

Abstract
Doxorubicin (DOX) has limited efficacy in colorectal cancer due to multi-drug resistance. Resveratrol (RES) and didox (DID) are polyhydroxyphenols with potential chemosensitizing effects. Herein, we assessed the chemomodulatory effects of RES and DID to DOX in colorectal cancer cells. Equitoxic combination of DOX with RES and DID in HCT 116 reduced the IC50 of DOX from 0.96 ± 0.02 μM to 0.52 ± 0.05 μM and 0.4 ± 0.06 μM, respectively. Similarly, combination of DOX with RES and DID in HT-29 decreased the IC50’s of DOX from 0.88 ± 0.03 μM to 0.47 ± 0.02 μM and 0.29 ± 0.04 μM, respectively. The expressions of p53 and Bax genes were markedly elevated in HCT 116 cells after exposure to DOX/DID. In HT-29 cells, the expression of Bcl-XL gene was significantly decreased after exposure to DOX/DID. In addition, combination of DOX with RES significantly increased the expression of Bax gene in HCT 116 cells. RES treatment induced significant S-phase arrest in DOX-treated HCT 116 cells, while DID induced G2/M- and S-phase arrest in HCT 116 and HT-29, respectively. Both RES and DID significantly enhanced the intracellular entrapment of DOX due to blocking the efflux activity of p-glycoprotein pump. In conclusion, RES and DID sensitize colorectal cancer cells to DOX via facilitating apoptosis and enhancing intracellular entrapment of DOX.
Colorectal cancer (CRC) is the third most commonly diagnosed cancer in males and the second in females with an estimated 1.4 million cases and 693.000 deaths occurring in 2012, accounting for 8% of all cancer related deaths1. Despite recent advances in chemotherapy, currently used anticancer molecules are unable to improve the prognosis of advanced or recurrent colorectal cancer, which remains incurable2.
The anthracycline, doxorubicin (DOX), is a widely used chemotherapy due to its efficacy in fighting wide range of cancers such as carcinomas, sarcomas and hematological neoplasias3,4. However, there are clinical limitations that arise from its susceptibility to multi-drug resistance5. Overexpression of ATP-dependent efflux pump p-glycoprotein (P-gp) and its related proteins is crucial to multidrug resistance (MDR) and chemotherapy failure in cancer treatment6. P-gp is encoded by MDRl gene; and is considered a member of the ATP-binding cassette (ABC) transporter superfamily. It is energy-dependent transporter pump that removes xenobiotics such as, DOX outward from cells and confer drug resistance in tumor cells7,8.
Compounds of natural origin are very rich source for leads with potential anticancer properties as well as chemomodulatory effects such as, P-gp inhibitors7,9,10,11,12,13,14,15. Resveratrol (RES) is naturally occurring plant antibiotic known as phytoalexin, found in various plants, nuts, fruits and especially abundant in grapes and red wine16,17. It has been extensively studied for its antioxidant, anti-aging and anti-inflammatory activities18,19,20,21,22,23. In addition, in vivo and in vitro studies showed that RES possesses potential anti-tumor activity against several malignancies24,25,26,27. According to our previous study, RES potentiates the cytotoxic properties of DOX in MCF-7, HeLa and HepG2 cells via P-gp inhibition and downregulation of MDR1 gene28.
Didox (DID) is a synthetic polyphenolic compound which shares important biochemical targets with RES29. It is potent inhibitor for ribonucleotide reductase enzyme which interferes with DNA synthesis and repair30. Ribonucleotide reductase enzyme has been considered potential target for cancer chemotherapy31. DID showed anti-tumor effects in a variety of experimental systems, and several human tumor xenografts32,33,34. It may exert its anti-tumor effect via the activation of various apoptosis pathways35. According to our previous work as well as other research groups, DID and RES improve the cytotoxic profile of different anticancer agents and protect from their toxic effects28,36,37,38,39.
DID and RES might be potential successful adjuvant candidates for combination with DOX33,40. Therefore, we investigated the potential improvement effects of RES and DID on DOX-anticancer properties and the possible underlying mechanisms in two colorectal cancer cell lines with different expression levels of MDR1 gene.
Results
RES and DID improve the cytotoxicity of DOX in colorectal cancer cells
To study the effect of RES and DID on the cytotoxic profile of DOX, the dose response curve of DOX alone was assessed relative to its combination with RES or DID in two colorectal cancer cell lines (Fig. 1A–D) (Table 1). In HCT 116 cells, DOX exerted gradient cytotoxic activity with increasing concentration; viability started to drop significantly (P < 0.05) at concentration of 0.3 μM. Cellular log kill was gradual in profile with IC50 of 0.96 ± 0.02 μM (Fig. 1A,B). Similarly, RES and DID single treatments exerted gradual cytotoxic activity with increasing concentration; viability started to drop significantly (P < 0.05) at concentrations of 10 μM and 100 μM, respectively. Both RES and DID have steep cellular log kill profile with IC50’s of 17.5 ± 02 μM and 105 ± 1.5 μM, respectively (Fig. 1A,B). Equitoxic combination of RES or DID with DOX significantly improved the cytotoxic profile of DOX (Fig. 1A,B). IC50’s of DOX after combination with RES and DID were significantly (P < 0.05) decreased from 0.96 ± 0.02 μM to 0.52 ± 0.05 μM and 0.4 ± 0.06 μM, respectively (Table 1). The calculated CI-values for DOX with RES and DID were 1.16 and 0.76, respectively. These CI-values are indicative of additive interaction of DOX with RES and synergistic interaction with DID in HCT 116 cell line (Table 1).
Figure 1. The effect of RES (A&C) and DID (B&D) on the dose response curve of DOX in HCT 116 (A&B) and HT-29 (C&D) colorectal cancer cell line.

Cells were exposed to serial dilution of DOX (long dashed lines), RES/DID (short dashed lines) or combination of RES/DID with DOX (solid lines) for 72h. Cell viability was determined using SRB-assay and data are expressed as mean ± SD (n = 3).
Table 1. Effect of RES and DID on the cytotoxicity of DOX in HCT 116 and HT-29 cell lines.
| IC50 against HCT 116 cells (μM) | IC50 against HT-29 cells (μM) | |
| DOX | 0.96 ± 0.02 | 0.88 ± 0.03 |
| RES | 17.5 ± 02 | 187.1 ± 4.7 |
| DID | 105 ± 1.5 | 501.6 ± 53 |
| DOX + RES | 0.52* ± 0.05 | 0.47* ± 0.02 |
| DOX + DID | 0.4* ± 0.06 | 0.29* ± 0.04 |
| CI-value (DOX + RES) | Additive/1.16 | Additive/1.02 |
| CI-value (DOX + DID) | Synergism/0.76 | Synergism/0.6 |
*Significantly different from DOX alone at p-value < 0.05.
Similarly in HT-29 cell line, DOX exerted gradient cytotoxic activity with increasing concentration; viability started to drop significantly (P < 0.05) at concentration of 0.3 μM (Fig. 1C,D). Cellular log kill was gradual in profile with IC50 of 0.88 ± 0.03 μM (Table 1). Also, RES and DID single treatments exerted gradual cytotoxic activity with increasing concentration; viability started to drop significantly (P < 0.05) at concentrations of 100 μM and 300 μM, respectively (Fig. 1C,D). Both RES and DID showed steeping cellular log kill with IC50’s of 187.1 ± 4.7 μM and 501.6 ± 53 μM, respectively. Equitoxic combination of RES and DID with DOX significantly improved the cytotoxic profile of DOX in HT-29 cell line (Fig. 1C,D). IC50’s of DOX after combination with RES and DID were significantly (P < 0.05) decreased from 0.88 ± 0.03 μM to 0.47 ± 0.02 μM and 0.29 ± 0.04 μM, respectively (Fig.1C,D) (Table 1). The calculated CI-values for DOX with RES and DID were 1.02 and 0.6, respectively. These CI-values are indicative of additive interaction for DOX with RES and synergistic interaction characteristics for DOX with DID in HT-29 cell line (Table 1).
RES and DID enhance DOX induced apoptosis in colorectal cancer cells
To understand the interaction characteristics of DOX with RES or DID, gene expression analysis for apoptosis key markers was assessed using real time PCR. In DOX-treated HCT 116, the apoptotic gene, Bax, was not significantly changed compared to the untreated cells. However, combination of RES or DID with DOX significantly increased the relative Bax gene expression by two fold compared to DOX-treated cells (Fig. 2A). On the other hand, Bcl2 antiapoptotic gene was not significantly changed in DOX treatment compared to the untreated cells. Surprisingly, combination of DID with DOX significantly (p < 0.05) increased the relative Bcl2 gene expression (Fig. 2A). Moreover, the expression of the antiapoptotic gene, Bcl-XL, was significantly decreased after treatment with RES alone compared to control cells. All other treatments did not show any significant change in the expression level of Bcl-XL compared to control cells (Fig. 2A). Interestingly, p53 level was significantly over expressed after exposure to combination of DID with DOX (Fig. 2A). This might be attributed to overweighing the total apoptotic signal relative to the anti-apoptotic signal in DOX/DID-treated HCT 116 cells. Accordingly, the effect of RES and DID on the intracellular concentration of active caspase-3 was assessed. In HCT 116, there was no significant change in active caspase-3 concentration in all single treatments. DID and DOX treatment alone induced apparently higher level of caspase-3; however did not reach a significant level (p < 0.05). Yet, combination of RES or DID with DOX increased the concentration of the active form of caspase-3 by 3.6 and 3.5 fold, respectively (Fig. 2C).
Figure 2.

Gene expression of Bax, Bcl2, Bcl-xl and p53 was determined in HCT 116 (A) and HT-29 (B) cells using RT-PCR after treatment with DOX, RES, DID or their combination for 24 h. Concentration of active caspase-3 was assessed in HCT 116 (C) and HT-29 (D) cells after treatment for 48 h. Data are expressed as mean ± S.D. (n = 3). *Significantly different from control group (p < 0.05). **Significantly different from DOX treatment alone (p < 0.05).
In HT-29 cells, the apoptotic gene, Bax, was significantly under expressed in all single and combined treatments (Fig. 2B). On the other hand, Bcl2 antiapoptotic gene was not significantly changed in all treatment groups compared to untreated cells (Fig. 2B). Moreover, the apoptotic gene, p53, was significantly under expressed upon single DID treatment. Combination of RES or DID with DOX had no effect on p53 expression (Fig. 2B). Nonetheless, the antiapoptotic gene, Bcl-XL, was not under expressed after single DOX treatment. However, combination of RES or DID with DOX significantly decreased the expression level of Bcl-XL (Fig. 2B). Accordingly, the effect of RES and DID on the intracellular concentration of active caspase-3 was assessed. DOX or DID treatment alone significantly increased the intracellular concentration of active caspase-3 compared to control cells. Yet, combination of DOX/DID further increased the intracellular concentration of active caspase-3 significantly higher than single DOX treatment (Fig. 2D).
RES, DID, DOX and their combination influence cell cycle distribution of colorectal cancer cells
DNA flow-cytometry was used to assess the effect of RES and DID alone or in combination with DOX on the cell cycle distribution of HCT 116 and HT-29 cell lines. In HCT 116, DOX showed no significant change in cell population in G0/G1-phase compared to the control cells, while both RES and DID decreased cell population in G0/G1-phase from 63.8 ± 0.9% to 45.4 ± 1.3% and 60.5 ± 0.3% respectively. Combination of DOX with RES and DID markedly decreased cell population in G0/G1-phase to 29.5 ± 1.8% and 38.8 ± 1.6% respectively compared to DOX alone (64.95 ± 0.4%) (Fig. 3A,C). In addition, DOX alone decreased cell population in S-phase from 27.8 ± 1.1% to 13 ± 0.8%. On the other hand, both RES and DID single treatments increased the S-phase population to 52.9 ± 1.2% and 31.0 ± 0.4% respectively (control cells in S-phase were 27.8 ± 0.9%). Combination of DOX with RES caused a marked increase in S-phase population to 49.6 ± 1.7%. on the other hand, DOX combination with DID showed significant decrease of S-phase population to 7.0 ± 0.7% compared to DOX treatment alone (13 ± 0.8%) (Fig. 3A,C). DOX treatment showed significant increase in G2/M-phase population from 7.4 ± 0.7% to 20.6 ± 0.7%. RES showed marked decrease in G2/M-phase cell population to 0.8 ± 0.1%. On the other hand, no marked change in G2/M-phase population was induced by treatment with DID compared to control cells. Co-treatment of DOX and RES had no significant change in the G2/M-phase population compared to DOX alone, while combination of DOX with DID induced a significant increase in the G2/M-phase (53.0 ± 3.1%) compared to DOX treatment alone (20.6 ± 0.7%) (Fig. 3A,C).
Figure 3.

HCT 116 (A) and HT-29 (B) cells were exposed to DOX, RES, DID and their combinations for 48 h. Cell cycle distribution was determined using DNA cytometry analysis and percentages of cell phases were plotted (C,D) compared to control cells; (n = 3).
In HT-29, DOX significantly increased cells in G0/G1-phase from 69.6 ± 1.0% to 82.3 ± 1.0%, while RES significantly decreased cell population in G0/G1-phase to 44.3 ± 0.7%. DID did not induce any significant change in G0/G1-phase cell population. Combination of DOX with RES and DID significantly decreased G0/G1 cell population to 77.0 ± 2.1% and 74.2 ± 0.4%, respectively compared to DOX treatment alone (82.3 ± 1.0%) (Fig. 3B,D). DOX alone abolished cells in S-phase from 29.4 ± 1.1% to 1.9 ± 0.5%. RES induced significant increase in S-phase cells from 29.4 ± 0.6% to 48.1 ± 0.8% compared to control cells. DID moderately decreased S-phase population to 20 ± 1.0%. Combination of DOX with RES or DID increased S-phase population from 1.9 ± 0.8% to 7.6 ± 0.5% and 11.4 ± 1.3%, respectively compared to DOX treatment alone (Fig. 3B,D). No significant change in cell population in G2/M-phase upon DOX treatment. However, significant increase in G2/M phase was noticed by RES (5.2 ± 0.7%), and DID (4.4 ± 0.6%) compared to control cells (0.1 ± 0.2%). Treatment with DOX, RES or DID significantly increased the apoptotic Pre-G phase from 0.9 ± 0.1% to 15.5 ± 1.5%, 2.4 ± 0.1 and 6.7 ± 0.5%, respectively. Combination of DOX with DID or RES did not induced further increase in the apoptotic Pre-G phase population compared to DOX treatment alone (Fig. 3B,D).
RES and DID increase cellular entrapment of DOX within colorectal cancer cells via inhibiting P-gp efflux pump activity without affecting MDR1 gene expression
P-glycoprotein (P-gp) is a plasma membrane efflux pump coded by MDR1 gene and highly abundant in colorectal cancer cells (MDR1 gene is highly expressed in HCT 116 cells and weakly expressed in HT-29 cells41). Attributed to its fluorescent properties, the intracellular concentration of DOX was determined after incubation with HCT 116 and HT-29 cells in the presence and absence of RES, DID and VRP (positive P-gp inhibitor). RES significantly increased the intracellular concentration of DOX by 202.1% and 68.8% in HCT 116 and HT-29 cells, respectively (Fig. 4A,B). Similarly, DID significantly increased the intracellular concentration of DOX by 94.8% and 85% in HCT 116 and HT-29 cells, respectively (Fig. 4A,B). The positive control P-gp blocker, VRP, resulted in increasing the intracellular concentration of DOX by 154.2% and 120% in HCT 116 and HT-29 cells, respectively (Fig. 4A,B). Inhibition of P-pgp activity might be mediated by inactivating P-gp related ATPase enzyme activity or via competitive covalent binding to P-gp molecules.
Figure 4.

The influence of RES and DID on the intracellular entrapment of DOX was assessed in HCT 116 (A) and HT-29 (B) cells and compared to positive control agent (VRP). The type of interaction between RES and DID with human recombinant P-gp molecules attached to ATPase subunit was assessed using ATP consumption assay in comparison to VRP and Sod vanadate positive control agents (C). Gene expression of MDR1 was assessed in HCT 116 (D) and HT-29 (E) cells using RT-PCR after treatment with DOX, RES, DID or their combination for 24 h. Data are expressed as mean ± S.D. (n = 3). *Significantly different from control group (p < 0.05).
To further investigate the sub-molecular interaction between RES and DID with P-gp molecules, ATP consumption assay was carried out on purified human recombinant P-gp molecules attached to ATPase enzyme. VRP (covalent P-gp inhibitor) and sodium vanadate (ATPase inhibitor) were used as positive controls; and are supposed to increase and decrease ATP consumptions, respectively compared to non-treated control (NTC). RES significantly decreased remaining ATP molecules by 29% compared to 31.5% for VRP (Fig. 4C). Interestingly, DID did not show any significant change in ATP consumption (Fig. 4C) compared to NTC.
Finally, the effect of RES and DID on the expression level of MDR1 gene was measured by real time PCR. In both HCT 116 and HT-29, none of the treatments under investigation showed any significant change in MDR1 gene expression (Fig. 4D,E). Together with the previous results (Fig. 4A–C), the ability of DID to enhance the intracellular entrapment of DOX within HCT 116 and HT-29 cells could be attributed to dual interaction with P-gp efflux pump via ATPase inhibition and covalent binding.
Discussion
The activity of P-glycoprotein (P-gp) is involved in multidrug resistance of CRC42,43. Doxorubicin (DOX) is a chemotherapeutic drug which has been widely used over the past four decades for the treatment of several humoral and solid neoplasias4. DOX might not be the optimum clinical choice for CRC which might be attributed to intrinsic resistance to DOX44. In this study, we explored the potential influence of RES and DID on the cytotoxic profile of DOX in two different CRC cells (HCT 116 and HT-29) and its associated mechanism of action/resistance.
According to our data, DID per se did not show any promising cytotoxicity in CRC cells under investigation (HCT 116 and HT-29). In the current work as well as in our previous publication, RES showed weak cytotoxicity against HCT 116 cells (IC50 17.5 μM)28. At low concentrations, RES decreased DOX-induced cytotoxicity in HCT 116. While at higher concentrations (above 2 μM), RES enhanced DOX-induced viability inhibition in a concentration dependent manner (Fig. 1). This finding is consistent with Chen et al.45 who demonstrated an opposing effect of RES at low vs. high concentrations. In the present study, RES showed additive while DID showed synergistic effect with DOX against HCT 116 and HT-29 cell lines (Table 1). Similarly, we showed synergistic interaction between RES and DOX or docetaxel in MCF-7 cell line and additive interactions against HeLa and HepG2 cells28. In addition, the combination of RES or DID with transtuzumab resulted in synergistic effect in MCF-7 and T47D cells36. Moreover, the combination of DID with DOX showed additive interaction in liver cancer cells39.
The effects of combining RES or DID with DOX on some apoptosis key markers (Bax, Bcl2, p53 and Bcl-XL) were further investigated. According to our observations, in HCT 116 cell line, RES enhanced DOX-induced Bax gene expression which might explain the significant increase in active caspase-346. On the other hand, DID significantly enhanced DOX-induced Bcl2, Bax and p53 gene expression. Yet, p53 promotes apoptosis through down-regulating the antiapoptotic Bcl2 and up-regulating the apoptotic Bax protein expression at transcriptional level47. Thus, p53 might be increased in attempt to down regulate the elevated level of Bcl2 gene expression (Fig. 2A,B). Moreover, the significant increase in p53 expression might explain the G2/M cell cycle arrest induced in DID/DOX-treated group48. This is consistent with the study of Zhang and coworkers who found that genistein, another polyphenolic compound, induces G2/M cell cycle arrest and apoptosis in p53-dependent manner49. Again, p53 can exert a direct pro-apoptotic function within mitochondrial pathway of apoptosis which is known as the circuitry of p53 death signaling50. In another study, DID resulted in DNA damage and p53 induction resulting in apoptosis for acute myeloid leukemia cells51. Furthermore, the over expression of p53 and Bax in DID/DOX-treated group might interpret the significant increase in active caspase-3 in HCT 11646,52. As Bcl-XL overexpression resulted in inhibition of caspase-3 activity53, in HT-29 cell line, the increase in caspase-3 activity in DID/DOX treated group might be attributed to the reduction in Bcl-XL expression.
DID was suggested previously to induce apoptosis in other cancer cell lines such as, multiple myeloma cells33. Moreover, DID synergizes the anti-tumor effect of temozolomide54, carmustine40 and cidofovir34 against malignant brain tumor cells, DAOY human medulloblastoma cells and Epstein-Barr virus (EBV)-positive nasopharyngeal carcinoma xenografts, respectively. DOX-induced cytotoxicity is partly attributed to the generation of reactive oxygen species and free radicals55. DID possesses robust antioxidant activity and potent free radical scavenging capability56. Despite possessing potent antioxidant properties, DID did not antagonize DOX-induced cytotoxicity herein as well as in our previous work against liver cancer cells39. On contrary, DID can be described to possess sensitizing role for DOX-induced cytotoxicity in CRC cells. This might be similar to the effect of trimidox, another ribonuclueotide reductase inhibitor, which exhibits both anticancer and potent antioxidant properties57. DID is known to be a ribonucleotide reductase enzyme inhibitor which might partly explain its anti-proliferative and chemosensitizing effects33. Similar to DID, RES has an antioxidant effect58 which did not prevent its potentiating effect to DOX-induced cytotoxicity in more than solid tumor type28. Many mechanisms have been suggested for RES induced cytotoxicity; yet, the exact mode of action is still controversial59.
Further illustration to the potential underlying chemosensitizing effects of RES and DID on DOX might be taken from cell cycle analysis. DOX induced cytotoxicity is suggested to be cell cycle dependent60. RES blocked the cell cycle progression in the S-phase in both cell lines which is also consistent with previous studies61. In HCT 116, RES showed very strong S-phase arrest in DOX-treated cells, while in HT-29 it exerts mild S-phase arrest compared with DOX-alone (Fig. 3). S-phase arrest generates higher cell population sensitive to S-phase specific agents such as DOX and would be partly the reason for potentiating DOX-induced cytotoxicity28. On the other hand, DID induced G2/M cell cycle arrest in DOX-treated HCT 116 cells which might add more evidence to the sensitizing effect of DID to DOX-induced cytotoxicity (Fig. 3). Mittal and coworkers demonstrated that the sensitizing effect of berberine to DOX-induced cytotoxicity was associated with a G2/M-phase arrest62. Moreover, DID/DOX combination induced significant increase in S-phase with reciprocal decrease in G0/G1-phase compared to DOX treatment alone in HT-29 (Fig. 3). S-phase arrest induced by the combination of DID with DOX may add an extra evidence to the sensitizing effect of DID to DOX in HT-29 cells63. In support of the importance of cell cycle arrest to DOX cytotoxicity, Ling and colleagues60 found that P388 cells synchronized in S and G2/M phases were more sensitive to DOX than cells in G1-phase. Strategies to increase G2/M arrest have also been associated with enhanced apoptosis64.
Although no effect was observed for either DID or RES on the expression level of MDR1 gene, both agents significantly decreased the P-gp pumping activity (Fig. 4). In our previous work and herein, we showed that RES via covalent binding inhibits P-gp activity in different solid tumor cell lines28. To the best of our knowledge, this is the first time to prove P-gp-inhibitory effect attributed to DID via dual P-gp covalent binding and ATPase inhibition. This may shed the light to a field for using DID in improving cellular pharmacokinetics of different antitumor drugs via increasing their intracellular concentrations thus enhancing their cell killing effect.
In conclusion, DID showed superior improvement to DOX-induced cytotoxicity in CRC cells compared to RES. This potentiating effect might be attributed to enhancing apoptosis, inducing cell cycle arrest and enhancing the cellular pharmacokinetics via P-gp inhibitory activity.
Materials and Methods
Chemicals and Drugs
Didox (DID) was generously gifted from Professor Howard L. Elford, Molecules for Health Inc., Richmond, VA, USA. Doxorubicin (DOX), resveratrol (RES), and sulpharodamine (SRB) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). RPMI-1640 media, fetal bovine serum and other cell culture materials were purchased from Lonza Group Ltd. (Basel, Switzerland). Other reagents were of the highest analytical grade.
Cell culture
Human colorectal cell lines, HCT 116 (ATCC®CCL-247) and HT-29 (ATCC®HTB-38), were obtained from the Vaccera (Giza, Egypt). Cells were maintained in RPMI-1640 supplemented with streptomycin (100 μg/mL); penicillin (100 units/mL) and heat-inactivated fetal bovine serum (10% v/v) in a humidified, 5% (v/v) CO2 atmosphere at 37 °C.
Cytotoxicity assays
The cytotoxicity of DOX, RES and DID were tested against HCT 116 and HT-29 cells by SRB assay as previously described65. Exponentially growing cells were collected using 0.25% Trypsin-EDTA and plated in 96-well plates at 1000–2000 cells/well. Cells were exposed to serial concentration of DOX, RES and DID for 72 h and subsequently fixed with TCA (10% w/v) for 1 h at 4 °C. After several washings, cells were exposed to 0.4% (w/v) SRB solution for 10 min in dark place and subsequently washed with 1% (v/v) glacial acetic acid. After drying overnight, Tris-HCl (50 mM, pH 7.4) was used to dissolve the SRB-stained cells and color intensity was measured at λmax of 540 nm and calculated as percent viability of control cells (cells exposed to drug free media)65.
Data analysis
The dose response curves of compounds were analyzed using 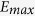 model (Eq. 1).
model (Eq. 1).
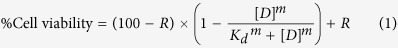 |
where R is the residual unaffected fraction (the resistance fraction); [D] is the drug concentration used; Kd is the drug concentration that produces 50% reduction of the maximum inhibition rate and m is a Hill-type coefficient. IC50 is defined as the drug concentration required to reduce absorbance to 50% of that of the control (i.e., Kd = IC50 when R = 0 and Emax = 100 − R)14.
Combination index (CI) was calculated as previously described66. Briefly, exponentially growing cells were exposed to equitoxic concentrations of RES and DOX or DID and DOX in 96-well plates for 72 h and subsequently subjected to SRB assay. CI was calculated from the formula:
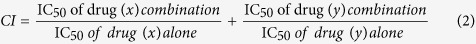 |
The nature of drug interaction is defined as synergism if CI > 0.8; antagonism if CI < 1.2; and additive if CI ranges from 0.8–1.2.
RNA extraction, Real time PCR analysis and quantification of gene expression
To assess the gene expression of Bcl2, Bax, Bcl-XL, p53 and MDR1 following treatment of cells with DOX, RES, DID and their combination, total RNA extraction from cells was performed using RNeasy Mini Kit® (Qiagen Inc. Valencia, CA, USA). Reverse transcription was undertaken to construct cDNA library from different treatments using High-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). The archived cDNA libraries were then subjected to quantitative real time PCR reactions using cyber green fluorophore (Fermentas Inc., Glen Burnie, MD, USA). Primer sequences were as follow: Bcl2 forward primer GGG-TAC-GAT-AAC-CGG-GAG-AT and reverse primer CTG-AAG-AGC-TCC-TCC-ACC-AC; Bax forward primer TCT-GAC-GGC-AAC-TTCAAC-TG and reverse primer TGG-GTG-TCC-CAA-AGT-AGG-AG; Bcl-XL forward primer GGC GGA TTT GAA TCT CTT TCT C and reverse primer TTA TAA TAG GGA TGG GCT CAA CC; p53 forward primer CCT-CAC-CAT-CAT-CAC-ACT-GG and reverse primer CTG-AGT-CAGGCC-CTT-CTG-TC; MDR1 forward primer GCT-GGG-AAG-ATC-GCT-ACT-GA and reverse primer GGT-ACC-TGC-AAA-CTC-TGA-GCA. GAPDH was used as reference housekeeping gene with forward primer TGC-ACC-ACC-AAC-TGC-TTAG and reverse primer GAT-GCA-GGG-ATG-ATG-TTC67. mdr1 Gene expression was normalized using the expression level of corresponding GAPDH gene. The rest of gene expressions in different treatments (single or combined) were expressed relative to its corresponding normalized level in control untreated cells (cells exposed to drug free media) and control treatment gene expression level was expressed as dotted line.
Assessment of active caspase-3 concentration in colorectal cell lines
To assess the effect of RES, DID, DOX and their combination on apoptosis, the active caspase-3 level was measured by using Quantikine® caspase-3 ELISA kit (R&D Systems, Inc. USA). Briefly, the cells were exposed to the predetermined IC50’s of test compounds (single or combined treatments) or drug free media (control group) for 24 h. Cells were harvested and washed with PBS, then incubated with the biotin-ZVKD-fmk inhibitor for 1 hour. Cells were transferred into the wells of a microplate pre-coated with a monoclonal antibody specific for caspase-3. Following a wash to remove any unbound substances, streptavidin conjugated to horseradish peroxidase was added to the wells and bound to the biotin on the inhibitor. Following a wash to remove any unbound Streptavidin-HRP, a substrate solution was added to the wells. The enzyme reaction yields a blue product that turned yellow when a Stop Solution was added. The optical density of each well was determined within 30 minutes, using a microplate reader set to 450 nm with a wavelength correction at 540 nm or 570 nm. The concentrations of active caspase-3 were calculated from a standard curve of constructed with known concentrations of active caspase-3. Caspase concentration was expressed as ng/mg protein. Proteins were determined spectrophotometrically by the method of Bradford using purified bovine serum albumin as a standard.
Analysis of cell cycle distribution
To determine the effect of DOX, RES, DID and their combinations on the cell cycle distribution in HCT 116 and HT-29 cell lines; cells were exposed to the predetermined IC50’s of test compounds (single or combination) for 24 h and compared to control cells (treated with drug free media). Cell cycle analysis was performed using the CycleTEST™ PLUS DNA Reagent Kit (Becton Dickinson Immunocytometry Systems, San Jose, California, USA). Control cells with known DNA content (PBMCs) were used as a reference point for determining the DI (DNA Index) for the test samples. Cells were stained with propodium iodide following the procedure provided by the kit and then run on the DNA cytometer. Cell cycle distribution was calculated using CELLQUEST software (Becton Dickinson Immunocytometry Systems, San Jose, California, USA).
Assessment of Pgp activity
To assess the effect of RES and DID on the efflux pumping activity of P-gp in tumor cell lines, the P-gp substrate, DOX, was used as a probe and verapamil (VRP) was used as standard p-gp blocker. Briefly, exponentially growing cells were plated in 6-well plates in plating density of 105 cells/well. Cells were exposed to DOX (10 μM), DOX (10 μM) and RES (10 μM), DOX (10 μM) and DID (10 μM) or DOX (10 μM) and VRP (10 μM) for 2 h at 37 °C and subsequently extracellular DOX containing media was washed trice with ice cold PBS. Intracellular DOX were extracted after cell lysis by incubation with SDS (2% w/v), saturated aqueous solution of ZnSO4 (100 μl), Acetonitril (500 μl) and acetone (500 μl) for 30 min at 37 °C. After centrifugation, supernatant was collected and DOX concentration was measured spectroflourometrically at λex/em 482/550 nm28,68.
Assessment of Pgp- ATPase activity
To reveal the mechanism of P-gp-inhibition induced by RES and DID, assessment of P-gp attached ATPase activity was performed using Pgp-Glo™ Assay Systems (Promega Corporation, Madison, Wisconsin, USA). Briefly, in 96-well plates, Pgp-Glo™ Assay Buffer was mixed with Na3VO4, VRP, RES, or DID. Recombinant human P-gp membrane fraction was incubated with the reaction mixture at 37 °C for about 5 minutes, and then the reaction was initiated by adding 10 μl of 25 mM MgATP and incubating for 40 minutes at 37 °C. An identical reaction mixture without drug treatment (NTC) was assayed in parallel. The reaction was stopped and the remaining un-consumed ATP was detected using luciferase firefly luminescent signal and ATP standard curve was plotted. The remaining concentrations of ATP were expressed as (p. mole/μg P-gp molecules). Sodium vanadate (Na3VO4) was used as selective inhibitor of P-gp related ATPase enzyme, and samples treated with Na3VO4 have greater luminescent signal than un-treated samples (NTC). In addition, VRP inhibits pgp molecules via covalent binding and competing with other substrates, hence, stimulating ATPase activity resulting in more consumption of ATP molecules compared to NTC.
Statistical analysis
Data are presented as mean ± SD. Analysis of variance (ANOVA) with LSD post hoc test was used for testing the significance using SPSS® for windows, version 17.0.0. p < 0.05 was taken as a cut off value for significance.
Additional Information
How to cite this article: Khaleel, S. A. et al. Didox and resveratrol sensitize colorectal cancer cells to doxorubicin via activating apoptosis and ameliorating P-glycoprotein activity. Sci. Rep. 6, 36855; doi: 10.1038/srep36855 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Author Contributions S.A.K. performed the experiments, collected the data, analyzed the data, performed the graphical and statistical analysis and wrote the manuscript; A.M.A. developed the research idea, designed the experiments, supervised the experiment execution, analyzed the data, and revised the manuscript; A.A.A. shared developing the research idea, followed up the experiments and revised the manuscript; A.B.A. shared developing the idea, supervised the experiments execution, supervised the data analysis and revised the manuscript.
References
- Torre L. A. et al. Global cancer statistics, 2012. CA: a cancer journal for clinicians 65, 87, doi: 10.3322/caac.21262 (2015). [DOI] [PubMed] [Google Scholar]
- Prados J. et al. Colon cancer therapy: recent developments in nanomedicine to improve the efficacy of conventional chemotherapeutic drugs. Anticancer Agents Med Chem 13, 1204 (2013). [DOI] [PubMed] [Google Scholar]
- Carvalho C. et al. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem 16, 3267 (2009). [DOI] [PubMed] [Google Scholar]
- Sartiano G. P., Lynch W. E. & Bullington W. D. Mechanism of action of the anthracycline anti-tumor antibiotics, doxorubicin, daunomycin and rubidazone: preferential inhibition of DNA polymerase alpha. The Journal of antibiotics 32, 1038 (1979). [DOI] [PubMed] [Google Scholar]
- Effenberger-Neidnicht K. & Schobert R. Combinatorial effects of thymoquinone on the anti-cancer activity of doxorubicin. Cancer Chemother Pharmacol 67, 867, doi: 10.1007/s00280-010-1386-x (2011). [DOI] [PubMed] [Google Scholar]
- Sun H. et al. Reversal of P-glycoprotein-mediated multidrug resistance by the novel tetrandrine derivative W6. J Asian Nat Prod Res 17, 638, doi: 10.1080/10286020.2015.1047772 (2015). [DOI] [PubMed] [Google Scholar]
- Abdallah H. M., Al-Abd A. M., El-Dine R. S. & El-Halawany A. M. P-glycoprotein inhibitors of natural origin as potential tumor chemo-sensitizers: A review. J Adv Res 6, 45, doi: 10.1016/j.jare.2014.11.008 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai G. M., Chen Y. N., Mickley L. A., Fojo A. T. & Bates S. E. P-glycoprotein expression and schedule dependence of adriamycin cytotoxicity in human colon carcinoma cell lines. International journal of cancer 49, 696 (1991) [DOI] [PubMed] [Google Scholar]
- Soliman F. M. et al. Cytotoxic activity of acyl phloroglucinols isolated from the leaves of Eucalyptus cinerea F. Muell. ex Benth. cultivated in Egypt. Scientific Reports 4, 5410, doi: 10.1038/srep05410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolba M. F. et al. Caffeic acid phenethyl ester synergistically enhances docetaxel and paclitaxel cytotoxicity in prostate cancer cells. IUBMB life 65, 716, doi: 10.1002/iub.1188 (2013). [DOI] [PubMed] [Google Scholar]
- Nawwar M. A. et al. Cytotoxic isoferulic acidamide from Myricaria germanica (Tamaricaceae). Plant Signal Behav 8, e22642, doi: 10.4161/psb.22642 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarif W. M. et al. Selective cytotoxic effects on human breast carcinoma of new methoxylated flavonoids from Euryops arabicus grown in Saudi Arabia. European journal of medicinal chemistry, 201, 66, doi: 10.1016/j.ejmech.2013.05.025 (2013). [DOI] [PubMed] [Google Scholar]
- Nawwar M. A. et al. Cytotoxic ellagitannins from Reaumuria vermiculata. Fitoterapia 83, 1256, doi: 10.1016/j.fitote.2012.06.007 (2012). [DOI] [PubMed] [Google Scholar]
- Mahmoud A. M., Al-Abd A. M., Lightfoot D. A. & El-Shemy H. A. Anti-cancer characteristics of mevinolin against three different solid tumor cell lines was not solely p53-dependent. J Enzyme Inhib Med Chem 27, 673, doi: 10.3109/14756366.2011.607446 (2012). [DOI] [PubMed] [Google Scholar]
- Aboul-Enein A. M. et al. Eichhornia crassipes (Mart) solms: from water parasite to potential medicinal remedy. Plant Signal Behav 6, 834 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero R. F., Garcia-Parrilla M. C., Puertas B. & Cantos-Villar E. Wine, resveratrol and health: a review. Natural product communications 4, 658 (2009). [PubMed] [Google Scholar]
- Chan M. M. Antimicrobial effect of resveratrol on dermatophytes and bacterial pathogens of the skin. Biochemical pharmacology 63, 99 (2002). [DOI] [PubMed] [Google Scholar]
- Brisdelli F., D’Andrea G. & Bozzi A. Resveratrol: a natural polyphenol with multiple chemopreventive properties. Current drug metabolism 10, 530 (2009). [DOI] [PubMed] [Google Scholar]
- Dobrzynska M. M. Resveratrol as promising natural radioprotector. A review. Rocz Panstw Zakl Hig 64, 255 (2013). [PubMed] [Google Scholar]
- Tresguerres I. F. et al. Resveratrol as anti-aging therapy for age-related bone loss. Rejuvenation research 17, 439, doi: 10.1089/rej.2014.1551 (2014). [DOI] [PubMed] [Google Scholar]
- Wu M. et al. Resveratrol delays polycystic kidney disease progression through attenuation of nuclear factor kappaB-induced inflammation. Nephrology, dialysis, transplantation (In press), doi: 10.1093/ndt/gfw058 (2016). [DOI] [PubMed] [Google Scholar]
- Chang G. R., Chen P. L., Hou P. H. & Mao F. C. Resveratrol protects against diet-induced atherosclerosis by reducing low-density lipoprotein cholesterol and inhibiting inflammation in apolipoprotein E-deficient mice. Iranian journal of basic medical sciences 18, 1063 (2015). [PMC free article] [PubMed] [Google Scholar]
- Abdel-Wahab B. A. & Abdel-Wahab M. M. Protective effect of resveratrol against chronic intermittent hypoxia-induced spatial memory deficits, hippocampal oxidative DNA damage and increased p47Phox NADPH oxidase expression in young rats. Behavioural brain research 305, 65, doi: 10.1016/j.bbr.2016.02.030 (2016). [DOI] [PubMed] [Google Scholar]
- Gomez L. S. et al. Resveratrol decreases breast cancer cell viability and glucose metabolism by inhibiting 6-phosphofructo-1-kinase. Biochimie 95, 1336, doi: 10.1016/j.biochi.2013.02.013 (2013). [DOI] [PubMed] [Google Scholar]
- Hao Y. Q. et al. Study of apoptosis related factors regulatory mechanism of resveratrol to human skin squamous cell carcinoma A431 xenograft in nude mice. Zhonghua yi xue za zhi 93, 464 (2013). [PubMed] [Google Scholar]
- Kaminski B. M., Steinhilber D., Stein J. M. & Ulrich S. Phytochemicals resveratrol and sulforaphane as potential agents for enhancing the anti-tumor activities of conventional cancer therapies. Curr Pharm Biotechnol 13, 137 (2012). [DOI] [PubMed] [Google Scholar]
- Liu P. et al. Resveratrol induces apoptosis of pancreatic cancers cells by inhibiting miR-21 regulation of BCL-2 expression. Clinical & translational oncology: official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 15, 741, doi: 10.1007/s12094-012-0999-4 (2013). [DOI] [PubMed] [Google Scholar]
- Al-Abd A. M. et al. Resveratrol enhances the cytotoxic profile of docetaxel and doxorubicin in solid tumour cell lines in vitro. Cell Prolif 44, 591, doi: 10.1111/j.1365-2184.2011.00783.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekeres T., Saiko P., Fritzer-Szekeres M., Djavan B. & Jager W. Chemopreventive effects of resveratrol and resveratrol derivatives. Ann N Y Acad Sci 1215, 89, doi: 10.1111/j.1749-6632.2010.05864.x (2011). [DOI] [PubMed] [Google Scholar]
- Elford H. L., Wampler G. L. & van’t Riet B. New ribonucleotide reductase inhibitors with antineoplastic activity. Cancer Res 39, 844 (1979). [PubMed] [Google Scholar]
- Fritzer-Szekeres M. et al. Iron binding capacity of didox (3,4-dihydroxybenzohydroxamic acid) and amidox (3,4-dihydroxybenzamidoxime) new inhibitors of the enzyme ribonucleotide reductase. Life sciences 61, 2231 (1997). [DOI] [PubMed] [Google Scholar]
- Inayat M. S. et al. Didox (a novel ribonucleotide reductase inhibitor) overcomes Bcl-2 mediated radiation resistance in prostate cancer cell line PC-3. Cancer Biol Ther 1, 539 (2002). [DOI] [PubMed] [Google Scholar]
- Raje N. et al. Didox, a ribonucleotide reductase inhibitor, induces apoptosis and inhibits DNA repair in multiple myeloma cells. Br J Haematol 135, 52, doi: 10.1111/j.1365-2141.2006.06261.x (2006). [DOI] [PubMed] [Google Scholar]
- Wakisaka N., Yoshizaki T., Raab-Traub N. & Pagano J. S. Ribonucleotide reductase inhibitors enhance cidofovir-induced apoptosis in EBV-positive nasopharyngeal carcinoma xenografts. Int J Cancer 116, 640, doi: 10.1002/ijc.21096 (2005). [DOI] [PubMed] [Google Scholar]
- Grusch M. et al. Activation of caspases and induction of apoptosis by novel ribonucleotide reductase inhibitors amidox and didox. Exp Hematol 29, 623 (2001). [DOI] [PubMed] [Google Scholar]
- Abdel-Latif G. A. et al. The chemomodulatory effects of resveratrol and didox on herceptin cytotoxicity in breast cancer cell lines. Scientific Reports 5, 12054, doi: 10.1038/srep12054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan J. et al. Oxidative stress in HIV demented patients and protection ex vivo with novel antioxidants. Neurology 60, 307 (2003). [DOI] [PubMed] [Google Scholar]
- Matsebatlela T. M., Anderson A. L., Gallicchio V. S., Elford H. & Rice C. D. 3,4-Dihydroxy-benzohydroxamic acid (Didox) suppresses pro-inflammatory profiles and oxidative stress in TLR4-activated RAW264.7 murine macrophages. Chemico-biological interactions 233, 95, doi: 10.1016/j.cbi.2015.03.027 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Abd A. M., Al-Abbasi F. A., Asaad G. F. & Abdel-Naim A. B. Didox potentiates the cytotoxic profile of doxorubicin and protects from its cardiotoxicity. Eur J Pharmacol 718, 361, doi: 10.1016/j.ejphar.2013.08.009 (2013). [DOI] [PubMed] [Google Scholar]
- Horvath Z. et al. Combination chemotherapy of BCNU and Didox acts synergystically in 9L glioma cells. Nucleosides, nucleotides & nucleic acids 23, 1531, doi: 10.1081/NCN-200027746 (2004). [DOI] [PubMed] [Google Scholar]
- Lee T. B., Park J. H., Min Y. D., Kim K. J. & Choi C. H. Epigenetic mechanisms involved in differential MDR1 mRNA expression between gastric and colon cancer cell lines and rationales for clinical chemotherapy. BMC gastroenterology 8, 33, doi: 10.1186/1471-230X-8-33 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard G. D., Fojo T. & Bates S. E. The role of ABC transporters in clinical practice. The oncologist 8, 411 (2003). [DOI] [PubMed] [Google Scholar]
- Liu Z. et al. Sinomenine sensitizes multidrug-resistant colon cancer cells (Caco-2) to doxorubicin by downregulation of MDR-1 expression. PLoS One 9, e98560, doi: 10.1371/journal.pone.0098560 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Nielsen D., Maare C. & Skovsgaard T. Cellular resistance to anthracyclines. Gen Pharmacol 27, 251 (1996). [DOI] [PubMed] [Google Scholar]
- Chen C., Jiang X., Zhao W. & Zhang Z. Dual role of resveratrol in modulation of genotoxicity induced by sodium arsenite via oxidative stress and apoptosis. Food Chem Toxicol 59, 8, doi: 10.1016/j.fct.2013.05.030 (2013). [DOI] [PubMed] [Google Scholar]
- Finucane D. M., Bossy-Wetzel E., Waterhouse N. J., Cotter T. G. & Green D. R. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. The Journal of biological chemistry 274, 2225 (1999). [DOI] [PubMed] [Google Scholar]
- Miyashita T. et al. Tumor suppressor p53 is a regulator of bcl-2 and bax gene expression in vitro and in vivo. Oncogene 9, 1799 (1994). [PubMed] [Google Scholar]
- Pucci B., Kasten M. & Giordano A. Cell cycle and apoptosis. Neoplasia (New York, NY) 2, 291 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. et al. Genistein induces G2/M cell cycle arrest and apoptosis via ATM/p53-dependent pathway in human colon cancer cells. Int J Oncol 43, 289, doi: 10.3892/ijo.2013.1946 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll U. M., Marchenko N. & Zhang X. K. p53 and Nur77/TR3 - transcription factors that directly target mitochondria for cell death induction. Oncogene 25, 4725, doi: 10.1038/sj.onc.1209601 (2006). [DOI] [PubMed] [Google Scholar]
- Cook G. J., Caudell D. L., Elford H. L. & Pardee T. S. The efficacy of the ribonucleotide reductase inhibitor Didox in preclinical models of AML. PLoS One 9, e112619, doi: 10.1371/journal.pone.0112619 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler M., Bossy-Wetzel E., Goldstein J. C., Fitzgerald P. & Green D. R. p53 induces apoptosis by caspase activation through mitochondrial cytochrome c release. The Journal of biological chemistry 275, 7337 (2000). [DOI] [PubMed] [Google Scholar]
- Hinz S. et al. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 19, 5486, doi: 10.1038/sj.onc.1203936 (2000). [DOI] [PubMed] [Google Scholar]
- Figul M., Soling A., Dong H. J., Chou T. C. & Rainov N. G. Combined effects of temozolomide and the ribonucleotide reductase inhibitors didox and trimidox in malignant brain tumor cells. Cancer Chemother Pharmacol 52, 41, doi: 10.1007/s00280-003-0611-2 (2003). [DOI] [PubMed] [Google Scholar]
- Cervantes A., Pinedo H. M., Lankelma J. & Schuurhuis G. J. The role of oxygen-derived free radicals in the cytotoxicity of doxorubicin in multidrug resistant and sensitive human ovarian cancer cells. Cancer Lett 41, 169 (1988). [DOI] [PubMed] [Google Scholar]
- Mayhew C. N. et al. In vivo and in vitro comparison of the short-term hematopoietic toxicity between hydroxyurea and trimidox or didox, novel ribonucleotide reductase inhibitors with potential anti-HIV-1 activity. Stem Cells 17, 345, doi: 10.1002/stem.170345 (1999). [DOI] [PubMed] [Google Scholar]
- Kanno S. et al. Preventive effect of trimidox on oxidative stress in U937 cell line. Biol Pharm Bull 30, 994 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang M., Zhou X. & Zhou K. Resveratrol inhibits human nasopharyngeal carcinoma cell growth via blocking pAkt/p70S6K signaling pathways. Int J Mol Med 31, 621, doi: 10.3892/ijmm.2013.1237 (2013). [DOI] [PubMed] [Google Scholar]
- Oi N. et al. Resveratrol, a red wine polyphenol, suppresses pancreatic cancer by inhibiting leukotriene A(4)hydrolase. Cancer Res 70, 9755, doi: 10.1158/0008-5472.CAN-10-2858 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling Y. H., el-Naggar A. K., Priebe W. & Perez-Soler R. Cell cycle-dependent cytotoxicity, G2/M phase arrest, and disruption of p34cdc2/cyclin B1 activity induced by doxorubicin in synchronized P388 cells. Mol Pharmacol 49, 832 (1996). [PubMed] [Google Scholar]
- Zhou R., Fukui M., Choi H. J. & Zhu B. T. Induction of a reversible, non-cytotoxic S-phase delay by resveratrol: implications for a mechanism of lifespan prolongation and cancer protection. British journal of pharmacology 158, 462, doi: BPH268 [pii]10.1111/j.1476-5381.2009.00268.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal A., Tabasum S. & Singh R. P. Berberine in combination with doxorubicin suppresses growth of murine melanoma B16F10 cells in culture and xenograft. Phytomedicine 21, 340, doi: 10.1016/j.phymed.2013.09.002 (2014). [DOI] [PubMed] [Google Scholar]
- Songgang L. et al. Somatostatin enhances the chemosensitivity of GBC-SD cell line to doxorubicin through arresting the cell cycle to S phase rather than through the P53/Bax-depended apoptosis way in vitro. Hepato-gastroenterology 56, 1253 (2009). [PubMed] [Google Scholar]
- Tyagi A. K., Singh R. P., Agarwal C., Chan D. C. & Agarwal R. Silibinin strongly synergizes human prostate carcinoma DU145 cells to doxorubicin-induced growth Inhibition, G2-M arrest, and apoptosis. Clin Cancer Res 8, 3512 (2002). [PubMed] [Google Scholar]
- Skehan P. et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 82, 1107 (1990). [DOI] [PubMed] [Google Scholar]
- Chou T. C. & Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Advances in enzyme regulation 22, 27 (1984). [DOI] [PubMed] [Google Scholar]
- Longo M. C., Berninger M. S. & Hartley J. L. Use of uracil DNA glycosylase to control carry-over contamination in polymerase chain reactions. Gene 93, 125 (1990). [DOI] [PubMed] [Google Scholar]
- Neyfakh A. A. Use of fluorescent dyes as molecular probes for the study of multidrug resistance. Experimental cell research 174, 168, doi: 0014-4827(88)90152-8 [pii] (1988). [DOI] [PubMed] [Google Scholar]