

Abstract
Antibody–drug conjugates (ADCs) represent an emerging new paradigm in cancer therapy. The approval of two ADCs has spurred considerable interest in this area of research, and over 55 ADCs are currently in clinical testing. In order to improve the clinical success rate of ADC therapy, all three components of the ADC: the antibody, linker, and payload have to be optimized. While considerable improvements have been made in antibody properties and target selection, medicinal chemistry efforts have lagged behind, and there is a significant need for innovation in linker design and payloads.
Background
The concept of antibody–drug conjugates (ADCs) evolved over 30 years ago from oncologists’ desire to improve the therapeutic index of anticancer drugs. Monoclonal antibodies that bind preferentially to tumor-associated antigens served as the means to selectively deliver a cytotoxic agent to the tumor. Thus, the ADC approach was envisioned as a means to lower the systemic toxicity of chemotherapy and achieve a higher dose in patients, resulting in greater efficacy. Early ADCs (1985–1995) sought to improve the tumor selectivity of clinically used anticancer drugs, such as doxorubicin and vinblastine.1 Lack of clinical success dampened enthusiasm in this approach and pharmaceutical companies exited the field. Analysis of the possible causes for the lack of success pointed to several factors, notable among them were the instability of the linkers that connected the antibody to the payload, and the modest potency of the cytotoxic agents. It has been estimated that ∼2 × 108 molecules of doxorubicin are required intracellularly to kill a cell, a number not achievable through antibody-mediated delivery due to moderate antigen expression (typically 1 × 105 to 1 × 106 antigens/cell) on the surface of tumor cells.
ADCs in Development
The next set of ADCs to enter the clinic incorporated purpose-developed cytotoxic agents that were ∼1000-fold more potent in vitro than doxorubicin and vinblastine. The first proof of concept with ADCs based upon a more potent payload was achieved with FDA approval in 2000 of gemtuzumab ozogamicin, for the treatment of acute myeloid leukemia. This ADC incorporated calicheamicin, a potent enediyne compound that causes double strand breaks in DNA. At the same time, compelling preclinical data with ADCs using potent tubulin polymerization inhibitors maytansinoids and auristatins were being reported.2 Despite the new data, most companies were still not ready to adopt the newer ADC technologies: in 2006, only three new ADCs commenced clinical trials (Figure 1). In 2010, the first ADC to be approved, gemtuzumab ozogamicin, was withdrawn from the market due to safety concerns. In the meantime, promising clinical data on the maytansinoid-based ADC, ado-trastuzumab emtansine (Kadcyla, T-DM1) targeting HER2, and the auristatin-based ADC, brentuximab vedotin (Adcetris) targeting CD30, were reported at scientific meetings and published in 2010.3,4 Currently, these are the only two ADCs to receive marketing authorization from the FDA. These two clinical success stories have revitalized the ADC field. New ADCs entering in the clinic saw a spike in 2011 (Figure 1).5 As of 2016, >55 ADCs, sponsored by 24 different major pharmaceutical or biotechnology companies, are in clinical testing. The overall success rate of the ADC approach for cancer treatment is still quite low, and at least 27 ADCs have been discontinued from clinical development. Thus, to become a mainstream option for cancer treatment, there is a need to improve the safety of ADCs and efficacy in more cancer types by optimizing each component: the antibody, the linker, and the cytotoxic compound.
Figure 1.
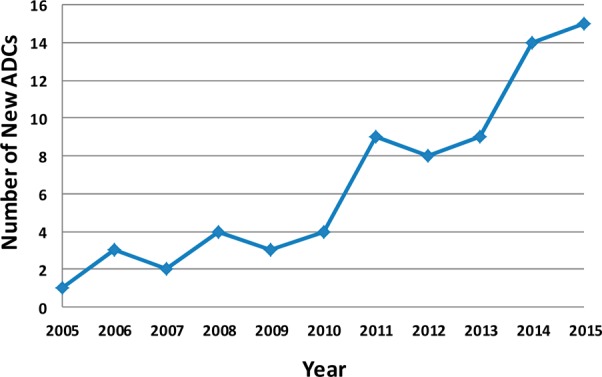
Number of new ADCs entering clinical testing each year.
The Biologists’ Contribution
There is considerable diversity in the antibodies and cell-surface antigens that are being targeted by ADCs currently in clinical evaluation. The diversity includes a broad range of tumor types (solid tumors and hematological malignancies), varying nature of the antigenic epitope (peptide, carbohydrate, glycoprotein, etc.), and antibodies with or without inherent functional activity. While HER2 is a popular target, with four different ADCs in Phase 1 clinical trials, there are antibodies to >40 distinct antigen targets in clinical evaluation as ADCs. Early ADCs to enter clinical testing elicited an immune response to the murine antibody component. With advances in antibody engineering, a majority of ADCs currently in the clinic contain “humanized” or “fully human” antibodies, and immunogenicity is rarely a limiting issue. Innovation to improve the biological properties of the antibody component of ADCs is continuing. Biparatopic antibodies that can bind two different nonoverlapping epitopes on the same target antigen, is one such example. A biparatopic antibody to HER2 was shown to cause receptor clustering, resulting in improved internalization, lysosomal trafficking, and degradation as compared to trastuzumab. An ADC of this antibody with a tubulysin-based microtubule inhibitor demonstrated good antitumor activity in some tumor xenograft models.6 Bispecific antibodies that can bind to two different antigens simultaneously offer a means of combining the binding specificity of two antibodies, thus targeting a wider population of antigen-expressing tumor cells. Nanobodies and other smaller molecular weight fragments of antibodies that have the potential for better tumor penetration are also being developed. Molecular biologists have also engineered antibodies to incorporate amino acids with functional groups that enable site-specific conjugation of a set number of molecules of a cytotoxic payload to provide homogeneous ADCs. The first innovation in this area described the incorporation of cysteine residues at specific sites on the antibody.7 Subsequently, a number of other site-specific conjugation approaches have been reported.8
A Challenge for the Medicinal Chemists
The only two FDA-approved ADCs use microtubule inhibitors (a maytansinoid and an auristatin) as the cytotoxic agent, spurring the advancement to the clinic of many more ADCs with payloads from these two classes. However, the success rate of ADCs of microtubule inhibitors is quite low as 24 of them appear to have been discontinued from clinical development, mainly due to insufficient efficacy. Nevertheless, medicinal chemists continue to design and synthesize tubulin agents, which are analogues of the parent auristatins (MMAE, MMAF) and maytansinoids (DM1, DM4), and in a few cases, tubulysins, a family of compounds that have similar mechanism of action and potency as the auristatins and maytansinoids. At least six companies have disclosed new auristatin analogues in patent applications or peer-reviewed publications.9 It is not clear whether any of these auristatin analogues offer a significant advantage over the parent compound or merely provide a proprietary position for the company. It is customary in the pharmaceutical industry to go after validated disease targets and develop compounds that differentiate from the lead molecule in the field. After the discovery that inhibition of HMG-CoA reductase could be an effective means of lowering plasma cholesterol in humans, the first such inhibitor, lovastatin, received FDA approval in 1987. Subsequently, six other statins that inhibit the same enzyme have been approved and commercially launched. However, cancer is not one disease that is treatable by a single class of compounds. There are >200 approved anticancer drugs, with diverse mechanisms of action. Why then is this type of diversity missing in the payloads used in ADCs? It is indeed easier to design analogues of known molecules. However, the low success rate of ADC can be attributed, at least in part, to the use of payloads with the same mechanism of cell killing for every antigen target and every type of cancer. Thus, ∼70% of the 50+ ADCs currently in clinical evaluation use inhibitors of tubulin polymerization as the payload, regardless of whether the disease type is known to be sensitive to compounds with that mechanism of action (Figure 2a).
Figure 2.
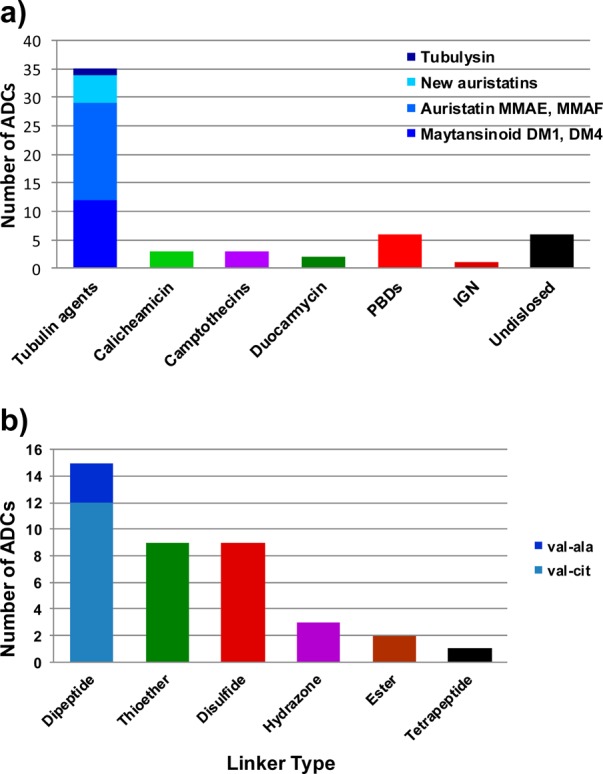
(a) Payload classes used in ADCs currently in clinical testing. (b) Linker classes used in ADCs currently in clinical testing.
There are now a growing number of ADCs that incorporate one class of DNA cross-linkers, the pyrrolobenzodiazepines (PBDs), as the cytotoxic payload. Cytotoxic agents that target DNA, either through direct interaction (e.g., doxorubicin, platinum-based compounds) or indirectly (topoisomerase inhibitors, antimetabolites such as 5-fluorouracil and gemcitabine) are widely used in cancer therapy, often in a front line setting. Given the sensitivity of cancer cells to DNA-interacting agents, there is a need for novel agents from this class for use as ADC payloads. There are indeed challenges to finding a cytotoxic molecule for use in ADCs that meets the unique requirements of high in vitro potency (IC50 in the subnanomolar range) and adequate aqueous solubility and stability. In addition, the structure of the chosen molecule should be amenable to chemical modification for linkage to antibodies. However, these are not insurmountable hurdles but may require scouring the literature for potent compounds or going back and analyzing the library of natural products or synthetic compounds in a company’s repository to identify those that meet these basic criteria. Surely, there were compounds, such as highly potent kinase inhibitors, that were deemed too toxic or suffered from low cell permeability, which could not be developed as small molecules. After all, maytansinoids and auristatins that are used in ADCs were derived from the potent parent compounds maytansine and dolastatin 10, both of which were too toxic on their own in the clinic to be therapeutically useful.
Another important element of ADC design that has not seen much diversity is the linker that connects the antibody to the cytotoxic molecule. Linkers serve the dual purpose of stably connecting the payload to the targeting agent during administration and circulation, and then efficiently and completely releasing the payload in the tumor microenvironment. Brentuximab vedotin uses a valine-citrulline dipeptide linker that is cleaved by cathepsin B, while ado-trastuzumab emtansine uses a thioether linkage that relies on lysosomal degradation of the antibody component to release the payload. Besides these two types of linkages, several ADCs employ disulfide linkers that can be reductively cleaved inside a cell to release the active payload. The linker choice in most of the ADCs currently in the clinic has been biased toward these three classes, most likely because they are the only ones that have been clinically validated (Figure 2b). Innovation in linker chemistry has the potential to significantly impact safety and efficacy of ADCs. Thus, linkers can be designed to release catabolites that are detoxified, for example, by oxidation in the liver, resulting in lower systemic toxicity. Peptide linkers that are selectively and more efficiently proteolyzed by lysosomal enzymes in cancer cells to release active catabolites could improve safety and antitumor activity. One such linker was recently described.10 Linker designs that release metabolites that are potent and cell-permeable could diffuse into and kill proximal bystander cells resulting in greater efficacy against tumors that express the antigen heterogeneously. Linkers that do not rely on lysosomal cleavage to release active drug may provide the opportunity to use antibodies that do not efficiently traffic through lysosomes.
The ADC field affords a unique opportunity for medicinal chemists to innovate to improve the antitumor activity and therapeutic index of ADCs and thus affect the therapeutic outcome for patients. With the large number of pharmaceutical and biotechnology companies actively involved in ADC research and development, there is expectation that chemists will contribute to advancement of the field. Recent publications on new payload and linkers, although still sparse, along with understanding of the mechanism of action of ADCs, are encouraging signs that chemistry efforts to optimize ADCs are being prioritized and that some progress is now being made.11,12
Acknowledgments
I thank Heather Donaghy of Hanson Wade for providing information from Beacon ADC for Figures 1 and 2.
Views expressed in this editorial are those of the author and not necessarily the views of the ACS.
The author declares the following competing financial interest(s): I am an employee and stockholder of ImmunoGen, Inc.
References
- Chari R. V. Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc. Chem. Res. 2008, 41, 98–107. 10.1021/ar700108g. [DOI] [PubMed] [Google Scholar]
- Chari R. V.; Miller M. L.; Widdison W. C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Krop I. E.; Beeram M.; Modi S.; Jones S. F.; Holden S. N.; Yu W.; Girish S.; Tibbitts J.; Yi J. H.; Sliwkowski M. X.; Jacobson F.; Lutzker S. G.; Burris H. A. Phase I study of trastuzumab-DM1, an HER2 antibody-drug conjugate, given every 3 weeks to patients with HER2-positive metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2698–2704. 10.1200/JCO.2009.26.2071. [DOI] [PubMed] [Google Scholar]
- Younes A.; Bartlett N. L.; Leonard J. P.; Kennedy D. A.; Lynch C. M.; Sievers E. L.; Forero-Torres A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. 10.1056/NEJMoa1002965. [DOI] [PubMed] [Google Scholar]
- Donaghy H. Personal communication. Hanson Wade, 2016. [Google Scholar]
- Li J. Y.; Perry S. R.; Muniz-Medina V.; Wang X.; Wetzel L. K.; Rebelatto M. C.; Hinrichs M. J.; Bezabeh B. Z.; Fleming R. L.; Dimasi N.; Feng H.; Toader D.; Yuan A. Q.; Xu L.; Lin J.; Gao C.; Wu H.; Dixit R.; Osbourn J. K.; Coats S. R. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. 10.1016/j.ccell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; Lu Y.; Meng Y. G.; Ng C.; Yang J.; Lee C. C.; Duenas E.; Gorrell J.; Katta V.; Kim A.; McDorman K.; Flagella K.; Venook R.; Ross S.; Spencer S. D.; Lee Wong W.; Lowman H. B.; Vandlen R.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Mallet W. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 2015, 26, 176–192. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderna A.; Doroski M.; Subramanyam C.; Porte A.; Leverett C. A.; Vetelino B. C.; Chen Z.; Risley H.; Parris K.; Pandit J.; Varghese A. H.; Shanker S.; Song C.; Sukuru S. C.; Farley K. A.; Wagenaar M. M.; Shapiro M. J.; Musto S.; Lam M. H.; Loganzo F.; O’Donnell C. J. Discovery of cytotoxic dolastatin 10 analogues with N-terminal modifications. J. Med. Chem. 2014, 57, 10527–10543. 10.1021/jm501649k. [DOI] [PubMed] [Google Scholar]
- Singh R.; Setiady Y. Y.; Ponte J.; Kovtun Y. V.; Lai K. C.; Hong E. E.; Fishkin N.; Dong L.; Jones G. E.; Coccia J. A.; Lanieri L.; Veale K.; Costoplus J. A.; Skaletskaya A.; Gabriel R.; Salomon P.; Wu R.; Qiu Q.; Erickson H. K.; Lambert J. M.; Chari R. V.; Widdison W. C. A New Triglycyl Peptide Linker for Antibody-Drug Conjugates (ADCs) with Improved Targeted Killing of Cancer Cells. Mol. Cancer Ther. 2016, 15, 1311–1320. 10.1158/1535-7163.MCT-16-0021. [DOI] [PubMed] [Google Scholar]
- Miller M. L.; Fishkin N. E.; Li W.; Whiteman K. R.; Kovtun Y.; Reid E. E.; Archer K. E.; Maloney E. K.; Audette C. A.; Mayo M. F.; Wilhelm A.; Modafferi H. A.; Singh R.; Pinkas J.; Goldmacher V.; Lambert J. M.; Chari R. V. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–1878. 10.1158/1535-7163.MCT-16-0184. [DOI] [PubMed] [Google Scholar]
- Puthenveetil S.; Loganzo F.; He H.; Dirico K.; Green M.; Teske J.; Musto S.; Clark T.; Rago B.; Koehn F.; Veneziale R.; Falahaptisheh H.; Han X.; Barletta F.; Lucas J.; Subramanyam C.; O’Donnell C. J.; Tumey L. N.; Sapra P.; Gerber H. P.; Ma D.; Graziani E. I. Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjugate Chem. 2016, 27, 1880–1888. 10.1021/acs.bioconjchem.6b00291. [DOI] [PubMed] [Google Scholar]