

Summary
Myeloid‐derived suppressor cells (MDSCs) have a wide spectrum of immunosuppressive activity; control of these cells is a new target for improving clinical outcomes in cancer patients. MDSCs originate from unusual differentiation of neutrophils or monocytes induced by inflammatory cytokines, including granulocyte‐colony stimulating factor (G‐CSF) and granulocyte–macrophage (GM)‐CSF. However, MDSCs are difficult to detect in neutrophil or monocyte populations because they are not uniform cells, resembling both neutrophils and monocytes; thus, they exist in a heterogeneous population. In this study, we investigated GPI‐80, a known regulator of Mac‐1 (CD11b/CD18) and associated closely with neutrophil maturation, to clarify this unusual differentiation. First, we demonstrated that the mean fluorescence intensity (MFI) of GPI‐80 and coefficient of variation (CV) of GPI‐80 were increased by treatment with G‐CSF and GM‐CSF, respectively, using a human promyelocytic leukaemia (HL60) cell differentiation model. To confirm the value of GPI‐80 as a marker of unusual differentiation, we measured GPI‐80 expression and MDSC functions using peripheral blood cells from metastatic renal cell carcinoma patients. The GPI‐80 CV was augmented significantly in the CD16hi neutrophil cell population, and GPI‐80 MFI was increased significantly in the CD33hi monocyte cell population. Furthermore, the GPI‐80 CV in the CD16hi population was correlated inversely with the proliferative ability of T cells and the GPI‐80 MFI of the CD33hi population was correlated with reactive oxygen species production. These results led us to propose that the pattern of GPI‐80 expression in these populations is a simple and useful marker for unusual differentiation, which is related to MDSC functions.
Keywords: GPI‐80/VNN2, MDSCs, myeloid cell differentiation, renal cell carcinoma
Introduction
Many studies have focused on the differentiation of neutrophils and monocytes under pathological conditions, because myeloid‐derived suppressor cells (MDSCs) are thought to result from unusual differentiation of these cells. MDSCs are regulatory innate immune cells present in many pathological conditions, including cancer 1. MDSCs are expanded by imbalanced production of growth factors and inflammatory cytokines 2, 3, and these cells can interrupt immunological therapy. In addition, MDSCs can prevent hyperactivation of immune responses 4. Therefore, detection of the unusual differentiation of myeloid cells is important to understand the immune status of individual patients 5.
MDSCs do not represent a defined subset of myeloid cells but, rather, a heterogeneous population of activated immature myeloid cells that have been prevented from differentiating fully into mature cells 6. In humans, MDSCs are defined most commonly as CD33+CD14–CD11b+ cells, and lack the expression of mature myeloid and lymphoid cell markers as well as human leucocyte antigen D‐related (HLA‐DR). In healthy individuals, immature myeloid cells comprise ∼0·5% of peripheral blood mononuclear cells 6. In contrast, a subset of mature human neutrophils (CD11cbrightCD62LdimCD11bbrightCD16bright) represents a unique circulating population of myeloid cells that is capable of suppressing human T cell proliferation 7. Thus, the expansion of a heterogeneous MDSC population is related to either the prevention or acceleration of myeloid cell differentiation, which is referred to as ‘unusual differentiation’ in this report.
Advanced studies using high‐dimensional analysis methods (such as mass spectra flow cytometry or single‐cell RNA sequencing) have uncovered the existence of five neutrophil subsets and spatiotemporal regulation of monocyte/macrophage differentiation 8, 9, 10. High‐dimensional analysis is a powerful tool to understand a complicated cell population. However, a biomarker reliant on high‐dimensional methodology is not a practical clinical test. To use informative high‐dimensional biomarkers for various clinical therapies, there is a need to convert high‐dimensional data to a defined and understandable dimension.
The detection of unusual neutrophils and monocytes using limited depth is very difficult. This is based on the fact that neutrophils and monocytes are basically heterogeneous, and MDSCs are also observed in a wide variety of cell types. In the case of renal cell carcinoma (RCC) patients, six types of MDSCs have been reported 11. To address this issue, we tried to scale the heterogeneity of the cells and investigate the relationship between the non‐uniformity of the cells and immune status in this study.
In mice, Gr‐1 (Ly6G, and Ly6C) and arginase I are used for the detection of MDSCs 12. Disappointingly, in humans, orthologues of Gr‐1 are not found 11, and arginase I is detected in normal neutrophils 13, 14. Furthermore, although arginase activity has been correlated with the function of MDSCs 15, 16, l‐arginine supplementation failed to demonstrate a clinical benefit 17. In this study, we propose to measure glycosylphosphatidylinositol‐anchored 80 kD protein/Vanin 2 (GPI‐80/VNN2), which is expressed highly on human neutrophils and not found in mice. GPI‐80 is suggested to work as a modulator of macrophage‐1 antigen (Mac‐1) (CD11b/CD18) function 18. Our previous studies also demonstrated that GPI‐80 expression is associated with myeloid maturation 19.
MDSCs have a wide spectrum of immunosuppressive activities, such as suppression of T cell proliferation and cytotoxicity, inhibition of natural killer (NK) cell activation and expansion of regulatory T cells 20. Furthermore, previous studies have implicated the involvement of MDSCs in the escape of tumours from immune suppression as well as anti‐angiogenic drug resistance 21. Therefore, suppression of MDSCs is a promising target for effective cancer immunotherapy 20. Indeed, for myeloid lineage redistribution, induced by sunitinib, in metastatic RCC (mRCC), the target of an oral anti‐angiogenic agent used for first‐line therapy can be regarded as a predictive marker for tumour regression 22. In this study, we demonstrate that the pattern of GPI‐80 expression is a useful marker for detection of unusual differentiation of myeloid cells in peripheral blood from mRCC patients. This marker will contribute towards the development of MDSC‐targeting therapies.
Materials and methods
Peripheral blood collection and manipulation
This study was approved by the Ethics Committee of Yamagata University, Faculty of Medicine. Peripheral blood (5–10 ml) was collected from mRCC patients and healthy volunteers after receiving informed consent, and heparinized through the addition of 5 U/ml low molecular‐weight heparin. The blood was collected from patients immediately before starting clinical therapies. These samples were kept at 20–25°C and used for experiments within 24 h. Of the included mRCC patients, 10 were male and two were female, with a mean age of 62·9 years (range = 38–78). Detailed characteristics of patients are presented in Table 1. Healthy volunteers included 10 men and one woman, with a mean age of 46·5 years (range = 45–54).
Table 1.
Patient characteristics (n = 12)
| Age, median (range, years) | 64·5 (44–78) |
|---|---|
| Gender, n (%) | |
| Male | 10 (83·3) |
| Female | 2 (16·7) |
| Histology, n (%) | |
| Clear cell | 7 (58·3) |
| Papillary | 2 (16·7) |
| Unknown | 3 (25·0) |
| T stage, n (%) | |
| T1a | 1 (8·3) |
| T1b | 1 (8·3) |
| T2a | 1 (8·3) |
| T2b | 1 (8·3) |
| T3a | 7 (58·3) |
| T3b | 0 (0) |
| T3c | 0 (0) |
| T4 | 1 (8·3) |
| N stage, n (%) | |
| N0 | 9 |
| N1 | 1 (8·3) |
| N2 | 2 (75·0) |
| M stage, n (%) | |
| M0 | 0 (0) |
| M1 | 12 (100) |
| Site of metastasis, n (%) | |
| Lung | 6 (50·0) |
| Liver | 3 (25·0) |
| Bone | 2 (16·7) |
| Lymph node | 5 (41·7) |
| Peritoneum | 1 (8·3) |
TNM (tumour node metastasis) stage was determined with reference to Unio Internationalis Contra Cancrum (UICC). The clinical stage of all patients was stage IV.
Differentiation of human promyelocytic leukaemia (HL60)
The human promyelocytic cell line, HL60, was obtained from the Health Science Research Resources Bank of the Human Science Foundation (Osaka, Japan). The cells were differentiated as described previously 19. In this study, 4 × 105 cells/ml were incubated with 1·25% dimethyl sulphoxide (DMSO) in RPMI‐1640 medium (Sigma‐Aldrich, St Louis, MO, USA) containing 10% fetal bovine serum, 50 U/ml penicillin G potassium and 50 μg/ml streptomycin sulphate at 37°C in an atmosphere of 5% CO2 in air with humidity. During incubation, various cytokines were added to the medium at day 0. Several cytokines were gifted from companies as follows: recombinant human (rh) interleukin (IL)‐1β was from Otsuka Pharmaceutical (Tokyo, Japan); rhIL‐6 was from Ajinomoto Co. (Tokyo, Japan); rh tumour necrosis factor (TNF)‐α was from Dainippon Sumitomo Pharma (Osaka, Japan); and rh granulocyte‐colony stimulating factor (G‐CSF) was from Chugai Pharmaceutical (Tokyo, Japan). In addition, rh granulocyte/macrophage (GM)‐CSF was purchased from Genzyme (Cambridge, MA, USA) and rhIL‐21 was from PeproTech (Rocky Hill, NJ, USA).
Cell surface staining
HL60 or whole blood cells were stained with antibodies as described previously 19, 23. Briefly, cells were aliquoted into microtubes (50 μl of 2 × 105 cells or blood per tube) and incubated with Fc Blocker (BioLegend, San Diego, CA, USA) for 5 min. After blocking of Fc receptors, cells were incubated with antibodies for 30 min on ice, and then treated with BD Phosflow lyse/fix buffer (0·5 ml; BD Biosciences, San Jose, CA, USA) for 10 min at 37°C to lyse red blood cells (RBCs) and fix white blood cells (WBCs). HL60 cells were fixed with lyse/fix buffer diluted in phosphate‐buffered saline (PBS). After washing with PBS, the cells were measured by flow cytometry using a fluorescence activated cell sorter (FACS)Canto II (BD Biosciences). For morphological observations, the cells were haemolysed using ammonium chloride buffer and then sorted using FACSAria (BD Biosciences). The data were analysed using FlowJo software version 6.2 (TreeStar, Ashland, OR, USA). The antibodies used in this study are as follows: fluorescein isothiocyanate (FITC)‐conjugated anti‐CD11b mAb (BEAR1), phycoerythrin (PE)‐conjugated anti‐CD71 mAb (YDJ.1.2.2), both from ImmunoTec (Marseille, France); PE‐conjugated anti‐CD16 mAb (3G8), FITC or PE‐conjugated anti‐CD14 mAb (MφP9), FITC‐conjugated anti‐CD64 mAb (clone 10.1) and allophycocyanin (APC)‐conjugated anti‐HLA‐DR mAb (G46‐6) from BD Biosciences; Pacific Blue‐conjugated anti‐CD3 mAb (UCHT1), FITC‐conjugated anti‐CD15 mAb (HI98), Brilliant violet 421‐conjugated anti‐CD33 mAb (WM53) and FITC‐conjugated anti‐CD62L mAb (DREG‐56) from BioLegend; APC‐conjugated anti‐latency‐associated polypeptide (LAP)‐1 [transforming growth factor (TGF)‐β1] mAb (#27232) from R&D Systems (Minneapolis, MN, USA); PE‐ or FITC‐conjugated anti‐GPI‐80 mAb (3H9) from MBL (Nagoya, Japan) or prepared originally as described previously 24, 25. The isotype‐matched control mAbs, immunoglobulin (Ig)G1 control (MOPC‐21) and IgG2a control (G155‐178), were obtained from BD Biosciences. The secondary antibodies, PE‐conjugated rabbit anti‐mouse Ig antibodies, were purchased from Dako (Glostrup, Denmark).
Phagocytosis assay
Phagocytosis assay was performed as described previously 23. Briefly, a cell suspension (0·2 ml) was mixed with FITC‐labelled Escherichia coli BioParticles (0·05 mg/ml; Life Technologies, Eugene, OR, USA) and incubated for 30 min at 37°C in microtubes. After incubation, 50 μl of the sample was fixed with immediately lyse/fix buffer (0·5 ml) and washed with PBS. The fixed cells were incubated with unlabelled anti‐GPI‐80 mAb (3H9) and stained with PE‐labelled rabbit anti‐mouse Ig antibody (Dako). The mean fluorescence intensity (MFI) of each sample was measured by flow cytometry (ec800; Sony, Tokyo, Japan).
Cell adhesion assay
Cell adhesion assays were performed as described previously 19. Cells (2 × 105 cells/0·2 ml/well) were incubated with or without 2 μM formyl‐methionyl‐leucyl‐phenylalanine (fMLP; Sigma‐Aldrich) for 30 min in 96‐well flat‐bottomed culture plates. After incubation, non‐adherent cells were aspirated and adherent cells were stained with 0·05% crystal violet solution containing 12% neutralized formaldehyde and 10% methanol for 30 min at room temperature, as described previously 25, 26. The stained cells were lysed using a 1% sodium dodecyl sulphate (SDS) solution, and the optical density (OD) at 595 nm was measured for each well using a microplate reader (Sunrise Remote; Tecan, Männedorf, Switzerland).
Measurement of reactive oxygen species (ROS)
ROS measurement was performed as described previously 23. Peripheral blood or differentiated HL60 cells were incubated with 5 μM CellRox Green (ThermoFisher Scientific, Carlsbad, CA, USA) for 30 min at 37°C and then fixed immediately with lyse/fix buffer at 37°C for 10 min. In this study, the cells were not stimulated using any reagents, such as phorbol 12‐myristate 13‐acetate, which means that spontaneous ROS production was measured. The fixed cells were stained with anti‐GPI‐80 mAb and analysed as described above.
Measurement of IL‐10‐producing cells
IL‐10‐producing cells in whole blood were detected using an IL‐10 Secretion Assay Detection Kit (Miltenyi Biotec, Bergish Gladbach, Germany), according to the manufacturer's instructions. In brief, peripheral blood (0·1 ml) was washed with RPMI‐1640 and then labelled with IL‐10 catch reagent. The labelled cells were incubated in 1 ml RPMI‐1640 with gentle horizontal rotation at 37°C for 45 min. After incubation, the cells were washed and stained with IL‐10 detection antibody and anti‐CD33, CD16 and GPI‐80 mAbs for 10 min at 4°C. After reactions with these antibodies, erythrocytes were lysed immediately and leucocytes were fixed using lyse/fix buffer (BD Bioscience), and fixed cells were analysed by flow cytometry (FACSCanto II). In this study, the blood samples were not stimulated using any reagents, such as lipopolysaccharide.
T cell proliferation assay
The T cell proliferation assay was performed as described previously 27. Mononuclear leucocytes (MNL) and polymorphonuclear cells (PMN) were separated from peripheral blood using Ficoll‐Paque. T cells in MNL samples were isolated using the BD IMag anti‐human CD3 (HIT3a) particle (BD Bioscience), according to the manufacturer's instructions. Isolated T cells (1 × 106 cells/ml) were stained with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; ThermoFisher Scientific, and stimulated with anti‐biotin MACSiBead Particles preloaded with biotinylated CD2, CD3 and CD28 [regulatory T cell (Treg) Suppression Inspector; Miltenyi Biotec]. Autologous PMNs were added at ratio of 1 : 1 (T cells: PMNs) and incubated for 7 days in RPMI‐1640 containing 10% autologous plasma in 96‐well flat‐bottomed culture plates. After incubation, the cells were stained with Pacific blue‐conjugated anti‐CD3 mAb (UCHT1), and CFSE fluorescence intensity was analysed, gating with CD3+ cells by flow cytometry.
Statistics
Data are displayed as the mean ± standard error (s.e.). Comparisons between two groups were performed using an unpaired Student's t‐test, and comparisons across multiple groups were analysed using a one‐way analysis of variance (anova) with a Bonferroni's post‐hoc test. The correlation was calculated with Spearman's rank test. These statistical analyses were performed using Prism software version 5.03 (GraphPad Software, San Diego, CA, USA). P‐values less than 0·05 were considered significant.
Results
GPI‐80 expression with neutrophil maturation in HL60 cells
To confirm the association between GPI‐80 expression and neutrophil differentiation, HL60 cells were differentiated with 1·25% DMSO for 4 days, and several cell surface differentiation markers were analysed by flow cytometry (Supporting information, Fig. 1, panels of flow cytometry analysis). Before differentiation, CD71 (transferrin receptor), which is a known marker of proliferating cells, was detected on almost all cells (Supporting information, Fig. 1, upper‐left panel). After differentiation, CD71 was down‐regulated (Supporting information, Fig. 1, lower‐left panel). In contrast, the expression of CD11b (Mac‐1α chain), a known myeloid cell marker, was found in a small population (< 10%) before the differentiation (Supporting information, Fig. 1, upper‐middle and left panels). After differentiation, more than half the cells became CD11b+ (Supporting information, Fig. 1, lower‐middle and left panels). CD16 (FcγR III), a known neutrophil marker, was detected in a small fraction of cells before differentiation (Supporting information, Fig. 1, upper‐middle and right panels). After differentiation, the proportion of CD16+ cells was increased in the population of CD11b+ cells (Supporting information, Fig. 1, lower‐middle panel). Before differentiation, GPI‐80 expression was not detected in cells (Supporting information, Fig. 1, upper‐right panel). Ten to 20% of CD16+ cells expressed GPI‐80 at 4 days differentiation (Supporting information, Fig. 1, lower‐right panel). These results showed that differentiation inhibited cell proliferation and increased the number of myeloid‐cell antigen‐positive cells, and that the expression of a neutrophil antigen (CD16) was present in myeloid antigen‐positive cells. Furthermore, GPI‐80+ cells were mainly present in the population of neutrophil antigen (CD16)‐positive cells, indicating that GPI‐80 expression was linked to neutrophil maturation.
To support these results, each cell subset was sorted and observed for cell morphology (Supporting information, Fig. 1, lower panels). The sorting gates were indicated as gates (a) to (f) in Supporting information, Fig. 1, lower panels. Gate (a), CD11b–CD16– cells, was from a specimen of promyelocytes, with cells exhibiting a large nucleus and blue‐stained cytosol. Gate (b), CD11b+ CD16– cells, consisted of myelocyte‐type cells, which have a brighter cytosol than that of promyelocytes. Gate (c) CD11b+CD16+ cells were of the metamyelocyte type, which show slight segmentation of nuclei and a very bright cytosol. Gate (d), CD16–GPI‐80– cells, was a mixture of promyelocytes and myelocytes. Gate (e), CD16–GPI‐80+ cells, represented a minor population with neutrophil morphology, such as segmented and condensed nuclei and a very bright cytosol. Gate (f), CD16+GPI‐80+ cells, consisted of cells with a similar shape to that of gate (e); the morphology was more indicative of a neutrophil, compared to that of gate (c). These morphological observations indicated that GPI‐80 expression was related to neutrophil maturation in HL60 cells.
Effect of cytokines on neutrophil maturation
Inflammatory cytokines are considered to inhibit neutrophil maturation and induce myeloid‐derived suppressor cells (MDSCs) 2. To clarify the effects of cytokines in a human neutrophil maturation model, we investigated if various inflammatory cytokines could inhibit neutrophil maturation in the HL60 model (Fig. 1). In this experiment, CD11b+CD16+ cells were mature neutrophils, and CD16+GPI‐80+ cells were more maturate than CD11b+CD16+ cells. It is known that all‐trans retinoic acid (ATRA) induces granulocytic differentiation but inhibits the maturation of HL60 cells, as shown in Fig. 1 19. Treatment with TNF‐α accelerated maturation, but also induced cell death (more than 60% of cells were dead, data not shown). Unexpectedly, there was no significant difference in maturation when cells were treated with IL‐1β and IL‐21 in this comparison group. Furthermore, IL‐6 and G‐CSF, believed to be inducers of MDSCs, slightly augmented maturation. Among the six inflammatory cytokines, only GM‐CSF inhibited maturation. These results suggested that inflammatory cytokines, except GM‐CSF, did not inhibit neutrophil maturation.
Figure 1.
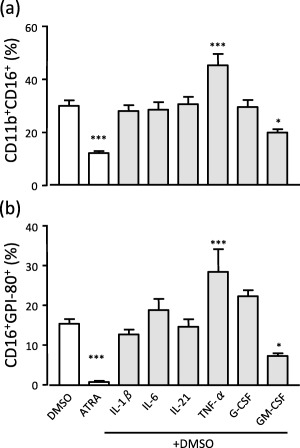
Effect of inflammatory cytokines on neutrophil differentiation. HL60 cells were incubated with 1·25% dimethylsulphoxide (DMSO) in the presence of interleukin (IL)‐1β (100 U/ml), IL‐6 (1 nM), IL‐21 (1 nM), tumour necrosis factor (TNF)‐α (100 U/ml), granulocyte‐colony stimulating factor (G‐CSF) (1 nM) and granulocyte–macrophage (GM)‐CSF (1 nM). The cells were also differentiated with 1 μM all‐trans retinoic acid (ATRA), used as a negative control for terminal maturation. After 4 days incubation, the percentages of viable (a) CD11b+CD16+ or (b) CD16+ glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) cells were analysed by flow cytometry. *P < 0·05; ***P < 0·001; compared to DMSO (n = 4).
Inhibition of phagocytosis and cell adhesion by GM‐CSF, but not by G‐CSF or GM‐CSF in combination with IL‐6
It is commonly assumed that IL‐6 is a factor of MDSC induction, and that blockade of IL‐6 signalling is a promising therapeutic strategy for some cancers 28, 29, 30. However, single stimulation with IL‐6 did not inhibit the neutrophil maturation in HL60 cells (Fig. 1). Thus, we speculated that the combination of IL‐6 with growth factors might be required to inhibit neutrophil maturation. To clarify the effects of growth factors, alone or in combination with IL‐6, on neutrophil differentiation, neutrophil functions were measured (Supporting information, Fig. 2). Phagocytosis was observed only in GPI‐80+ cells (Supporting information, Fig. 2a). The addition of G‐CSF (+G‐CSF) or the combination of IL‐6 and G‐CSF (+G‐CSF + IL‐6) during the differentiation augmented phagocytosis significantly compared to that of a vehicle treatment (Supporting information, Fig. 2b, left side). The combination of IL‐6 and G‐CSF (+G‐CSF + IL‐6) did not show an additive effect compared to the addition of G‐CSF (+G‐CSF) alone (Supporting information, Fig. 2b, left side). Neither GM‐CSF (+GM‐CSF) nor the combination of IL‐6 and GM‐CSF (+GM‐CSF + IL‐6) augmented phagocytosis (Supporting information, Fig. 2b, right side). Cell adhesion, which was induced by fMLP stimulation, was also augmented significantly by the addition of G‐CSF, but not by the addition of GM‐CSF alone or in combination with IL‐6 (Supporting information, Fig. 2c). We also sorted GPI‐80+ cells after differentiation in the presence of these cytokine combinations, and observed morphology. However, there were no significant differences in the neutrophil features of morphology (data not shown). These results suggested that G‐CSF augmented these neutrophilic functions, whereas IL‐6 had no effects. In contrast, GM‐CSF inhibited these functions strongly.
Augmentation of ROS production in GPI‐80+ cells by both G‐CSF and GM‐CSF
ROS are known as effector molecules required for the inhibition of T cell proliferation by MDSCs 7, 16. We measured ROS production in differentiated HL60 cells, which were treated with inflammatory cytokines (Supporting information, Fig. 2d). In contrast to phagocytosis results, ROS production was increased slightly but significantly by the addition of both G‐CSF (+G‐CSF) and GM‐CSF (+GM‐CSF) compared to that of the control (vehicle). These results suggested that ROS production was augmented by the addition of GM‐CSF, in contrast to its functions in phagocytosis and cell adhesion.
Increased coefficient of variation (CV) of GPI‐80 expression after treatment with a combination of G‐CSF with GM‐CSF
We have demonstrated that GPI‐80 expression is useful to monitor neutrophil maturation, as described above. Furthermore, G‐CSF augments neutrophil maturation, whereas GM‐CSF inhibits it. Therefore, we tried to reconstitute conditions required for the expansion of MDSCs by treating HL60 cells with a combination of G‐CSF and GM‐CSF, and subsequently analysing GPI‐80 expression (Fig. 2).
Figure 2.
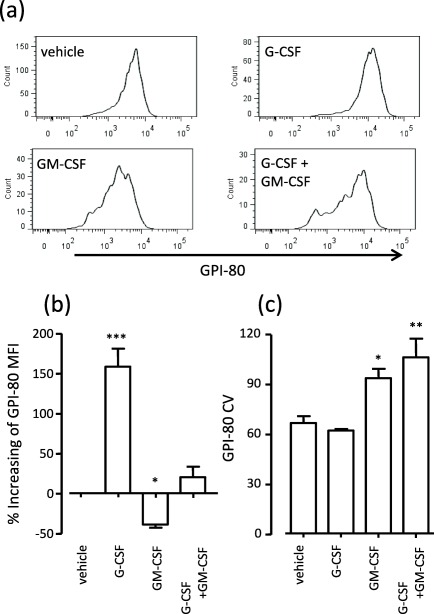
Increasing coefficient of variation (CV) of glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression through treatment with a combination of granulocyte‐colony stimulating factor (G‐CSF) with granulocyte–macrophage (GM)‐CSF. HL60 cells were differentiated by 1·25% DMSO in the presence of 1 nM G‐CSF or GM‐CSF, or a combination of G‐CSF and GM‐CSF for 4 days. The differentiated cells were measured by flow cytometry, and the mean fluorescence intensity (MFI) and coefficient of variation (CV) of GPI‐80 in CD16+ cells were analysed. Percentage of increase in GPI‐80 MFI was calculated as follows: % increase of GPI‐80 MFI = [(GPI‐80 MFI treated with G‐CSF, GM‐CSF or combination) – (GPI‐80 MFI treated with vehicle)/(GPI‐80 MFI treated with vehicle] × 100. CV was calculated automatically as robust CV by FlowJo software using the following formula: CV = 100 × ½ (intensity at 84·13 percentile – intensity at 15·87 percentile)/median. *P < 0·05; **P < 0·01; ***P < 0·001 compared to vehicle (n = 4).
HL60 cells were differentiated by 1·25% DMSO in the presence of G‐CSF, GM‐CSF or a combination of G‐CSF and GM‐CSF; GPI‐80 expression was then measured in differentiated cells (CD16+ cells), as shown in Fig. 2a. The histogram patterns of GPI‐80 expression were drastically different for each treatment (Fig. 2a). Indeed, the MFI of GPI‐80 was increased significantly by G‐CSF and decreased significantly by GM‐CSF (Fig. 2b). However, there was no significant difference between the vehicle‐treated group and that treated with a combination of G‐CSF and GM‐CSF, even after calculating the percentage increase. The GPI‐80 MFI may have been counteracted by a mixture of the increasing effect of G‐CSF and the decreasing effect of GM‐CSF.
To present the variation of GPI‐80 (for example, the variation of neutrophil maturation), we analysed the coefficient of variation (CV) for GPI‐80 (Fig. 2c). The addition of GM‐CSF and the combination of G‐CSF and GM‐CSF augmented the GPI‐80 CV significantly. These results suggested that the increasing GPI‐80 MFI was indicative of augmentation of neutrophil maturation (dominant in the G‐CSF‐treated group) and that the increasing GPI‐80 CV was indicative of unusual neutrophil maturation (dominant in the GM‐CSF‐ or G‐CSF and GM‐CSF‐treated groups). Therefore, we speculated that the unbalancing effect of cytokines was elevated with MDSC differentiation and may be detected by the GPI‐80 MFI or CV parameters.
Simple separation of monocytic and neutrophilic cell types in peripheral blood using CD33 and CD16
To analyse whole blood, we first determined the simple parameters to separate myeloid cells from peripheral blood. CD33 is a well‐used myeloid cell marker used to detect human MDSCs 11. HL60 cells also express CD33 abundantly before and after differentiation (data not shown). To separate the neutrophilic cell type from the monocytic cell type, peripheral blood cells were stained with CD16 as described previously 23. Representative analysis is shown in Fig. 3. According to morphological observation of the sorted cells, the CD33hi population comprised mainly monocytic cells. This population also expressed CD14 (data not shown). The CD16hi population comprised mainly neutrophilic cells. CD11b has also been used to detect human MDSCs, as described previously 11. Thus, we also confirmed CD11b expression on CD33hi and CD16hi populations (Supporting information, Fig. 4, right‐side histogram). However, CD11b did not discriminate between the monocytic cell type (CD33hi) and the neutrophilic cell type (CD16hi). In contrast, there was clearly differential expression of GPI‐80 between CD33hi and CD16hi cells (Supporting information, Fig. 4, lower‐side histogram). These observations demonstrated that the marking set, consisting of CD33, CD16 and GPI‐80, was useful for the separation of myeloid cells.
Figure 3.
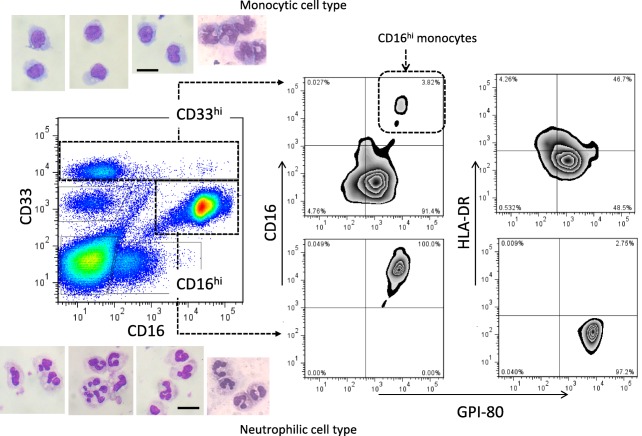
Representative analysis of the myeloid cell population in human peripheral blood. Whole blood cells were stained with CD33, CD16, human leucocyte antigen D‐related (HLA‐DR) and glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80). The population of CD33hi (monocytic cell type, upper side) or CD16hi (neutrophilic cell type, lower side) were sorted, and then stained with May–Giemsa as shown in each respective photograph. The photographs are representative of four independent experiments. Bar = 10 μm. The expression of CD16 (middle panels) and HLA‐DR (right side panels) was compared to that of GPI‐80 in the CD33hi and CD16hi cell population. [Colour figure can be viewed at wileyonlinelibrary.com]
CD16 is also a monocyte marker, in particular of non‐classical and intermediate monocytes 31, 32. The non‐classical monocyte subset represents a new nomenclature of monocyte subsets, which are associated with various diseases such as infections and tumours. In this study, the monocytic cell population was gated by the CD33hi population (Fig. 3). This CD33hi population included classical monocytes (CD14++CD16–), intermediate monocytes (CD14++CD16+) and non‐classical monocytes (CD14+CD16++). We have investigated previously the GPI‐80 expression on monocytes in peripheral blood 33. The GPI‐80 expression on monocytes was detected in both CD14++ and CD14+ subpopulations, suggesting that all three monocytic subpopulations express GPI‐80.
The GPI‐80+ population on monocytes was detected mainly on the CD11bhi, Fc receptors+ and HLA class IIlo population, which is superior in terms of phagocytosis and ROS production but inferior in antigen presentation (neutrophil‐like functions). With regard to the non‐classical monocyte subset (CD14+CD16++), down‐regulation of HLA‐DR expression and up‐regulation of CD11b expression were induced by bacterial infections 32. Thus, the expression of GPI‐80 might be augmented in non‐classical monocyte subsets during systemic inflammation. Indeed, GPI‐80 expression correlated strongly with CD16 expression in the CD33hi monocytic population (Supporting information, Fig. 6), and CD16hi cells in the CD33hi population also exhibited high GPI‐80 expression (Fig. 3).
Augmentation of percentage and CV of GPI‐80 in CD16hi neutrophilic cell type from mRCC patients
To verify our hypothesis (increased GPI‐80 MFI and GPI‐80 CV suggests a pathological imbalance of MDSC‐related cytokines), we analysed peripheral blood from mRCC patients and healthy volunteers. In peripheral blood from patients, the percentage and CV of GPI‐80 in CD16hi cells were elevated significantly compared to those of healthy volunteers (Fig. 4).
Figure 4.
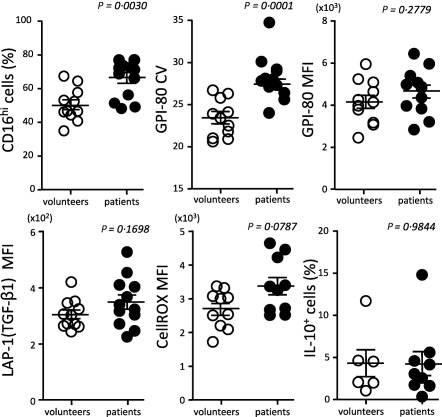
Augmentation of CD16hi cell percentage and glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80)+ coefficient of variation (CV) in CD16hi cell population from metastatic renal cell carcinoma (mRCC) patients. Neutrophil maturation markers [GPI‐80 CV and GPI‐80 mean fluorescence intensity (MFI)] and immunosuppressive molecules {latency‐associated peptide (LAP)‐1 (transforming growth factor (TGF)‐β1] MFI, CellROX MFI and interleukin (IL)‐10+ cells} were measured in CD16hi cell (neutrophilic cell type) populations from the peripheral blood from healthy volunteers and mRCC patients.
IL‐10, TGF‐β and ROS produced by MDSCs are considered to be effector molecules for the suppression of T cell proliferation and the induction of Tregs 12. We therefore measured TGF‐β expression (LAP‐1 MFI), ROS production (CellROX MFI) and IL‐10‐producing cells (IL‐10+ cells) in the CD16hi cell population. There were no significant differences in these effectors between patients and healthy volunteers. These results suggested that the immunoregulatory functions of the CD16hi population were not enhanced clearly in the peripheral blood.
Augmentation of GPI‐80 MFI, CD16 MFI, TGF‐β expression and ROS production in monocytic cell types from mRCC patients
In the CD33hi monocytic population, GPI‐80 MFI and CD16 MFI were increased significantly in patients compared to those parameters in healthy volunteers (Fig. 5a). The increases in GPI‐80 and CD16 expression were similar to the results from G‐CSF‐treated HL60 cells. Therefore, these observations suggested that the monocytic population from patients had neutrophilic features.
Figure 5.
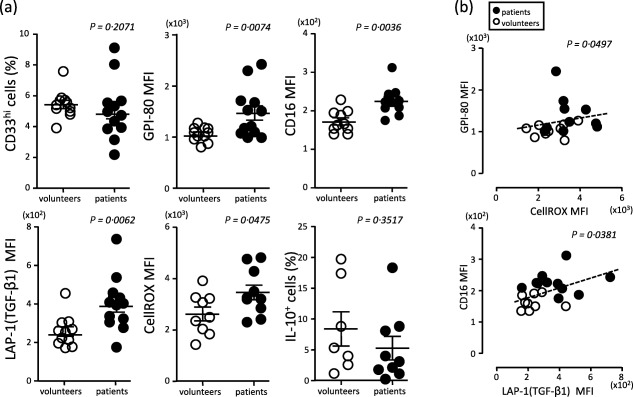
Increase in CD16 mean fluorescence intensity (MFI) and glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80)+ MFI in CD33hi cell populations from metastatic renal cell carcinoma (mRCC) patients. (a) Neutrophil maturation markers (GPI‐80 MFI and CD16 MFI) and immunosuppressive molecules {[latency‐associated peptide (LAP)‐1 (transforming growth factor (TGF)‐β1] MFI, CellROX MFI and IL‐10+ cells} were measured in CD33hi cell (monocytic cell type) populations from peripheral blood of healthy volunteers and mRCC patients. (b) Correlation between GPI‐80 MFI and reactive oxygen species (ROS) production (upper figure) or between CD16 MFI and LAP‐1 (TGF‐β1) MFI (lower figure). Open circles indicate the results from healthy volunteers and closed circles show the results from mRCC patients.
Both TGF‐β1 expression (LAP‐1 MFI) and ROS production (CellROX MFI) in the CD33hi monocytic population from mRCC patients were elevated significantly compared to those of healthy volunteers (Fig. 5a). However, there was no significant difference in IL‐10 production between patients and healthy volunteers. Interestingly, the GPI‐80 MFI was correlated significantly with ROS production, and the CD16 MFI was also correlated significantly with LAP‐1 MFI (Fig. 5b). The GPI‐80 MFI and CD16 MFI were also correlated significantly with each other (P = 0·0008, Supporting information, Fig. 6). These results suggested that GPI‐80 expressing CD33hi cells may have immunoregulatory functions related to ROS production and TGF‐β1 expression, as described in previous reports 16, 34.
Relationship between GPI‐80 CV and T cell proliferation
To verify the immunosuppressive effect of the neutrophilic cell type population from patients in this study, we examined the relationship between increasing GPI‐80 CV and inhibition of T cell proliferation (Fig. 6). Predictably, the results from patient samples showed a higher GPI‐80 CV and a lower ratio of proliferating T cells, compared to results from healthy volunteers. An increase in neutrophilic cell variation (higher GPI‐80 CV) was observed significantly in the patients (Fig. 4). Therefore, higher GPI‐80 CV in patients indicates a high potency of suppression of proliferating T cells. Furthermore, a correlation was also observed between intermediate levels of GPI‐80 CV and intermediate levels of suppression of proliferating T cells in healthy volunteers. These observations suggest that the correlation between GPI‐80 CV and suppression of proliferating T cells is found in physiological and pathological conditions.
Figure 6.
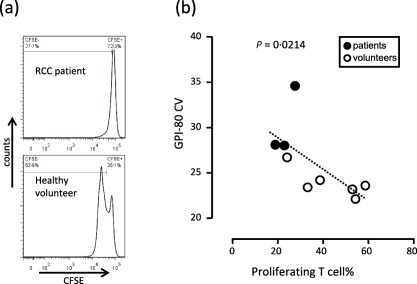
Inverse correlation between glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80)+ coefficient of variation (CV) and T cell proliferation. (a) Representative histograms of carboxyfluorescein diacetate succinimidyl ester (CFSE)‐labelled CD3+ T cell proliferation assay, analysed by flow cytometry, are shown. The upper panel is the result from patients and the lower panel is from healthy volunteers. (b) GPI‐80 CV in CD16hi population in peripheral blood was measured (vertical axis) and CFSE‐labelled T cells were incubated with autologous polymorphonuclear leucocytes [polymorphonuclear cells (PMN), mainly CD16hi population]; the ratio of T cells to PMN was adjusted to 1 : 1. After incubation, the percentage of proliferating T cells was measured (horizontal axis). The results from the patients are indicated as closed circles and the results from healthy volunteers are shown as open circles.
Based on these observations, we summarized the association between reported MDSC markers and GPI‐80 expression in Supporting information, Table 1. The observations of GPI‐80 CV in CD16hi and GPI‐80 MFI in CD33hi populations were applied to all reported MDSC subsets.
Discussion
We have demonstrated that a disturbance or augmentation in neutrophilic maturation was accompanied by an increase in GPI‐80 CV or GPI‐80 MFI, respectively. Indeed, increases in the GPI‐80 CV in the neutrophilic cell population and GPI‐80 MFI in the monocytic cell population were observed in mRCC patients. Furthermore, the GPI‐80 CV was correlated inversely with the proliferative ability of T cells, whereas GPI‐80 MFI was correlated with ROS production. These results allow us to propose that the GPI‐80 CV combined with the GPI‐80 MFI in each cell population is a simple and useful marker for analysis of unusual myeloid cell differentiation associated with MDSC function.
HL60 cells can also differentiate to a monocyte linage 35. Therefore, it was necessary to verify GPI‐80 expression on HL60 cells undergoing monocytic differentiation. Previously, we examined the expression of GPI‐80 on HL60 cells that were differentiated towards a monocytic lineage using activating vitamin D and phorbol 12‐myristate 13‐acetate 19. GPI‐80 expression was not detected on monocytic‐differentiated HL60 cells. Furthermore, monoblastic cell lines such as U937 and THP‐1 did not express GPI‐80 before and after treatment with DMSO. These findings suggest that GPI‐80 expression was restricted to HL60 cells treated with DMSO.
The GPI‐80+ population of DMSO‐treated HL60 cells was analysed with various antigens, as in a previous study 19. The GPI‐80+ population included CD71–CD11+CD16+ cells, and expressed the urokinase‐type plasminogen activator receptor (uPAR, CD87) highly. Furthermore, we also measured monocytic antigens (CD14, CD64 and HLA‐DR) on HL60 cells after differentiation induced by DMSO (Supporting information, Fig. 3a–c). CD14 expression was increased by the addition of G‐CSF, similar to GPI‐80 and CD16 expression. However, the expression of CD64 and HLA‐DR did not increase on DMSO‐treated HL60 cells, even with the addition of G‐CSF. These results suggested that GPI‐80 was detected in the population of CD16+ and CD14+ HL60 cells, which is similar to the mixed feature of neutrophils, intermediate monocytes and non‐classical monocytes, but not to that of classical monocytes, as described previously 31, 32. Thus, GPI‐80+ cells were CD14+CD16+HLA‐DR–CD64– HL60 cells induced by G‐CSF.
CD15, CD16 and CD33 are often used for the separation of myeloid populations 36, 37. GPI‐80 expression was compared to expression of other markers (Supporting information, Fig. 4). The CD33hi monocyte population (red gate) consisted of a CD16− and CD16hi subpopulation. The CD16–CD33lo population was considered an eosinophil population (green gate), whereas the CD16hiCD33lo population was the neutrophil population (blue gate). The levels of GPI‐80 expression were clearly different in each of the three populations. The CD16hiCD33lo population (neutrophils) showed the highest expression of GPI‐80, and the CD16–CD33lo population (green = eosinophils) showed the lowest GPI‐80 expression among the three populations. The CD33hi population (red = monocytes) showed intermediate GPI‐80 expression. CD15 staining showed similar results to CD16 staining. GPI‐80 expression was very low on the CD33– lymphoid cell population (black gate). Furthermore, the CD16+ NK cell population also showed very low GPI‐80 expression (Supporting information, Fig. 5). These observations indicate that GPI‐80 is expressed selectively on the myeloid cell population and that GPI‐80 expression is highest in neutrophilic cells.
We measured the amount of IL‐6, TNF‐α, G‐CSF and GM‐CSF in plasma, as these cytokines are known as candidate inducers of MDSCs 2, 3, 12. However, these cytokines were either virtually undetectable or not elevated significantly in the plasma from mRCC patients in this study (data not shown). Although these cytokines certainly regulate MDSC expansion in bone marrow, they may be at undetectable levels or they might fluctuate transiently in plasma samples. Thus, measurements of GPI‐80 in these neutrophilic and monocytic cell populations appear to be a more sensitive assay than the measurement of these cytokines to detect unusual myeloid cell differentiation.
MDSCs are defined by their inhibitory function on T cell proliferation. In this study, we found a correlation between an increase in GPI‐80 CV and a decrease in T cell proliferation. However, augmentation by inhibitory effector molecules, such as TGF‐β, ROS and IL‐10, was not observed in neutrophilic cell populations. One possibility is that orchestration of these molecules might be associated with inhibitory functions. Another possibility is that the sensitivity of the proliferation assay might be higher than that of the detection assay for these effector molecules. In either case, the measurement of GPI‐80 CV in neutrophilic cells is a sensitive assay, in addition to the T cell proliferation assay.
Scaling the heterogeneity of neutrophils using the GPI‐80 CV has the advantage of analysing unknown cell subsets expressing unknown effector molecules or unknown surface antigens. The concept of scaling the heterogeneity has already been used for the analysis of complex systems such as the gut microbiota or for assessing biodiversity 38, 39. This study proposes that the CV of flow cytometry analysis is a convenient biomarker for the diversity of cell subsets.
A previous study reported that IL‐6 was correlated with MDSCs in cancer patients 40. In the HL60 model system, IL‐6 augmented GPI‐80 MFI slightly but significantly in the presence of GM‐CSF (data not shown). Recently, targeted anti‐IL‐6 mAb therapy has been used not only for inflammatory diseases, but also for cancer 28, 41. Monitoring of GPI‐80 MFI in CD33hi populations in the peripheral blood of cancer patients could be used to predict the clinical outcome of anti‐IL‐6 mAb therapy.
In this study, spontaneous production of ROS and precursors of TGF‐β were elevated significantly in the monocytic cell population of mRCC patients. Although IL‐10 is known to be an MDSC effector molecule of immunosuppression 12, there was no significant difference in IL‐10‐producing cells in the peripheral blood of patients and healthy volunteers. IL‐10 production from MDSCs may not occur in peripheral blood. Monocytic MDSCs have the ability to differentiate into tumour‐associated macrophages (TAM) or granulocytic MDSCs after migration to tumour tissues in mice 42, 43. Thus, it is possible that IL‐10 production is induced after the migration into tumour tissues in humans.
Blood samples used in this study were collected from mRCC patients immediately before treatment. We also collected blood samples from mRCC patients after various clinical treatments. The administration of molecular‐targeting drugs to patients sometimes induced haematotoxicity, and induced an elevation in GPI‐80 CV. Therefore, accurate evaluation of unusual myeloid differentiation was difficult during drug administration. However, several cases exhibited a decrease in GPI‐80 CV after therapy compared to the GPI‐80 CV value before therapy (Kato and Takeda, unpublished results). A follow‐up study of GPI‐80 expression will clarify the relationship between efficacy of therapies and pattern of GPI‐80 expression.
Up‐ or down‐regulation of various cell surface antigens including MDSC markers are induced by both cell differentiation and cell activation. The clarification of the relationship between MDSCs and activated neutrophils is important to understand the heterogeneity of MDSCs. Conversely, our group has established two different monoclonal antibodies to GPI‐80, named 3H9 and 4D4 44. In this study we used 3H9, which recognizes peptide of GPI‐80. Previously, we demonstrated that there are two populations of GPI‐80 that differ in their ability to bind 4D4, which recognizes carbohydrate moieties. The 4D4‐binding form might regulate Mac‐1‐dependent neutrophil adhesion, and might be converted subsequently to a 4D4‐unrecognized form during neutrophil activation. It is of interest to compare the characteristics of these two populations based on the recognition of the two different anti‐GPI‐80 antibodies. In future, the ratio of the 4D4‐recognized form to the 3H9‐recognized form might be a useful indicator for MDSCs associated with neutrophil activation.
Herein we propose the identification of unusual myeloid differentiation by analysing the ectopic expression and CV of the neutrophil maturation molecule, GPI‐80. This analysis method could be useful for the clinical detection of a new cell subset in heterogeneous myeloid cell populations. In other words, the change in deviation of a known single parameter indicates the appearance of unknown cell subsets, which is composed of non‐uniform groups or heterogeneous single cells. This viewpoint might complement high‐dimensional analysis.
Disclosure
None.
Supporting information
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Association between reported myeloid‐derived suppressor cell (MDSC) markers and glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression
Fig. S1. Glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) was expressed in mature human promyelocytic leukaemia (HL60) cells. HL60 cells were differentiated with 1·25% dimethylsulphoxide (DMSO) for 4 days. CD11b, CD16, CD71 and GPI‐80 expression on undifferentiated (upper panels, day 0) or differentiated HL60 cells (lower panels, day 4) was analysed by flow cytometry. The differentiated cells were sorted from the indicated gated area (a–f). The sorted cells were stained with May–Giemsa and are shown in photographs. (a–f) The lower panels correspond to gated areas (a–f), respectively. Data are representative of four independent experiments. Bar = 10 μm.
Fig. S2. Effects of granulocyte‐colony stimulating factor (G‐CSF) and granulocyte–macrophage GM‐CSF, alone or in combination with interleukin (IL)‐6, on neutrophil functions. Human promyelocytic leukaemia (HL60) cells were differentiated by 1·25% dimethylsulphoxide (DMSO) in the presence of 1 nM G‐CSF or GM‐CSF, or a combination of 1 nM IL‐6 and G‐CSF or GM‐CSF for 4 days. After differentiation, the cells were used for functional assays as below: (a) phagocytosis assay. Differentiated cells were incubated with BioParticle (30 min). For background staining, cells were fixed immediately after the addition of BioParticle (0 min). After incubations, the cells were fixed and stained with anti‐ glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) monoclonal antibody (mAb) and phycoerythrin (PE)‐conjugated rabbit anti‐mouse immunoglobulin (Ig) antibodies. The cells were measured by flow cytometry and the mean fluorescence intensity (MFI) of BioParticle in the GPI‐80+ cell subset was analysed. Representative data are shown in (a) and the results are presented in (b). (c) Cell adhesion assay in response to N‐formyl‐methionyl‐leucyl‐phenylalanine (fMLP). The differentiated cells were incubated with (closed column) or without (open column) 2 μM of fMLP for 1 h. After incubation, the adherent cells were stained with crystal violet solution and optical density (OD) was measured at 595 nm. (d) Reactive oxygen species (ROS) production. The differentiated cells were incubated with CellROX green for 30 min to detect ROS production. After the incubations, the cells were fixed and stained with anti‐GPI‐80 mAb as described above. The cells were analysed by flow cytometry, and the MFI of CellROX green was measured in both GPI‐80– (closed column) and GPI‐80+ (open column) cell subsets. *P < 0·05; **P < 0·01; ***P < 0·001; n.s. = non‐significant; compared to vehicle (n = 4–5).
Fig. S3. The relationship between glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression and monocytic markers, CD14, CD64 and human leucocyte antigen D‐related (HLA‐DR) in differentiated human promyelocytic leukaemia (HL60) cells. HL60 cells were differentiated with 1·25% dimethylsulphoxide (DMSO) in the presence of 1 nM granulocyte‐colony stimulating factor (G‐CSF) or granulocyte–macrophage (GM)‐CSF, or a combination of G‐CSF and GM‐CSF for 4 days. After differentiation, the expression of GPI‐80, CD14, CD64 and HLA‐DR on the cells was measured by flow cytometry. The cells were analysed with antibodies targeting (a) GPI‐80 (vertical axis) and CD14 (horizontal axis), (b) HLA‐DR (vertical axis) and CD14 (horizontal axis) and (c) GPI‐80 (vertical axis) and CD64 (horizontal axis). Treatments with cytokines (alone or in combination) are indicated in each panel. Data are representative of three independent experiments.
Fig. S4. Representative analysis of glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression on various cell populations in human peripheral blood. Whole blood cells were stained with CD11b, CD16, CD15, CD33 and GPI‐80. The blood cells were separated by CD16 and CD33 expression into four populations as shown, and included the CD33hi population (red gate), CD16–CD33lo population (green gate), CD16hiCD33lo population (blue gate) and CD33– population (black gate) (left side panels). To compare CD16 staining with CD15 staining, the cells were also separated by CD15 and CD33 into four populations as shown, which included the CD33hi population (red gate), CD15–CD33lo population (green gate), CD15hiCD33lo population (blue gate) and CD33– population (black gate) (middle panels). The expression of GPI‐80 in each population was compared in the overlay histogram (under open arrow). The histogram colours correspond to the gate colours. The expression of CD11b in the CD33hi population (grey filled histogram) was compared to that in the CD16hiCD33lo or CD15hiCD33lo population (open line) on the right side of the histogram (indicated as dashed arrow to right side panel).
Fig. S5. Glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression on lymphocytes in human peripheral blood. Whole blood cells were stained with CD16, CD33, human leucocyte antigen D‐related (HLA‐DR) and GPI‐80. The blood cells were separated into CD33–CD16– lymphocytes (lower left side panel) and CD33–CD16+ natural killer (NK) cells (lower right side panel). These cell populations were analysed for HLA‐DR (vertical axis) and GPI‐80 (horizontal axis) expression.
Fig. S6. Correlation between glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) mean of fluorescence intensity (MFI) and CD16 MFI in CD33hi monocytic cell population. Open circles indicate the results from healthy volunteers and closed circles show the results from metastatic renal cell carcinoma (mRCC) patients.
Acknowledgements
This work was supported by a Grant‐in‐Aid for Scientific Research from the Japan Society for the Promotion of Science (no. 15K10574). The authors would like to thank Ms Chihiro Watanabe and Ms Naomi Abe for microscopic observation of myeloid cell morphology.
References
- 1. Gabrilovich DI, Ostrand‐Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 2012; 12:253–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Condamine T, Gabrilovich DI. Molecular mechanisms regulating myeloid‐derived suppressor cell differentiation and function. Trends Immunol 2011; 32:19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Trikha P, Carson WE III. Signaling pathways involved in MDSC regulation. Biochim Biophys Acta 2014; 1846:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ochando JC, Chen SH. Myeloid‐derived suppressor cells in transplantation and cancer. Immunol Res 2012; 54:275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Diaz‐Montero CM, Finke J, Montero AJ. Myeloid‐derived suppressor cells in cancer: therapeutic, predictive, and prognostic implications. Semin Oncol 2014; 41:174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gabrilovich DI, Nagaraj S. Myeloid‐derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009; 9:162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pillay J, Kamp VM, van Hoffen E et al A subset of neutrophils in human systemic inflammation inhibits T cell responses through Mac‐1. J Clin Invest 2012; 122:327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Becher B, Schlitzer A, Chen J et al High‐dimensional analysis of the murine myeloid cell system. Nat Immunol 2014; 15:1181–9. [DOI] [PubMed] [Google Scholar]
- 9. Bain CC, Bravo‐Blas A, Scott CL et al Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 2014; 15:929–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol 2015; 17:34–40. [DOI] [PubMed] [Google Scholar]
- 11. Solito S, Marigo I, Pinton L, Damuzzo V, Mandruzzato S, Bronte V. Myeloid‐derived suppressor cell heterogeneity in human cancers. Ann NY Acad Sci 2014; 1319:47–65. [DOI] [PubMed] [Google Scholar]
- 12. Ostrand‐Rosenberg S, Sinha P. Myeloid‐derived suppressor cells: linking inflammation and cancer. J Immunol 2009; 182:4499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Munder M, Mollinedo F, Calafat J et al Arginase I is constitutively expressed in human granulocytes and participates in fungicidal activity. Blood 2005; 105:2549–56. [DOI] [PubMed] [Google Scholar]
- 14. Jacobsen LC, Theilgaard‐Monch K, Christensen EI, Borregaard N. Arginase 1 is expressed in myelocytes/metamyelocytes and localized in gelatinase granules of human neutrophils. Blood 2007; 109:3084. [DOI] [PubMed] [Google Scholar]
- 15. Zea AH, Rodriguez PC, Atkins MB et al Arginase‐producing myeloid suppressor cells in renal cell carcinoma patients: a mechanism of tumor evasion. Cancer Res 2005; 65:3044–8. [DOI] [PubMed] [Google Scholar]
- 16. Vasquez‐Dunddel D, Pan F, Zeng Q et al STAT3 regulates arginase‐I in myeloid‐derived suppressor cells from cancer patients. J Clin Invest 2013; 123:1580–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gey A, Tadie JM, Caumont‐Prim A et al Granulocytic myeloid‐derived suppressor cells inversely correlate with plasma arginine and overall survival in critically ill patients. Clin Exp Immunol 2015; 180:280–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nitto T, Onodera K. Linkage between coenzyme A metabolism and inflammation: roles of pantetheinase. J Pharmacol Sci 2013; 123:1–8. [DOI] [PubMed] [Google Scholar]
- 19. Takeda Y, Fu J, Suzuki K et al Expression of GPI‐80, a β2‐integrin‐associated glycosylphosphatidylinositol‐anchored protein, requires neutrophil differentiation with dimethyl sulfoxide in HL‐60 cells. Exp Cell Res 2003; 286:199–208. [DOI] [PubMed] [Google Scholar]
- 20. Albeituni SH, Ding C, Yan J. Hampering immune suppressors: therapeutic targeting of myeloid‐derived suppressor cells in Cancer. Cancer J 2013; 19:490–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Finke J, Ko J, Rini B, Rayman P, Ireland J, Cohen P. MDSC as a mechanism of tumor escape from sunitinib mediated anti‐angiogenic therapy. Int Immunopharmacol 2011; 11:856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. van Cruijsen H, van der Veldt AA, Vroling L et al Sunitinib‐induced myeloid lineage redistribution in renal cell cancer patients: CD1c+ dendritic cell frequency predicts progression‐free survival. Clin Cancer Res 2008; 14:5884–92. [DOI] [PubMed] [Google Scholar]
- 23. Takeda Y, Nara H, Araki A, Asao H. Human peripheral neutrophils express functional IL‐21 receptors. Inflammation 2014; 37:1521–32. [DOI] [PubMed] [Google Scholar]
- 24. Ohtake K, Takei H, Watanabe T et al A monoclonal antibody modulates neutrophil adherence while enhancing cell motility. Microbiol Immunol 1997; 41:67–72. [DOI] [PubMed] [Google Scholar]
- 25. Suzuki K, Watanabe T, Sakurai S et al A novel glycosylphosphatidyl inositol‐anchored protein on human leukocytes: a possible role for regulation of neutrophil adherence and migration. J Immunol 1999; 162:4277–84. [PubMed] [Google Scholar]
- 26. Yakuwa N, Inoue T, Watanabe T, Takahashi K, Sendo F. A novel neutrophil adherence test effectively reflects the activated state of neutrophils. Microbiol Immunol 1989; 33:843–52. [DOI] [PubMed] [Google Scholar]
- 27. Brandau S, Trellakis S, Bruderek K et al Myeloid‐derived suppressor cells in the peripheral blood of cancer patients contain a subset of immature neutrophils with impaired migratory properties. J Leukoc Biol 2011; 89:311–7.] [DOI] [PubMed] [Google Scholar]
- 28. Trikha M, Corringham R, Klein B, Rossi JF. Targeted anti‐interleukin‐6 monoclonal antibody therapy for cancer: a review of the rationale and clinical evidence. Clin Cancer Res 2003; 9:4653–65. [PMC free article] [PubMed] [Google Scholar]
- 29. Guo Y, Xu F, Lu T, Duan Z, Zhang Z. Interleukin‐6 signaling pathway in targeted therapy for cancer. Cancer Treat Rev 2012; 38:904–10. [DOI] [PubMed] [Google Scholar]
- 30. Tanaka T, Kishimoto T. The biology and medical implications of interleukin‐6. Cancer Immunol Res 2014; 2:288–94. [DOI] [PubMed] [Google Scholar]
- 31. Maffei R, Bulgarelli J, Fiorcari S et al The monocytic population in chronic lymphocytic leukemia shows altered composition and deregulation of genes involved in phagocytosis and inflammation. Haematologica 2013; 98:1115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wong KL, Yeap WH, Tai JJ, Ong SM, Dang TM, Wong SC. The three human monocyte subsets: implications for health and disease. Immunol Res 2012; 53:41–57. [DOI] [PubMed] [Google Scholar]
- 33. Sendo D, Takeda Y, Ishikawa H, Sendo F, Araki Y. Localization of GPI‐80, a beta2‐integrin‐associated glycosylphosphatidyl‐inositol anchored protein, on strongly CD14‐positive human monocytes. Immunobiology 2003; 207:217–21. [DOI] [PubMed] [Google Scholar]
- 34. Bierie B, Moses HL. Transforming growth factor β (TGF‐β) and inflammation in cancer. Cytokine Growth Factor Rev 2010; 21:49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chatterjee D, Han Z, Mendoza J et al Monocytic differentiation of HL‐60 promyelocytic leukemia cells correlates with the induction of Bcl‐xL. Cell Growth Differ 1997; 8:1083–9. [PubMed] [Google Scholar]
- 36. Gopinath R, Nutman TB. Identification of eosinophils in lysed whole blood using side scatter and CD16 negativity. Cytometry 1997; 30:313. [PubMed] [Google Scholar]
- 37. van Lochem EG, van der Velden VH, Wind HK, te Marvelde JG, Westerdaal NA, van Dongen JJ. Immunophenotypic differentiation patterns of normal hematopoiesis in human bone marrow: reference patterns for age‐related changes and disease‐induced shifts. Cytometry B Clin Cytom 2004; 60:1–13. [DOI] [PubMed] [Google Scholar]
- 38. Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet‐induced extinctions in the gut microbiota compound over generations. Nature 2016; 529:212–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Faith DP. Biodiversity and evolutionary history: useful extensions of the PD phylogenetic diversity assessment framework. Ann N Y Acad Sci 2013; 1289:69–89. [DOI] [PubMed] [Google Scholar]
- 40. Mundy‐Bosse BL, Young GS, Bauer T et al Distinct myeloid suppressor cell subsets correlate with plasma IL‐6 and IL‐10 and reduced interferon‐α signaling in CD4(+) T cells from patients with GI malignancy. Cancer Immunol Immunother 2011; 60:1269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tawara K, Oxford JT, Jorcyk CL. Clinical significance of interleukin (IL)‐6 in cancer metastasis to bone: potential of anti‐IL‐6 therapies. Cancer Manag Res 2011; 3:177–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Youn JI, Kumar V, Collazo M et al Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol 2013; 14:211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wynn TA. Myeloid‐cell differentiation redefined in cancer. Nat Immunol 2013; 14:197–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nitto T, Takeda Y, Yoshitake H, Sendo F, Araki Y. Structural divergence of GPI‐80 in activated human neutrophils. Biochem Biophys Res Commun 2007; 359:227–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting information may be found in the online version of this article at the publisher's web‐site:
Table S1. Association between reported myeloid‐derived suppressor cell (MDSC) markers and glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression
Fig. S1. Glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) was expressed in mature human promyelocytic leukaemia (HL60) cells. HL60 cells were differentiated with 1·25% dimethylsulphoxide (DMSO) for 4 days. CD11b, CD16, CD71 and GPI‐80 expression on undifferentiated (upper panels, day 0) or differentiated HL60 cells (lower panels, day 4) was analysed by flow cytometry. The differentiated cells were sorted from the indicated gated area (a–f). The sorted cells were stained with May–Giemsa and are shown in photographs. (a–f) The lower panels correspond to gated areas (a–f), respectively. Data are representative of four independent experiments. Bar = 10 μm.
Fig. S2. Effects of granulocyte‐colony stimulating factor (G‐CSF) and granulocyte–macrophage GM‐CSF, alone or in combination with interleukin (IL)‐6, on neutrophil functions. Human promyelocytic leukaemia (HL60) cells were differentiated by 1·25% dimethylsulphoxide (DMSO) in the presence of 1 nM G‐CSF or GM‐CSF, or a combination of 1 nM IL‐6 and G‐CSF or GM‐CSF for 4 days. After differentiation, the cells were used for functional assays as below: (a) phagocytosis assay. Differentiated cells were incubated with BioParticle (30 min). For background staining, cells were fixed immediately after the addition of BioParticle (0 min). After incubations, the cells were fixed and stained with anti‐ glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) monoclonal antibody (mAb) and phycoerythrin (PE)‐conjugated rabbit anti‐mouse immunoglobulin (Ig) antibodies. The cells were measured by flow cytometry and the mean fluorescence intensity (MFI) of BioParticle in the GPI‐80+ cell subset was analysed. Representative data are shown in (a) and the results are presented in (b). (c) Cell adhesion assay in response to N‐formyl‐methionyl‐leucyl‐phenylalanine (fMLP). The differentiated cells were incubated with (closed column) or without (open column) 2 μM of fMLP for 1 h. After incubation, the adherent cells were stained with crystal violet solution and optical density (OD) was measured at 595 nm. (d) Reactive oxygen species (ROS) production. The differentiated cells were incubated with CellROX green for 30 min to detect ROS production. After the incubations, the cells were fixed and stained with anti‐GPI‐80 mAb as described above. The cells were analysed by flow cytometry, and the MFI of CellROX green was measured in both GPI‐80– (closed column) and GPI‐80+ (open column) cell subsets. *P < 0·05; **P < 0·01; ***P < 0·001; n.s. = non‐significant; compared to vehicle (n = 4–5).
Fig. S3. The relationship between glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression and monocytic markers, CD14, CD64 and human leucocyte antigen D‐related (HLA‐DR) in differentiated human promyelocytic leukaemia (HL60) cells. HL60 cells were differentiated with 1·25% dimethylsulphoxide (DMSO) in the presence of 1 nM granulocyte‐colony stimulating factor (G‐CSF) or granulocyte–macrophage (GM)‐CSF, or a combination of G‐CSF and GM‐CSF for 4 days. After differentiation, the expression of GPI‐80, CD14, CD64 and HLA‐DR on the cells was measured by flow cytometry. The cells were analysed with antibodies targeting (a) GPI‐80 (vertical axis) and CD14 (horizontal axis), (b) HLA‐DR (vertical axis) and CD14 (horizontal axis) and (c) GPI‐80 (vertical axis) and CD64 (horizontal axis). Treatments with cytokines (alone or in combination) are indicated in each panel. Data are representative of three independent experiments.
Fig. S4. Representative analysis of glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression on various cell populations in human peripheral blood. Whole blood cells were stained with CD11b, CD16, CD15, CD33 and GPI‐80. The blood cells were separated by CD16 and CD33 expression into four populations as shown, and included the CD33hi population (red gate), CD16–CD33lo population (green gate), CD16hiCD33lo population (blue gate) and CD33– population (black gate) (left side panels). To compare CD16 staining with CD15 staining, the cells were also separated by CD15 and CD33 into four populations as shown, which included the CD33hi population (red gate), CD15–CD33lo population (green gate), CD15hiCD33lo population (blue gate) and CD33– population (black gate) (middle panels). The expression of GPI‐80 in each population was compared in the overlay histogram (under open arrow). The histogram colours correspond to the gate colours. The expression of CD11b in the CD33hi population (grey filled histogram) was compared to that in the CD16hiCD33lo or CD15hiCD33lo population (open line) on the right side of the histogram (indicated as dashed arrow to right side panel).
Fig. S5. Glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) expression on lymphocytes in human peripheral blood. Whole blood cells were stained with CD16, CD33, human leucocyte antigen D‐related (HLA‐DR) and GPI‐80. The blood cells were separated into CD33–CD16– lymphocytes (lower left side panel) and CD33–CD16+ natural killer (NK) cells (lower right side panel). These cell populations were analysed for HLA‐DR (vertical axis) and GPI‐80 (horizontal axis) expression.
Fig. S6. Correlation between glycosylphosphatidylinositol‐anchored 80 kD protein (GPI‐80) mean of fluorescence intensity (MFI) and CD16 MFI in CD33hi monocytic cell population. Open circles indicate the results from healthy volunteers and closed circles show the results from metastatic renal cell carcinoma (mRCC) patients.