

Abstract
As part of our efforts to develop new classes of tubulin inhibitor payloads for antibody–drug conjugate (ADC) programs, we developed a tubulysin ADC that demonstrated excellent in vitro activity but suffered from rapid metabolism of a critical acetate ester. A two-pronged strategy was employed to address this metabolism. First, the hydrolytically labile ester was replaced by a carbamate functional group resulting in a more stable ADC that retained potency in cellular assays. Second, site-specific conjugation was employed in order to design ADCs with reduced metabolic liabilities. Using the later approach, we were able to identify a conjugate at the 334C position of the heavy chain that resulted in an ADC with considerably reduced metabolism and improved efficacy. The examples discussed herein provide one of the clearest demonstrations to-date that site of conjugation can play a critical role in addressing metabolic and PK liabilities of an ADC. Moreover, a clear correlation was identified between the hydrophobicity of an ADC and its susceptibility to metabolic enzymes. Importantly, this study demonstrates that traditional medicinal chemistry strategies can be effectively applied to ADC programs.
Keywords: Antibody−drug conjugate (ADC), tubulysin, hydrophobic interaction chromatograph (HIC), plasma stability
Targeted delivery of cytotoxic agents directly into tumor cells has long been speculated as a potential breakthrough for the treatment of cancer. This speculation was transformed into reality with the FDA approval of the first antibody–drug conjugate (ADC), gemtuzumab ozogamicin (Mylotarg), which utilizes a DNA damaging agent as a payload.1 While regulatory concerns have precluded its broad utility,2 a second generation of ADCs has emerged that takes advantage of potent tubulin binding agents that interfere with cell division machinery. Maytansinoid and auristatin payloads have been particularly useful in this regard, as evidenced by the clinical and regulatory successes of two recently approved ADCs, ado-trastuzumab emtansine (Kadcyla) and brentuximab vedotin (Adcetris).1 The approval of these ADCs has sparked tremendous interest in the development of additional tubulin binding agents that may offer advantages over existing payloads and may be useful in the treatment of tumors that come to these molecules.
Indeed a number of new classes of tubulin binding agents have been described in recent years and several have been evaluated as potential ADC payloads.5 One such payload that has received increasing amounts of interest is tubulysin, a class of structurally related tetrapeptides produced by myxobacteria.6 Like auristatins such as MMAE, tubulysins bind to the vinca binding site of tubulin and exhibit exceptionally potent cytotoxic activity against a variety of cancer cells lines, including breast, colon, lung, ovarian, and prostate. However, unlike auristatins and maytansines, tubulysins have been shown to retain their potency in MDR1 (ABCB1) expressing cell lines.7 Like other microtubule inhibitors, tubulysins have not shown promise as stand-alone agents due to their extreme toxicity. This has prompted a flurry of work to harness the potency of tubulysin through targeting it to specific tissues, thereby perhaps avoiding some of the side effects associated with tubulin inhibition.8−10 Building upon these efforts, we wish to report herein the development and optimization of a series of antibody conjugates of a synthetic tubulysin payload linked via a maleimide glycine linker.
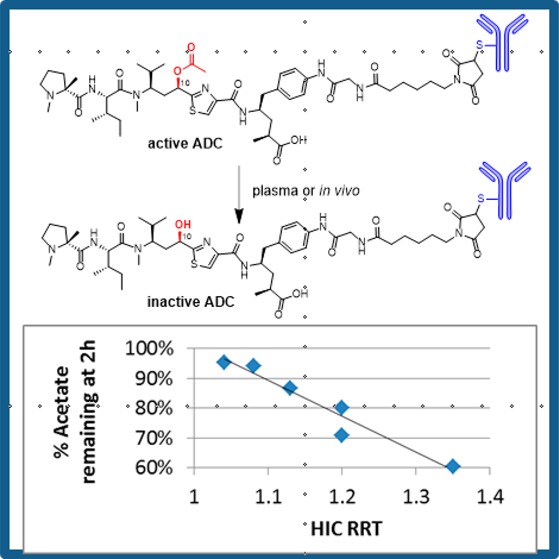
Harnessing the potency of a cytotoxic agent as an ADC requires that the molecule have a suitable “handle” for the attachment of a linker moiety serving to anchor the payload to the antibody while the ADC is in circulation. While tubulysin 1 has a C-terminal carboxylate and a phenolic OH that could be linked to an antibody via an ester linkage, numerous reports have recently suggested that plasma esterases may prematurely cleave such a linkage thereby resulting in payload release in circulation (Figure 1).11,12 This, of course, would be undesirable, perhaps resulting in inadvertent toxicity and reduced efficacy. Therefore, the focus of our efforts was to introduce a reactive amine handle, a functional group well-known to provide stable ADCs via an amide linkage. The full scope of our efforts to introduce this handle while retaining the payload potency will be reported in a separate publication, but for the purposes of the present study we selected compound 2(13) as a potential ADC payload containing an aniline handle for attachment to a linker.
Figure 1.

Structure of tubulysin payloads.
Compound 2 exhibited low nanomolar potency against a variety of cancer cell lines (Table 1), and therefore, we proceeded to explore linkers by which we could attach the payload to an antibody. After exploring a variety of linkers, we settled on the simple mcGly (maleimide-caproyl glycine) linker. Thus, LP1 was prepared according to the route shown in Scheme 1. In short, payload 2 was coupled with preactivated N-Boc glycine to provide the glycyl intermediate. Removal of the Boc protecting group followed by reaction of resulting amine with maleimide caproyl succinimide ester 6 gave the desired LP1, which was conjugated to partially reduced trastuzumab using previously described methodology14 resulting in ADC1 (Table 2). The resulting ADC had an average drug antibody ratio (DAR) of 4.4 and was shown to possess excellent potency against N87 and BT474 cells, both of which express high levels of Her2. The ADC had slightly lower activity against a moderate expressing cell line (MDA-MB-453) and nearly 1000-fold reduced potency against a non-Her2 expressing cell line (HT-29). A negative control (nontargeted) ADC was also generated by the same method (ADC2) and showed no activity against the tested cell lines (Table 2). Combined, this data demonstrated that ADC1 effectively delivers the desired payload specifically to cells expressing the cognate antigen.
Table 1. In Vitro Cytotoxicity of Representative Payloadsa.
| compd | R | N87 IC50 (nM) | BT474 IC50 (nM) | MDA-MB-453 IC50 (nM) | HT29 IC50 (nM) |
|---|---|---|---|---|---|
| 2 | OAc | 1.1 | 1.0 | 0.79 | 0.76 |
| 3 | OH | >100 | >100 | >100 | >100 |
| 4 | OC(O)NHEt | 1.9 | 0.99 | 1.6 | 1.5 |
Reported IC50 is the mean of 2–13 independent determinations.
Scheme 1. Structure and Synthesis of LP1 and LP2.

Reagents and conditions: (a) N-Boc glycine, HATU, DIPEA, rt; (b) TFA, rt; (c) 6, DIPEA, rt.
Table 2. In Vitro Cytotoxicity of Tubulysin ADCsa.

| compda | mAb | LP | conj. methodb | DAR | N87 (+++) IC50 (ng/mL) | BT474 (+++) IC50 (ng/mL) | MDA-MB-453 (++) IC50 (ng/mL) | HT29 (−) IC50 (ng/mL) | Relative HIC retention (RRT) | %OAc cleavage in mouse plasma @ 2 h @ 74 h | %OAc cleavage @ 72 h after dosing in mice (3 mpk) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ADC1 | Tras | LP1 | A | 4.4 | 14 | 25 | 64 | 16,000 | NA | 13% | 89%d | 83% |
| ADC2 | Neg8.8 | LP1 | A | 4.0 | >30,000 | >30,000 | >30,000 | >30,000 | NA | NA | NA | NA |
| ADC3 | Tras-114C | LP2 | B | 1.9 | >60,000 | >60,000 | >60,000 | >60,000 | NA | NA | NA | NA |
| ADC4 | Tras | LP3 | A | 4.0 | 100 | 16 | 26 | >60,000 | NA | 0%c | 0%c | 0%c |
| ADC5 | Tras-392C | LP1 | B | 2.0 | 52 | nd | nd | >60,000 | 1.13 | 4% | 26% | NA |
| ADC6 | Tras-334C | LP1 | B | 2.0 | 36 | 32 | 370 | >60,000 | 1.04 | 5% | 6% | 0% |
| ADC7 | Tras-347C | LP1 | B | 2.0 | 53 | 45 | 11,000 | >60,000 | 1.20 | 20% | 64% | NA |
| ADC8 | Tras-443C | LP1 | B | 2.0 | 49 | 26 | 74 | >60,000 | 1.35 | 40% | 80% | NA |
| ADC9 | Tras-388C | LP1 | B | 2.0 | 49 | 31 | 880 | >60,000 | 1.20 | 29% | 68% | NA |
| ADC10 | Tras-kappa-183C | LP1 | B | 2.0 | 42 | 21 | 23 | >60,000 | 1.08 | 6% | 75% | NA |
Relative antigen expression is classified as high (+++), medium (++), and low (−). Reported IC50 is the mean of 2–13 independent determinations.
See experimental details.
No carbamate cleavage was observed.
50 h.
Incubation of ADC1 with mouse liver crude lysosomal fraction or with human liver S9 fraction under conditions optimized for lysosomal enzyme activity4 resulted in the formation of cysteine-linked payload 5 (Scheme 2, Figure S1). Only trace amounts of free payload were observed after 18 h, strongly suggesting that the mcGly linker is not rapidly cleaved by proteolysis in the presence of these enzymes. Interestingly, no acetate cleavage was observed with these enzyme systems (vide infra). Compound 5 was shown to bind tubulin with approximately the same affinity as the corresponding free payload, 2 (Table S4). Thus, we believe that this ADC functions as a classical “non-cleavable” ADC, delivering its payload by complete ADC catabolism rather than by a specific linker cleavage event.
Scheme 2. Forced ADC Catabolism.

ADC1 was evaluated in a N87 gastric cancer xenograft study (dosed qdx4) where it exhibited nearly complete tumor regression at 10 mpk, partial regression at 3 mpk, and modestly delayed growth at 1 mpk (Figure 4A). Ligand-binding assay (LBA) based pharmacokinetic (PK) experiments showed that the total antibody (tAb) clearance was typical for that of an ADC, exhibiting a T1/2 of 149 h and a clearance of 0.48 mL/h·kg in female nu/nu mice. However, immunocapture LCMS of the PK samples showed that the ADC was rapidly forming a new species with a MW of approximately 42 Da lower than the parent (Figure S2). This mass is consistent with cleavage of the acetate ester, a process that has precedent in recent studies of a spliceostatin ADC.11 Unlike spliceostatin ADCs, in which cleavage resulted in no loss of potency, the tubulysin acetate is reported to be critical for payload cytotoxicity.15 Indeed, we found that payload 3 was >100-fold less active than parent payload 2 (Table 1) and had 10–30× lower potency in a tubulin binding assay (Table S4). A recent crystal structure of tubulysin M bound to tubulin may provide insight into this loss of potency. While neither of the acetate oxygens directly interact with tubulin, the methyl group of the acetate fits nicely into a hydrophobic pocket created by Thr-223, Thr-221, and Pro325.17 The loss of this interaction may be a key determinant of the reduced potency of this payload.
Figure 4.

Efficacy in an N87 xenograft study for ADC1 (4A), ADC4 (B), and ADC6 (C) dosed Q4dx4. (D) Comparison of the efficacy of each ADC along with a nontargeted control ADC at the 3mpk dose.
Although cytotoxicity of a free payload is dependent upon both the functional activity (tubulin binding) and the permeability of the payload, ADCs do not typically rely on payload permeability for their in vitro potency. Rather, the payload is delivered directly into the cell via antigen-mediated uptake. Therefore, in spite of poor cytotoxicity and tubulin binding, we found it prudent to evaluate the potency of an ADC containing payload 3. As such, LP2 was prepared following the method outlined in Scheme 1 and conjugated to trastuzumab-A114C providing ADC3 (Table 2). This ADC was inactive in all cell lines evaluated, thereby clearly demonstrating that the metabolite of ADC1 is an inactive species.
The esterase-mediated inactivation of ADC1 took place far more rapidly in mouse than in rat (Figure S2), consistent with reports that mice have considerably higher esterase activity than rat and cyno.11 However, the fact that the vast majority of currently available efficacy models utilize mice clearly precludes the use of this conjugate in a therapeutic program. Therefore, it was necessary to solve the metabolic problem in mice prior to evaluating this payload for safety in higher species. With this in mind, we set about addressing the metabolic issue using two approaches: (1) replacement of the ester with a stable isostere and (2) attachment of the LP to antibody sites3 that may sterically or electronically prevent esterase-mediated recognition of the LP.19
In the first approach, we took advantage of the historical precedent for using nonhydrolyzable ester isosteres to block metabolism while maintaining the favorable molecular interactions required for target binding. As such, we found that carbamate-containing payload 4 regained significant cytotoxic activity as compared to the corresponding alcohol 3 (Table 1). As a result, we pursued the synthesis of the corresponding linker payload (LP3, Scheme 3) beginning with the previously reported Boc-protected amine 7, which was reduced, coupled with glycine, and deprotected with TFA to give compound 8. This compound, in turn, was allowed to react with the previously reported PFF-ester 9,13 deprotected with diethylamine, and coupled with maleimide caproyl succinimide ester 6 to provide target LP3 in 52% yield. Conjugation with trastuzumab gave ADC4, which exhibited in vitro activity only slightly lower than that observed with parent ADC1 (Table 2). Moreover, as expected, mouse plasma stability studies indicated that no carbamate cleavage occurred after 72 h of incubation, compared to 83% acetate cleavage of the corresponding ester conjugate (Table 2). Thus, we hypothesized that the increased stability of ADC4 would offset the slightly reduced cytotoxicity (vide infra).
Scheme 3. Synthesis of Payloads 4 and LP3.

Reagents and conditions: (a) Pd/C, H2; (b) HATU, N-Fmoc glycine, DIPEA, rt; (c) TFA, reflux; (d) 9, DIPEA, rt; (e) diethylamine, rt; (f) 6, DIPEA, rt.
In a second approach to address the acetate metabolism, we hypothesized that the placement of the linker-payload at different locations on the antibody may impact the ability of the plasma esterase to hydrolyze the acetate of the tubulysin ADC. While conceptually intuitive, this approach has only limited precedent in the literature, primarily focused on the site-dependent proteolytic cleavage of a ValCit linkage in mouse plasma.20 We reasoned that a similar site-dependent effect may impede payload metabolism of the tubulysin ADC. Thus, a series of trastuzumab cysteine engineered variants were carefully selected based upon criteria such as solvent exposure, predicted structural stability, lack of interference with key binding partners such as FcγR and FcRN, and resistance to retro-Michael mediated payload loss16 (unpublished work). The selected mutants (Figure 2) were expressed transiently in transfected HEK-293 cells and were isolated by protein A capture. The six resulting antibodies were conjugated to LP1 using slight modifications of previously described chemistry.
Figure 2.
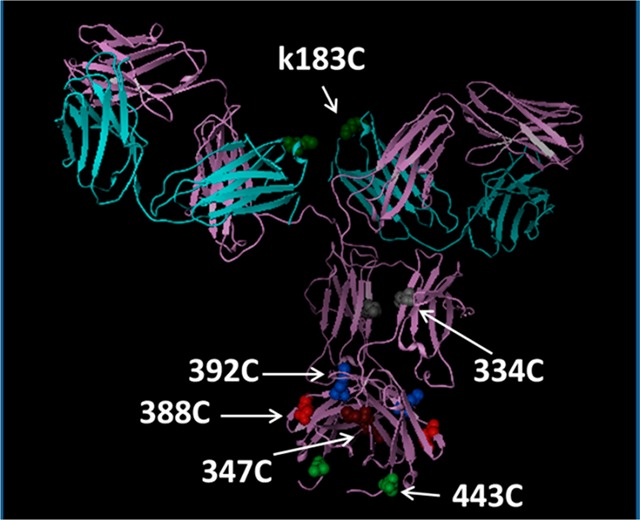
Modeled structure of trastuzumab illustrating the selected cysteine engineered variants.
In order to identify the site variants most likely to impart steric hindrance, thereby blocking ADC metabolism, we employed a chromatographic method that is widely used in the ADC field for the calculation of DAR: Hydrophobic interaction chromatography (HIC). HIC is a nondenaturing LC method that separates proteins based on their affinity to a hydrophobic stationary phase.22 ADCs typically have a rightward shift of the major peak as compared to their corresponding naked mAb due to the hydrophobicity of the LP. For the purpose of comparing various ADCs, the HIC retention time was normalized by dividing the ADC retention time by the retention time of a naked antibody (trastuzumab) resulting in a “relative retention time” (RRT) for each ADC. Attachment of LP1 to the six trastuzumab mutants above resulted in a set of conjugates that had various HIC RRTs (Table 2). We reasoned that in order to interact with the HIC stationary phase, the LP must be able to “reach” out into bulk solvent. Therefore, the HIC retention of various site-specific ADCs may be a crude method of comparing the steric accessibility of the LP toward metabolic enzymes.
Indeed, in the examples outlined herein (Table 2, Figure 3) we see a distinct correlation between the HIC retention of the ADC and the rate of cleavage of the tubulysin acetate ester in mouse plasma. The three ADCs with the lowest HIC retention, the 392C, 334C, and kappa-183C mutants (ADC5, ADC6, and ADC10, respectively), also display the lowest rate of acetate hydrolysis. ADC6 (at the 334C site) was found to be particularly stable in mouse plasma (<10% acetate loss in 74 h) and to have a HIC shift only marginally higher than the naked antibody (RRT = 1.04). Only trace amounts of acetate cleavage (<10%) were observed even after 1 week of incubation in mouse plasma. As can be seen in Figure 2, the 334C site is buried in the cavity between the two Fc chains in close proximity to the glycosyl group at Asn297. The steric hindrance provided by Fc arms and the glycosylation appears to diminish the accessibility of the culprit enzymes and thereby provide significant protection from enzymatic degradation. The remaining site mutants (ADC7–ADC9) have higher HIC shifts, as might be expected due to their more exposed positions (Figure 2) and are rapidly metabolized in mouse plasma. A plot of the %OAc loss at 2 h vs the HIC retention (Figure 3) shows a very clear correlation between ADC hydrophobicity and metabolism (R2 = 0.93). The 2 h time point was selected for this analysis because this is a good approximation of the initial rate of hydrolysis. At later time points other factors may play a role in the rate of acetate cleavage, including concentration of the remaining substrate, stability of the esterase, and the conformational stability of the ADC at 37 °C. Importantly, all the site mutant ADCs retain their potency against high Her2 expressing cell lines while having minimal to no effect on Her2 negative cell lines (Table 2). It should be noted that the 2–3-fold loss in potency of ADC5–ADC10 compared to ADC1 is easily rationalized by the fact that the DAR of the site-specific conjugates is ∼1/2 that of the hinge-conjugated ADC1.
Figure 3.
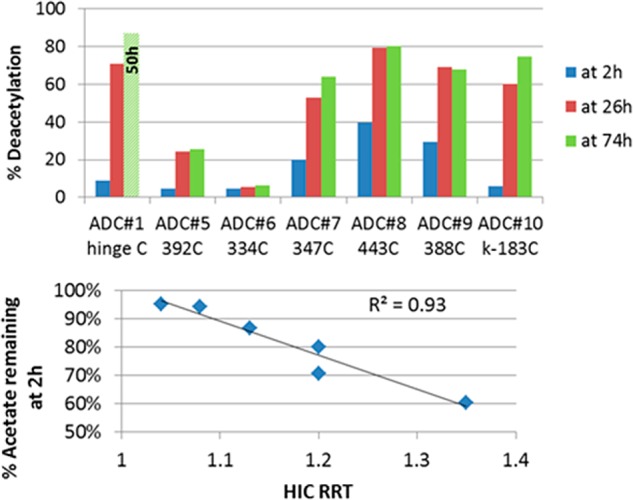
Deacetylation rate of various ADCs (top) has a strong correlation with ADC hydrophobicity (bottom).
Armed now with two approaches to address the acetate cleavage liability, ADC6 and ADC4 were evaluated in an N87 xenograft model to determine if the stabilization of the acetate would result in an improvement in potency. As can be seen in Figure 4B, the acetate isostere ADC (ADC4) had approximately equivalent efficacy to ADC1 in spite of the reduced in vitro potency. More significantly, ADC6 had improved efficacy as compared to conjugate ADC1 despite delivering only 1/2 of the amount of payload at a given dosage (Figure 4C). In fact, in spite of the reduced DAR and reduced in vitro cytotoxicity, the 3 mpk dose of ADC6 showed roughly equivalent efficacy to the 10 mpk dose of ADC1. A comparison of ADC1, ADC4, and ADC6 at a 3 mpk dose clearly shows the impact of the improved stability (Figure 4D). Blood samples were removed 72 h after the final 3mpk doses of ADC4, ADC6, and ADC1 in order to verify that the in vitro improvement in payload stability translated to the in vivo system. ADC4 and ADC6 exhibited little or no loss of the acetate (or the corresponding carbamate) from the ADC as compared to ∼83% acetate loss for ADC1 (Table 2). While partial maleimide ring opening was observed in both ADCs, very little (<10%) payload was lost via retro-Michael mediated deconjugation during the 72 h of in vivo exposure.16,18 This data strongly suggests that the improvement in efficacy observed for ADC6 is driven largely by the mitigation of payload metabolism imparted by the site of conjugation.
The advancement of a small molecule therapeutic program and an ADC therapeutic program may at first glance seem to be very different. ADCs are typically dosed by IV and have in vivo half-lives of days to weeks, while small molecule therapeutics are typically taken orally and have half-lives measured in hours. The cellular activity of an ADC does not rely on compound permeability, as it does for small molecules, but rather the active agent is delivered directly into the cell via endocytosis. ADCs rely on cellular metabolism to release the active moiety, while small molecule therapeutics are typically adversely impacted by any sort of cellular processing.
In spite of these stark differences, the work presented herein is a reminder and a demonstration that the ADC scientist and the small molecule scientist have much in common. During the optimization of our tubulysin ADC program, we encountered a metabolism issue that precluded its advancement into therapeutic programs. The acetate ester of tubulysin ADC1 was being quickly metabolized resulting in the rapid formation of inactive ADC2 (Figure 3). In order to address this metabolism issue, we employed a strategy that should be familiar to most small molecule medicinal chemists. First, we attempted to block the metabolism by the introduction of a commonly used ester isostere. While the resulting ADC (ADC4) proved to be potent and selective, it did not offer an improvement in efficacy as compared to the parent (Figure 4A vs Figure 4B). Second, we attempted to block the metabolism of the acetate by attachment of the payload to sterically occluded sites on the antibody. While this approach may at first seem foreign to many small molecule chemists, it is highly analogous to the commonly employed practice of sterically blocking a site of metabolism, for instance, by the introduction of a nearby tert-butyl group or fluorine atom. In order to identify an appropriate site of attachment, it was necessary to rely on plasma stability studies in order to triage the samples being selected for in vivo evaluation. Fascinatingly, we found that there appears to be a tight correlation between the HIC retention of the tubulysin ADCs and their rate of metabolism by plasma esterases. We believe that this correlation is driven by the fact that HIC retention provides a useful approximation of the steric occlusion of a given site. In other words, conjugation at sites that are “protected” from bulk solvent due to their site of attachment results in ADCs in which the payload cannot effectively interact with the HIC stationary phase, translating into low retention times. In contrast, conjugation at sites that are highly exposed provides ADCs in which the linker-payload is free to interact with the HIC stationary phase, resulting in high retention times. The strong correlation displayed above suggests that HIC retention time may serve as a useful guide to address potential metabolic liabilities that may be encountered in other ADC programs.
The improved metabolic stability of ADC6 resulted in a conjugate with approximately 2-fold improved efficacy in spite of drug-loading that was one-half of the parent ADC1 (Figure 4A vs C). Additionally, the metabolic improvements observed in plasma translated effectively to the in vivo system. ADC6 retained its acetate ester upon in vivo dosing in contrast to ADC1, which was rapidly metabolized in vivo. This is a clear demonstration that the site of attachment can effectively modulate ADC metabolism. This adds to a growing body of evidence that site specific ADCs can provide significant advantages over conventional conjugations (to hinge cysteine and lysine) that result in mixtures of ADC species.
Pioneering work first reported in 2008 by Junutula and colleagues paved the way for the advancement of site-specific ADCs for the treatment of cancer.23 Since that time, a variety of enzymatic and chemical approaches have been used to generate ADCs loaded at specifically defined sites on the antibody backbone.3,24 These conjugation approaches have resulted in ADCs with improved PK,19 decreased metabolism,19,25 and improved safety.23 Based on these results, the ADC community has slowly but inexorably shifted from conventional lysine and hinge-cysteine conjugations to site-specific conjugations.3 However, as this shift has been taking place, there have been very few reports that address the underlying biophysical properties that might be driving such improvements thereby allowing researchers to proactively select beneficial sites for up-and-coming ADC programs. We believe that the present work may begin the process of addressing this gap by providing evidence that the metabolism of site-specific conjugates is correlated with chromatographic retention on a HIC stationary phase. The HIC retention, in turn, is likely driven largely by the solvent exposure of particular sites of conjugation, a feature that in the future may be interrogated by protein modeling prior to undertaking synthetic efforts. This work serves to complement the emerging research showing the HIC retention may correlate with total antibody exposure in various animal models.21 Together, these trends strongly suggest that the ADC scientist should pay careful attention to ADC hydrophobicity during the discovery process.
In summary, we have described the optimization of the in vivo activity of a tubulysin ADC. We believe that the examples provided herein are an important demonstration that traditional medicinal chemistry strategy can be effectively applied in the development of ADC programs. Moreover, we clearly show that site of conjugation should be a tool in the arsenal of every ADC scientist. Additionally, we provide an example showing the translation of an ADC biophysical property (HIC retention) to in vitro and in vivo metabolism and ultimately to improved efficacy of the resulting ADC. This study is a timely reminder that the principles of small molecule medicinal chemistry can be effectively applied even to biotherapeutic programs such as ADCs.
Acknowledgments
The authors would like to that Hans Peter Gerber and Puja Sapra for their leadership and support and WeiDong Ding for performing the tubulin binding assays.
Biography
Nathan Tumey received his Ph.D. in organic chemistry from Duke University, NC. He spent about 10 years as a small-molecule medicinal chemist in the biotech and pharmaceutical industry before transitioning to antibody−drug conjugate (ADC) research in 2011. He is currently an associate research fellow at Pfizer where he leads the conjugation chemistry team as they pursue new conjugation, linker, and payload technology for use in up-and-coming ADC programs. Nathan’s primary research interests include ADC metabolism and PK, new conjugation technologies, and bioanalytical strategies for ADC characterization.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00195.
Experimental description; data for tubulin binding, ADC catabolism, immunocapture LCMS; raw HIC traces (PDF)
Author Present Address
† SBP Discovery, La Jolla, California 92037, United States.
Author Present Address
‡ Solstice Biologics, San Diego, California 92121, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Thomas A.; Teicher B. A.; Hassan R. Antibody–drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254. 10.1016/S1470-2045(16)30030-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hear C.; Rubnitz J. E. Recent research and future prospects for gemtuzumab ozogamicin: could it make a comeback?. Expert Rev. Hematol. 2014, 7, 427–429. 10.1586/17474086.2014.924849. [DOI] [PubMed] [Google Scholar]
- Akkapeddi P.; Azizi S.; Freedy A. M.; Cal P. M. S. D.; Gois P. M. P.; Bernardes G. J. L. Construction of homogeneous antibody–drug conjugates using site-selective protein chemistry. Chem. Sci. 2016, 7, 2954. 10.1039/C6SC00170J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessire A. J.; Ballard T. E.; Charati M.; Cohen J. D.; Green M. E.; Lam M.; Loganzo F.; Nolting B.; Pierce B.; Puthenveetil S.; Roberts L.; Schildknegt K.; Subramanyam C. Determination of antibody-Drug-Conjugate Released Payload Species Using Directed In Vitro Assays and Mass Spectrometric Interrogation. Bioconjugate Chem. 2016, 10.1021/acs.bioconjchem.6b00192. [DOI] [PubMed] [Google Scholar]
- Klute K.; Nackos E.; Tasaki S.; Nguyen D. P.; Bander N. H.; Tagawa S. T. Microtubule inhibitor-based antibody-drug conjugates for cancer therapy. OncoTargets Ther. 2014, 7, 2227. 10.2147/OTT.S46887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray B. C.; Peterson M. T.; Fecik R. A. Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat. Prod. Rep. 2015, 32, 654–662. 10.1039/C4NP00036F. [DOI] [PubMed] [Google Scholar]
- Kaur G.; Hollingshead M.; Holbeck S.; Schauer-Vukasinovic V.; Camalier R. F.; Domling A.; Agarwal S. Biological evaluation of tubulysin A: a potential anticancer and antiangiogenic natural product. Biochem. J. 2006, 396, 235–242. 10.1042/BJ20051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J. A.; Dorton R.; Dawson A.; Vetzel M.; Parker N.; Nicoson J. S.; Westrick E.; Klein P. J.; Wang Y.; Vlahov I. R.; Leamon C. P. In vivo structural activity and optimization studies of folate-tubulysin conjugates. Mol. Pharmaceutics 2009, 6, 1518–1525. 10.1021/mp900086w. [DOI] [PubMed] [Google Scholar]
- Kularatne S. A.; Venkatesh C.; Santhapuram H. K.; Wang K.; Vaitilingam B.; Henne W. A.; Low P. S. Synthesis and biological analysis of prostate-specific membrane antigen-targeted anticancer prodrugs. J. Med. Chem. 2010, 53, 7767–7777. 10.1021/jm100729b. [DOI] [PubMed] [Google Scholar]
- Li J. Y.; Perry S. R.; Muniz-Medina V.; Wang X.; Wetzel L. K.; Rebelatto M. C.; Hinrichs M. J.; Bezabeh B. Z.; Fleming R. L.; Dimasi N.; Feng H.; Toader D.; Yuan A. Q.; Xu L.; Lin J.; Gao C.; Wu H.; Dixit R.; Osbourn J. K.; Coats S. R. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. 10.1016/j.ccell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Tumey L. N.; Rago B.; Han X. In vivo biotransformations of antibody-drug conjugates. Bioanalysis 2015, 7, 1649–1664. 10.4155/bio.15.84. [DOI] [PubMed] [Google Scholar]
- Dokter W.; Ubink R.; van der Lee M.; van der Vleuten M.; van Achterberg T.; Jacobs D.; Loosveld E.; van den Dobbelsteen D.; Egging D.; Mattaar E.; Groothuis P.; Beusker P.; Coumans R.; Elgersma R.; Menge W.; Joosten J.; Spijker H.; Huijbregts T.; de Groot V.; Eppink M.; de Roo G.; Verheijden G.; Timmers M. Preclinical profile of the HER2-targeting ADC SYD983/SYD985: introduction of a new duocarmycin-based linker-drug platform. Mol. Cancer Ther. 2014, 13, 2618–2629. 10.1158/1535-7163.MCT-14-0040-T. [DOI] [PubMed] [Google Scholar]
- Leverett C. A. Submitted, 2016.
- Stefano J. E.; Busch M.; Hou L.; Park A.; Gianolio D. A. Micro- and mid-scale maleimide-based conjugation of cytotoxic drugs to antibody hinge region thiols for tumor targeting. Methods Mol. Biol. (N. Y., NY, U. S.) 2013, 1045, 145–171. 10.1007/978-1-62703-541-5_9. [DOI] [PubMed] [Google Scholar]
- Patterson A. W.; Peltier H. M.; Sasse F.; Ellman J. A. Design, synthesis, and biological properties of highly potent tubulysin D analogues. Chem. - Eur. J. 2007, 13, 9534–9541. 10.1002/chem.200701057. [DOI] [PubMed] [Google Scholar]
- Tumey L. N.; Charati M.; He T.; Sousa E.; Ma D.; Han X.; Clark T.; Casavant J.; Loganzo F.; Barletta F.; Lucas J.; Graziani E. I. Mild Method for Succinimide Hydrolysis on ADCs: Impact on ADC Potency, Stability, Exposure, and Efficacy. Bioconjugate Chem. 2014, 25 (10), 1871. 10.1021/bc500357n. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Benz F. W.; Wu Y.; Wang Q.; Chen Y.; Chen X.; Li H.; Zhang Y.; Zhang R.; Yang J. Structural Insights into the Pharmacophore of Vinca Domain Inhibitors of Microtubules. Mol. Pharmacol. 2016, 89, 233–242. 10.1124/mol.115.100149. [DOI] [PubMed] [Google Scholar]
- Wei C.; Zhang G.; Clark T.; Barletta F.; Tumey L. N.; Rago B.; Hansel S.; Han X. Where Did the Linker-Payload Go? A Quantitative Investigation on the Destination of the Released Linker-Payload from an Antibody-Drug Conjugate with a Maleimide Linker in Plasma. Anal. Chem. 2016, 88 (9), 4979. 10.1021/acs.analchem.6b00976. [DOI] [PubMed] [Google Scholar]
- Strop P.; Liu S.-H.; Dorywalska M.; Delaria K.; Dushin R. G.; Tran T.-T.; Ho W.-H.; Farias S.; Casas M. G.; Abdiche Y.; Zhou D.; Chandrasekaran R.; Samain C.; Loo C.; Rossi A.; Rickert M.; Krimm S.; Wong T.; Chin S. M.; Yu J.; Dilley J.; Chaparro-Riggers J.; Filzen G. F.; O’Donnell C. J.; Wang F.; Myers J. S.; Pons J.; Shelton D. L.; Rajpal A. Location Matters: Site of Conjugation Modulates Stability and Pharmacokinetics of Antibody Drug Conjugates. Chem. Biol. 2013, 20, 161–167. 10.1016/j.chembiol.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Dorywalska M.; Strop P.; Melton-Witt J. A.; Hasa-Moreno A.; Farias S. E.; Galindo Casas M.; Delaria K.; Lui V.; Poulsen K.; Loo C.; Krimm S.; Bolton G.; Moine L.; Dushin R.; Tran T. T.; Liu S. H.; Rickert M.; Foletti D.; Shelton D. L.; Pons J.; Rajpal A. Effect of attachment site on stability of cleavable antibody drug conjugates. Bioconjugate Chem. 2015, 26, 650–659. 10.1021/bc5005747. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Bovee T. D.; Doronina S. O.; Burke P. J.; Hunter J. H.; Neff-LaFord H. D.; Jonas M.; Anderson M. E.; Setter J. R.; Senter P. D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. 10.1038/nbt.3212. [DOI] [PubMed] [Google Scholar]
- Ouyang J. Drug-to-antibody ratio (DAR) and drug load distribution by hydrophobic interaction chromatography and reversed phase high-performance liquid chromatography. Methods Mol. Biol. (N. Y., NY, U. S.) 2013, 1045, 275–283. 10.1007/978-1-62703-541-5_17. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; Lu Y.; Meng Y. G.; Ng C.; Yang J.; Lee C. C.; Duenas E.; Gorrell J.; Katta V.; Kim A.; McDorman K.; Flagella K.; Venook R.; Ross S.; Spencer S. D.; Wong W. L.; Lowman H. B.; Vandlen R.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Mallet W. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26 (8), 925. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-specific antibody-drug conjugates: the nexus of bioorthogonal chemistry, protein engineering, and drug development. Bioconjugate Chem. 2015, 26, 176. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F.; Lu Y.; Manibusan A.; Sellers A.; Tran H.; Sun Y.; Phuong T.; Barnett R.; Hehli B.; Song F.; De Guzman M. J.; Ensari S.; Pinkstaff J. K.; Sullivan L. M.; Biroc S. L.; Cho H.; Schultz P. G.; Di Joseph J.; Dougher M.; Ma D.; Dushin R.; Leal M.; Tchistiakova L.; Feyfant E.; Gerber H.-P.; Sapra P. A general approach to site-specific antibody drug conjugates. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 1766–1771. 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.