

Abstract
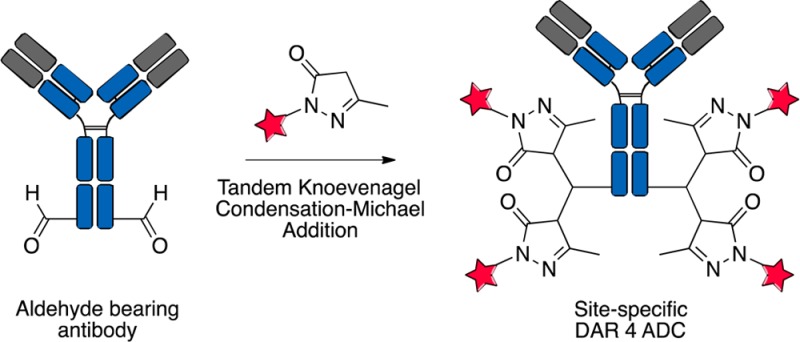
Expanded ligation techniques are sorely needed to generate unique linkages for the growing field of functionally enhanced proteins. To address this need, we present a unique chemical ligation that involves the double addition of a pyrazolone moiety with an aldehyde-labeled protein. This ligation occurs via a tandem Knoevenagel condensation–Michael addition. A pyrazolone reacts with an aldehyde to generate an enone, which undergoes subsequent attack by a second pyrazolone to generate a bis-pyrazolone species. This rapid and facile ligation technique is performed under mild conditions in the absence of catalyst to generate new architectures that were previously inaccessible via conventional ligation reactions. Using this unique ligation, we generated three site-specifically labeled antibody–drug conjugates (ADCs) with an average of four drugs to one antibody. The in vitro and in vivo efficacies along with pharmacokinetic data of the site-specific ADCs are reported.
Keywords: Antibody−drug conjugate (ADC), site-specific bioconjugation, bivalent drugs, tandem reactions
Functionally enhanced proteins are of growing promise in the therapeutic arena.1,2 The lynch pin holding the field together is the ligation chemistry that unites the protein with its payload. Historically, protein modification has relied on the functionalization of native residues, such as Glu, Asp, Lys, and Cys. However, the abundance of these residues within a protein results in nonspecific and promiscuous conjugation of the payload of interest.3 In order to access new and expanded utilities, new conjugation chemistries must be developed to broaden the available conjugation chemistry tool kit, especially outside the realm of native amino acids, the use of which often results in stochastic mixtures of labeled proteins.
Antibody–drug conjugates (ADCs) are a field that will directly benefit from the intense exploration of new conjugation techniques. ADCs combine the targeted specificity and long circulating half-life of an antibody with the lethal potency of a cytotoxic chemotherapy. First-generation ADCs are prepared using conventional ligation chemistries resulting in heterogeneous mixtures containing average drug-to-antibody ratios (DARs) ranging from zero to eight.4 These mixtures contain unique chemical entities that exhibit differing potencies, pharmacokinetics (PK), and toxicities.5 The complexities in activity, analysis, and manufacturing associated with ADCs synthesized by conventional ligation have created a strong push toward using site-specific chemistries that promise homogeneous entities with well-defined, monodisperse DARs and payload placement.
Site-specific bioconjugation can be implemented with a bioorthogonal handle on which chemistry can be performed.2 Aldehydes are an attractive option for handles due to their ability to selectively react with nucleophiles in the presence of diverse sets of functionality. Our group utilizes an aldehyde in the context of the formylglycine (fGly) amino acid residue. The fGly is produced through the highly selective oxidation of a cysteine residue found within a specific pentapeptide consensus sequence by formylglycine-generating enzyme (FGE).6 The fGly-containing protein can then be modified using aldehyde-specific chemistries (Scheme 1A).7,8
Scheme 1. Overview of the Tandem-Knoevenagel Condensation–Michael Addition Ligation Using a Pyrazolone Nucleophile.
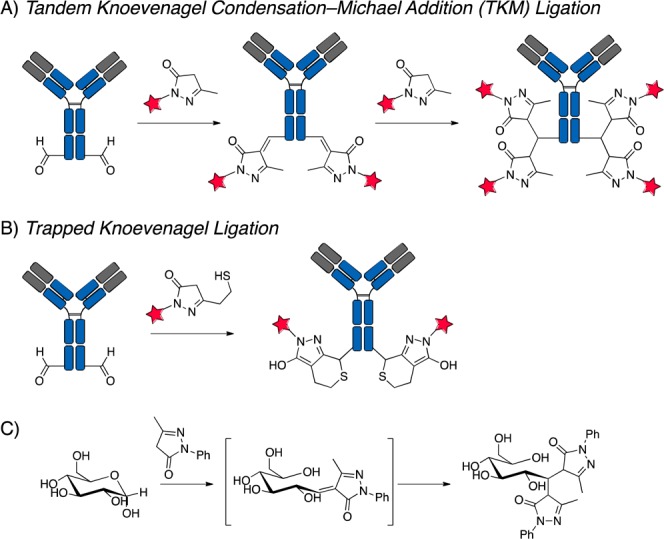
(A) Generation of a DAR 4 site-specific ADC via first addition of pyrazolone via Knoevenagel condensation, then subsequent second addition of pyrazolone via Michael addition. (B) Previously published trapped-Knoevenagel ligation. (C) Double addition of pyrazolone to reducing sugar via a tandem Knoevenagel condensation–Michael addition.
The value of site-specific chemistry and the connections used to create the bioconjugates is becoming more apparent as it has been shown that ADCs generated using these methods possess increased therapeutic indices and increased therapeutic activities.9,10 In addition to these significant advances, there is mounting literature that demonstrates that payload placement is an increasingly important design parameter.11−13
A key design component of an ADC is the average DAR. In a seminal publication, Hamblett et al.5 showed that the in vivo efficacy of a DAR 4 species was equal to that of a DAR 8 species due to improved PK. Recent evidence has shown that even DAR 8 species, if sufficiently hydrophilic, can exhibit potent in vivo efficacy and good clearance properties.14 This observation has opened the door to a reexamination of higher DAR species13 especially in the context of targets with low antigen copy number where delivery of more drug/antibody could potentially enhance therapeutic outcomes.
The potency effects of DAR 4 species in conjunction with the compelling site-specific ADC literature suggests that it would be of significant interest to examine site-specific ADCs with higher average DARs. While there exist means to form homogeneous DAR 4 species using the interchain disulfides of a native antibody, the payload placement is restricted to cysteine locations that do not significantly interfere with endogenous cysteines involved in disulfide bonding.15 Ideally, one would vary the site at which the payloads are placed allowing for insight into structure–activity relationships.
There are at least two straightforward approaches to generate DAR 4 species in a site-specific manner. First, incorporation of two separate conjugation sites, which collectively would allow for a DAR 4 species with the payload located at two distinct amino acid residue positions.13 Second, chemical synthesis of a linker that contains two cleavable payloads attached to a single conjugatable group, which would allow for well-defined, monodisperse DAR 4 species at one amino acid residue (tag-site). We envisioned a third, less obvious approach employing a tandem process based on two additions of linker-drug to the same aldehyde using the same nucleophile as shown in Scheme 1A.
In this letter, we present a simple and mild one-pot conjugation chemistry that allows access to well-defined, monodisperse, and site-specific DAR 4 ADCs via a tandem Knovenagel condensation–Michael addition (TKM). We then examined the impact of payload placement at three different sites on the antibody.
Previously, we presented a trapped-Knoevenagel ligation where a stabilized carbanion originating from a pyrazolone reacts with an aldehyde to form an olefin. This olefin is then subject to Michael addition via a pendant thiol (Scheme 1B). Work performed by Honda et al.16 (Scheme 1C) showed that when pyrazolones react with reducing sugars under mild conditions, a double addition product is formed via an initial Knoevenagel condensation followed by rapid Michael addition by a second pyrazolone moiety. The Michael addition is proposed to be faster than the Knoevenagel condensation, as only double addition products are formed. The extensive work done on carbohydrate derivitization using pyrazolones under mild conditions emboldened us to try using this pyrazolone double addition as an aldehyde-based conjugation chemistry. We postulated that we could harness this rapid second addition via a tandem Knovenagel condensation–Michael addition to form homogeneous site-specific ADCs with a DAR of 4 under physiological conditions.
In order to achieve a DAR 4 ADC, it is important to use a cleavable construct because a noncleavable double addition adduct would result in a DAR 2 ADC with the payload being a bivalent drug. With this in mind we chose to synthesize compound 8, which would release compound 5 as our cytotoxic payload. As shown in Figure S1, the valine-citrulline dipeptide is cleaved by cathepsin B releasing the p-aminobenzylcarbamate (PABC) self-immolative spacer, which undergoes a 1,6-elimination to liberate carbon dioxide and cytotoxic compound 5.17
The pyrazolone acid 2 was synthesized in two steps from ethyl acetoacetate and the ethyl hydrazinoacetate hydrochloride salt.8 In two steps, this was converted to compound 3 (Scheme 2A). The maytansinoid payload was synthesized starting from previously reported compound 4, which was coupled to Fmoc-N-methyl-γ-aminobutyric acid followed by deprotection to afford compound 5 in 73% yield. Amine 5 was then reacted with the commercially available peptide 6, and the resulting carbamate was treated with piperidine to afford free amine 7 in 59% yield over two steps. Coupling of this to the pyrazolone acid 3 in 48% yield provided the desired pyrazolone construct 8 containing the cleavable maytansine moiety (Scheme 2B).
Scheme 2. Synthesis of Building Blocks and Conjugation Construct.

(A) Synthetic route to pyrazolone acid construct (3). (B) Synthetic route to pyrazolone conjugation construct (8).
As there have been several papers highlighting the importance of site-specificity with ADCs,10−13,20 we sought to explore this new conjugation technique in that context. In order to assess the impact of payload placement, we used three differentially tagged antibodies bearing aldehydes either at the heavy chain C-terminus (CT), in the CH1 domain (CH1), or at the hinge region (H) (Figure 1A). The antibodies all had the same variable region, which was directed against an undisclosed epitope found on liquid tumors. The fGly-containing antibodies were conjugated to compound 8 at 15 mg/mL and 8.3 drug/antibody equivalents for 16 h at 37 °C in 50 mM Na-citrate/50 mM NaCl, pH 7.2 or 6.0 in 5% DMA and 0.085% Triton X-100. There was no observable loss of protein or increase in aggregate under the conjugation conditions. At pH 6.0, the fastest initial pyrazolone addition occurred at the CT, followed by the CH1 position, and finally the hinge region (Figure S2A). The same trend existed for the rate of the second addition of the pyrazolone via Michael addition (Figure S2B). Presumably, this trend is a function of sterics at the site of the aldehyde. The more accessible CT has the fastest rate of addition, and all second additions are slowed due to the increased steric density resulting from the first addition. Interestingly, at all conjugation sites the rates of first and second addition were retarded slightly at pH 7.2 relative to pH 6.0 (Figure S2A,B). Mass spectrometric analysis confirmed the double addition of compound 8 on the tagged antibody heavy chain (Figure S3). Additionally, a control experiment showed that no reaction occurred on an untagged antibody (Figure S4).
Figure 1.
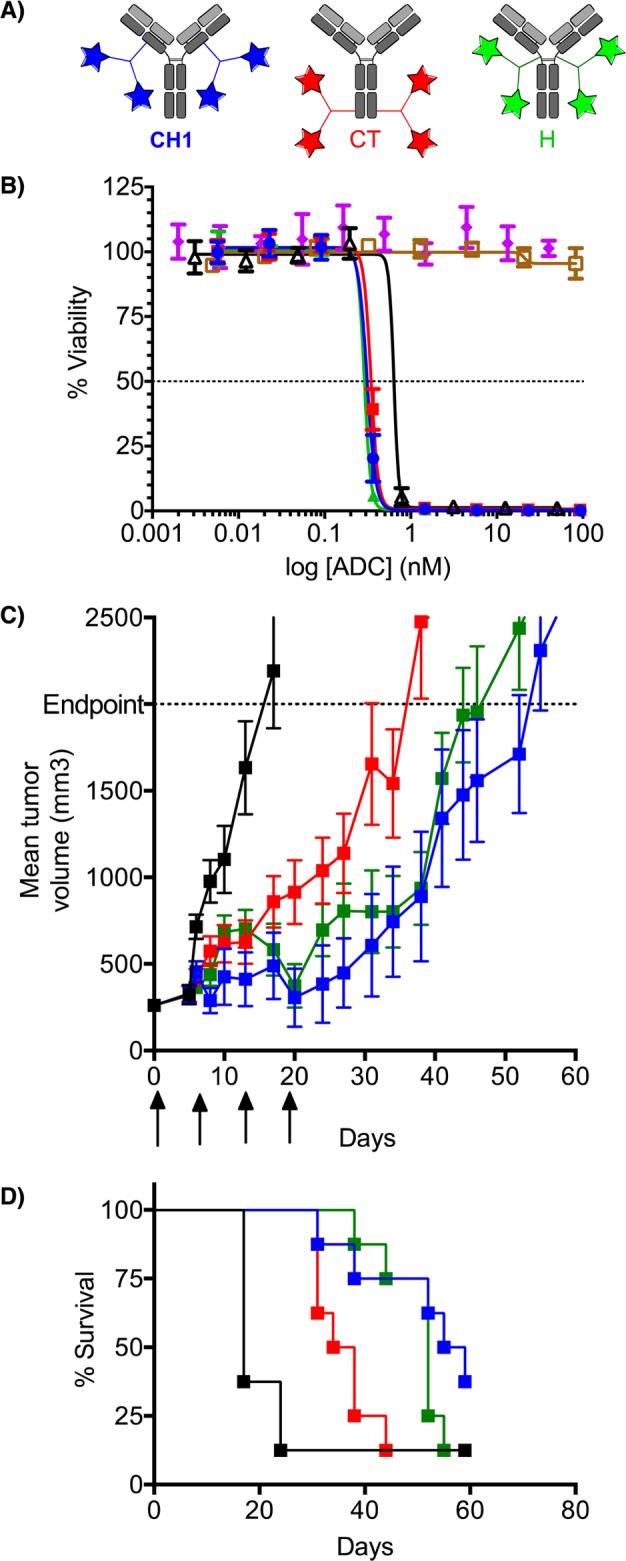
Site-specific DAR 4 ADCs generated using the TKM ligation display high potency in in vitro and in vivo tumor models. (A) ADCs with payloads placed at CH1 (blue), CT (red), and Hinge (green). (B) In vitro cytotoxicity. Cells were treated with varying concentrations of maytansine (black), compound 5 (purple), antigen-targeted ADCs conjugated at the CH1 (blue), CT (red), or Hinge (green) positions, and an isotype control ADC (gold). Error bars = SD; n = 9. (C,D) The in vivo efficacy of ADCs bearing payloads at the CH1 (blue), CT (red), or hinge (green) positions was compared to the vehicle-treated negative control group. Mean tumor volume ((C) error bars = SEM) and survival (D) are shown.
The three ADCs were examined for in vitro potency against an antigen-positive cell line (antigen copy number ∼10000–40000/cell); free maytansine was included as a positive control, and an isotype anitbody conjugated to compound 8 at the CT was included as a negative control. As shown in Figure 1B, all antigen-targeting ADCs exhibited potent dose-dependent toxicity with IC50 values of 74, 66, and 61 pM (antibody) for the CT-, CH1-, and hinge-tagged constructs, respectively, as compared to 207 pM for the natural product maytansine. Compound 5 displayed no activity. The low IC50 values demonstrate the efficient internalization of the ADC and the effective release of the cleavable payload. The isotype control exhibited no effect on cell growth at the doses administered, highlighting the antigen specific response and the chemical stability of the TKM ligation linkage. We did not observe meaningful differentiation among the three payload placements with respect to in vitro activity.
Next, we examined the TKM ADCs in an in vivo efficacy study in mice bearing the antigen-expressing WSU-DLCL2 xenograft. The ADCs were dosed intravenously at 10 mg/kg every 4 days for a total of four doses. ADCs bearing the payload at the CH1, the hinge (H), and the CT position exhibited 77, 73, and 60% tumor growth inhibition, respectively, as compared to the vehicle control group at day 15 (Figure 1C). After the last dose at day 12, the tumors in mice treated with CT-tagged ADC (red) began to regrow immediately, whereas the tumors in the mice dosed with the other ADCs did not begin to regrow for another 10 days. This disparity is reflected in the survival curves (Figure 1D) and the resulting tumor growth delay (TGD) values: 115, 106, and 57% TGD for groups treated with ADCs conjugated at the CH1, hinge, or CT sites, respectively.
Until recently,7,8 oximes were the default conjugation technique used with carbonyl-labeled proteins. The major drawbacks of oxime ligation are the slow rate of reaction and the low pH requirement (pH 4.6) for the conjugation to occur. This limits the oxime ligation utility, as not all proteins are stable under these conditions.18 While there have been advancements in oxime formation catalysts that shift the pH closer to neutral,19 the oxime is subject to hydrolysis and has limited serum stability.7 The TKM ligation is performed under physiological conditions in citrate buffer (pH 7.2) and creates a C–C bond that is not subject to hydrolysis.
In an effort to understand the in vivo efficacy differences observed among the payload locations, we conducted a PK study in rats. Previous data from our group has shown that payload conjugation to an inserted aldehyde tag need not markedly change the basic PK properties of an antibody.12 The total antibody half-life for the CT DAR 4 ADC was the shortest at 4.1 days, while the CH1 and H were markedly better at 12.0 and 11.7 days, respectively. The two payload locations that resulted in the strongest in vivo efficacy, CH1 and H, also were the most stable in circulation, with total ADC half-lives of 5.8 and 5.2 days, respectively. Regarding the difference between conjugate and antibody half-lives, a small molecule model system showed that the newly formed C–C bonds were stable at 37 °C, pH 7.4 over 4 days (Figure S5), indicating that this connection likely persisted in vivo. By contrast, we have previously observed hydrolysis of maytansine to release maytansinol;12 this may account for the differentiation between total antibody and total ADC.
In a demonstration of the importance of site placement, the CT conjugate had an ADC half-life of only 1.2 days (Figure 2). The rapid clearance of this conjugate could explain its relatively poor in vivo efficacy and could be a consequence of placing four hydrophobic maytansine payloads close to one another, thus drastically increasing the local hydrophobicity of the molecule. Intriguingly, the HIC retention times, which are a proxy for hydrophobicity of the overall construct, correspond closely to the PK outcomes,13,14 with the CT-tagged conjugate demonstrating both the longest HIC retention time and the worst PK performance (Table 1).
Figure 2.
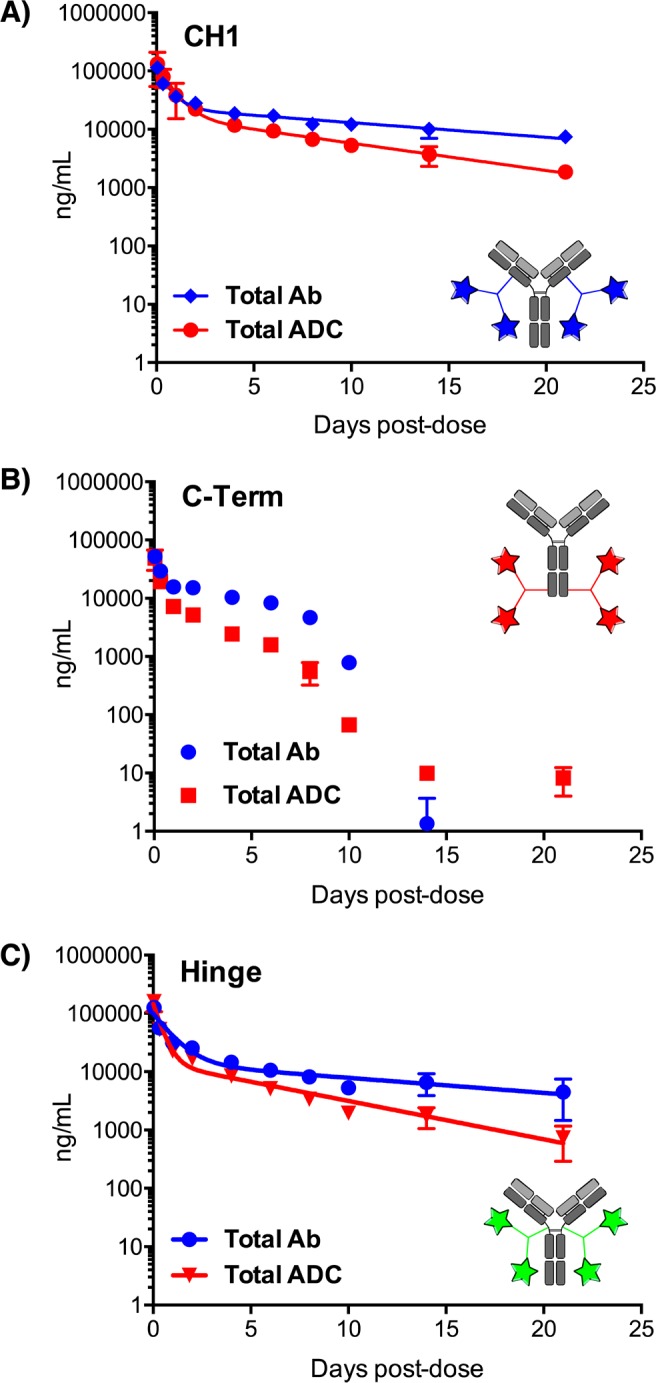
Pharmacokinetic stability of the site-specific DAR 4 ADCs varies with placement of the payload. Rats were dosed with 3 mg/kg of site-specific ADC conjugated to the (A) CH1, (B) CT, or (C) Hinge sites.
Table 1. Examination of the Effect of Payload Placement on Antibody Half-Life, ADC Half-Life, and ADC Retention Time Using a Hydrophobic Interaction Column (HIC).
| payload placement | half-life total Ab (days) | half-life ADC (days) | HIC retention time (min) |
|---|---|---|---|
| CH1 | 12 ± 2.0 | 5.8 ± 0.6 | 8.17 |
| Hinge | 11.7 ± 4.0 | 5.2 ± 0.9 | 7.64 |
| C-Term | 4.1 ± 0.6 | 1.2 ± 0.3 | 9.01 |
Here, we have developed a unique new bioconjugation technique that allows access to well-defined, monodisperse, and site-specific DAR 4 ADCs via a tandem Knoevenagel condensation–Michael addition, where the pyrazolone nucleophile reacts with an aldehyde to form a reactive enone. This Michael acceptor is then subject to attack by a second molecule of the same pyrazolone to form the double addition species. This ligation occurs under neutral pH in the absence of catalyst, minimizing perturbation of essential amino acid residues in the variable region of the antibody (e.g., deamidation of asparagine residues that might occur with maleidimide chemistries). We have examined this new TKM ligation in the context of site-specific payload placement of DAR 4 ADCs. Three different DAR 4 ADCs were synthesized with the payloads placed at the CT, CH1, and hinge regions. In vivo efficacy and PK data revealed that the location of the payload affects both PK and efficacy. These observations are corroborated by studies from Strop et al. showing that site-specific high DAR species prepared via multiple tag-sites have poor PK when hydrophobic payloads are placed in high spatial concentration.13 We envision that this unique ligation chemistry, which affords ready access to bivalent molecules, will prove valuable both for ADC generation and to the bioconjugation community as a whole.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00253.
Five figures, experimental details, and characterization data for new compounds (PDF)
The authors declare the following competing financial interest(s): All authors are employees of Catalent Pharma Solutions.
Supplementary Material
References
- Sievers E. L.; Senter P. D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-Specific Antibody–Drug Conjugates: the Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26 (2), 176. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephanopoulos N.; Francis M. B. Choosing an Effective Protein Bioconjugation Strategy. Nat. Chem. Biol. 2011, 7 (12), 876–884. 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- Kim M. T.; Chen Y.; Marhoul J.; Jacobson F. Statistical Modeling of the Drug Load Distribution on Trastuzumab Emtansine (Kadcyla), a Lysine-Linked Antibody Drug Conjugate. Bioconjugate Chem. 2014, 25 (7), 1223–1232. 10.1021/bc5000109. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M. C.; Lenox J. S.; Cerveny C. G.; Kissler K. M.; Bernhardt S. X.; Kopcha A. K.; Zabinski R. F.; Meyer D. L.; Francisco J. A. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Rabuka D.; Rush J. S.; deHart G. W.; Wu P.; Bertozzi C. R. Site-Specific Chemical Protein Conjugation Using Genetically Encoded Aldehyde Tags. Nat. Protoc. 2012, 7 (6), 1052–1067. 10.1038/nprot.2012.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Kudirka R.; Albers A. E.; Barfield R. M.; de Hart G. W.; Drake P. M.; Jones L. C.; Rabuka D. Hydrazino-Pictet-Spengler Ligation as a Biocompatible Method for the Generation of Stable Protein Conjugates. Bioconjugate Chem. 2013, 24 (6), 846–851. 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- Kudirka R.; Barfield R. M.; McFarland J.; Albers A. E.; de Hart G. W.; Drake P. M.; Holder P. G.; Banas S.; Jones L. C.; Garofalo A. W.; Rabuka D. Generating Site-Specifically Modified Proteins via a Versatile and Stable Nucleophilic Carbon Ligation. Chem. Biol. 2015, 22, 293. 10.1016/j.chembiol.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; Lu Y.; Meng Y. G.; Ng C.; Yang J.; Lee C. C.; Duenas E.; Gorrell J.; Katta V.; Kim A.; McDorman K.; Flagella K.; Venook R.; Ross S.; Spencer S. D.; Lee Wong W.; Lowman H. B.; Vandlen R.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Mallet W. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 2008, 26 (8), 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Pillow T. H.; Tien J.; Parsons-Reponte K. L.; Bhakta S.; Li H.; Staben L. R.; Li G.; Chuh J.; Fourie-O’Donohue A.; Darwish M.; Yip V.; Liu L.; Leipold D. D.; Su D.; Wu E.; Spencer S. D.; Shen B.-Q.; Xu K.; Kozak K. R.; Raab H.; Vandlen R.; Lewis Phillips G. D.; Scheller R. H.; Polakis P.; Sliwkowski M. X.; Flygare J. A.; Junutula J. R. Site-Specific Trastuzumab Maytansinoid Antibody–Drug Conjugates with Improved Therapeutic Activity Through Linker and Antibody Engineering. J. Med. Chem. 2014, 57 (19), 7890–7899. 10.1021/jm500552c. [DOI] [PubMed] [Google Scholar]
- Strop P.; Liu S.-H.; Dorywalska M.; Delaria K.; Dushin R. G.; Tran T.-T.; Ho W.-H.; Farias S.; Casas M. G.; Abdiche Y.; Zhou D.; Chandrasekaran R.; Samain C.; Loo C.; Rossi A.; Rickert M.; Krimm S.; Wong T.; Chin S. M.; Yu J.; Dilley J.; Chaparro-Riggers J.; Filzen G. F.; O’Donnell C. J.; Wang F.; Myers J. S.; Pons J.; Shelton D. L.; Rajpal A. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20 (2), 161–167. 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Drake P. M.; Albers A. E.; Baker J.; Banas S.; Barfield R. M.; Bhat A. S.; de Hart G. W.; Garofalo A. W.; Holder P.; Jones L. C.; Kudirka R.; McFarland J.; Zmolek W.; Rabuka D. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014, 25 (7), 1331–1341. 10.1021/bc500189z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strop P.; Delaria K.; Foletti D.; Witt J. M.; Hasa-Moreno A.; Poulsen K.; Casas M. G.; Dorywalska M.; Farias S.; Pios A.; Lui V.; Dushin R.; Zhou D.; Navaratnam T.; Tran T.-T.; Sutton J.; Lindquist K. C.; Han B.; Liu S.-H.; Shelton D. L.; Pons J.; Rajpal A. Site-Specific Conjugation Improves Therapeutic Index of Antibody Drug Conjugates with High Drug Loading. Nat. Biotechnol. 2015, 33 (7), 694–696. 10.1038/nbt.3274. [DOI] [PubMed] [Google Scholar]
- Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-LaFord H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D.; Lyon R. P. Reducing Hydrophobicityof Homogeneous Antibody-Drug Conjugates Improvespharmacokinetics Andtherapeutic Index. Nat. Biotechnol. 2015, 33, 733. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Badescu G.; Bryant P.; Bird M.; Henseleit K.; Swierkosz J.; Parekh V.; Tommasi R.; Pawlisz E.; Jurlewicz K.; Farys M.; Camper N.; Sheng X.; Fisher M.; Grygorash R.; Kyle A.; Abhilash A.; Frigerio M.; Edwards J.; Godwin A. Bridging Disulfides for Stable and Defined Antibody Drug Conjugates. Bioconjugate Chem. 2014, 25 (6), 1124–1136. 10.1021/bc500148x. [DOI] [PubMed] [Google Scholar]
- Kakehi K.; Ueda M.; Suzuki S.; Honda S. Determination of Hyaluronic Acid by High-Performance Liquid Chromatography of the Oligosaccharides Derived Therefrom as 1-(4-Methoxy)Phenyl-3-Methyl-5-Pyrazolone Derivatives. J. Chromatogr. 1993, 630 (1–2), 141–146. 10.1016/0021-9673(93)80449-I. [DOI] [PubMed] [Google Scholar]
- Dubowchik G. M.; Firestone R. A.; Padilla L.; Willner D.; Hofstead S. J.; Mosure K.; Knipe J. O.; Lasch S. J.; Trail P. A. Cathepsin B-Labile Dipeptide Linkers for Lysosomal Release of Doxorubicin From Internalizing Immunoconjugates: Model Studies of Enzymatic Drug Release and Antigen-Specific in Vitro Anticancer Activity. Bioconjugate Chem. 2002, 13 (4), 855–869. 10.1021/bc025536j. [DOI] [PubMed] [Google Scholar]
- Liu J.; Hanne J.; Britton B. M.; Shoffner M.; Albers A. E.; Bennett J.; Zatezalo R.; Barfield R.; Rabuka D.; Lee J.-B.; Fishel R. An Efficient Site-Specific Method for Irreversible Covalent Labeling of Proteins with a Fluorophore. Sci. Rep. 2015, 5, 16883. 10.1038/srep16883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisalli P.; Kool E. T. Importance of orthoProton Donors in Catalysis of Hydrazone Formation. Org. Lett. 2013, 15 (7), 1646. 10.1021/ol400427x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers A. E.; Garofalo A. W.; Drake P. M.; Kudirka R.; de Hart G. W.; Barfield R. M.; Baker J.; Banas S.; Rabuka D. Exploring the effects of linker composition on site-specifically modified antibody-drug conjugates. Eur. J. Med. Chem. 2014, 88, 1–7. 10.1016/j.ejmech.2014.08.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.