

Abstract

Pyrrolobenzodiazepine dimers are an emerging class of warhead in the field of antibody–drug conjugates (ADCs). Tesirine (SG3249) was designed to combine potent antitumor activity with desirable physicochemical properties such as favorable hydrophobicity and improved conjugation characteristics. One of the reactive imines was capped with a cathepsin B-cleavable valine-alanine linker. A robust synthetic route was developed to allow the production of tesirine on clinical scale, employing a flexible, convergent strategy. Tesirine was evaluated in vitro both in stochastic and engineered ADC constructs and was confirmed as a potent and versatile payload. The conjugation of tesirine to anti-DLL3 rovalpituzumab has resulted in rovalpituzumab-tesirine (Rova-T), currently under evaluation for the treatment of small cell lung cancer.
Keywords: Tesirine, antibody−drug conjugates, talirine, SG3249, Rova-T
Since their discovery in 1963,1,2 pyrrolobenzodiazepines (PBDs) have been studied for their antitumor properties. Both naturally occurring monomer anthramycin, and synthetic dimer SG2000 have undergone clinical evaluation as stand-alone agents.3 Until recently,4 however, this more potent class of molecule remained under-exploited as a source of warhead for antibody–drug conjugates (ADCs).
Antibody–drug conjugates combine the potency of a cytotoxic warhead with the selectivity of an antibody to enable targeted killing of tumor cells. Most ADCs currently in clinical evaluation are based on antimitotic warheads such as auristatin and maytansine.5
Given their potency and mode of action as sequence-selective cross-linking agents, we hypothesized the ADC field would benefit from the employment of PBDs as a new class of warhead. A collaboration between Spirogen and Seattle Genetics led to the development of SGN-CD33A, an ADC under clinical investigation for the treatment of acute myeloid leukemia, featuring the PBD payload SGD-1910 or talirine.6,7 We aimed to improve on the design of SGD-1910 by lowering hydrophobicity while maintaining potency (Figure 1).
Figure 1.
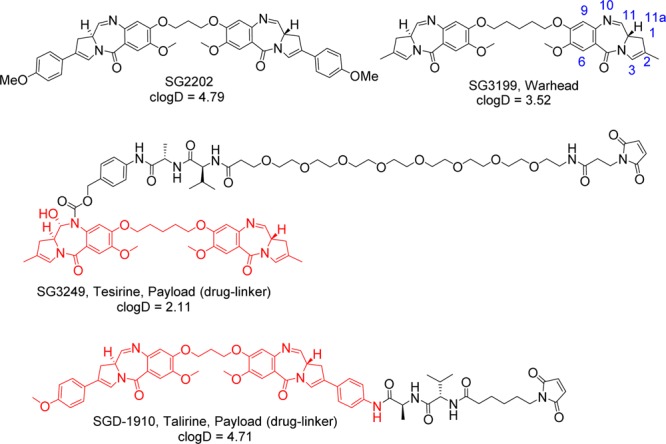
Structures of SG2202 and ADC warhead SG3199. LogD values calculated at pH 7.4. Structures of payloads (drug-linker), SG3249 (tesirine), and SGD-1910 (talirine). Red coding denotes released warhead. Conventional ring numbering shown on SG3199.
We also investigated trapping one of the reactive imine moieties in its carbinolamine form by incorporating the N10 nitrogen as part of a cathepsin B-cleavable valine-alanine carbamate prodrug linker.8 Finally, a discrete PEG8 spacer was inserted to further enhance the solubility properties of the molecule. This design resulted in the payload SG3249 (tesirine).
The warhead component of tesirine, SG3199 (Figure 1), was based on rational design principles. The C2 aryls, responsible both for the high potency and poor aqueous solubility of SG2202 and analogues,9,10 were replaced with simpler methyl groups. The expected drop in potency was offset by the introduction of a 5-carbon tether between the two monomers. Both 3-carbon and 5-carbon tether linked PBDs are isohelical with the DNA helix, but the 5-carbon tether is more flexible and has greater opportunities for contact with the minor groove.11,12 SG3199 was tested in a panel of cancer cell lines and retained picomolar activity in vitro (Table 1).
Table 1. In Vitro Activity of Warhead SG3199.
| cell
line IC50a |
||||
|---|---|---|---|---|
| compd | K562 | NCIN87 | BT474 | SKBR3 |
| SG3199 | 150 pM | 20 pM | 1 nM | 320 pM |
IC50 values (mean of three independent experiments) were determined using CellTiter96 (MTS) following 96 h incubation.
Rather than following a linear route as used in the synthesis of SG2202 or SG2000 (SJG-136),13 we decided to follow a convergent synthetic route, allowing both the left-hand (linker) and right-hand (imine) sides of the unsymmetrical dimer to be made in parallel, followed by late-stage dimerization. Monomeric routes have been developed previously by Howard et al.12 and Kamal et al.14 Recently, Kolakowski et al.15 have reported the application of a convergent strategy to the synthesis of an unsymmetrical C2-aryl substituted PBD. In contrast to these other strategies, we selected N-alloc and O-TBS as the N10-imine and C11-hydroxy protecting groups for mild and efficient deprotection in the final stages of the synthesis. Both groups are resistant to base and mild acid but can be orthogonally removed when required.
Controlled nitration of benzylvanillin afforded intermediate 2 (Scheme 1).16 This was followed by an exchange of protecting groups at the phenolic position. TIPS was selected for its relatively high resistance to acid and base, with the option of selective phenolic silyl deprotection in the presence of aliphatic silyl ethers. Clean and efficient Pinnick oxidation of benzaldehyde 4 gave acid 5. Amide bond formation with hydroxyproline derivative 6(11) gave alcohol 7 (see Supporting Information), which was oxidized using the reactive TEMPO/TCCA combination. Thermodynamic treatment of compound 8(17,18) with triflic anhydride gave triflate 9 in 78% yield (Scheme 2). This was followed by a sp2–sp3 Suzuki coupling to install a methyl group at the C2 position. Activated conditions developed by Mu and Gibbs,19 employing silver oxide and triphenylarsine, were required for efficient coupling. Optimization of the reaction conditions was necessary to avoid competing triflate reduction; the resulting impurity 10 having similar chromatographic retention properties to the desired compound 11. When the same conditions were investigated with homologous ethyl boronic acid, a much larger proportion of reduced material 10 was found to contaminate the mixture. The nitro group was reduced with zinc and dilute formic acid in ethanol. These conditions were mild enough to leave both the primary TBS ether and the internal unsaturation intact.
Scheme 1. Synthesis and Coupling of A and C Rings.

Reagents and conditions: (a) HNO3, 12 °C, 95%; (b) TFA, 85 °C, 50%; (c) TIPSCl, imidazole, 100 °C, 88%; (d) NaClO2, NaH2PO4, H2O2, THF, −78 °C to rt, 100%; (e) DCC, HOBt, Et3N, DCM, −10 °C to rt, 90%; (f) TEMPO, TCCA, DCM, 100%.
Scheme 2. Synthesis of Monomeric Phenol 20 and 21.

Reagents and conditions: (a) Tf2O, 2,6-lutidine, DCM, −45 °C, 78%; (b) MeB(OH)2, Ag2O, AsPh3, Pd(PhCN)2Cl2, 70 °C, 70%; (c) Zn (30 equiv), HCOOH/EtOH 5/95, 30 °C, 80%; (d) Alloc-Cl, pyridine, DCM, −78 °C to rt, 100%; (e) triphosgene, Et3N, THF, 5 °C, then Alloc-Val-Ala-PAB-OH, Et3N, THF, 40 °C, 50%; (f) AcOH/MeOH/THF/water 7/1/1/2, rt, 71%, 15, 80%, 16; (g) DMSO, (COCl)2, Et3N, DCM, −78 °C to rt, 66%, 17, 60%, 18; (h) TBS-OTf, 2,6-lutidine, DCM, 0 °C, 85%, 19, 65%, 20; (i) LiOAc, DMF/water, 95/5, rt, 100%, 21, 100%, 22.
At this key juncture in the synthesis, carbamates 13 and 14 were formed from 12, either by treatment with alloc chloroformate or through the initial formation of an isocyanate20 followed by addition of alloc-Val-Ala-para-aminobenzylalcohol.21 The two resulting monomeric intermediates 13 and 14 were subjected to the same sequence of reactions beginning with mild acid hydrolysis of the primary TBS and ring-closing oxidation under Swern conditions. Protection of the 11-hydroxy group is critical at this point to allow further chemistry to proceed under basic or acidic conditions while preserving the 11a stereochemistry.22 The use of highly reactive TBS triflate23 was necessary to achieve efficient secondary alcohol protection. Finally, mild and orthogonal deprotection of the phenolic TIPS in the presence of aliphatic TBS was achieved with LiOAc in wet DMF24 to provide both the right-hand and left-hand side monomers 21 and 22.
Williamson ether chemistry, proceeding via iodopentane derivatization of alloc-protected monomer 21 and subsequent reaction with monomer 22, provided dimer 24 in 86% yield (Scheme 3). Removal of 11-hydroxy TBS with TBAF resulted in partial racemization at C11a (detectable by LC and optical rotation analyses of SG3249: Figure 2, Table 2). Buffering the fluoride solution with acetic acid was found to prevent racemization.25,26
Scheme 3. Final Steps, Synthesis of SG3249 (Tesirine).

Reagents and conditions: (a) 1,5-diiodopentane, K2CO3, acetone, 60 °C, 90%; (b) 22, K2CO3, acetone, 65 °C, 86%; (c) TBAF/AcOH, THF, rt, 80%; (d) Pd(PPh3)4, pyrrolidine, DCM, rt, 100%; (e) Mal-dPEG8-acid, EDCI, DCM, rt, 73%.
Figure 2.
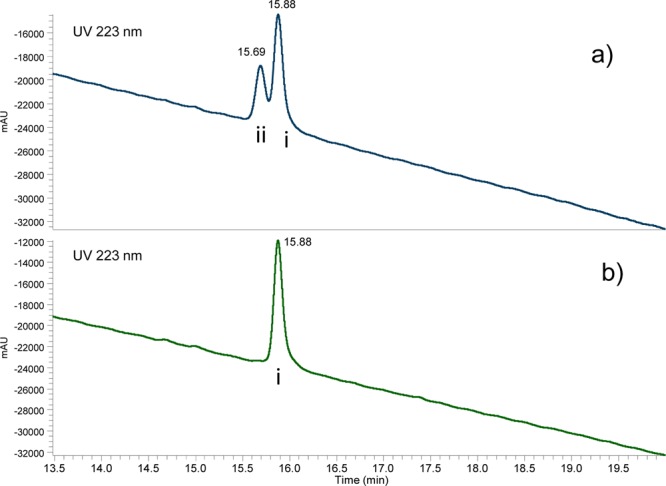
UPLC of SG3249: (a) unbuffered TBAF used in Scheme 3, step c; (b) buffered TBAF. Ace Excel 2 C18-AR (2 μm, 3.0 mm × 100 mm), 40 °C, 20 mM ammonium formate (pH = 4)/acetonitrile, 25/75 to 55/45 over 30 min, 0.6 mL/min. Peaks i and ii in (a) diastereoisomers of SG3249. Peak i in (b) pure SG3249.
Table 2. SG3249 Optical Rotationa.
| SG3249 batch | optical rotation | TBS cleavage |
|---|---|---|
| unbuffered | +118° (c = 0.43) | TBAF |
| buffered | +262° (c = 0.56) | TBAF/AcOH 1/1 |
Both alloc groups were rapidly and efficiently removed with Pd(0) and pyrrolidine as an allyl scavenger,27 in a Tsuji–Trost reaction.28 The absence of pyrrolidine scavenging during the deprotection resulted in the formation of N10-allylated impurities. The resulting compound 26 was immediately used in the next step without further purification. Traces of palladium were effectively removed, both by chromatography of the payload SG3249 and purification of the final ADC. Interestingly, reversible macrocycle 27 was found to form upon standing in solution with the valine amino group adding across the PBD imine of 26 (NMR and structure in Supporting Information). However, we found that both 26 and 27 could be fully consumed under final coupling conditions with EDCI and Mal-dPEG8-Acid to form SG3249 in 73% yield. The overall yield for the synthesis was 0.54% from benzylvanillin over 30 steps.
Despite its length, the synthetic route described has shown a number of advantages such as robustness on scale, modularity of monomeric building blocks, and mild and efficient final steps. The same route was used to provide clinical grade material and is continuously being optimized.
To explore its potential as a payload, SG3249 was conjugated to trastuzumab (Herceptin) in a stochastic fashion and to an engineered version featuring two reactive cysteine positions (site-specific Herceptin-SG3249) (Table 3). Finally, SG3249 was conjugated to a control, nonbinding, engineered antibody.
Table 3. ADC Conjugation Properties and GI50 in SKBR3a.
| ADC | DAR | yield (%) | process aggregation (% dimer) | GI50 (ng/mL) |
|---|---|---|---|---|
| Her-SG3249 | 2.51 | 86 | 2.81 | 1.74 |
| site-specific Her-SG3249 | 1.81 | 89 | 3.93 | 2.62 |
| IgG control mAb-SG3249 | 1.79 | 87 | 3.11 | 1101 |
SG3249 conjugation to all antibodies was highly efficient, as illustrated by a readily tunable stochastic ADC average drug-to-antibody ratio (DAR of 2.5 in this instance; the DAR can be tuned by varying the amount of TCEP reducing agent) and site-specific ADC DAR of 1.8 (90% conjugation efficiency). SG3249 was found readily soluble in the 10% DMSO aqueous conjugation buffer (50 mg/L, see Supporting Information, solubility limit not determined). In contrast, SGD-1910, bearing C2-aryls and devoid of PEG linker, had to be conjugated in 50% propylene glycol.6 SG3249 low hydrophobicity (logD7.4 = 2.11) resulted in low levels of aggregation (usually <5%). As a result, the conjugation process reproducibly delivered high monomeric purity ADCs in high yields, on microgram to gram scale.
Gratifyingly, both the stochastic and site-specific Herceptin-SG3249 ADCs were active in the ng/mL range against HER2-expressing human breast cancer cell line SKBR3 (Figure 3, Table 3). Both HER2-targeted ADCs were greater than 2 orders of magnitude more potent than the control non-HER2-binding ADC, indicating target specificity. The residual activity of the nontargeted control ADC is likely due to nonspecific uptake (such as clathrin-mediated endocytosis) at high assay concentrations. Linker stability studies will be undertaken to further address this point and will appear in subsequent publications. Preliminary evidence tends to show good linker stability when SG3249 is conjugated at buried positions within the antibody structure.
Figure 3.
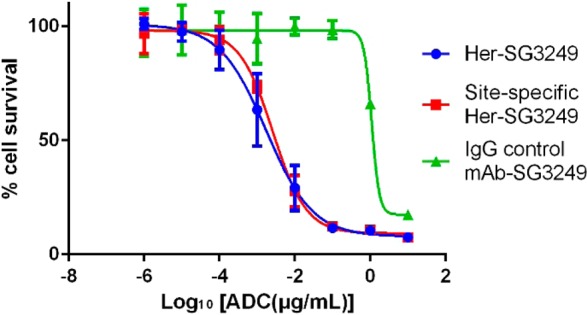
Growth inhibition curves in human SKBR3 cells for SG3249 ADCs.
In conclusion, SG3249 possesses an attractive set of properties: potency, synthetic route scalability, low hydrophobicity contributing to low aggregation, cathepsin B-cleavable linker, and efficient maleimide conjugation.
The in vivo evaluation of SG3249 ADCs will be described elsewhere. SG3249 has been conjugated with rovalpituzumab. The resulting ADC has recently successfully completed a phase I clinical trial for the treatment of small cell lung cancer.29,30 Initial data from rovalpituzumab-tesirine (Rova-T) is encouraging31 and should pave the way for a phase II trial. In addition, SG3249 forms the payload component of ADCT-301, which is currently in a phase I trial for CD25-positive hematological malignancies.32,33
The pyrrolobenzodiazepine dimer class of warheads is finding increasing utility in the ADC arena based on their versatility and complementary mode of action to antimitotic tubulin binders.
Acknowledgments
The authors wish to thank Stephen Gregson for editing the manuscript and providing valuable scientific feedback. The authors also wish to thank Krisztina Radi for providing SG3249 UPLC data and Gyoung Dong Kang and Conor Barry for providing HRMS data. Christian Noti and colleagues are acknowledged for their NMR work on SG3249 and macrocycle 27. Michael Torgov is acknowledged for his contribution to the development of SG3249 analytical conditions.
Glossary
ABBREVIATIONS
- ADC
antibody–drug conjugate
- PBD
pyrrolobenzodiazepine
- DLL3
delta-like 3
- Her
herceptin
- HER2
human epidermal growth factor receptor 2
- PABC
para-aminobenzylcarbamate
- DAR
drug-to-antibody ratio
- TBAF
tetrabutylammonium fluoride
- DCM
dichloromethane
- Mal
maleimide
- dPEG
discrete poly ethylene glycol
- EDCI
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- TFA
trifluoroacetic acid
- TIPS
triisopropyl silyl
- DCC
dicyclohexylcarbodiimide
- HOBt
1-hydroxybenzotriazole
- TEMPO
2,2,6,6-tetramethylpiperidine 1-oxyl
- TCCA
trichloroisocyanuric acid
- TBS
tert-butyldimethylsilyl
- alloc
allyloxycarbonyl
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00062.
Author Present Address
† Stemcentrx, Inc., 450 East Jamie Court, South San Francisco, California 94080, United States.
This study was supported by Spirogen, a member of AstraZeneca group of companies.
The authors declare the following competing financial interest(s): A.T., L.M., N.P., L.A., D.W., J.H., and P.H. are Spirogen employees. J.L. is a Stemcentrx employee.
Supplementary Material
References
- Leimgruber W.; Stefanovic V.; Schenker F.; Karr A.; Berger J. Isolation and characterization of anthramycin, a new antitumor antibiotic. J. Am. Chem. Soc. 1965, 87 (24), 5791–3. 10.1021/ja00952a050. [DOI] [PubMed] [Google Scholar]
- Tendler M. D.; Korman S. ’Refuin’: A non-cytotoxic carcinostatic compound proliferated by a thermophilic actinomycete. Nature 1963, 199, 501. 10.1038/199501a0. [DOI] [PubMed] [Google Scholar]
- Hartley J. A. The development of pyrrolobenzodiazepines as antitumour agents. Expert Opin. Invest. Drugs 2011, 20 (6), 733–44. 10.1517/13543784.2011.573477. [DOI] [PubMed] [Google Scholar]
- Kung Sutherland M. S.; Walter R. B.; Jeffrey S. C.; Burke P. J.; Yu C.; Kostner H.; Stone I.; Ryan M. C.; Sussman D.; Lyon R. P.; Zeng W.; Harrington K. H.; Klussman K.; Westendorf L.; Meyer D.; Bernstein I. D.; Senter P. D.; Benjamin D. R.; Drachman J. G.; McEarchern J. A. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122 (8), 1455–63. 10.1182/blood-2013-03-491506. [DOI] [PubMed] [Google Scholar]
- Chari R. V.; Miller M. L.; Widdison W. C. Antibody-drug conjugates: an emerging concept in cancer therapy. Angew. Chem., Int. Ed. 2014, 53 (15), 3796–827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Jeffrey S. C.; Burke P. J.; Lyon R. P.; Meyer D. W.; Sussman D.; Anderson M.; Hunter J. H.; Leiske C. I.; Miyamoto J. B.; Nicholas N. D.; Okeley N. M.; Sanderson R. J.; Stone I. J.; Zeng W.; Gregson S. J.; Masterson L.; Tiberghien A. C.; Howard P. W.; Thurston D. E.; Law C. L.; Senter P. D. A potent anti-CD70 antibody-drug conjugate combining a dimeric pyrrolobenzodiazepine drug with site-specific conjugation technology. Bioconjugate Chem. 2013, 24 (7), 1256–63. 10.1021/bc400217g. [DOI] [PubMed] [Google Scholar]
- A safety study of SGN-CD33A in AML patients. https://www.clinicaltrials.gov/ct2/show/NCT01902329.
- Jeffrey S. C.; Nguyen M. T.; Andreyka J. B.; Meyer D. L.; Doronina S. O.; Senter P. D. Dipeptide-based highly potent doxorubicin antibody conjugates. Bioorg. Med. Chem. Lett. 2006, 16 (2), 358–362. 10.1016/j.bmcl.2005.09.081. [DOI] [PubMed] [Google Scholar]
- Howard P. W.; Chen Z.; Gregson S. J.; Masterson L. A.; Tiberghien A. C.; Cooper N.; Fang M.; Coffils M. J.; Klee S.; Hartley J. A.; Thurston D. E. Synthesis of a novel C2/C2′-aryl-substituted pyrrolo[2,1-c][1,4]benzodiazepine dimer prodrug with improved water solubility and reduced DNA reaction rate. Bioorg. Med. Chem. Lett. 2009, 19 (22), 6463–6. 10.1016/j.bmcl.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Hartley J. A.; Hamaguchi A.; Coffils M.; Martin C. R. H.; Suggitt M.; Chen Z.; Gregson S. J.; Masterson L. A.; Tiberghien A. C.; Hartley J. M.; Pepper C.; Lin T. T.; Fegan C.; Thurston D. E.; Howard P. W. SG2285, a Novel C2-Aryl-Substituted Pyrrolobenzodiazepine Dimer Prodrug That Cross-links DNA and Exerts Highly Potent Antitumor Activity. Cancer Res. 2010, 70 (17), 6849–6858. 10.1158/0008-5472.CAN-10-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregson S. J.; Howard P. W.; Gullick D. R.; Hamaguchi A.; Corcoran K. E.; Brooks N. A.; Hartley J. A.; Jenkins T. C.; Patel S.; Guille M. J.; Thurston D. E. Linker length modulates DNA cross-linking reactivity and cytotoxic potency of C8/C8′ ether-linked C2-exo-unsaturated pyrrolo[2,1-c][1,4]benzodiazepine (PBD) dimers. J. Med. Chem. 2004, 47 (5), 1161–74. 10.1021/jm030897l. [DOI] [PubMed] [Google Scholar]
- Howard P. W.; Kang G.-D.. Preparation, DNA crosslinking reactivity and antiproliferative activity of pyrrolobenzodiazepine dimers. WO2005085259A2, 2005.
- Gregson S. J.; Howard P. W.; Hartley J. A.; Brooks N. A.; Adams L. J.; Jenkins T. C.; Kelland L. R.; Thurston D. E. Design, synthesis, and evaluation of a novel pyrrolobenzodiazepine DNA-interactive agent with highly efficient cross-linking ability and potent cytotoxicity. J. Med. Chem. 2001, 44 (5), 737–48. 10.1021/jm001064n. [DOI] [PubMed] [Google Scholar]
- Kamal A.; Ramesh G.; Laxman N.; Ramulu P.; Srinivas O.; Neelima K.; Kondapi A. K.; Sreenu V. B.; Nagarajaram H. A. Design, synthesis, and evaluation of new non-cross-Linking pyrrolobenzodiazepine dimers with efficient DNA binding ability and potent antitumor activity. J. Med. Chem. 2002, 45 (21), 4679–4688. 10.1021/jm020124h. [DOI] [PubMed] [Google Scholar]
- Kolakowski R. V.; Young T. D.; Howard P. W.; Jeffrey S. C.; Senter P. D. Synthesis of a C2-aryl-pyrrolo[2,1-c][1,4]benzodiazepine monomer enabling the convergent construction of symmetrical and non-symmetrical dimeric analogs. Tetrahedron Lett. 2015, 56 (30), 4512–4515. 10.1016/j.tetlet.2015.05.116. [DOI] [Google Scholar]
- Althuis T. H.; Hess H. J. Synthesis and identification of the major metabolites of prazosin formed in dog and rat. J. Med. Chem. 1977, 20 (1), 146–9. 10.1021/jm00211a031. [DOI] [PubMed] [Google Scholar]
- Howard P. W.Preparation of pyrrolobenzodiazepine dimers and their conjugates with cell binding agents, especially pyrrolobenzodiazepine dimer drug linker antibody conjugates containing dipeptide linkers, and their use for treating proliferative diseases, particularly cancer. WO2014057074A1.
- Howard P. W.; Tiberghien A.. Process for the preparation of intermediates useful for the synthesis of pyrrolobenzodiazepine dimers. WO2013053872A1.
- Mu Y.; Gibbs R. A. Coupling of isoprenoid triflates with organoboron nucleophiles: Synthesis of all-trans-geranylgeraniol. Tetrahedron Lett. 1995, 36 (32), 5669–5672. 10.1016/00404-0399(50)11193-. [DOI] [Google Scholar]
- Masterson L. A.; Spanswick V. J.; Hartley J. A.; Begent R. H.; Howard P. W.; Thurston D. E. Synthesis and biological evaluation of novel pyrrolo[2,1-c][1,4]benzodiazepine prodrugs for use in antibody-directed enzyme prodrug therapy. Bioorg. Med. Chem. Lett. 2006, 16 (2), 252–6. 10.1016/j.bmcl.2005.10.017. [DOI] [PubMed] [Google Scholar]
- Howard P. W.; Masterson L.; Tiberghien A.; Flygare J. A.; Gunzner J. L.; Polakis P.; Polson A.; Raab H. E.; Spencer S. D.. Preparation of pyrrolobenzodiazepine dimers and conjugates, especially pyrrolobenzodiazepine dimer drug linker conjugates containing peptide linkers, and their use for treating proliferative diseases including cancer. WO2011130598A1.
- Wells G.; Martin C. R.; Howard P. W.; Sands Z. A.; Laughton C. A.; Tiberghien A.; Woo C. K.; Masterson L. A.; Stephenson M. J.; Hartley J. A.; Jenkins T. C.; Shnyder S. D.; Loadman P. M.; Waring M. J.; Thurston D. E. Design, synthesis, and biophysical and biological evaluation of a series of pyrrolobenzodiazepine-poly(N-methylpyrrole) conjugates. J. Med. Chem. 2006, 49 (18), 5442–61. 10.1021/jm051199z. [DOI] [PubMed] [Google Scholar]
- Corey E. J.; Cho H.; Rucker C.; Hua D. H. Studies with trialkylsilyltriflates: new syntheses and applications. Tetrahedron Lett. 1981, 22 (36), 3455–3458. 10.1016/S0040-4039(01)81930-4. [DOI] [Google Scholar]
- Wang B.; Sun H. X.; Sun Z. H. LiOAc-catalyzed chemoselective deprotection of aryl silyl ethers under mild conditions. J. Org. Chem. 2009, 74 (4), 1781–4. 10.1021/jo802472s. [DOI] [PubMed] [Google Scholar]
- Kocienski P. J.Protecting Groups, 3rd ed.; Thieme: 2005; p 208. [Google Scholar]
- Terauchi T.; Tanaka T.; Terauchi T.; Morita M.; Kimijima K.; Sato I.; Shoji W.; Nakamura Y.; Tsukada T.; Tsunoda T.; Hayashi G.; Kanoh N.; Nakata M. Formal total synthesis of altohyrtin C (spongistatin 2). Part 2: Construction of fully elaborated ABCD and EF fragments. Tetrahedron Lett. 2003, 44 (42), 7747–7751. 10.1016/j.tetlet.2003.08.083. [DOI] [Google Scholar]
- Deziel R. Mild palladium (O)-catalyzed deprotection of allyl esters. A useful application in the synthesis of carbapenems and other B-lactam derivatives. Tetrahedron Lett. 1987, 28 (38), 4371–4372. 10.1016/S0040-4039(00)96512-2. [DOI] [Google Scholar]
- Kurti L.; Czako B.. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier Academic Press: 2005; pp 458–9. [Google Scholar]
- SC16LD6.5 in recurrent small cell lung cancer. https://clinicaltrials.gov/ct2/show/NCT01901653.
- Saunders L. R.; Bankovich A. J.; Anderson W. C.; Aujay M. A.; Bheddah S.; Black K.; Desai R.; Escarpe P. A.; Hampl J.; Laysang A.; Liu D.; Lopez-Molina J.; Milton M.; Park A.; Pysz M. A.; Shao H.; Slingerland B.; Torgov M.; Williams S. A.; Foord O.; Howard P.; Jassem J.; Badzio A.; Czapiewski P.; Harpole D. H.; Dowlati A.; Massion P. P.; Travis W. D.; Pietanza M. C.; Poirier J. T.; Rudin C. M.; Stull R. A.; Dylla S. J. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 2015, 7 (302), 302ra136. 10.1126/scitranslmed.aac9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietanza M. C.; Spigel D.; Bauer T. M.; Ready N. E.; Glisson B. S.; Morgensztern D.; Robert F.; Salgia R.; Kochendorfer M.; Patel M.; Strickland D. K.; Govindan R.; Burris H. A.; Rudin C. M.; Dylla S. Safety, activity, and response durability assessment of single agent rovalpituzumab tesirine, a delta-like protein 3 (DLL3)-targeted antibody drug conjugate (ADC), in small cell lung cancer (SCLC). Eur. J. Cancer 2015, 51, S712. 10.1016/S0959-8049(16)31931-1. [DOI] [Google Scholar]
- Study of ADCT-301 in patients with relapsed or refractory Hodgkin and non-Hodgkin lymphoma. https://www.clinicaltrials.gov/ct2/show/NCT02432235.
- Flynn M.; Berkel P.; Zammarchi F.; Levy J.; Tiberghien A.; Masterson L.; D’Hooge F.; Adams L.; Williams D.; Howard P.; Hartley J., Pre-Clinical Activity of Adct-301, a novel pyrrolobenzodiazepine (PBD) dimer-containing antibody drug conjugate (ADC) targeting CD25-expressing hematological malignancies. In 56th ASH Annual Meeting and Exposition, San Francisco, 2014; p 4491.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.