less than 1% of the study population) [79]. The importance of an association of stroke with waist-to-hip ratio should not be diminished because this measure of obesity may be better than BMI. A waist-to-hip ratio provides an indication of fat distribution [80] and abdominal obesity, which has been widely linked with cardiovascular risk [81]. As with obesity, not all studies agree that IR increases the risk of stroke. A recent report from the Rotterdam Study suggests that in non-diabetic elderly patients IR does not correlate with stroke risk [82].
4.2. The Obesity Paradox
The effects of obesity on stroke risk and stroke outcome are not the same. There are a preponderance of studies showing that obesity increases stroke risk [75], but only one suggests that obesity increases post-stroke morbidity [83]. This study showed that patients with a higher BMI had longer hospital stays and were more likely to need residential care and than lean patients. This was not a function of stroke severity, which suggests the obese patients may be more likely to have in-hospital complications [83]. However, many other studies find that the opposite is true. Obese and overweight patients are more likely to survive a stroke [84–88] and have less morbidity and better outcomes than lean patients [89]. The potential reasons for this finding are several fold and have been reviewed recently by others [90]. One important consideration is that the majority of the studies base their stratification of patients on BMI. It might be more prudent to consider measures that more accurately describe the body fat content and distribution such as waist-to-hip ratio [91]. It is also important to look beyond the basics to consider the types of strokes and the demographic of the population having them. For example, in the study by Ovibagele et al [87] the obese stroke patients were younger than the normal weight patients, and there was a higher percentage of women and a lower number of smokers in the obese group. The obese patients were also more likely to have small lacunar strokes, which in general have better prognosis. Therefore, there may be an inherent selection bias in the published studies. Additional trials and prospective studies will be needed to unravel the findings described above.
While obesity may increase the odds of post-stroke survival it also interferes with standard stroke therapies. Tissue plasminogen activator (tPA) is the only FDA approved drug for the treatment of strokes. tPA produces recanalization of the artery by breaking down the blood clot. Recent studies suggest that tPA is not as effective in obese patients as it is in normal controls. In a study of Japanese patients with MCA occlusions only 57% of the patients receiving tPA had effective recanalization. There were significantly more obese patients in the non-recanalized group than in the group where tPA was effective [92]. It is important to note that this was a rather small trial, but it raises some interesting questions. The authors propose that an increase in plasminogen activator inhibitor-1 in the obese patients limits the effectiveness of the tPA treatment [93]. There is a literature to support this hypothesis [94] but it has not been directly tested.
4.3. Animal Studies of Ischemic Stroke and Obesity
The studies showing that obesity increases stroke risk in humans have not been replicated in rodents. This may be because it is difficult to assess stroke risk in rodent models. The only model that has spontaneous strokes that would allow for the analysis of stroke risk is the stroke prone spontaneously hypertensive rat (SHRSP), but these rats are not obese and they have malignant hypertension. The SHRSP fatty (fa/fa) rat [95] could potentially provide a model of stroke risk, but at present there are no studies supporting this idea. The studies in the literature relating to stroke and obesity in rodents have therefore focused on stroke severity.
Earlier we described our own finding of inward MCA remodeling in HF-fed rats [53]. In the same study we assed the outcome of permanent focal cerebral ischemia. We found that after 24 hours of ischemia the HF-fed rats had significantly larger infarcts than the control rats [53]. The infarct size in permanent ischemia is modulated by the ability of the collateral arteries around the infarct to dilate and increase perfusion to the ischemic tissue [96–98]. Therefore, the increased ischemic damage observed in the HF-fed rats suggests that obesity may impair the ability of the collateral arteries to dilate in response to ischemia.
When cerebral ischemia is induced in OZR only adult hypertensive rats have an increase in the area damaged by the stroke. Young normotensive OZR do not differ in infarct size from lean LZR [27]. In these rats MCA remodeling was only present in the older hypertensive OZR. The authors studied the effects of blood pressure lowering on the outcome of ischemia in the OZR. Reducing the blood pressure normalized the damage caused by cerebral ischemia. The authors attribute this to an effect of chronic blood pressure lowering, because the acute post-stroke increases in blood pressure were similar in all the groups studied. These studies used a model of ischemia with reperfusion injury. The pathogenesis of this model is different from permanent ischemia models in that oxidative stress and reactive oxygen species generation contribute significantly to the damage observed. Interestingly the glycemic response to ischemia was different in the OZR compared to the LZR. The obese rats were hyperglycemic before the induction of ischemia, the authors state that this is not normal for this age group and is likely an effect of anesthesia. This presents the possibility that the OZR have an enhanced hyperglycemic response to anesthesia compared to the LZR. The hyperglycemia was maintained and increased in magnitude during the ischemic period and on into reperfusion. The hydrochlorothiazide treatment did not affect this response suggesting that blood pressure is an independent determinant of infarct size in the OZR [54].
Similar increases in ischemic injury have been observed in obese ob/ob mice [94, 99]. Ob/ob mice exhibit an inflammatory and prothrombotic phenotype post-stroke. This effect occurs independently of hypertension, hyperglycemia and hypercholesterolemia and may exacerbate the damage caused by ischemia. After 30 minutes of ischemia followed by 4 hours of reperfusion Terao et al reported an increase in the number of leukocytes and platelets adhering in the cerebral venules. The authors also observed increased BBB breakdown in the ob/ob mice 24 hours after the induction of ischemia; the increased edema associated with this could exacerbate the ischemic insult. Circulating monocyte chemoattractant protein-1 (MCP-1) and interleukin-6 were increased in the plasma from ob/ob mice after ischemia. Blocking antibodies to MCP-1, but not interleukin-6, reduced the damage caused by the stroke [99]. These studies present the possibility that MCP-1 could be a therapeutic target for stroke.
Other studies using the ob/ob mice have focused on statins as potential stroke therapies. Rosuvastatin reduced the damage caused by 60 minutes of MCAO followed by 24 hours of reperfusion in ob/ob mice. The duration of the statin therapy was not sufficient to lower cholesterol suggesting the beneficial effect is a pleiotropic effect of the statin. From this study the effect does not appear to be related to an increase in cerebral blood flow. Interestingly, there was more hemorrhagic transformation in the ob/ob mice and this was not improved by statin therapies and was not related to the infarct size [100]. The reason for the increased hemorrhagic transformation is not clear but it may relate to changes in the vasculature post-stroke. Occluded MCAs loose their ability to generate tone and to regulate blood flow [101, 102]. If the loss of tone is faster or more pronounced in the ob/ob mice it could increase hemorrhage at reperfusion. Other studies of ob/ob mice have shown increased hemorrhagic transformation after 40 minutes of ischemia followed by reperfusion. In this case the authors attribute the increased hemorrhage and BBB breakdown to increased expression and activation of MMP-9 in the brain of the ob/ob mice [103].
5. THE EFFECTS OF OBESITY ON DEMENTIA AND COGNITIVE DECLINE
5.1. Human Studies
The structural and functional changes observed in cerebral arteries with obesity have the potential to increase cerebral vascular resistance and reduce cerebral blood flow. Cerebral blood flow is reduced in obese patients [1–3], and this could lead to dementia and cognitive decline [104]. Mild cerebral hypoperfusion impairs learning and memory by reducing protein synthesis and synaptic plasticity [105]. Moderate to severe hypoperfusion reduces ATP synthesis and impairs neuronal action potential generation [106]. Impaired cerebral blood flow also causes edema formation and white matter lesions. It can also cause accumulation of proteins in the brain, including β-amyloid and phosphorylated tau. These proteins are associated with Alzheimer’s disease, which is the leading cause of dementia [107]. Vascular injury is the second most common cause of dementia and vascular cognitive impairments can range from mild cognitive impairment to severe vascular dementia [108].
The evidence linking obesity to dementia and impaired cognitive function has been recently described in a review [109] and a meta-analysis [110]. The later reports a U shaped association between BMI and dementia, with both underweight and overweight individuals being at risk. The risk of obesity-associated dementia was higher in women than in men [110]. The associations between obesity and dementia risk are strongest when the incidence of obesity at midlife (40–55 years) was compared with dementia [104, 111–115]. The effects of BMI and cognitive function in the elderly have been more difficult to define; this may be because BMI may not be a good measure of obesity in elderly patients. Lean body mass falls with age [116], and this can cause misleading associations between weight and dementia. However, a recent study showed that obese and overweight elderly individuals have a worse performance in neuropsychological tests than their lean counterparts. The results of this study were adjusted for several confounding factors including waist circumference, and medications that have known CNS effects [117].
The link between obesity and dementia has been suggested to occur independently of co-morbid conditions such as hypertension and diabetes [104, 111]. However, studies suggest that hypertension and hypercholesterolemia might exacerbate the risk of dementia and Alzheimer’s disease [104]. Other studies also suggest that obesity and hypertension have cumulative effects on cognitive decline. Surprisingly, this study showed that the effects of hypertension and obesity on learning and memory were only present in men [118]. This finding is at odds with other studies including those in the meta-analysis described above [110] which indicate that women are at an increased risk of dementia.
More recent studies have focused on analysis of brain volume to better define the association between obesity and dementia. Reduced total brain and hippocampal volumes, and increased white matter hyperintensities and infarcts are important predictors of dementia [119–122]. A recent study attempted to define the effects of visceral and subcutaneous adipose tissue on these measures of brain aging. BMI, waist circumference, visceral and subcutaneous adipose tissue mass were inversely associated with total brain volume. These associations all occurred independently of other vascular risk factors. The association between brain volume and visceral adipose tissue was the strongest, suggesting that abdominal obesity may be the most important predictor of brain atrophy [123]. Abdominal obesity and visceral fat are better predictors of vascular risk than total body fat [81, 114]. However, the authors failed to show a link between any measure of body fat and white matter hyperintensities or infarcts suggesting that, at least in this population at this time point, obesity and cerebrovascular disease are not associated [123]. The authors propose that the link between obesity and total brain volume is partly due to the increased inflammation associated with obesity, but they also recognize the potential involvement of diabetes and IR in the cognitive decline.
5.2. Rodent Studies of Dementia
From the work of Winocur and Greenwood there is little doubt that feeding rats a HF diet causes a decline in their cognitive function, this appears to be true for both high saturated and unsaturated fat diets [124, 125]. In a study from McNeilly et al [126], rats with adult onset obesity showed cognitive impairment in tests that identified cortical dysfunction, but there was no association between obesity and impaired hippocampal function. Interestingly, a recent study in mice suggests that obesity may only produce deficits in hippocampal function if it begins early in life [127]. Given the increasing prevalence of juvenile obesity in the human population [128] it would seem important that we endeavor to understand the differences in the development of obesity associated dementia with age.
Recent studies have attempted to identify the mechanisms responsible for obesity-associated cognitive impairment. We will focus on the mechanisms that have a potential vascular component; however, it is important to note that none of the vascular effects proposed here have been tested directly. Recent studies suggest that the IR associated with obesity increases the cognitive deficits observed in obese rats [126]. As described above insulin is a dilator in cerebral arteries [39], it would be interesting to investigate if the IR observed in rats fed a HF diet changes cerebral blood flow or functional hyperemia. Other studies suggest inflammation is a potential mediator of the obesity induced cognitive decline. Cognitively impaired obese mice have elevated levels of inflammatory markers including TNF-alpha, interleukin-6 and MCP-1. These mice also exhibit increased neuroinflammation as evidenced by increased expression of astrocyte and microglia specific markers [129]. Several studies have linked inflammation to cardiovascular diseases [130].
Male rats consuming a HF diet exhibit reduced hippocampal neurogenesis. These rats also have increased serum corticosterone levels and previous studies suggest this could be responsible for inhibiting hippocampal neurogenesis [131, 132]. The results observed in the female rats were quiet different, it appears that control female rats have lower neurogenesis than control males, and the HF diet in the females caused an insignificant reduction in neurogenesis [133]. This suggests there could be significant sex differences in the mechanisms through which obesity induces dementia. An increase in circulating corticosterone could also reduce cerebral blood flow because corticosteroids impair the function of eNOS by inhibiting tetrahydrobioptern production [134].
6. CONCLUSIONS
There appears to be little doubt that obesity impairs cerebral artery function in a manner that could reduce cerebral blood flow. The most clearly defined impairment is the reduction in NO mediated dilation, but insulin signaling and K+ channel function are also impaired in obese rats. When one links the impaired dilation with the inward remodeling process which reduces the lumen diameter of the cerebral arteries, the potential for chronic hypoperfusion of the brain becomes clear. Although we have made great strides in understanding the effect of obesity on the cerebral vasculature in recent years, there are still several issues that need to be given consideration. The first is how best to separate the effects of prolonged obesity from the increase in blood pressure that is invariably associated with it. We also must begin to consider other segments of the cerebral vascular tree. The published studies have focused on the larger cerebral arteries and limited attention has been paid to the penetrating and parenchymal arterioles. The function of these arteries may be particularly important for studies of dementia in obese rats. We also need to better consider the population we are attempting to model; this will require that we study older animals and that we consider both sexes.
Acknowledgments
This study was funded by the American Heart Association, PWP is a recipient of an AHA pre-doctoral fellowship and AMD has received an AHA Established Investigator Award (0840122N). The authors thank Daniel Bollman for producing the image of the brain and cerebral arteries in (Fig. 1).
ABBREVIATIONS
- BBB
Blood brain barrier
- BMI
Body mass index
- COX
Cyclooxygenase
- EDHF
Endothelium derived hyperpolarizing factor
- eNOS
Endothelial nitric oxide synthase
- HF
High fat
- IR
Insulin resistance
- L-NAME
L-NG-nitroarginine methyl ester
- LZR
Lean Zucker rat
- MCA
Middle cerebral artery
- MCP-1
Monocyte chemoattractant 1
- NO
Nitric oxide
- OZR
Obese Zucker rat
- SOD
Superoxide dismutase
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
The author contributions are as follows: Anne Dorrance, conceived the review plan and wrote and edited the manuscript. Paulo Pires and Nusrat Matin both wrote significant sections of the manuscript.
References
- 1.Alosco ML, Spitznagel MB, Raz N, et al. Obesity interacts with cerebral hypoperfusion to exacerbate cognitive impairment in older adults with heart failure. Cerebrovascular diseases extra. 2012;2(1):88–98. doi: 10.1159/000343222. Epub 2012/12/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Willeumier KC, Taylor DV, Amen DG. Elevated BMI is associated with decreased blood flow in the prefrontal cortex using SPECT imaging in healthy adults. Obesity (Silver Spring) 2011;19(5):1095–7. doi: 10.1038/oby.2011.16. Epub 2011/02/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selim M, Jones R, Novak P, Zhao P, Novak V. The effects of body mass index on cerebral blood flow velocity. Clin Auton Res. 2008;18(6):331–8. doi: 10.1007/s10286-008-0490-z. Epub 2008/08/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cipolla MJ. The Cerebral Circulation. San Rafael (CA): 2009. [PubMed] [Google Scholar]
- 5.Moody DM, Bell MA, Challa VR. Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: an anatomic study. AJNR American journal of neuroradiology. 1990;11(3):431–9. Epub 1990/05/01. [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura N, Schaffer CB, Friedman B, Lyden PD, Kleinfeld D. Penetrating arterioles are a bottleneck in the perfusion of neocortex. Proc Natl Acad Sci U S A. 2007;104(1):365–70. doi: 10.1073/pnas.0609551104. Epub 2006/12/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen Z, Bonvento G, Lacombe P, Hamel E. Serotonin in the regulation of brain microcirculation. Progress in neurobiology. 1996;50(4):335–62. doi: 10.1016/s0301-0082(96)00033-0. Epub 1996/11/01. [DOI] [PubMed] [Google Scholar]
- 8.Hamel E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol. 2006;100(3):1059–64. doi: 10.1152/japplphysiol.00954.2005. Epub 2006/02/10. [DOI] [PubMed] [Google Scholar]
- 9.Dirnagl U. Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci. 2012;1268:21–5. doi: 10.1111/j.1749-6632.2012.06691.x. Epub 2012/09/22. [DOI] [PubMed] [Google Scholar]
- 10.Nag S, Kapadia A, Stewart DJ. Review: molecular pathogenesis of blood-brain barrier breakdown in acute brain injury. Neuropathol Appl Neurobiol. 2011;37(1):3–23. doi: 10.1111/j.1365-2990.2010.01138.x. Epub 2010/10/16. [DOI] [PubMed] [Google Scholar]
- 11.Faraci FM. Protecting against vascular disease in brain. Am J Physiol Heart Circ Physiol. 2011;300(5):H1566–82. doi: 10.1152/ajpheart.01310.2010. Epub 2011/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Faraci FM, Heistad DD. Regulation of large cerebral arteries and cerebral microvascular pressure. Circ Res. 1990;66(1):8–17. doi: 10.1161/01.res.66.1.8. [DOI] [PubMed] [Google Scholar]
- 13.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. Epub 2009/08/12. [DOI] [PubMed] [Google Scholar]
- 14.Szasz T, Webb RC. Perivascular adipose tissue: more than just structural support. Clin Sci (Lond) 2012;122(1):1–12. doi: 10.1042/CS20110151. Epub 2011/09/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aghamohammadzadeh R, Heagerty AM. Obesity-related hypertension: epidemiology, pathophysiology, treatments, and the contribution of perivascular adipose tissue. Annals of medicine. 2012;44(Suppl 1):S74–84. doi: 10.3109/07853890.2012.663928. Epub 2012/06/22. [DOI] [PubMed] [Google Scholar]
- 16.Gu P, Xu A. Interplay between adipose tissue and blood vessels in obesity and vascular dysfunction. Reviews in endocrine & metabolic disorders. 2013 doi: 10.1007/s11154-012-9230-8. Epub 2013/01/04. [DOI] [PubMed] [Google Scholar]
- 17.Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation. Cerebrovascular and brain metabolism reviews. 1990;2(2):161–92. Epub 1990/01/01. [PubMed] [Google Scholar]
- 18.Talman WT, Nitschke Dragon D. Neuronal nitric oxide mediates cerebral vasodilatation during acute hypertension. Brain Res. 2007;1139:126–32. doi: 10.1016/j.brainres.2007.01.008. Epub 2007/02/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duchemin S, Boily M, Sadekova N, Girouard H. The complex contribution of NOS interneurons in the physiology of cerebrovascular regulation. Frontiers in neural circuits. 2012;6:51. doi: 10.3389/fncir.2012.00051. Epub 2012/08/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamner JW, Tan CO, Lee K, Cohen MA, Taylor JA. Sympathetic control of the cerebral vasculature in humans. Stroke. 2010;41(1):102–9. doi: 10.1161/STROKEAHA.109.557132. Epub 2009/12/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hamner JW, Tan CO, Tzeng YC, Taylor JA. Cholinergic control of the cerebral vasculature in humans. J Physiol. 2012;590(Pt 24):6343–52. doi: 10.1113/jphysiol.2012.245100. Epub 2012/10/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koller A, Toth P. Contribution of flow-dependent vasomotor mechanisms to the autoregulation of cerebral blood flow. J Vasc Res. 2012;49(5):375–89. doi: 10.1159/000338747. Epub 2012/06/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faraci FM, Baumbach GL, Heistad DD. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl. 1989;7(4):S61–4. discussion S5. Epub 1989/09/01. [PubMed] [Google Scholar]
- 24.Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol. 2002;283(6):H2260–7. doi: 10.1152/ajpheart.00634.2002. Epub 2002/10/22. [DOI] [PubMed] [Google Scholar]
- 25.Geary GG, Krause DN, Duckles SP. Estrogen reduces mouse cerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2000;279(2):H511–9. doi: 10.1152/ajpheart.2000.279.2.H511. Epub 2000/08/03. [DOI] [PubMed] [Google Scholar]
- 26.Cipolla MJ, Porter JM, Osol G. High glucose concentrations dilate cerebral arteries and diminish myogenic tone through an endothelial mechanism. Stroke. 1997;28(2):405–10. doi: 10.1161/01.str.28.2.405. discussion 10–1. Epub 1997/02/01. [DOI] [PubMed] [Google Scholar]
- 27.Osmond JM, Mintz JD, Dalton B, Stepp DW. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension. 2009;53(2):381–6. doi: 10.1161/HYPERTENSIONAHA.108.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Katakam PV, Domoki F, Snipes JA, Busija AR, Jarajapu YP, Busija DW. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2009;296(2):R289–98. doi: 10.1152/ajpregu.90656.2008. Epub 2008/11/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howitt L, Sandow SL, Grayson TH, Ellis ZE, Morris MJ, Murphy TV. Differential effects of diet-induced obesity on BKCa {beta}1-subunit expression and function in rat skeletal muscle arterioles and small cerebral arteries. Am J Physiol Heart Circ Physiol. 2011;301(1):H29–40. doi: 10.1152/ajpheart.00134.2011. Epub 2011/05/04. [DOI] [PubMed] [Google Scholar]
- 30.You J, Johnson TD, Marrelli SP, Bryan RM., Jr Functional heterogeneity of endothelial P2 purinoceptors in the cerebrovascular tree of the rat. Am J Physiol. 1999;277(3 Pt 2):H893–900. doi: 10.1152/ajpheart.1999.277.3.H893. Epub 1999/09/14. [DOI] [PubMed] [Google Scholar]
- 31.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol. 2005;288(2):R522–30. doi: 10.1152/ajpregu.00655.2004. Epub 2004/10/30. [DOI] [PubMed] [Google Scholar]
- 32.Schwaninger RM, Sun H, Mayhan WG. Impaired nitric oxide synthase-dependent dilatation of cerebral arterioles in type II diabetic rats. Life Sci. 2003;73(26):3415–25. doi: 10.1016/j.lfs.2003.06.029. Epub 2003/10/24. [DOI] [PubMed] [Google Scholar]
- 33.Erdos B, Snipes JA, Miller AW, Busija DW. Cerebrovascular dysfunction in Zucker obese rats is mediated by oxidative stress and protein kinase C. Diabetes. 2004;53(5):1352–9. doi: 10.2337/diabetes.53.5.1352. Epub 2004/04/28. [DOI] [PubMed] [Google Scholar]
- 34.Drouin A, Thorin-Trescases N, Hamel E, Falck JR, Thorin E. Endothelial nitric oxide synthase activation leads to dilatory H2O2 production in mouse cerebral arteries. Cardiovasc Res. 2007;73(1):73–81. doi: 10.1016/j.cardiores.2006.10.005. Epub 2006/11/23. [DOI] [PubMed] [Google Scholar]
- 35.Karagiannis J, Reid JJ, Darby I, Roche P, Rand MJ, Li CG. Impaired nitric oxide function in the basilar artery of the obese Zucker rat. J Cardiovasc Pharmacol. 2003;42(4):497–505. doi: 10.1097/00005344-200310000-00007. Epub 2003/09/26. [DOI] [PubMed] [Google Scholar]
- 36.Erdos B, Snipes JA, Tulbert CD, Katakam P, Miller AW, Busija DW. Rosuvastatin improves cerebrovascular function in Zucker obese rats by inhibiting NAD(P)H oxidase-dependent superoxide production. Am J Physiol Heart Circ Physiol. 2006;290(3):H1264–70. doi: 10.1152/ajpheart.00804.2005. Epub 2005/11/15. [DOI] [PubMed] [Google Scholar]
- 37.Katakam PV, Wappler EA, Katz PS, et al. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol. 2013;33(4):752–9. doi: 10.1161/ATVBAHA.112.300560. Epub 2013/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katakam PV, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;292(2):R920–6. doi: 10.1152/ajpregu.00520.2006. Epub 2006/09/30. [DOI] [PubMed] [Google Scholar]
- 39.Katakam PV, Domoki F, Lenti L, et al. Cerebrovascular responses to insulin in rats. J Cereb Blood Flow Metab. 2009;29(12):1955–67. doi: 10.1038/jcbfm.2009.177. Epub 2009/09/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen AF, Chen DD, Daiber A, et al. Free radical biology of the cardiovascular system. Clin Sci (Lond) 2012;123(2):73–91. doi: 10.1042/CS20110562. Epub 2012/03/30. [DOI] [PubMed] [Google Scholar]
- 41.Katakam PV, Snipes JA, Steed MM, Busija DW. Insulin-induced generation of reactive oxygen species and uncoupling of nitric oxide synthase underlie the cerebrovascular insulin resistance in obese rats. J Cereb Blood Flow Metab. 2012;32(5):792–804. doi: 10.1038/jcbfm.2011.181. Epub 2012/01/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorlach C, Wahl M. Bradykinin dilates rat middle cerebral artery and its large branches via endothelial B2 receptors and release of nitric oxide. Peptides. 1996;17(8):1373–8. doi: 10.1016/s0196-9781(96)00223-9. [DOI] [PubMed] [Google Scholar]
- 43.Kontos HA, Wei EP, Kukreja RC, Ellis EF, Hess ML. Differences in endothelium-dependent cerebral dilation by bradykinin and acetylcholine. Am J Physiol. 1990;258(5 Pt 2):H1261–6. doi: 10.1152/ajpheart.1990.258.5.H1261. Epub 1990/05/01. [DOI] [PubMed] [Google Scholar]
- 44.Erdos B, Miller AW, Busija DW. Impaired endothelium-mediated relaxation in isolated cerebral arteries from insulin-resistant rats. Am J Physiol Heart Circ Physiol. 2002;282(6):H2060–5. doi: 10.1152/ajpheart.01124.2001. Epub 2002/05/11. [DOI] [PubMed] [Google Scholar]
- 45.Erdos B, Miller AW, Busija DW. Alterations in KATP and KCa channel function in cerebral arteries of insulin-resistant rats. Am J Physiol Heart Circ Physiol. 2002;283(6):H2472–7. doi: 10.1152/ajpheart.00516.2002. Epub 2002/10/22. [DOI] [PubMed] [Google Scholar]
- 46.Feihl F, Liaudet L, Levy BI, Waeber B. Hypertension and microvascular remodelling. Cardiovasc Res. 2008;78(2):274–85. doi: 10.1093/cvr/cvn022. [DOI] [PubMed] [Google Scholar]
- 47.Mulvany MJ. Small artery remodelling in hypertension: causes, consequences and therapeutic implications. Med Biol Eng Comput. 2008;46(5):461–7. doi: 10.1007/s11517-008-0305-3. [DOI] [PubMed] [Google Scholar]
- 48.Mulvany MJ. Small artery remodelling in hypertension. Basic & clinical pharmacology & toxicology. 2012;110(1):49–55. doi: 10.1111/j.1742-7843.2011.00758.x. Epub 2011/07/08. [DOI] [PubMed] [Google Scholar]
- 49.Heagerty AM, Aalkjaer C, Bund SJ, Korsgaard N, Mulvany MJ. Small artery structure in hypertension. Dual processes of remodeling and growth. Hypertension. 1993;21(4):391–7. doi: 10.1161/01.hyp.21.4.391. [DOI] [PubMed] [Google Scholar]
- 50.Mulvany MJ, Baumbach GL, Aalkjaer C, et al. Vascular remodeling. Hypertension. 1996;28(3):505–6. [PubMed] [Google Scholar]
- 51.Baumbach GL, Heistad DD. Remodeling of cerebral arterioles in chronic hypertension. Hypertension. 1989;13(6 Pt 2):968–72. doi: 10.1161/01.hyp.13.6.968. [DOI] [PubMed] [Google Scholar]
- 52.Smith AD, Brands MW, Wang MH, Dorrance AM. Obesity-induced hypertension develops in young rats independently of the renin-angiotensin-aldosterone system. Exp Biol Med (Maywood) 2006;231(3):282–7. doi: 10.1177/153537020623100307. [DOI] [PubMed] [Google Scholar]
- 53.Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res. 2009;78(1):100–6. doi: 10.1016/j.mvr.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osmond JM, Mintz JD, Stepp DW. Preventing increased blood pressure in the obese Zucker rat improves severity of stroke. Am J Physiol Heart Circ Physiol. 2010;299(1):H55–61. doi: 10.1152/ajpheart.01111.2009. Epub 2010/04/27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stepp DW, Pollock DM, Frisbee JC. Low-flow vascular remodeling in the metabolic syndrome X. Am J Physiol Heart Circ Physiol. 2004;286(3):H964–70. doi: 10.1152/ajpheart.00836.2003. [DOI] [PubMed] [Google Scholar]
- 56.Hajdu MA, Heistad DD, Baumbach GL. Effects of antihypertensive therapy on mechanics of cerebral arterioles in rats. Hypertension. 1991;17(3):308–16. doi: 10.1161/01.hyp.17.3.308. [DOI] [PubMed] [Google Scholar]
- 57.Clozel JP, Kuhn H, Hefti F. Effects of cilazapril on the cerebral circulation in spontaneously hypertensive rats. Hypertension. 1989;14(6):645–51. doi: 10.1161/01.hyp.14.6.645. Epub 1989/12/01. [DOI] [PubMed] [Google Scholar]
- 58.Chillon JM, Baumbach GL. Effects of an angiotensin-converting enzyme inhibitor and a beta-blocker on cerebral arterioles in rats. Hypertension. 1999;33(3):856–61. doi: 10.1161/01.hyp.33.3.856. [DOI] [PubMed] [Google Scholar]
- 59.Dupuis F, Atkinson J, Liminana P, Chillon JM. Captopril improves cerebrovascular structure and function in old hypertensive rats. Br J Pharmacol. 2005;144(3):349–56. doi: 10.1038/sj.bjp.0706001. Epub 2005/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rigsby CS, Ergul A, Portik Dobos V, Pollock DM, Dorrance AM. Effects of spironolactone on cerebral vessel structure in rats with sustained hypertension. Am J Hypertens. 2011;24(6):708–15. doi: 10.1038/ajh.2011.20. Epub 2011/02/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rigsby CS, Pollock DM, Dorrance AM. Spironolactone improves structure and increases tone in the cerebral vasculature of male spontaneously hypertensive stroke-prone rats. Microvasc Res. 2007;73(3):198–205. doi: 10.1016/j.mvr.2006.12.001. Epub 2007/01/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dorrance AM, Rupp NC, Nogueira EF. Mineralocorticoid receptor activation causes cerebral vessel remodeling and exacerbates the damage caused by cerebral ischemia. Hypertension. 2006;47(3):590–5. doi: 10.1161/01.HYP.0000196945.73586.0d. [DOI] [PubMed] [Google Scholar]
- 63.Whaley-Connell A, Johnson MS, Sowers JR. Aldosterone: role in the cardiometabolic syndrome and resistant hypertension. Prog Cardiovasc Dis. 2010;52(5):401–9. doi: 10.1016/j.pcad.2009.12.004. Epub 2010/03/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Briones AM, Nguyen Dinh Cat A, Callera GE, et al. Adipocytes produce aldosterone through calcineurin-dependent signaling pathways: implications in diabetes mellitus-associated obesity and vascular dysfunction. Hypertension. 2012;59(5):1069–78. doi: 10.1161/HYPERTENSIONAHA.111.190223. Epub 2012/04/12. [DOI] [PubMed] [Google Scholar]
- 65.MacKenzie SM, Huda SS, Sattar N, Fraser R, Connell JM, Davies E. Depot-specific steroidogenic gene transcription in human adipose tissue. Clin Endocrinol (Oxf) 2008;69(6):848–54. doi: 10.1111/j.1365-2265.2008.03262.x. [DOI] [PubMed] [Google Scholar]
- 66.Northcott CA, Fink GD, Garver H, et al. The Development of Hypertension and Hyperaldosteronism in a Rodent Model of Life-Long Obesity. Endocrinology. 2012 doi: 10.1210/en.2011-1176. Epub 2012/02/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sokolova IA, Manukhina EB, Blinkov SM, Koshelev VB, Pinelis VG, Rodionov IM. Rarefication of the arterioles and capillary network in the brain of rats with different forms of hypertension. Microvasc Res. 1985;30(1):1–9. doi: 10.1016/0026-2862(85)90032-9. Epub 1985/07/01. [DOI] [PubMed] [Google Scholar]
- 68.St-Pierre P, Bouffard L, Papirakis ME, Maheux P. Increased extravasation of macromolecules in skeletal muscles of the Zucker rat model. Obesity (Silver Spring) 2006;14(5):787–93. doi: 10.1038/oby.2006.91. Epub 2006/07/21. [DOI] [PubMed] [Google Scholar]
- 69.Dell’Omo G, Penno G, Pucci L, Mariani M, Del Prato S, Pedrinelli R. Abnormal capillary permeability and endothelial dysfunction in hypertension with comorbid Metabolic Syndrome. Atherosclerosis. 2004;172(2):383–9. doi: 10.1016/j.atherosclerosis.2003.11.013. Epub 2004/03/17. [DOI] [PubMed] [Google Scholar]
- 70.Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL. Measuring the Global Burden of Disease and Risk Factors, 1990–2001. In: Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL, editors. Global Burden of Disease and Risk Factors. Washington (DC): 2006. [Google Scholar]
- 71.Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008;371(9624):1612–23. doi: 10.1016/S0140-6736(08)60694-7. Epub 2008/05/13. [DOI] [PubMed] [Google Scholar]
- 72.Warlow C, Sudlow C, Dennis M, Wardlaw J, Sandercock P. Stroke. Lancet. 2003;362(9391):1211–24. doi: 10.1016/S0140-6736(03)14544-8. Epub 2003/10/22. [DOI] [PubMed] [Google Scholar]
- 73.Krafft PR, Bailey EL, Lekic T, et al. Etiology of stroke and choice of models. Int J Stroke. 2012;7(5):398–406. doi: 10.1111/j.1747-4949.2012.00838.x. Epub 2012/06/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howells DW, Porritt MJ, Rewell SS, et al. Different strokes for different folks: the rich diversity of animal models of focal cerebral ischemia. J Cereb Blood Flow Metab. 2010;30(8):1412–31. doi: 10.1038/jcbfm.2010.66. Epub 2010/05/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Strazzullo P, D’Elia L, Cairella G, Garbagnati F, Cappuccio FP, Scalfi L. Excess body weight and incidence of stroke: meta-analysis of prospective studies with 2 million participants. Stroke. 2010;41(5):e418–26. doi: 10.1161/STROKEAHA.109.576967. Epub 2010/03/20. [DOI] [PubMed] [Google Scholar]
- 76.Kurth T, Gaziano JM, Berger K, et al. Body mass index and the risk of stroke in men. Arch Intern Med. 2002;162(22):2557–62. doi: 10.1001/archinte.162.22.2557. Epub 2002/12/03. [DOI] [PubMed] [Google Scholar]
- 77.Mottillo S, Filion KB, Genest J, et al. The metabolic syndrome and cardiovascular risk a systematic review and meta-analysis. J Am Coll Cardiol. 2010;56(14):1113–32. doi: 10.1016/j.jacc.2010.05.034. Epub 2010/09/25. [DOI] [PubMed] [Google Scholar]
- 78.Lindenstrom E, Boysen G, Nyboe J. Lifestyle factors and risk of cerebrovascular disease in women. The Copenhagen City Heart Study. Stroke. 1993;24(10):1468–72. doi: 10.1161/01.str.24.10.1468. Epub 1993/10/01. [DOI] [PubMed] [Google Scholar]
- 79.Lapidus L, Bengtsson C, Larsson B, Pennert K, Rybo E, Sjostrom L. Distribution of adipose tissue and risk of cardiovascular disease and death: a 12 year follow up of participants in the population study of women in Gothenburg, Sweden. Br Med J (Clin Res Ed) 1984;289(6454):1257–61. doi: 10.1136/bmj.289.6454.1257. Epub 1984/11/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rheaume C, Leblanc ME, Poirier P. Adiposity assessment: explaining the association between obesity, hypertension and stroke. Expert review of cardiovascular therapy. 2011;9(12):1557–64. doi: 10.1586/erc.11.167. Epub 2011/11/23. [DOI] [PubMed] [Google Scholar]
- 81.Despres JP. Abdominal obesity and cardiovascular disease: is inflammation the missing link? The Canadian journal of cardiology. 2012;28(6):642–52. doi: 10.1016/j.cjca.2012.06.004. Epub 2012/08/15. [DOI] [PubMed] [Google Scholar]
- 82.Wieberdink RG, Koudstaal PJ, Hofman A, Witteman JC, Breteler MM, Ikram MA. Insulin resistance and the risk of stroke and stroke subtypes in the nondiabetic elderly. American journal of epidemiology. 2012;176(8):699–707. doi: 10.1093/aje/kws149. Epub 2012/10/05. [DOI] [PubMed] [Google Scholar]
- 83.Razinia T, Saver JL, Liebeskind DS, Ali LK, Buck B, Ovbiagele B. Body mass index and hospital discharge outcomes after ischemic stroke. Arch Neurol. 2007;64(3):388–91. doi: 10.1001/archneur.64.3.388. [DOI] [PubMed] [Google Scholar]
- 84.Towfighi A, Ovbiagele B. The impact of body mass index on mortality after stroke. Stroke. 2009;40(8):2704–8. doi: 10.1161/STROKEAHA.109.550228. [DOI] [PubMed] [Google Scholar]
- 85.Vemmos K, Ntaios G, Spengos K, et al. Association between obesity and mortality after acute first-ever stroke: the obesity-stroke paradox. Stroke. 2011;42(1):30–6. doi: 10.1161/STROKEAHA.110.593434. Epub 2010/12/04. [DOI] [PubMed] [Google Scholar]
- 86.Kim BJ, Lee SH, Jung KH, Yu KH, Lee BC, Roh JK. Dynamics of obesity paradox after stroke, related to time from onset, age, and causes of death. Neurology. 2012;79(9):856–63. doi: 10.1212/WNL.0b013e318266fad1. Epub 2012/08/17. [DOI] [PubMed] [Google Scholar]
- 87.Ovbiagele B, Bath PM, Cotton D, Vinisko R, Diener HC. Obesity and recurrent vascular risk after a recent ischemic stroke. Stroke. 2011;42(12):3397–402. doi: 10.1161/STROKEAHA.111.624957. Epub 2011/10/01. [DOI] [PubMed] [Google Scholar]
- 88.Olsen TS, Dehlendorff C, Petersen HG, Andersen KK. Body mass index and poststroke mortality. Neuroepidemiology. 2008;30(2):93–100. doi: 10.1159/000118945. Epub 2008/03/01. [DOI] [PubMed] [Google Scholar]
- 89.Doehner W, Schenkel J, Anker SD, Springer J, Audebert HJ. Overweight and obesity are associated with improved survival, functional outcome, and stroke recurrence after acute stroke or transient ischaemic attack: observations from the TEMPiS trial. Eur Heart J. 2013;34(4):268–77. doi: 10.1093/eurheartj/ehs340. Epub 2012/10/19. [DOI] [PubMed] [Google Scholar]
- 90.Scherbakov N, Dirnagl U, Doehner W. Body weight after stroke: lessons from the obesity paradox. Stroke. 2011;42(12):3646–50. doi: 10.1161/STROKEAHA.111.619163. Epub 2011/10/01. [DOI] [PubMed] [Google Scholar]
- 91.Katsnelson M, Rundek T. Obesity paradox and stroke: noticing the (fat) man behind the curtain. Stroke. 2011;42(12):3331–2. doi: 10.1161/STROKEAHA.111.632471. Epub 2011/10/01. [DOI] [PubMed] [Google Scholar]
- 92.Deguchi I, Ohe Y, Fukuoka T, et al. Relationship of obesity to recanalization after hyperacute recombinant tissue-plasminogen activator infusion therapy in patients with middle cerebral artery occlusion. J Stroke Cerebrovasc Dis. 2012;21(3):161–4. doi: 10.1016/j.jstrokecerebrovasdis.2011.11.003. Epub 2012/01/31. [DOI] [PubMed] [Google Scholar]
- 93.Abe K. An emerging topic on obesity, arterial endothelial function and thrombolysis. J Stroke Cerebrovasc Dis. 2012;21(3):159–60. doi: 10.1016/j.jstrokecerebrovasdis.2012.02.010. Epub 2012/03/24. [DOI] [PubMed] [Google Scholar]
- 94.Nagai N, Van Hoef B, Lijnen HR. Plasminogen activator inhibitor-1 contributes to the deleterious effect of obesity on the outcome of thrombotic ischemic stroke in mice. Journal of thrombosis and haemostasis: JTH. 2007;5(8):1726–31. doi: 10.1111/j.1538-7836.2007.02631.x. Epub 2007/06/29. [DOI] [PubMed] [Google Scholar]
- 95.Hiraoka-Yamamoto J, Nara Y, Yasui N, Onobayashi Y, Tsuchikura S, Ikeda K. Establishment of a new animal model of metabolic syndrome: SHRSP fatty (fa/fa) rats. Clin Exp Pharmacol Physiol. 2004;31(1–2):107–9. doi: 10.1111/j.1440-1681.2004.03962.x. Epub 2004/02/06. [DOI] [PubMed] [Google Scholar]
- 96.Coyle P, Heistad DD. Blood flow through cerebral collateral vessels in hypertensive and normotensive rats. Hypertension. 1986;8(6 Pt 2):II67–71. doi: 10.1161/01.hyp.8.6_pt_2.ii67. Epub 1986/06/01. [DOI] [PubMed] [Google Scholar]
- 97.Coyle P, Heistad DD. Blood flow through cerebral collateral vessels one month after middle cerebral artery occlusion. Stroke. 1987;18(2):407–11. doi: 10.1161/01.str.18.2.407. [DOI] [PubMed] [Google Scholar]
- 98.Heistad DD, Mayhan WG, Coyle P, Baumbach GL. Impaired dilatation of cerebral arterioles in chronic hypertension. Blood Vessels. 1990;27(2–5):258–62. doi: 10.1159/000158817. [DOI] [PubMed] [Google Scholar]
- 99.Terao S, Yilmaz G, Stokes KY, Ishikawa M, Kawase T, Granger DN. Inflammatory and injury responses to ischemic stroke in obese mice. Stroke. 2008;39(3):943–50. doi: 10.1161/STROKEAHA.107.494542. [DOI] [PubMed] [Google Scholar]
- 100.Mayanagi K, Katakam PV, Gaspar T, Domoki F, Busija DW. Acute treatment with rosuvastatin protects insulin resistant (C57BL/6J ob/ob) mice against transient cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(12):1927–35. doi: 10.1038/jcbfm.2008.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cipolla MJ, Curry AB. Middle cerebral artery function after stroke: the threshold duration of reperfusion for myogenic activity. Stroke. 2002;33(8):2094–9. doi: 10.1161/01.str.0000020712.84444.8d. Epub 2002/08/03. [DOI] [PubMed] [Google Scholar]
- 102.Coulson RJ, Chesler NC, Vitullo L, Cipolla MJ. Effects of ischemia and myogenic activity on active and passive mechanical properties of rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2002;283(6):H2268–75. doi: 10.1152/ajpheart.00542.2002. [DOI] [PubMed] [Google Scholar]
- 103.McColl BW, Rose N, Robson FH, Rothwell NJ, Lawrence CB. Increased brain microvascular MMP-9 and incidence of haemorrhagic transformation in obese mice after experimental stroke. J Cereb Blood Flow Metab. 2010;30(2):267–72. doi: 10.1038/jcbfm.2009.217. Epub 2009/10/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kivipelto M, Ngandu T, Fratiglioni L, et al. Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol. 2005;62(10):1556–60. doi: 10.1001/archneur.62.10.1556. Epub 2005/10/12. [DOI] [PubMed] [Google Scholar]
- 105.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature reviews Neuroscience. 2004;5(5):347–60. doi: 10.1038/nrn1387. Epub 2004/04/22. [DOI] [PubMed] [Google Scholar]
- 106.Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutrition reviews. 2010;68(Suppl 2):S74–87. doi: 10.1111/j.1753-4887.2010.00352.x. Epub 2010/12/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nature reviews Neuroscience. 2011;12(12):723–38. doi: 10.1038/nrn3114. Epub 2011/11/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dichgans M, Zietemann V. Prevention of vascular cognitive impairment. Stroke. 2012;43(11):3137–46. doi: 10.1161/STROKEAHA.112.651778. Epub 2012/09/01. [DOI] [PubMed] [Google Scholar]
- 109.Smith E, Hay P, Campbell L, Trollor JN. A review of the association between obesity and cognitive function across the lifespan: implications for novel approaches to prevention and treatment. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2011;12(9):740–55. doi: 10.1111/j.1467-789X.2011.00920.x. Epub 2011/10/13. [DOI] [PubMed] [Google Scholar]
- 110.Anstey KJ, Cherbuin N, Budge M, Young J. Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obesity reviews: an official journal of the International Association for the Study of Obesity. 2011;12(5):e426–37. doi: 10.1111/j.1467-789X.2010.00825.x. Epub 2011/02/26. [DOI] [PubMed] [Google Scholar]
- 111.Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Jr, Yaffe K. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005;330(7504):1360. doi: 10.1136/bmj.38446.466238.E0. Epub 2005/05/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rosengren A, Skoog I, Gustafson D, Wilhelmsen L. Body mass index, other cardiovascular risk factors, and hospitalization for dementia. Arch Intern Med. 2005;165(3):321–6. doi: 10.1001/archinte.165.3.321. Epub 2005/02/16. [DOI] [PubMed] [Google Scholar]
- 113.Gorospe EC, Dave JK. The risk of dementia with increased body mass index. Age Ageing. 2007;36(1):23–9. doi: 10.1093/ageing/afl123. Epub 2006/11/25. [DOI] [PubMed] [Google Scholar]
- 114.Fox CS, Massaro JM, Hoffmann U, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116(1):39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. Epub 2007/06/20. [DOI] [PubMed] [Google Scholar]
- 115.Debette S, Seshadri S, Beiser A, et al. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology. 2011;77(5):461–8. doi: 10.1212/WNL.0b013e318227b227. Epub 2011/08/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Baumgartner RN, Heymsfield SB, Roche AF. Human body composition and the epidemiology of chronic disease. Obes Res. 1995;3(1):73–95. doi: 10.1002/j.1550-8528.1995.tb00124.x. Epub 1995/01/01. [DOI] [PubMed] [Google Scholar]
- 117.Benito-Leon J, Mitchell AJ, Hernandez-Gallego J, Bermejo-Pareja F. Obesity and impaired cognitive functioning in the elderly: a population-based cross-sectional study (NEDICES) Eur J Neurol. 2013 doi: 10.1111/ene.12083. Epub 2013/01/18. [DOI] [PubMed] [Google Scholar]
- 118.Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham heart study. Int J Obes Relat Metab Disord. 2003;27(2):260–8. doi: 10.1038/sj.ijo.802225. Epub 2003/02/15. [DOI] [PubMed] [Google Scholar]
- 119.Jack CR, Jr, Petersen RC, Xu YC, et al. Hippocampal atrophy and apolipoprotein E genotype are independently associated with Alzheimer’s disease. Ann Neurol. 1998;43(3):303–10. doi: 10.1002/ana.410430307. Epub 1998/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jack CR, Jr, Shiung MM, Weigand SD, et al. Brain atrophy rates predict subsequent clinical conversion in normal elderly and amnestic MCI. Neurology. 2005;65(8):1227–31. doi: 10.1212/01.wnl.0000180958.22678.91. Epub 2005/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Prins ND, van Dijk EJ, den Heijer T, et al. Cerebral white matter lesions and the risk of dementia. Arch Neurol. 2004;61(10):1531–4. doi: 10.1001/archneur.61.10.1531. Epub 2004/10/13. [DOI] [PubMed] [Google Scholar]
- 122.Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348(13):1215–22. doi: 10.1056/NEJMoa022066. Epub 2003/03/28. [DOI] [PubMed] [Google Scholar]
- 123.Debette S, Beiser A, Hoffmann U, et al. Visceral fat is associated with lower brain volume in healthy middle-aged adults. Ann Neurol. 2010;68(2):136–44. doi: 10.1002/ana.22062. Epub 2010/08/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiology of aging. 2005;26(Suppl 1):46–9. doi: 10.1016/j.neurobiolaging.2005.09.003. Epub 2005/10/13. [DOI] [PubMed] [Google Scholar]
- 125.Greenwood CE, Winocur G. High-fat diets, insulin resistance and declining cognitive function. Neurobiology of aging. 2005;26(Suppl 1):42–5. doi: 10.1016/j.neurobiolaging.2005.08.017. Epub 2005/11/01. [DOI] [PubMed] [Google Scholar]
- 126.McNeilly AD, Williamson R, Sutherland C, Balfour DJ, Stewart CA. High fat feeding promotes simultaneous decline in insulin sensitivity and cognitive performance in a delayed matching and non-matching to position task. Behavioural brain research. 2011;217(1):134–41. doi: 10.1016/j.bbr.2010.10.017. Epub 2010/10/27. [DOI] [PubMed] [Google Scholar]
- 127.Boitard C, Etchamendy N, Sauvant J, et al. Juvenile, but not adult exposure to high-fat diet impairs relational memory and hippocampal neurogenesis in mice. Hippocampus. 2012;22(11):2095–100. doi: 10.1002/hipo.22032. Epub 2012/05/18. [DOI] [PubMed] [Google Scholar]
- 128.Rocchini AP. Childhood obesity and a diabetes epidemic. N Engl J Med. 2002;346(11):854–5. doi: 10.1056/NEJM200203143461112. [DOI] [PubMed] [Google Scholar]
- 129.Morrison CD, Pistell PJ, Ingram DK, et al. High fat diet increases hippocampal oxidative stress and cognitive impairment in aged mice: implications for decreased Nrf2 signaling. Journal of neurochemistry. 2010;114(6):1581–9. doi: 10.1111/j.1471-4159.2010.06865.x. Epub 2010/06/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Frontiers in physiology. 2012;3:128. doi: 10.3389/fphys.2012.00128. Epub 2012/05/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ambrogini P, Orsini L, Mancini C, Ferri P, Barbanti I, Cuppini R. Persistently high corticosterone levels but not normal circadian fluctuations of the hormone affect cell proliferation in the adult rat dentate gyrus. Neuroendocrinology. 2002;76(6):366–72. doi: 10.1159/000067581. Epub 2003/02/05. [DOI] [PubMed] [Google Scholar]
- 132.Heine VM, Maslam S, Joels M, Lucassen PJ. Prominent decline of newborn cell proliferation, differentiation, and apoptosis in the aging dentate gyrus, in absence of an age-related hypothalamus-pituitary-adrenal axis activation. Neurobiology of aging. 2004;25(3):361–75. doi: 10.1016/S0197-4580(03)00090-3. Epub 2004/05/05. [DOI] [PubMed] [Google Scholar]
- 133.Lindqvist A, Mohapel P, Bouter B, et al. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol. 2006;13(12):1385–8. doi: 10.1111/j.1468-1331.2006.01500.x. Epub 2006/11/23. [DOI] [PubMed] [Google Scholar]
- 134.Mitchell BM, Dorrance AM, Mack EA, Webb RC. Glucocorticoids decrease GTP cyclohydrolase and tetrahydrobiopterin-dependent vasorelaxation through glucocorticoid receptors. J Cardiovasc Pharmacol. 2004;43(1):8–13. doi: 10.1097/00005344-200401000-00002. Epub 2003/12/12. [DOI] [PubMed] [Google Scholar]