

Abstract
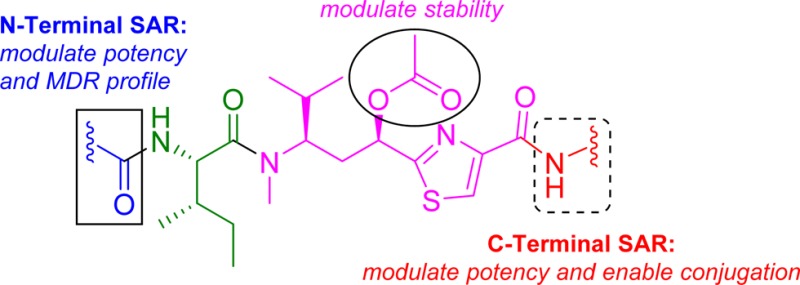
The tubulysin class of natural products has attracted much attention from the medicinal chemistry community due to its potent cytotoxicity against a wide range of human cancer cell lines, including significant activity in multidrug-resistant carcinoma models. As a result of their potency, the tubulysins have become an important tool for use in targeted therapy, being widely pursued as payloads in the development of novel small molecule drug conjugates (SMDCs) and antibody–drug conjugates (ADCs). A structure-based and parallel medicinal chemistry approach was applied to the synthesis of novel tubulysin analogues. These efforts led to the discovery of a number of novel and potent cytotoxic tubulysin analogues, providing a framework for our simultaneous report, which highlights the discovery of tubulysin-based ADCs, including use of site-specific conjugation to address in vivo stability of the C-11 acetate functionality.
Keywords: Tubulysin, microtubule inhibitors, antibody−drug conjugates, structure-based drug design, structure−activity relationships, rapid overlay of chemical structures, parallel medicinal chemistry
The tubulysins (Figure 1), originally isolated from the broth of myxobacteria strains by Höfle and co-workers,1,2 have drawn considerable attention from the synthetic and medicinal chemistry community3−5 due to their structural architecture and potent cytotoxic activity against a wide variety of cancers. To date 14 different tubulysins have been reported and their common structural characteristics comprise N-methyl-d-pipecolic acid (Mep), l-isoleucine (Ile), and tubuvaline (Tuv) units, which also contains a secondary alcohol or acetate at C-11. Additionally, all natural products contain an unusual N,O acetal and either a tubutyrosine (Tut) (tubulysin A) or tubuphenylalanine (Tup) functionality (tubulysins D, H, U, and V) at the C-termini.1 Further, it has been reported6 that the N,O-acetal can be replaced by a simple alkyl group to provide N-14-desacetoxytubulysin H (1) without loss in potency (Figure 1).
Figure 1.
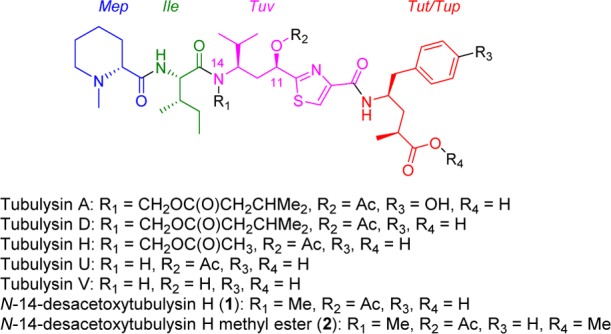
Natural and unnatural tubulysins.
The mechanism of action of tubulysins involves potent microtubule inhibition (MTIs), causing rapid disintegration of the cytoskeleton and mitotic machinery of dividing cells and leading to apoptosis.1,2,7,8 In addition to their excellent cytotoxic activity against a number of human cancer lines, the tubulysins are also shown to exhibit potent activity against multidrug-resistant (MDR) carcinoma4,8 cell lines. In particular, this activity against MDR cells, as well as widespread activity against many cancer types,1,2,8 has made tubulysins attractive payloads for selective targeting of cancer cells via small molecule drug conjugates (SMDCs)9 and antibody–drug conjugates (ADCs).4,10−12 We applied a combination of structure-based design (guided by the tubulin-bound structures of other vinca and peptide-binding inhibitors13,14) and parallel medicinal chemistry approaches for the discovery of novel and structurally simplified tubulysin analogues. We also designed a number of analogues which address in vivo liability of the labile C-11 acetate, a common structural feature of all tubulysins. Herein, we describe the details of the design, synthesis, and structure–activity relationship of these novel tubulysin analogues.
In order to explore N-terminal tubulysin SAR, a library was designed to probe whether a variety of amino acid monomers in our collection could replace the Mep residue. A shape-based molecular overlay method using ROCS (Rapid Overlay of Chemical Structures)15 was employed for the library design. A query molecule, ideally in the tubulin-bound conformation, is required for this method to work. Since coordinates from solved structures of tubulin-bound tubulysin analogues were unavailable to us, our first objective was to generate a model for the bound conformation of 1 and 2, the most potent tubulysin analogues in our assays at that time (Table 1). We achieved this by overlaying 1, using ROCS, on to our previously reported structure of tubulin-bound auristatin analogue 3 (PDB: 4X20).14 The stereochemical differences between the corresponding backbone Cαs (Figure 2A), especially the first and the third residue (C13 in tubulysin), does have an impact on the overlay and perhaps their interactions with tubulin. The conformational flexibility of the tubulysin peptide and the consequent challenges in reproducing the extended bioactive conformation resulted in only the truncated N-terminal tubulysin trimer yielding an optimal overlay (Figure 2B). Hence we used the overlaid N-terminal trimer (common to both 1 and 2) as a surrogate for the putative bound conformation to design our N-terminal tubulysin library. A diverse list of both cyclic and acyclic monomers were selected for the library synthesis based on 3D shape similarity to the query and knowledge-based evaluation using our auristatin N-termini SAR.14 Simultaneously, we kept in mind the need for a linker handle, which would be necessary for synthesis of the final ADC. There were two consistent observations during the library design using shape-based molecular overlay. First, the amide bond between Ile and Tuv residues in tubulysins appeared to be trans as compared to the cis configuration14 between corresponding Val and Dil residues in the tubulin-bound auristatin (Figure 2B). Second, the N-terminal backbone stereochemical differences between the compound classes (Figure 2A,B) seem to have a direct impact on the position of the terminal N atom, especially in compounds with cyclic monomers 17f (Figure 2C) and 17g (Figure 2D).
Table 1. Cytotoxicity Evaluation of N-Terminal Tubulysin Library Compounds.
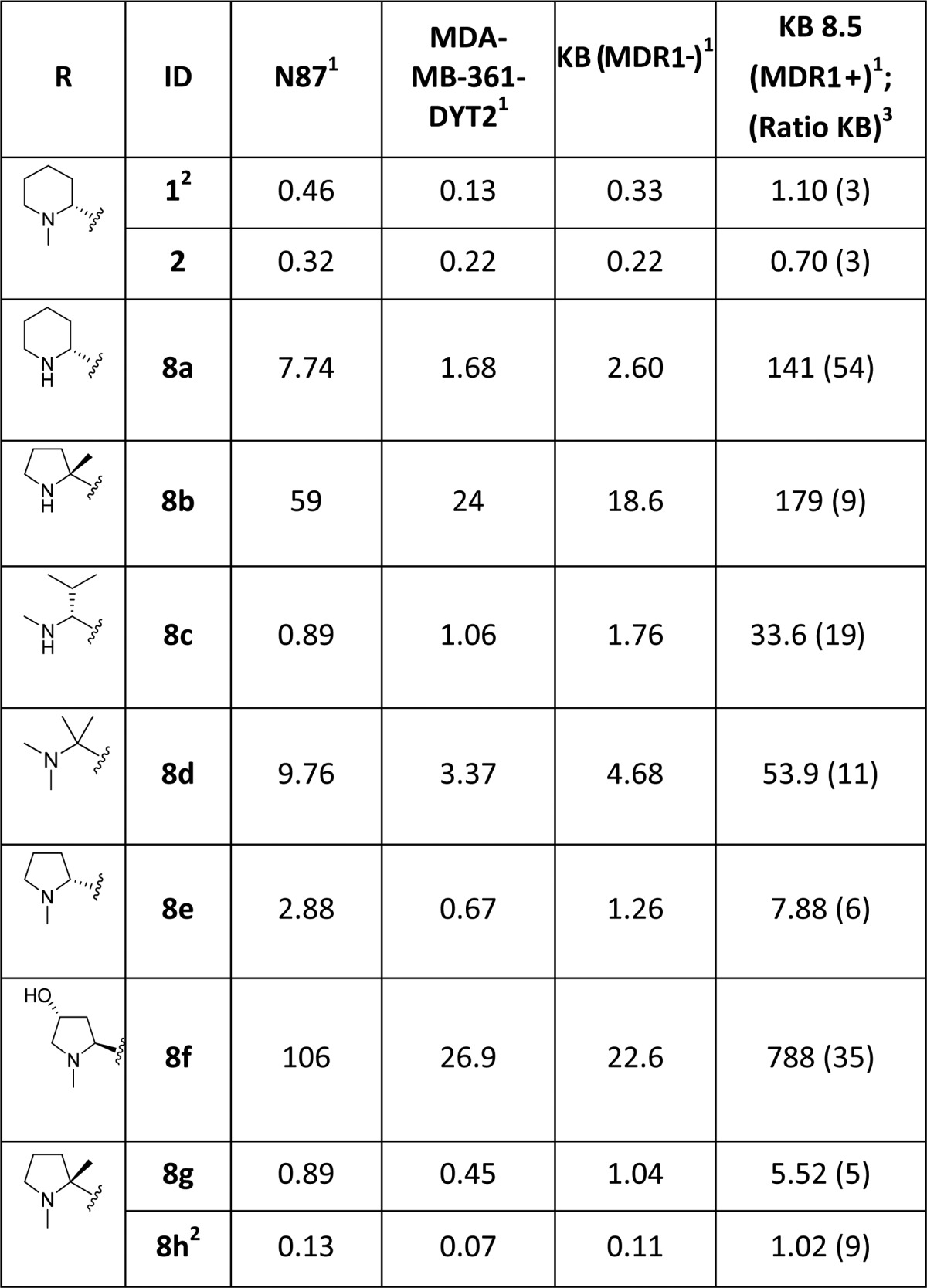
Reported IC50 (nM) is the mean of 2–13 independent determinations.
Carboxylic acid (Tup) C-terminus.
Relative resistance ratio: KB 8.5/KB.
Figure 2.
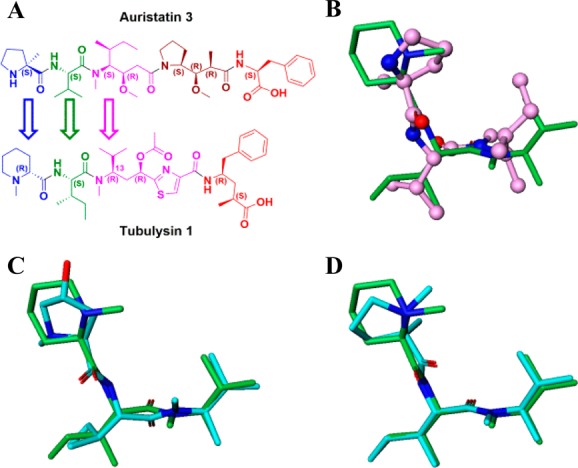
Tubulin-bound structure of auristatin 3 (rendered in pink tubes in B) was used to generate a bound model of truncated N-terminal trimer of 1 (rendered in green tubes in B–D). Computational shape-based molecular overlays of representative compounds 17f and 17g (rendered in cyan tubes) are shown in C and D, respectively.
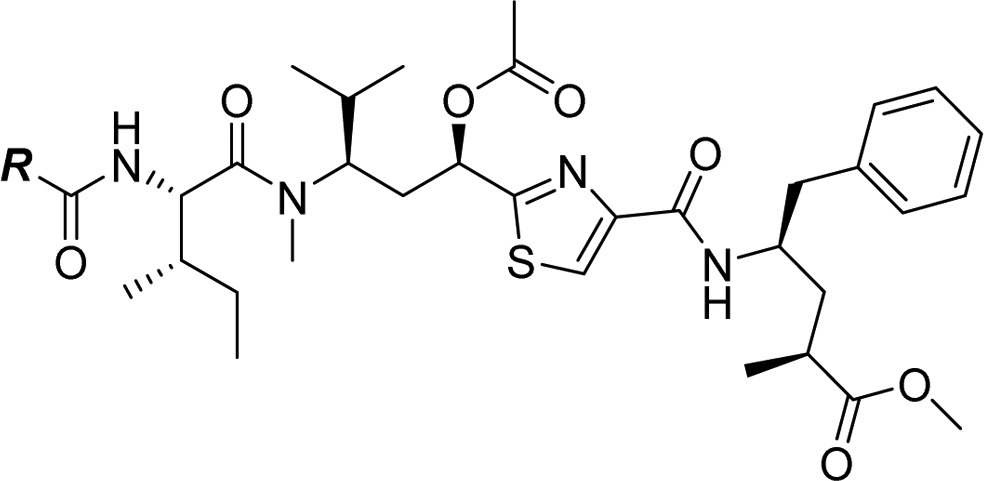
Our initial plans for SAR exploration of tubulysins focused on the N- and C-termini, which would be synthetically achievable via a one-step coupling of a corresponding acid or amine, respectively, with advanced intermediates 5/6 or 4 (Figure 3).16 It is also important to note that for synthetic simplicity we decided to proceed with the truncated N-14-Me analogue in the Tuv unit. We selected ester 4 rather than the corresponding natural acid-containing Tup C-terminus for exploring N-termini SAR, primarily to minimize possible diminished activity in a cell-based assay, known to be reliant on permeability.14 The N-termini modified library (analogues 8a–g) was generated by coupling of the amine 4, prepared from known intermediate 7,17 with selected Boc- or Fmoc-protected amino acids and removal of the protecting group (Scheme 1).
Figure 3.

Key intermediates for synthesis of modified tubulysin analogues.
Scheme 1. Synthesis of N-Terminal Library.

N87 cells (liver metastasis from gastric carcinoma) and MDA-MB-361-DYT2 cells (breast carcinoma) were employed for cytotoxic evaluation of tubulysin analogues.18 The cytotoxicity in parental KB (MDR1−) human epidermoid carcinoma cell lines and the P-glycoprotein (MDR1/ABCB1) expressing drug-resistant KB 8.5 (MDR1+) cell lines was used to measure cytotoxic activity in the presence of the MDR1 drug transporter.
SAR for the modified N-termini analogues are shown in Table 1. A majority of secondary amine-containing analogues (8a–c), designed with an objective of N-termini linker attachment, exhibited diminished potency compared to lead tubulysins 1 and 2. One exception was the N-Me-valine analogue 8c, which displayed activity similar to that of 2 in all cell lines excluding the MDR1-expressing KB 8.5 cell line. At this point, with the goal of maintaining potency and the essential activity against MDR1-expressing cell lines, we considered a variety of tertiary amines to identify optimal N-terminal substitution. Tertiary amine-containing analogues 8d–g were generally much more potent and, in many instances, led to analogues with potency equal or better than 2. Compound 8f, containing a secondary alcohol as a potential handle for linker attachment,19 led to an analogue with diminished activity. Another interesting observation was that replacement of Mep in 2 with pyrrolidine typically led to 3–5× loss in potency; however, the potency could be regained by use of an α-methyl pyrrolidine N-terminus (8e vs 8g). Removing a chiral center from the N-terminus (8d) led to substantial loss of potency. This is quite intriguing and clearly demonstrates a divergence of SAR between auristatins and tubulysins, as such substituents have yielded very potent auristatins.14 Overall, tertiary amine 8g, containing an α-methyl tertiary amine designed based on our auristatin SAR,14 provided the best balance of potency against a variety of cell lines, including the MDR1 expressing KB 8.5 line. It should be noted that 8g was predicted to overlay better with compound 2, especially the position of the terminal N atom, than compound 8f (Figure 2C vs D), thus giving support for our design approach. The Tup (acid) variant of 8g, 8h, displayed excellent potency, similar to the tubulysin analogue 1, likely due to improved permeability of these compounds.
In order to achieve our ultimate goal, employment of our newly discovered tubulysin analogue 8i as an ADC payload, we required a handle for attaching a linker. As such, compound 11 was designed, containing a C-terminal aniline handle for linker installation and further generation of a variety of conjugates. Synthesis of analogue 11 was completed by coupling of pentafluorophenyl (PFP) ester 5 with known Tup aniline 10(20) (Scheme 2), enabling further generation of potent ADCs as detailed in the accompanying paper by Tumey et al.21 In this case, analogue 11 proved to be very potent, albeit showing some attenuation in potency compared to parent compound 8i, paralleling similar reports by Cheng20 and Gingipalli.22 Compound 11 also demonstrated much better MDR profile than the clinically used auristatin payload MMAE14 (KB = 0.28 nM, KB 8.5 = 17.7 nM; KB 8.5/KB = 63). However, a liability we observed with ADCs derived from 11 was rapid hydrolysis of the C-11 acetate to the corresponding alcohol in vivo.21 This phenomenon, coupled with the substantially diminished potency of alcohol 12, led us to design analogues with improved stability in vivo (Table 2).
Scheme 2. Synthesis of Compound 11, Containing a C-Termini Linker Handle.

Table 2. Cytotoxicity Evaluation of C-11 Modified Tubulysins.
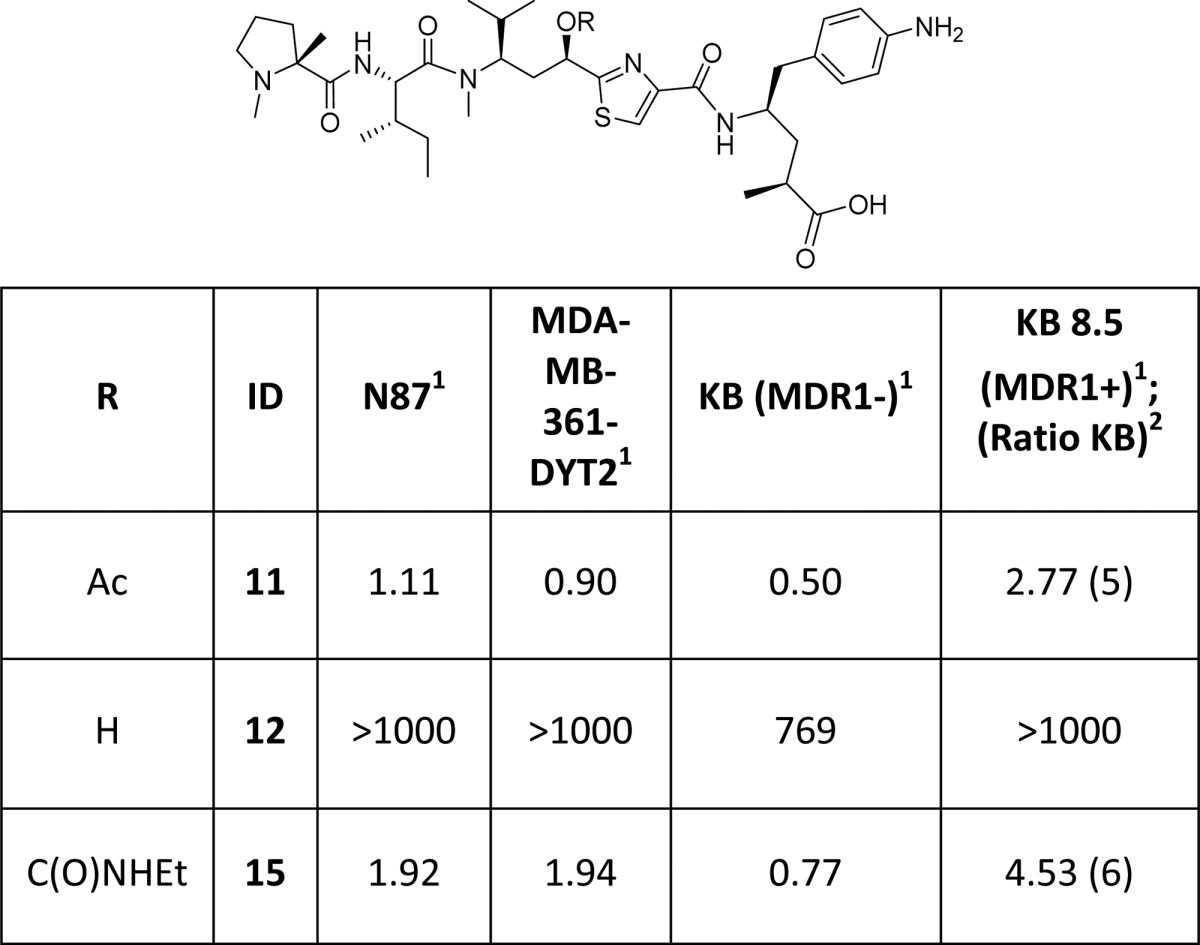
Reported IC50 (nM) is the mean of 2–13 independent determinations.
Relative resistance ratio: KB 8.5/KB.
Our design was aided by a high resolution (2.5 Å) cocrystal structure of 11 with tubulin23 (Figure 424). In this cocrystal structure, the amide bond between Ile and Tuv in 11 is trans, thus confirming the observations we made during our library design. Furthermore, it is conceivable that the acetate is involved in a van der Waals interaction with Thr 221 and Thr 223 (from the β-subunit, loop between Helices H6 and H7) and Pro 325 (from the α subunit – Helix H10). Therefore, considering traditional acetate isosteres,25 we proposed a simple substitution of the C-11 O–Ac with a carbamate moiety,11 which we expected would provide improved stability while maintaining the putative interactions between the binding site residues and O–Ac. Toward this end, alcohol 13 was converted to the carbamate analogue 15 (Scheme 3). Analogous to related work,11 compound 15 maintained similar potency compared to the acetate 11 (Table 2), and ADCs derived from 15 not only demonstrated comparable in vitro cytotoxicity but also improved efficacy in vivo compared to ADCs derived from 11 (see accompanying paper by Tumey et al).21
Figure 4.
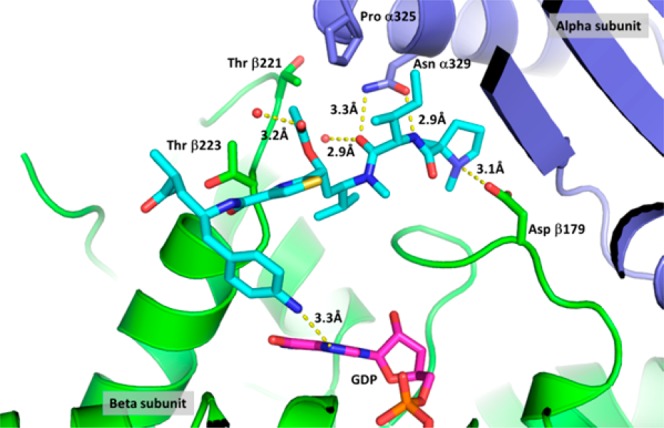
Cocrystal structure of 11 with the T2R-TTL tubulin construct, highlighting the interactions between 11 and the α,β subunits of tubulin.
Scheme 3. Synthesis of Carbamate Analogue 15.

Our C-terminal SAR exploration utilized both the α-methyl pyrrolidine (R1 = A) and Mep (R1 = B) N-termini. We were especially interested in identifying compounds equipotent to 11 while employing simplified C-terminal monomers with less synthetic investment than the traditional Tup. Using the tubulin-bound conformation of 11 (Figure 4), we first evaluated monomers using shape-based molecular overlay26 and visual inspection, keeping in mind the need for a linker handle, which might be necessary for ADC synthesis. Synthesis of the C-terminal modified library was carried out by reaction of the PFP ester 5 or 6(27) with the corresponding amines (Scheme 4).
Scheme 4. Synthesis of C-Termini Modified Tubulysins.

Activity of the C-terminal modified tubulysins is shown in Table 3. Replacement of the Tup substituent in 11 (α-methyl pyrrolidine (R1 = A) N-termini) with simple heterocyclic amines (compounds 16b–d) and the Doe amine (16a), inspired by our work with the auristatin class of compounds and known auristatin MMAD,14 led to a 10–50× drop in potency (vs 11). However, a majority of this potency could be restored by switching to the Mep N-terminus, present in the naturally occurring tubulysins (compounds 17a–d), providing a benzotriazole analogue within 5× potency of compounds 1 and 2. Keeping the benzyl functional group of Tup constant, we explored other monomers putatively replacing only the terminal acid portion in Tup with polar functional groups such as alcohols (16d/16f/16g/16d), sulfonamides (16e/17e), and an amine (17f) or amide (17g). Once again, majority of the compounds with the α-methyl pyrrolidine N-terminus displayed weaker activity than the corresponding Mep N-terminus. The truncated alkyl ester 17d and methyl sulfonamide 17e, both containing potential linker handles, as well as morpholine analogue 17f, were equipotent to 1 and 11. Shape-based molecular overlays25 indicate that these replacements could interact with the side chain of residue Arg B278. While these analogues represent some of the most simplified variations of Tup reported to date, the work reported herein further suggests that tubulysin SAR is more tolerant at the C-terminus than the N-terminus.
Table 3. Cytotoxicity Evaluation of C-Terminal Modified Tubulysins.
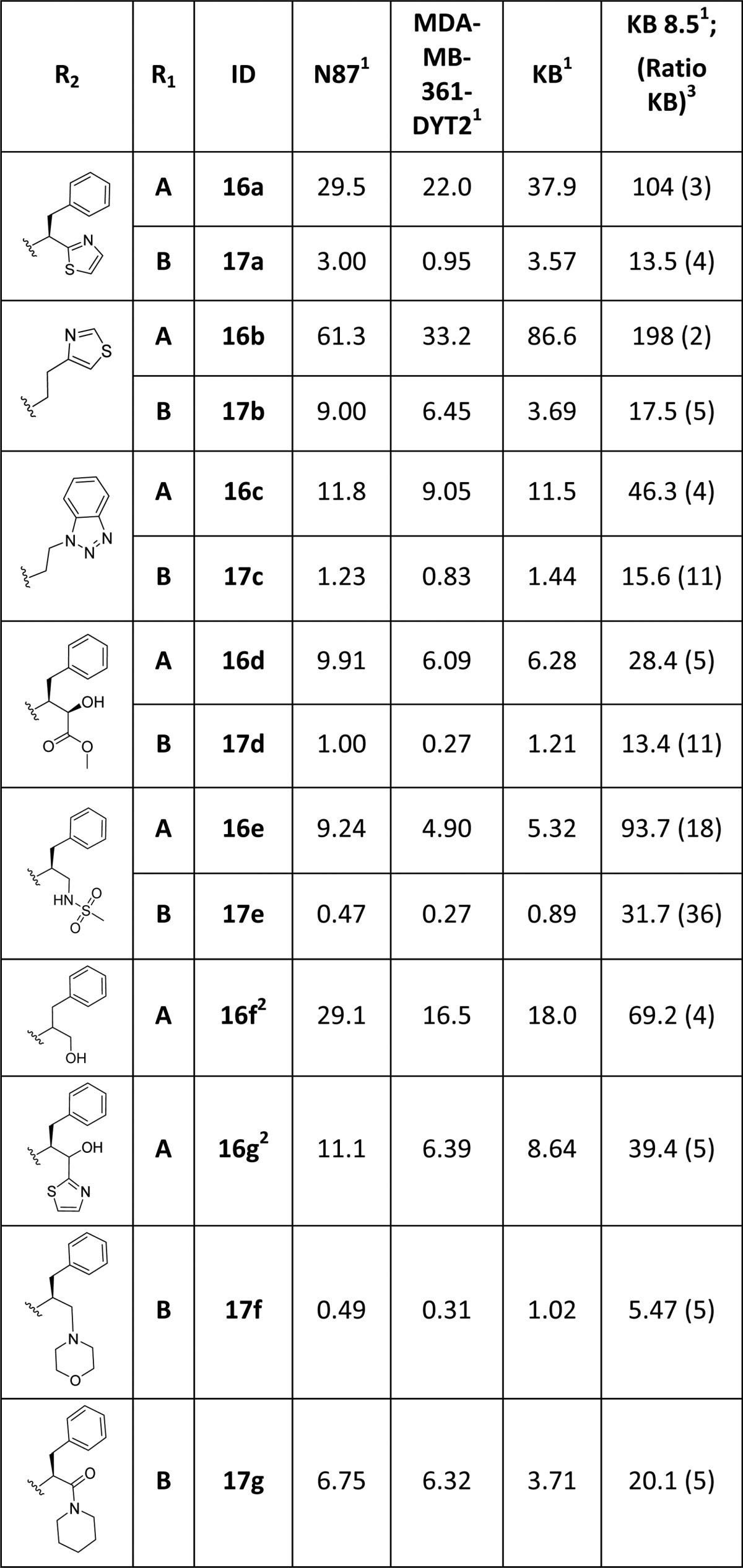
Reported IC50 (nM) is the mean of 2–13 independent determinations.
Tested as a 1:1 mixture of diastereomers.
Relative resistance ratio: KB 8.5/KB.
In conclusion, our studies have helped identify and understand novel N- and C-terminal modalities that, along with C-11 acetate modifications, appear to play a key role in the potency and in vivo stability of tubulysin analogues. Our efforts led to identification of potent tubulysin analogue 11, and ADCs derived from 11 showed excellent potency in vitro and good efficacy in vivo.21 A high resolution cocrystal structure of 11 with tubulin was obtained and utilized for designing analogues with improved stability, eliminating C-11 acetate hydrolysis in vivo. The crystal structure also enabled design and synthesis of a number of potent and novel tubulysin analogues with simplified C-terminus substituents. Development of ADCs from these analogues are underway and will be reported in due course.
Acknowledgments
The authors wish to thank Bhagyashree Khunte, Greg Ciszewski, and Xiaochun Wang for purification support and Xidong Feng for Mass spec and HRMS support. This research used resources at the Industrial Macromolecular Crystallography Association Collaborative Access Team (IMCA-CAT) beamline 17-ID, supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Glossary
ABBREVIATIONS
- Ac
acetate
- ADC
antibody–drug conjugate
- Boc
t-butoxycarbonyl
- DCM
dichloromethane
- DIPEA
diisopropylethyl amine
- DMAP
dimethylamino pyridine
- DMF
N,N-dimethylformamide
- DMSO
dimethyl sulfoxide
- Et
ethyl
- Fmoc
9-fluorenylmethoxycarbonyl
- HATU
1- [bis(dimethylamino) methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- HOAt
1-hydroxy-7-azabenzotriazole
- HPLC
high pressure liquid chromatography
- Ile
l-isoleucine
- mc
maleimido-caproyl
- mcValCitPABC
maleimido-caproyl-valine-citrulline-p-amino benzyl carbamate
- MDR
multidrug resistant
- Mep
d-pipecolic acid
- MMAD
monomethyl auristatin D
- MTI
microtubulin inhibitor
- NMR
nuclear magnetic resonance
- PDB
protein data bank
- PFP
pentafluorophenyl
- ROCS
rapid overlay of chemical structures
- SAR
structure–activity relationship
- SMDC
small molecule drug conjugate
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
- Tup
tubuphenylalanine
- Tut
tubutyrosone
- Tuv
tubavaline
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.6b00274.
Experimental methods, full cytotoxicity data including standard deviations (Tables S1–S3), X-ray data (Table S4), and Figures S1 (an overview of cocrystal structure of tubulin-bound 11; PDB accession code 5KX5) and S2 (C-terminal overlay) (PDF)
Author Present Address
§ SBP Discovery, La Jolla, California 92037, United States.
Author Present Address
∥ Solstice Biologics, San Diego, California 92121, United States.
Author Present Address
⊥ Avidity Biosciences, La Jolla, California 92037, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Sasse F.; Steinmetz H.; Heil J.; Hofle G.; Reichenbach H. Tubulysins, new cytostatic peptides from myxobacteria acting on microtubuli. Production, isolation, physico-chemical and biological properties. J. Antibiot. 2000, 53, 879–885. 10.7164/antibiotics.53.879. [DOI] [PubMed] [Google Scholar]
- Steinmetz H.; Glaser N.; Herdtweck E.; Sasse F.; Reichenbach H.; Hoefle G. Isolation, crystal and solution structure determination, and biosynthesis of tubulysins - powerful inhibitors of tubulin polymerization from Myxobacteria. Angew. Chem., Int. Ed. 2004, 43, 4888–4892. 10.1002/anie.200460147. [DOI] [PubMed] [Google Scholar]
- Xu X.; Friestad G. K.; Lei Y. Recent Advances in the Synthesis of Tubulysins. Mini-Rev. Med. Chem. 2013, 13, 1572–1578. 10.2174/13895575113139990063. [DOI] [PubMed] [Google Scholar]
- Murray B. C.; Peterson M. T.; Fecik R. A. Chemistry and biology of tubulysins: antimitotic tetrapeptides with activity against drug resistant cancers. Nat. Prod. Rep. 2015, 32, 654–662. 10.1039/C4NP00036F. [DOI] [PubMed] [Google Scholar]
- Nicolaou K. C.; Yin J.; Mandal D.; Erande R. D.; Klahn P.; Jin M.; Aujay M.; Sandoval J.; Gavrilyuk J.; Vourloumis D. Total Synthesis and Biological Evaluation of Natural and Designed Tubulysins. J. Am. Chem. Soc. 2016, 138, 1698–1708. 10.1021/jacs.5b12557. [DOI] [PubMed] [Google Scholar]
- Wang Z.; McPherson P. A.; Raccor B. S.; Balachandran R.; Zhu G.; Day B. W.; Vogt A.; Wipf P. Structure-activity and high-content imaging analyses of novel tubulysins. Chem. Biol. Drug Des. 2007, 70, 75–86. 10.1111/j.1747-0285.2007.00541.x. [DOI] [PubMed] [Google Scholar]
- Khalil M. W.; Sasse F.; Luensdorf H.; Elnakady Y. A.; Reichenbach H. Mechanism of action of tubulysin, an antimitotic peptide from myxobacteria. ChemBioChem 2006, 7, 678–683. 10.1002/cbic.200500421. [DOI] [PubMed] [Google Scholar]
- Kaur G.; Hollingshead M.; Holbeck S.; Schauer-vukasinovic V.; Camalier R. F.; Doemling A.; Agarwal S. Biological evaluation of tubulysin A: a potential anticancer and antiangiogenic natural product. Biochem. J. 2006, 396, 235–242. 10.1042/BJ20051735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy J. A.; Dorton R.; Dawson A.; Vetzel M.; Parker N.; Nicoson J. S.; Westrick E.; Klein P. J.; Wang Y.; Vlahov I. R.; Leamon C. P. In Vivo Structural Activity and Optimization Studies of Folate-Tubulysin Conjugates. Mol. Pharmaceutics 2009, 6, 1518–1525. 10.1021/mp900086w. [DOI] [PubMed] [Google Scholar]
- Li J. Y.; Perry S. R.; Muniz-Medina V.; Wang X.; Wetzel L. K.; Rebelatto M. C.; Hinrichs M. J. M.; Bezabeh B. Z.; Fleming R. L.; Dimasi N.; Feng H.; Toader D.; Yuan A. Q.; Xu L.; Lin J.; Gao C.; Wu H.; Dixit R.; Osbourn J. K.; Coats S. R. A Biparatopic HER2-Targeting Antibody-Drug Conjugate Induces Tumor Regression in Primary Models Refractory to or Ineligible for HER2-Targeted Therapy. Cancer Cell 2016, 29, 117–129. 10.1016/j.ccell.2015.12.008. [DOI] [PubMed] [Google Scholar]
- Cong Q.; Cheng H.; Gangwar S.. Preparation of antimitotic compounds structurally related to tubulysins and their conjugates for targeted delivery and their use for treating cancers. 2014-US14177376.
- Burke P. J.; Hamilton J. Z.; Pires T. A.; Setter J. R.; Hunter J. H.; Cochran J. H.; Waight A. B.; Gordon K. A.; Toki B. E.; Emmerton K. K.; Zeng W.; Stone I. J.; Senter P. D.; Lyon R. P.; Jeffrey S. C. Development of Novel Quaternary Ammonium Linkers for Antibody–Drug Conjugates. Mol. Cancer Ther. 2016, 15, 938. 10.1158/1535-7163.MCT-16-0038. [DOI] [PubMed] [Google Scholar]
- Cormier A.; Marchand M.; Ravelli R. B. G.; Knossow M.; Gigant B. Structural insight into the inhibition of tubulin by vinca domain peptide ligands. EMBO Rep. 2008, 9, 1101–1106. 10.1038/embor.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maderna A.; Doroski M.; Subramanyam C.; Porte A.; Leverett C. A.; Vetelino B. C.; Chen Z.; Risley H.; Parris K.; Pandit J.; Varghese A. H.; Shanker S.; Song C.; Sukuru S. C. K.; Farley K. A.; Wagenaar M. M.; Shapiro M. J.; Musto S.; Lam M.-H.; Loganzo F.; O’Donnell C. J. Discovery of Cytotoxic Dolastatin 10 Analogues with N-Terminal Modifications. J. Med. Chem. 2014, 57, 10527–10543. 10.1021/jm501649k. [DOI] [PubMed] [Google Scholar]
- Hawkins P. C. D.; Skillman A. G.; Nicholls A. Comparison of Shape-Matching and Docking as Virtual Screening Tools. J. Med. Chem. 2007, 50, 74–82. 10.1021/jm0603365. [DOI] [PubMed] [Google Scholar]
- Full synthetic details for the synthesis of these intermediates are provided in the Supporting Information.
- Ellman J. A.; Patterson A. W.; Peltier H.. Tubulysin D analogs for the treatment of proliferative disorders. 2008-US70677.
- For a recent report describing the analysis of tubulysin analogies using a cell-free tubulin binding affinity assay, see ref (12). A manuscript highlighting the development and use of an alternate cell-free tubulin binding assay is in progress and will be reported in due course.
- Kolakowski R. V.; Haelsig K. T.; Emmerton K. K.; Leiske C. I.; Miyamoto J. B.; Cochran J. H.; Lyon R. P.; Senter P. D.; Jeffrey S. C. The Methylene Alkoxy Carbamate Self-Immolative Unit: Utilization for the Targeted Delivery of Alcohol-Containing Payloads with Antibody–Drug Conjugates. Angew. Chem., Int. Ed. 2016, 55, 7948. 10.1002/anie.201601506. [DOI] [PubMed] [Google Scholar]
- Cheng H.; Cong Q.; Gangwar S.; Zhang Q.. Antiproliferative compounds structurally related to tubulysins, conjugates thereof for targeted delivery, methods of preparation, and uses thereof. 2010-US846493.
- Tumey L. N.; Leverett C. A.; Vetelino B. C.; Li F.; Rago B.; Han X.; Loganzo F.; Musto S.; Bai G.; Sukuru S. C. K.; Graziani E. I.; Puthenveetil S.; Casavant J.; Ratnayake A. S.; Marquette K.; Hudson S.; Doppalapudi V. R.; Stock J. R.; Tchistiakova L.; Bessire A. J.; Clark T.; Lucas J.; Hosselet C.; O’Donnell C. J.; Subramanyam C. Optimization of Tubulysin Antibody-Drug-Conjugates: A Case Study in Addressing ADC Metabolism. ACS Med. Chem. Lett. 2016, 10.1021/acsmedchemlett.6b00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingipalli L.; Toader D.; Wang F.. Tubulysin derivatives. 2015-US25235.
- Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Diaz J. F.; Altmann K.-H.; Steinmetz M. O. Molecular Mechanism of Action of Microtubule-Stabilizing Anticancer Agents. Science 2013, 339, 587–590. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- See Supporting Information (Figure S1) for additional information
- Meanwell N. A. The Influence of Bioisosteres in Drug Design: Tactical Applications to Address Developability Problems. Top. Med. Chem. 2013, 9, 283–381. 10.1007/7355_2013_29. [DOI] [Google Scholar]
- See Supporting Information (Figure S2) for additional information.
- Jackson D. Y.; Ha E.; Probst G. D.. Antibody-drug conjugates, compositions and methods of use. 2014-US41420.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.