

Summary
Apolipoprotein E (ApoE) deficiency promoted an exacerbation of autoimmune arthritis in mice by inducing proinflammatory immune responses. In this study we analysed the contribution of hypercholesterolaemia and/or the absence of ApoE anti‐inflammatory properties, unrelated to its function in the control of cholesterol metabolism, towards the acceleration of arthritis in these mutant animals. The induction and severity of collagen type II‐induced arthritis (CIA) were compared for B10.RIII wild‐type (WT), B10.RIII.ApoE +/–, B10.RIII.ApoE –/– and B10.RIII.low‐density lipoprotein receptor (LDLR –/–) mice with different concentrations of circulating ApoE and cholesterol. A 50–70% reduction in serum levels of ApoE was observed in heterozygous B10.RIII.ApoE +/– mice in comparison to B10.RIII.WT, although both strains of mice exhibited similar circulating lipid profiles. This ApoE reduction was associated with an increased CIA severity that remained lower than in homozygous B10.RIII.ApoE –/– mice. An important rise in circulating ApoE concentration was observed in hypercholesterolaemic B10.RIII.LDLR –/– mice fed with a normal chow diet, and both parameters increased further with an atherogenic hypercholesterolaemic diet. However, the severity of CIA in B10.RIII.LDLR –/– mice was similar to that of B10.RIII.WT controls. In conclusion, by comparing the evolution of CIA between several strains of mutant mice with different levels of serum ApoE and cholesterol, our results demonstrate that both hypercholesterolaemia and ApoE regulate the intensity of in‐vivo systemic autoimmune responses.
Keywords: ApoE, collagen type II‐induced arthritis, hypercholesterolaemia, LDLR
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease resulting in joint inflammation and destruction. A strong association between RA and an increased risk of cardiovascular disease (CVD), due to accelerated atherosclerosis, has been established 1, 2. In this regard, different studies show that dyslipidaemia is highly prevalent in patients with RA and may be present at least 10 years before the onset of the disease 3, 4, 5. However, a relationship between an altered lipid profile in plasma and the augmented incidence of CVD in RA patients has not been clearly proven and is still the subject of intense debate 6, 7.
Conversely, dyslipidaemia can trigger or potentiate already existing inflammatory responses and thereby enhance the severity of RA. Oxidized low‐density lipoproteins (oxLDL) that accumulate in the macrophages of the arterial intima during hypercholesterolaemia are good activators of Toll‐like receptor 4 (TLR‐4), acting as endogenous danger‐associated molecular patterns (DAMP) 8. Also, it has been reported that, in T cells from systemic lupus erythematosus patients, dyslipidaemia may potentiate antigen receptor signalling through the increase of glycosphingolipid synthesis and their incorporation into membrane lipid rafts 9. Accordingly, a relationship between hypercholesterolaemia and RA severity has been established 10, although it has not been confirmed by others 11.
Experimental animal models of RA in association with dyslipidaemia constitute excellent tools to explore some of above‐mentioned questions. In this regard, we and others have shown recently that ApoE (ApoE –/–) deficiency exacerbates the development of collagen type II (col II)‐induced autoimmune arthritis (CIA) in B10.RIII mice in association with enhanced T helper type 1 (Th1) and Th17 inflammatory responses 12 or the semi‐chronic K/BxN serum transfer‐induced inflammatory arthritis through the potentiation of innate immune responses 13. Accordingly, these experimental models can be used to explore whether the accelerated arthritis observed in ApoE –/– is related to the hypercholesterolaemia characteristic of these mutant mice and/or to the lack of some of the previously identified anti‐inflammatory properties of ApoE, that are unrelated to its function in the control of cholesterol metabolism 14. In the present study, we have explored these questions comparing the severity of CIA between mice expressing different amounts of ApoE in the context of either normal or altered circulating cholesterol levels.
Materials and methods
Mice
C57BL/6.ApoE –/– (B6.ApoE –/–, H‐2b) and B10.RIII (H‐2r) mice were purchased from Charles River (Barcelona, Spain) and Harlan Iberica (Barcelona, Spain), respectively. B6 mice deficient in LDL receptor (B6.LDLR –/–) were kindly provided by Dr Jorge Joven, Unitat de Recerca Biomèdica, Hospital Universitari Sant Joan, Institut d'Investigació Sanitària Pere Virgili, Universitat Rovira i Virgili, Reus, Spain. B10.RIII.ApoE –/– mice were obtained from our animal facilities as described recently 12 and intercrossed with B10.RIII wild‐type (B10.RIII.WT) mice to obtain B10.RIII.ApoE –/–, B10.RIII.ApoE +/– and B10.RIII.WT littermates. B10.RIII.LDLR –/–mice were produced in our animal facilities by back‐crossing B6.LDLR –/– mice with B10.RIII mice for seven back‐cross generations. In the second back‐cross generation, H‐2r/r mice were selected by flow cytometry using specific monoclonal antibodies (mAbs) against H‐2b and H‐2r (BD Biosciences, Madrid, Spain). In the last back‐cross generation, male and female heterozygous mice were intercrossed and B10.RIII.LDLR –/– and B10.RIII.WT littermates were selected by polymerase chain reaction (PCR) of genomic DNA extracted from mouse tails. Mice were fed ad libitum with a normal chow diet (NCD) or an atherogenic hypercholesterolaemic diet (HCD) (10·8% total fat, 0·75% cholesterol, S4892‐E010; Ssniff Spezialdiäten GmbH, Soest, Germany) and bled from the retro‐orbital plexus 4 weeks after immunization. All animal care and experimental procedures were carried out according to institutional guidelines and approved by the Universidad de Cantabria Institutional Laboratory Animal Care and Use Committee (ref. 2014/12).
Induction and assessment of arthritis
Bovine col II (MD Bioproducts, Zürich, Switzerland), dissolved at a concentration of 2 mg/ml in 0·05 M acetic acid, was emulsified with complete Freund's adjuvant (CFA) containing 4 mg/ml of Mycobacterium tuberculosis (MD Bioproducts). For the induction of CIA, 8–12‐week‐old male mice were immunized once at the base of the tail with 150 µg of antigen in a final volume of 150 µl. A clinical evaluation of arthritis severity was performed as described previously 15.
Radiological studies were performed using a CCX Rx ray source of 70 Kw with an exposition of 90 ms (Trophy Irix X‐Ray System; Kodak Spain, Madrid, Spain) and the Trophy RVG Digital Imagining system, as described previously 15. Radiological images were scale‐graded according to the presence of four different radiological lesions (1: soft tissue swelling, 2: juxta‐articular osteopenia due to alterations in bone density, 3: joint space narrowing or disappearance and 4: bone surface irregularities due to marginal erosions and/or periosteal new bone formation). The extension of every individual lesion in each paw (local: affecting one digit or one joint in the carpus; diffuse: affecting two or more digits and/or two or more joints in the carpus) was graded from 0 to 2 as follows: 0: absence; 1: local; 2: diffuse.
Flow cytometry studies
Spleen cells from B10.RIII.WT, B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice were stimulated in vitro with concanavalin A (Sigma‐Aldrich, St Louis, MO, USA) at a concentration of 5 µg/ml or with plastic‐bound anti‐CD3 (1 µg/well) and anti‐CD28 (0·5 µg/well) mAbs (αCD3/αCD28). The induction of the CD69 and CD25 activation markers in CD4+ cells was analysed after 3 and 12 h of stimulation by flow cytometry using commercially labelled antibodies (Biolegend, London, UK).
Two‐month‐old B10.RIII.WT, B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice were injected intraperitoneally (i.p.) with 10 µg of lipopolysaccharide (LPS) (Sigma‐Aldrich) or with phosphate‐buffered saline (PBS) as a control. Peritoneal cells were harvested 2 days later and the M1 [CD11b+ F4/80+ major histocompatibility complex (MHC)‐II+] and M2 (CD11b+ F4/80+ CD206+) macrophages characterized by flow cytometry. Cells were analysed in a fluorescence activated cell sorter (FACS)Canto II flow cytometer using FACSDiva software (BD Biosciences).
Serological studies
Total cholesterol (TC), high‐density lipoprotein‐cholesterol (HDL‐c), LDL/very low‐density lipoprotein (VLDL)‐c and triglyceride levels in serum samples were determined using an autoanalyser (Biosystems SA, Barcelona, Spain) following the manufacturer's instructions. Serum levels of immunoglobulin (Ig)G1 and IgG2a anti‐col II antibodies were measured by enzyme‐linked immunosorbent assay (ELISA), as described previously 12. Results were expressed in titration units/ml (U/ml) in reference to a standard curve obtained from a serum pool of col II‐CFA‐immunized dark brown Agouti (DBA)/1 mice. Circulating levels of ApoE were determined by ELISA, as described previously 16. Briefly, microtitre plates (Maxisorp Nunc‐immuno plates; ThermoFisher Scientific, Waltham, MA, USA) were coated with 0·1 µg/ml of WUE‐4 (mouse anti‐human ApoE mAb; Novus Biologicals, Madrid, Spain) and the assay was developed with goat anti‐mouse ApoE (Merck Millipore, Madrid, Spain), followed by a biotinylated rabbit anti‐goat antibody (Vector Laboratories Burlingame, CA, USA) and streptavidin–alkaline phosphatase (BD Biosciences). Results were expressed in µg/ml in reference to a standard curve obtained with purified mouse ApoE (a kind gift of Dr Karl Weisgraber, Gladstone Institute of Neurological Disease, University of California San Francisco, CA, USA).
Mouse HDL from each group was isolated by sequential ultracentrifugation at 100 000 g for 24 h at a density of 1·063–1·21 g/ml. HDL composition, including total and free cholesterol, triglycerides and phospholipids, was determined by commercial methods adapted to the Hitachi 917 autoanalyser (Roche Diagnostics, Rotkreuz, Switzerland). HDL protein concentrations were determined by the bicinchoninic acid method (Thermo Scientific, Rockford, IL, USA). The ability of HDL to protect against LDL oxidation was determined on an assay in which human LDL (0·1 mM phospholipids) was oxidized alone with 2·5 µM CuSO4 or in the presence of equal concentrations of HDL phospholipids from each experimental group (0·1 mM phospholipids). The oxidation kinetics were followed through continuous monitoring of the formation of conjugated diene at 37ºC for 4 h 17. The kinetics of LDL in the LDL + HDL incubations were calculated by subtracting the kinetics of HDL incubated without LDL. The lag phases were calculated and the results represented as relative lag phase to the LDL kinetics oxidized without HDL. 17. Paraoxonase (PON)‐1 was measured using phenylacetate as substrate 17.
Cytokine expression
The expression of mRNAs encoding for interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α, IL‐6, transforming growth factor (TGF)‐β and IL‐10 cytokines was explored in the paws before col II immunization and 8 weeks after using quantitative real time reverse transcriptase PCR (RT–qPCR). After skin removal, the paws were kept frozen at −70ºC until processing. Total RNA from powdered paws was obtained by TRIzol extraction (Invitrogen, ThermoFisher Scientific). One µg of the isolated RNA was used for cDNA synthesis with a RT–PCR kit (Bio‐Rad Laboratories, Madrid, Spain), according to the manufacturer's instructions. RT–qPCR was performed on a StepOne Plus real time PCR instrument (Applied Biosystems, ThermoFisher Scientific) using specific TaqMan expression assays and an universal PCR Master Mix (Applied Biosystems, ThermoFisher Scientific). Results (in triplicate) were normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression and measured in parallel for each sample.
Statistical analysis
The differences between the two groups were analysed by a two‐tailed Student's t‐ or two‐sample Mann–Whitney U‐tests. Probability values < 0·05 were considered significant.
Results
The severity of CIA in ApoE mutant mice correlates directly with the levels of circulating ApoE
We and others have reported recently the development of an exacerbated inflammatory arthritis in mice deficient in ApoE 12, 13. To analyse whether the enhanced disease in these mutant mice was related to the hypercholesterolaemia and/or to the absence of ApoE, we compared the development of CIA between B10.RIII.WT, B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice. Serum ApoE levels and lipid profiles were first analysed in the different experimental groups. The levels of circulating ApoE in heterozygous B10.RIII.ApoE +/– mice were approximately one‐third/half of those found in B10.RIII.WT, both before and after col II immunization (Fig. 1a). A significant reduction in the levels of circulating TC and HDL‐c, but not of LDL/VLDL‐c and triglycerides, was observed in B10.RIII.WT mice in association with the development of CIA (Fig. 1b; P < 0·05 in both cases). Despite the reduced ApoE concentration, the levels of circulating TC, HDL‐c, LDL/VLDL‐c and triglycerides in B10.RIII.ApoE +/– before immunization were normal and similar to those of non‐immunized B10.RIII.WT mice (Fig. 1b). Again, reduced levels of circulating TC and HDL‐c (P < 0·05 and P < 0·01, respectively) were detected in B10.RIII.ApoE +/– after immunization with col II, and these levels remained similar to those of immunized B10.RIII.WT controls (Fig. 1b). As expected, ApoE was undetectable in sera from non‐immunized and col II‐immunized B10.RIII.ApoE –/– mice, along with the presence of an abnormal lipid profile characterized by increased levels of triglycerides, TC and LDL/VLDL‐c (P < 0·001 in all cases) and normal levels of HDL‐c (Fig. 1a,b), in comparison to both B10.RIII.WT and B10.RIII.ApoE +/– mice.
Figure 1.
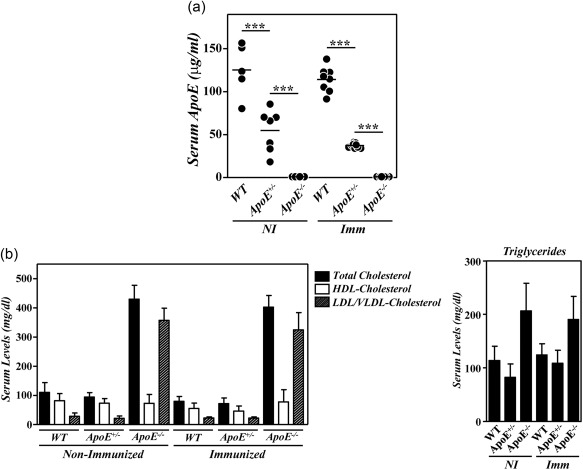
Serum lipid profiles and apolipoprotein E (ApoE) levels in ApoE mutant mice during collagen type II‐induced arthritis (CIA) development. (a) Serum levels of ApoE in B10.RIII.wild‐type (WT), B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice before (NI) and 8 weeks after (Imm) induction of CIA determined by enzyme‐linked immunosorbent assay (ELISA). (b) Serum levels of total cholesterol, high‐density lipoprotein cholesterol (HDLc), very low‐density lipoprotein/low‐density lipoprotein cholesterol (VLDL/LDLc) and triglycerides in B10.RIII.wild‐type (WT), B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice before (NI) and 8 weeks after (Imm) induction of CIA. Representative results of three independent experiments are expressed as the mean ± standard deviation (eight to 10 animals/group). Statistical differences are indicated as follows: ***P < 0·005.
As described previously 12, the severity of CIA in B10.RIII.ApoE –/– mice was higher than in B10.RIII.WT mice (Fig. 2). In inverse correlation with serum levels of ApoE, the clinical severity of CIA in B10.RIII.ApoE +/– mice was also significantly higher than in B10.RIII.WT controls but lower than in B10.RIII.ApoE –/– mice (Fig. 2a). This was confirmed by analysing different radiological signs associated with bone and cartilage damage. The extent of joint narrowing or disappearance of the interosseous spaces, reflecting cartilage loss, and of bone irregularities, secondary to periosteal new bone formation and/or marginal articular erosions, was higher in B10.RIII.ApoE –/– than in B10.RIII.ApoE +/– mice (Fig. 2b,c). Although the degree of soft tissue swelling and of juxta‐articular osteopenia was also slightly larger in B10.RIII.ApoE –/– than in B10.RIII.ApoE +/– mice, these increases were not statistically significant. The severity of all radiological signs was significantly lower in B10.RIII.WT controls than in B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice (Fig. 2b,c).
Figure 2.
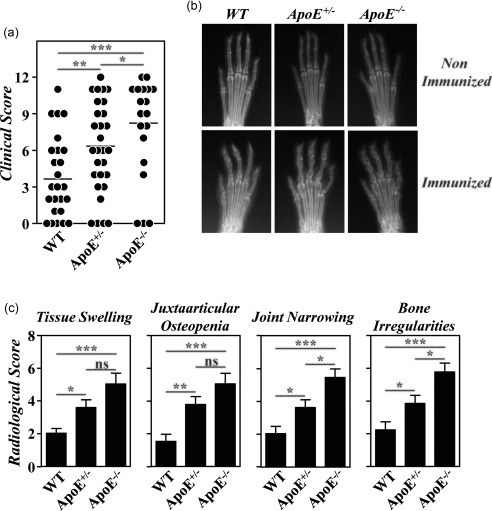
Development of collagen type II‐induced arthritis (CIA) in apolipoprotein E (ApoE) mutant mice; 8–12‐week‐old B10.RIII.wild‐type (WT), B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice were immunized with collagen type II‐complete Freund's adjuvant (col II‐CFA). (a) Clinical severity of CIA in individual mice 8 weeks after immunization with col II. Bars represent the mean values. (b) Representative radiological images of the front paws from B10.RIII.WT, B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice before and 8 weeks after immunization with col II. (c) Severity score of individual radiological signs, expressed as the mean ± standard deviation (seven to 10 mice/group), 8 weeks after immunization. Bars represent the mean values. Results from (a) to (c) are representative of four independent experiments. Statistical differences are indicated as follows: n.s. = non‐significant; *P < 0·05; **P < 0·01; ***P < 0·005.
The accelerated disease in B10.RIII.ApoE –/– mice was shown to correlate with qualitative changes in anti‐col II humoral immune responses and with an enhanced gene expression of arthritogenic cytokines in the paws 12. To explore whether similar abnormalities also accounted for the aggressive disease progression in heterozygous B10.RIII.ApoE +/– mice, circulating levels of IgG1 and IgG2a anti‐col II antibodies were first compared in the different experimental groups before CIA induction and 4 weeks after. As reported 12, the circulating levels of IgG1 anti‐col II antibodies were reduced significantly in immunized B10.RIII.ApoE –/– mice in comparison to both B10.RIII.WT and B10.RIII.ApoE +/– mice (Fig. 3a). Although the levels of IgG1 anti‐col II antibodies also tended to decrease in immunized B10.RIII.ApoE +/– in comparison to B10.RIII.WT controls, these differences did not reach statistical significance (Fig. 3a; P = 0·07). No changes in the titres of serum IgG2a anti‐col II antibodies were observed between the different experimental groups (Fig. 3a).
Figure 3.
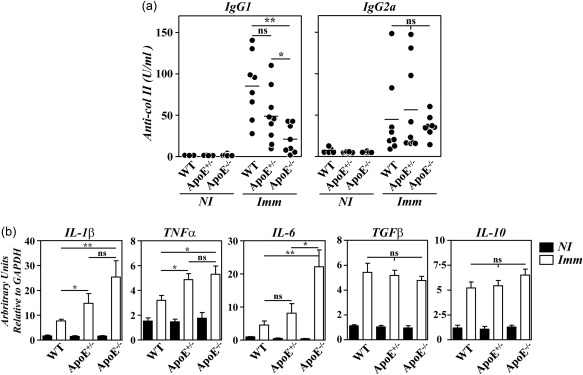
Anti‐collagen type II (col II) humoral responses and cytokine gene expression in apolipoprotein E (ApoE) mutant mice. (a) Serum levels of immunoglobulin (Ig)G1 and IgG2a anti‐col II antibodies in individual mice before (NI) and 4 weeks after (Imm) immunization with col II. Bars represent the mean values. Results are representative of two independent experiments. (b) Analysis by reverse transcription–quantitative polymerase chain reaction (RT–qPCR) of interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α and IL‐6 gene expression in the paws of B10.RIII.wild‐type (WT), B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice before (NI; closed bars) and 8 weeks after (Imm; open bars) immunization with col II. Representative results from one of three independent experiments (six to eight animals/group) are expressed as the mean ± standard deviation fold change of each cytokine relative to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression measured in parallel in each sample. Statistical differences are indicated as follows: n.s. = non‐significant; *P < 0·05; **P < 0·01.
A significantly increased expression of arthritogenic IL‐1β, TNF‐α and IL‐6 mRNAs was observed in the paws of B10.RIII.ApoE –/– in comparison to B10.RIII.WT mice 8 weeks after immunization with col II‐CFA (Fig. 3b). In B10.RIII.ApoE +/– mice, the expression of IL‐1β and TNF‐α, but not IL‐6, mRNAs in the paws was also augmented during CIA in comparison to B10.RIII.WT mice (Fig. 3b). No changes in the expression of the anti‐inflammatory cytokines TGF‐β and IL‐10 were observed among the different groups of immunized mice (Fig. 3b).
Different studies have indicated that ApoE influenced the in‐vitro activation of T cells and the macrophage polarization into proinflammatory M1 cells 18, 19. We then assessed whether the enhanced CIA observed in B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice correlated with an abnormal T cell activation and/or an enhanced M1 macrophage polarization. No differences in the kinetics of CD69 and CD25 induction were observed in CD4+ cells from the different strains of mice after concanavalin A or αCD3/αCD28 stimulation (Fig. 4a). However, an altered peritoneal macrophage polarization, characterized by an increased M1 polarization and a reduced M2 polarization, was observed in B10.RIII.ApoE –/– mice 48 h after i.p. injection of LPS in comparison to B10.RIII.WT controls (Fig. 4b). Again, an intermediate phenotype was observed in heterozygous B10.RIII.ApoE +/– mice after LPS treatment (Fig. 4b).
Figure 4.
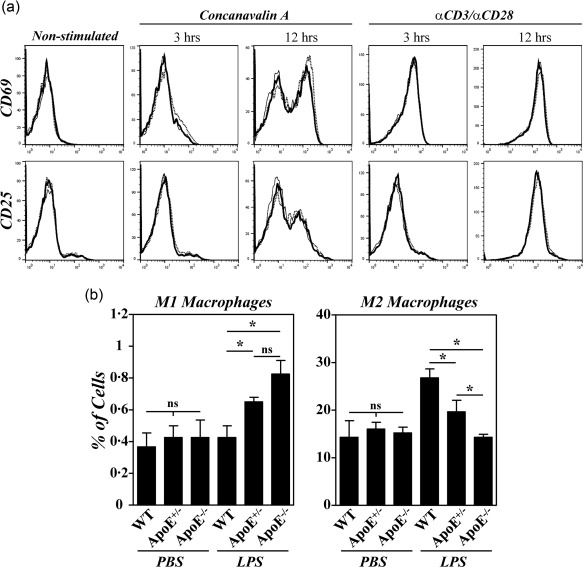
Effects of apolipoprotein E (ApoE) deficiency on the activation of CD4+ cells and in M1/M2 macrophage polarization. (a) Spleen cells from B10.RIII.wild‐type (WT), B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice were stimulated in vitro with concanavalin A or with αCD3/αCD28, and the kinetics of CD69 and CD25 induction was analysed by flow cytometry 3 and 12 h afterwards. Results are expressed as overlapping representative histograms of B10.RIII.WT (thin line), B10.RIII.ApoE +/– (dotted line) and B10.RIII.ApoE –/– (thick line) mice in one of two independent experiments. (b) B10.RIII.WT, B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice were injected with phosphate‐buffered saline (PBS) or lipopolysaccharide (LPS) and the percentages of M1 and M2 macrophages in the peritoneal cavity analysed by flow cytometry 48 h later. Representative results of two independent experiments are expressed as the mean ± standard deviation (three to four animals/group). Statistical differences are indicated as follows: n.s. = non‐significant; *P < 0·05.
Functional properties of HDL in ApoE mutant mice
ApoE is a component of HDL particles 20. In addition, chronic inflammation has been shown to alter the anti‐inflammatory properties of HDLs transforming them into proinflammatory molecules 20, 21. Therefore, we next explored whether the reduction or absence of ApoE and/or the exacerbated CIA observed in B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice induced functional changes in these lipoproteins. An assessment of the lipid and protein composition of HDLs obtained from each mouse genotype revealed significant increases in the distribution of free and esterified cholesterol as well as an increase in the percentage of proteins and a reduction in PON‐1 activity in B10.RIII.ApoE –/– mice (Table 1). With the exception of a moderate but significant increase in the percentage of proteins, the biochemical composition of HDLs from B10.RIII.ApoE +/– mice was similar to that of B10.RIII.WT mice (Table 1).
Table 1.
High‐density lipoprotein (HDL) composition and anti‐oxidant activity in ApoE mutant mice
| HDL characteristic | B10.RIII.WT | B10.RIII.ApoE +/– | B10.RIII.ApoE –/– |
|---|---|---|---|
| Esterified cholesterol (%) | 18·0 ± 3·5 | 15·4 ± 3·1 | 9·8 ± 3·4*† |
| Free cholesterol (%) | 2·6 ± 0·8 | 3·2 ± 0·8 | 6·5 ± 1·3*† |
| Phospholipids (%) | 27·9 ± 6·2 | 31·1 ± 4·8 | 29·6 ± 5·8 |
| Triglycerides (%) | 8·1 ± 3·8 | 5·3 ± 1·8 | 7·5 ± 1·5 |
| Protein (%) | 43·3 ± 0·4 | 45·0 ± 1·3* | 46·6 ± 2·3* |
| PON1 activity (µmol/ml.min) | 59·3 ± 1·7 | 58·8 ± 4·5 | 45·9 ± 4·9*† |
| LDL oxidation protection (%) | 175·2 ± 8·8 | 185·1 ± 49·3 | 167·9 ± 12·1 |
HDL was isolated from plasma by sequential ultracentrifugation at 100 000 g for 24 h at a density of 1·063–1·21 g/ml, and lipids and protein were determined. Values are expressed as relative (%) chemical composition and correspond to HDL preparations isolated from four pooled samples of four to six mice in each group. Serum plasma arylesterase activity was measured using phenylacetate as substrate and ethylenediamine tetraacetic acid (EDTA)‐sensitive plasma arylesterase (PON1) activity was calculated by subtracting the EDTA‐resistant arylesterase (five animals/group). Human low‐density lipoprotein (LDL) was incubated with 2·5 µM CuSO4 in the presence or absence of purified HDLs (0·1 mM phospholipids) from each mouse strain (three purified pools of HDL/group). The percentage of protection of LDL oxidation is expressed as the mean ± standard deviation (s.d.). Values are mean ± s.d. *P < 0·05 versus B10.RIII.WT mice. † P < 0·05 versus B10.RIII.ApoE +/– mice. ApoE = apolipoprotein E.
The ability of HDLs, purified from serum pools of col II immunized B10.RIII.WT, B10.RIII.ApoE +/– or B10.RIII.ApoE –/– mice, to inhibit the oxidation of human LDLs in the presence of CuSO4 was then analysed. Despite the biochemical differences in the composition of HDLs between B10.RIII.ApoE –/– mice and B10.RIII.WT and B10.RIII.ApoE +/– mice, these particles showed a similar anti‐oxidant capacity in all the experimental groups (Table 1).
Lack of exacerbation of CIA in hypercholesterolaemic B10.RIII.LDLR–/– mice in association with the increase in serum ApoE levels
Although our present observations in B10.RIII.ApoE +/– mice are compatible with an immunosuppressive activity of ApoE in vivo, independently of its role in cholesterol metabolism, these results cannot exclude formally the participation of hypercholesterolaemia in the aggravation of CIA in B10.RIII.ApoE –/– mice. To explore this issue further we used B10.RIII.LDLR–/– mice. As reported previously 22, these mice exhibited increased levels of TC and LDL/VLDL‐c in sera when fed with a NCD and these levels were even higher under a HCD (Fig. 5a: P < 0·001 in all cases). These abnormal circulating lipid profiles of B10.RIII.LDLR–/– mice were similar before and after CIA induction (Fig. 5a). In association with the presence of hypercholesterolaemia, very high levels of circulating ApoE were found in B10.RIII.LDLR–/– mice fed with NCD and these levels increased with the HCD, both before and after CIA induction (Fig. 5b). A significant reduction in circulating TC and HDL‐c but not LDL/VLDL‐c concentrations was again observed in B10.RIII.WT mice fed with a NCD, but not with a HCD, during the development of CIA (Fig. 5a). In these WT mice, the type of diet had no effect on serum levels of ApoE either before or after col II immunization (Fig. 5b).
Figure 5.
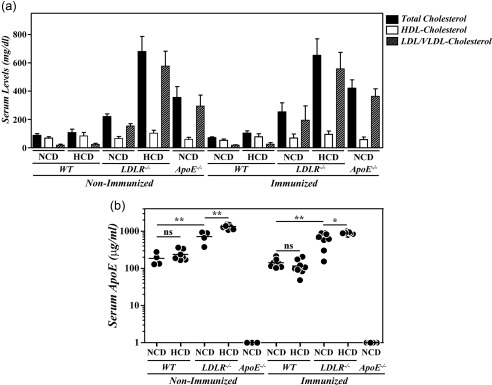
Serum lipid profiles and apolipoprotein E (ApoE) levels in B10.RIII. low‐density lipoprotein receptor (LDLR–/–) mice during collagen type II‐induced arthritis (CIA) development. (a) Serum levels of total cholesterol, high‐density lipoprotein cholesterol (HDLc) and very low‐density lipoprotein/low‐density lipoprotein cholesterol (VLDL/LDLc) in B10.RIII.wild‐type (WT), B10.RIII.LDLR –/– and B10.RIII.ApoE –/– mice fed with a normal chow diet (NCD) or an atherogenic hypercholesterolaemic diet (HCD) before and 8 weeks after induction of CIA. Representative results of two independent experiments are expressed as the mean ± standard deviation (eight to 10 animals/group). (b) Serum levels of ApoE in B10.RIII.WT, B10.RIII.LDLR –/– and B10.RIII.ApoE –/– mice fed with an NCD or an HCD before and 8 weeks after induction of CIA determined by enzyme‐linked immunosorbent assay (ELISA). Statistical differences are indicated as follows: n.s. = non‐significant; *P < 0·05; **P < 0·01.
Regardless of the diet received, no significant differences in the severity of clinical and radiological signs were observed between B10.RIII.LDLR–/– and B10.RIII.WT mice after CIA induction that were lower than in B10.RIII.ApoE –/– mice (Fig. 6a–c). Similarly, paw expression of IL‐1β, TNF‐α and IL‐6 mRNAs was significantly lower in B10.RIII.LDLR–/– and B10.RIII.WT mice than in B10.RIII.ApoE –/– mice 8 weeks after col II immunization (Fig. 6d). Again, the expression of TGF‐β and IL‐10 was similar in the different groups of mice after CIA induction (data not shown).
Figure 6.
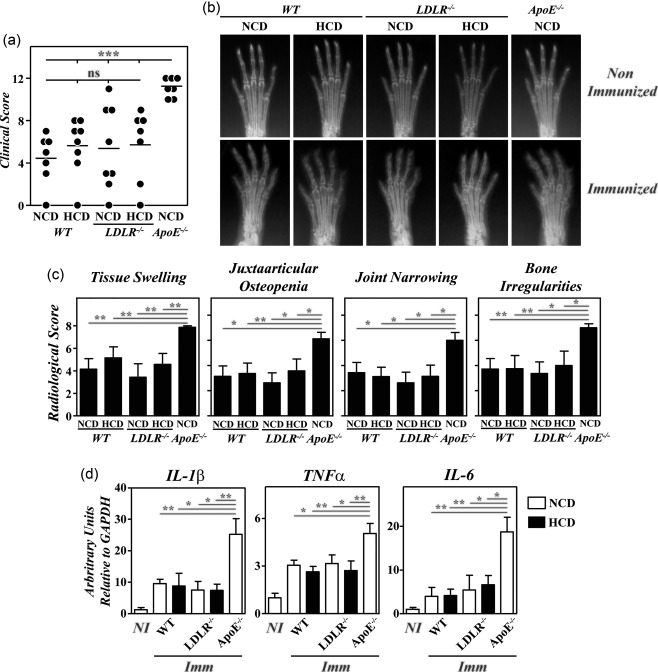
Lack of exacerbation of collagen type II‐induced arthritis (CIA) in B10.RIII. low‐density lipoprotein receptor (LDLR–/–) mice; 8–12‐week‐old B10.RIII.wild‐type (WT), B10.RIII.LDLR –/– and B10.RIII.apolipoprotein E (ApoE –/–) mice fed with a normal chow diet (NCD) or an atherogenic hypercholesterolaemic diet (HCD) were immunized with collagen type II‐complete Freund's adjuvant (col II‐CFA). (a) Clinical severity of CIA in individual mice 8 weeks after immunization with col II. Bars represent the mean values. (b) Representative radiological images of the front paws from mice before immunization with col II and 8 weeks afterwards. (c) Severity score of individual radiological signs, expressed as the mean ± standard deviation (n = 6–8 mice/group) 8 weeks after immunization. Bars represent the mean values. Results from (a) to (c) are representative of three independent experiments. (d) Expression of interleukin (IL)‐1β, tumour necrosis factor (TNF)‐α and IL‐6 mRNAs by reverse transcription–quantitative polymerase chain reaction (RT–qPCR) in the paws of the mice fed with NCD (open bars) or HCD (closed bars) 8 weeks after (Imm) immunization with col II. For each cytokine analysis, a mixed group of non‐immunized (NI) mice is included for comparison. Representative results from one of three independent experiments (five to nine animals/group) are expressed as mean ± standard deviation fold change of each cytokine relative to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression measured in parallel in each sample. Statistical differences are indicated as follows: n.s. = non‐significant; *P < 0·05; **P < 0·01; ***P < 0·005.
Discussion
The deficiency of ApoE has been shown recently to exacerbate disease severity in two experimental models of autoimmune arthritis in mice by inducing inflammatory immune responses 12, 13. However, the underlying mechanisms by which an ApoE deficiency promotes such immunological abnormalities have not been clarified. Using either normocholesterolaemic or hypercholesterolaemic mice with different concentrations of circulating ApoE, we have demonstrated here that both the hypercholesterolaemia and the deficiency in ApoE co‐operate towards the worsening of CIA in B10.RIII.ApoE –/– mice.
While a strong association between RA and increased risk of atherosclerosis and CVD has been established 1, 2, there exist controversies regarding the importance of dyslipidaemia in this association 3, 4, 5, 6, 7. Furthermore, some studies indicate that abnormal lipid profiles in sera, defined as high levels of TC and triglycerides and lower HDL‐c levels, were present in more than 50% of patients with RA before or after disease diagnosis 3, 4, 5. In contrast, studies show that a significant fraction of RA patients exhibit decreased levels of TC and LDL‐c and normal values of HDL‐c, reflecting what has been called the lipid paradox in RA 6, 7. Although the reasons for these discrepancies have not been determined exactly, they can be related to differences in the inflammatory status of the patients and/or in their dietary habits among the different studies. In this regard, we show here that the development of CIA in both B10.RIII.WT and B10.RIII.ApoE +/– mice is associated with a reduction in serum levels of TC and HDL‐c, but not of LDL/VLDL‐c and triglycerides, when fed using NCD but not HCD. Furthermore, inflammation or infection has been reported to alter lipid profiles in sera, as observed in mice with sepsis 23.
Previous observations demonstrate that small amounts of ApoE are sufficient to normalize plasma cholesterol levels and to inhibit atherosclerosis in mice 24, 25. In our present study, we show an aggravation of CIA in B10.RIII.ApoE +/– mice having 30–50% of the circulating ApoE observed in WT mice but normal cholesterol profiles. Independently of these dose‐dependent differences this may be related to the degree of systemic inflammation in each experimental model; both studies highlight the anti‐inflammatory role of ApoE in vivo that is unrelated to its activity in the control of cholesterol metabolism. The anti‐inflammatory activity of ApoE has been demonstrated in several studies. ApoE‐containing lipoproteins are very efficient in suppressing mitogen‐induced proliferative responses of T lymphocytes by reducing the production of IL‐2 [18]. ApoE also regulates the TLR‐4‐ and TLR‐3‐mediated production of IL‐12 26 and prevents the LPS‐induced production of cytokines and subsequent death in rodents 27. Furthermore, an ectopic ApoE expression in macrophages and monocytes from ApoE–/– mice suppresses nuclear factor‐κB‐mediated inflammation by enhancing miR‐146a levels 28. In this regard, our results indicate that the partial or total ApoE deficiency modifies the polarization of macrophages after a potent inflammatory insult in vivo but has no effect on the in‐vitro activation of CD4+ cells. The anti‐inflammatory capacity of ApoE appears to be isoform‐dependent, and animals expressing the E4 allele have greater inflammatory responses 29. Interestingly, one study has shown an association between the ApoE4 genotype and bone loss in human RA 30, although this has not been confirmed by other authors 31. The similarities observed in the anti‐col II antibody responses and in the pattern of cytokine expression in the paws during CIA development between B10.RIII.ApoE +/– and B10.RIII.ApoE –/– mice suggest that common mechanisms are responsible for the accelerated diseases in both strains of mice.
We show here that the biochemical composition of HDL particles differs between B10.RIII.ApoE –/– and WT controls, with an increase and reduction in the amount of free and esterified cholesterol in HDLs from mutant mice, respectively. Although the molecular basis of this alteration is unknown, it can be related to changes in the expression and/or function of the lecithin : cholesterol acyltransferase, an enzyme involved in the conversion of phosphatidylcholine and cholesterol into cholesteryl ester and lysophosphatidylcholine in plasma and other biological fluids 32. However, these differences do not modify their ability to protect against LDL oxidation. This, together with the fact that the biochemical composition of HDLs and their in‐vitro anti‐oxidant activity are similar in B10.RIII.ApoE +/– and B10.RIII.WT mice, suggests strongly that the exacerbation of arthritis observed in ApoE mutant mice is largely independent of HDL function. However, as our HDL experiments have been performed in a cell‐free system, we cannot completely rule out some direct proinflammatory effects of HDLs in B10.RIII.ApoE –/– and B10.RIII.ApoE +/– mice.
The link between cholesterol homeostasis and immune system activation has been documented extensively in recent years (reviewed in 33). To analyse directly the importance of hypercholesterolaemia in the control of CIA severity, we have employed B10.RIII.LDLR–/– mice. However, the increase in the levels of circulating cholesterol in these mice also promotes an important rise in serum ApoE concentration that, according to previous studies, probably reflects a liver X receptor‐dependent adaptive response to cholesterol overload 34. In agreement with the observations in B10.RIII.ApoE +/– mice, the high levels of ApoE in B10.RIII.LDLR–/– mice could initially anticipate a reduction in the severity of CIA in these animals. Nevertheless, the absence of such protection is compatible with a role for hypercholesterolaemia as an additional worsening factor for CIA severity in B10.RIII.ApoE –/– mice. In this regard, oxLDL and cholesterol crystals can act as DAMPs in the macrophages that infiltrate the arterial intima during hypercholesterolaemia, activating TLR‐4 and NOD‐like receptor family, pyrin domain‐containing 3 (NLRP3) inflammasome signalling pathways, respectively 8, 35. Also, hypercholesterolaemia can increase the content of lipid rafts in the plasma membrane, potentiating antigen receptor signalling in T cells 9.
ApoE binds to members of the LDLR family, including the LDLR, LDLR‐related protein 1, VLDL receptor (VLDLR) and APOE receptor 2 36 and several of these receptors have been involved in the anti‐inflammatory effect of ApoE 19, 37. The fact that the severity of CIA in B10.RIII.LDLR–/– mice is lower than in B10.RIII.ApoE –/– mice indicates that LDLR is not the main receptor by which ApoE modulates CIA severity. Additional experiments are required to clarify the receptor/s involved in this protective activity of ApoE. In summary, our present results underline the important role played by ApoE and cholesterol in the regulation of inflammatory responses during the development of inflammatory arthritis and highlight the importance of these factors as potential relevant targets for the control of autoimmune disorders.
Disclosure
The authors declare no disclosures.
Author contributions
P. A. performed the majority of experiments, analysed the data and wrote the manuscript, F. G. carried out arthritis experiments in B10.RIII.ApoE +/– mice, M. I., J. J. A. and E. T. performed the titration of anti‐col II antibodies, the RNA isolation from the paws and the RT–qPCR studies, F. B.‐V. and J. C. E.‐G. purified HDL from serum pools and characterized their biochemical properties and anti‐oxidative activities in vitro, B. L. performed the cholesterol profiles in sera and J. M. and R. M. conceived and designed the experiments, analysed the data and wrote the manuscript.
Acknowledgements
We thank Dr Jorge Joven, Unitat de Recerca Biomèdica, Hospital Universitari Sant Joan, Institut d'Investigació Sanitària Pere Virgili, Universitat Rovira i Virgili, Reus, Spain for the B6.LDLR –/– mice and Dr Karl Weisgraber, Gladstone Institute of Neurological Disease, University of California San Francisco, CA, USA for the purified mouse ApoE. We also thank María Aramburu, Natalia Cobo and Iván Gómez for their technical assistance. This work was supported by grants from the Spanish Ministerio de Economía y Competitividad to J. M. (SAF2012‐34059) and R. M. (SAF2014‐55088‐R), which were co‐funded by the European Regional Development Fund. M. I. was supported partially by a grant from the Spanish Ministerio de Economía y Competitividad (IPT2011‐1527‐010000) associated with fibrostatin SL.
References
- 1. John H, Kitas G. Inflammatory arthritis as a novel risk factor for cardiovascular disease. Eur J Intern Med 2012; 23:575–9. [DOI] [PubMed] [Google Scholar]
- 2. Young A, Koduri G, Batley M et al Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatology 2007; 46:350–7. [DOI] [PubMed] [Google Scholar]
- 3. Dessein PH, Joffe BI, Veller MG et al Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J Rheumatol 2005; 32:435–42. [PubMed] [Google Scholar]
- 4. Park YB, Lee SK, Lee WK et al Lipid profiles in untreated patients with rheumatoid arthritis. J Rheumatol 1999; 26:1701–4. [PubMed] [Google Scholar]
- 5. van Halm VP, Nielen MM, Nurmohamed MT et al Lipids and inflammation: serial measurements of the lipid profile of blood donors who later developed rheumatoid arthritis. Ann Rheum Dis 2007; 66:184–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Myasoedova E, Crowson CS, Kremers HM et al Lipid paradox in rheumatoid arthritis: the impact of serum lipid measures and systemic inflammation on the risk of cardiovascular disease. Ann Rheum Dis 2011; 70:482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bag‐Ozbek A, Giles JT. Inflammation, adiposity, and atherogenic dyslipidaemia in rheumatoid arthritis: is there a paradoxical relationship? Curr Allergy Asthma Rep 2015; 15:497 [DOI] [PubMed] [Google Scholar]
- 8. Imai Y, Kuba K, Neely GG et al Identification of oxidative stress and Toll‐like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008; 133:235–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McDonald G, Deepak S, Miguel L et al Normalizing glycosphingolipids restores function in CD4+ T cells from lupus patients. J Clin Invest 2014; 124:712–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lazarevic MB, Vitic J, Mladenovic V, Myones BL, Skosey JL, Swedler WI. Dyslipoproteinemia in the course of active rheumatoid arthritis. Semin Arthritis Rheum 1992; 22:172–8. [DOI] [PubMed] [Google Scholar]
- 11. Daoussis D, Panoulas VF, Antonopoulos I et al Cardiovascular risk factors and not disease activity, severity or therapy associate with renal dysfunction in patients with rheumatoid arthritis. Ann Rheum Dis 2010; 69:517–21. [DOI] [PubMed] [Google Scholar]
- 12. Postigo J, Genre F, Iglesias M et al Exacerbation of type II collagen‐induced arthritis in apolipoprotein E‐deficient mice in association with the expansion of Th1 and Th17 cells. Arthritis Rheum 2011; 63:971–80. [DOI] [PubMed] [Google Scholar]
- 13. Archer AM, Saber R, Rose S et al ApoE deficiency exacerbates the development and sustainment of a semi‐chronic K/BxN serum transfer‐induced arthritis model. J Transl Med 2016; 14:170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raffai RL. Apolipoprotein E regulation of myeloid cell plasticity in atherosclerosis. Curr Opin Lipidol 2012; 23:471–8. [DOI] [PubMed] [Google Scholar]
- 15. González J, Tamayo E, Santiuste I et al CD4+CD25+ T cell‐dependent inhibition of autoimmunity in transgenic mice overexpressing human Bcl‐2 in T lymphocytes. J Immunol 2007; 178:2778–86. [DOI] [PubMed] [Google Scholar]
- 16. Hirsch‐Reinshagen V, Donkin J, Stukas S et al LCAT synthesized by primary astrocytes esterifies cholesterol on glia‐derived lipoproteins. J Lipid Res 2009; 50:885–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Escolà‐Gil JC, Chen X, Julve J et al Hepatic lipase‐ and endothelial lipase‐deficiency in mice promotes macrophage‐to‐feces RCT and HDL antioxidant properties. Biochim Biophys Acta 2013; 1831:691–7. [DOI] [PubMed] [Google Scholar]
- 18. Kelly ME, Clay MA, Mistry MJ, Hsieh‐Li H‐M, Harmony JAK. Apolipoprotein E inhibition of proliferation of mitogen‐activated T lymphocytes: production of interleukin 2 with reduced biological activity. Cell Immunol 1994; 159:124–39. [DOI] [PubMed] [Google Scholar]
- 19. Baitsch D, Bock HH, Engel T et al Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler Thromb Vasc Biol 2011; 31:1160–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhu X, Parks JS. New roles of HDL in inflammation and hematopoiesis. Annu Rev Nutr 2012; 32:161–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McMahon M, Grossman J, FitzGerald J et al Proinflammatory high‐density lipoprotein as a biomarker for atherosclerosis in patients with systemic lupus erythematosus and rheumatoid arthritis. Arthritis Rheum 2006; 54:2541–9. [DOI] [PubMed] [Google Scholar]
- 22. Osuga J, Yonemoto M, Yamada N et al Cholesterol lowering in low density lipoprotein receptor knockout mice overexpressing apolipoprotein E. J Clin Invest 1998; 102:386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alvarez C, Ramos A. Lipids, lipoproteins, and apoproteins in serum during infection. Clin Chem 1986; 32:142–5. [PubMed] [Google Scholar]
- 24. Linton MF, Atkinson JB, Fazio S. Prevention of atherosclerosis in apolipoprotein E‐deficient mice by bone marrow transplantation. Science 1995; 267:1034–7. [DOI] [PubMed] [Google Scholar]
- 25. Zhu Y, Bellosta S, Langer C et al Low‐dose expression of a human apolipoprotein E transgene in macrophages restores cholesterol efflux capacity of apolipoprotein E‐deficient mouse plasma. Proc Natl Acad Sci USA 1998; 95:7585–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ali K, Middleton M, Puré E, Rader DJ. Apolipoprotein E suppresses the type I inflammatory response in vivo . Circ Res 2005; 97:922–7. [DOI] [PubMed] [Google Scholar]
- 27. Van Oosten M, Rensen PC, Van Amersfoort ES et al Apolipoprotein E protects against bacterial lipopolysaccharide‐induced lethality. A new therapeutic approach to treat gram‐negative sepsis. J Biol Chem 2001; 276:8820–4. [DOI] [PubMed] [Google Scholar]
- 28. Li K, Ching D, Luk FS, Raffai RL. Apolipoprotein E enhances microRNA‐146a in monocytes and macrophages to suppress nuclear factor‐κB‐driven inflammation and atherosclerosis. Circ Res 2015; 117:e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lynch JR, Tang W, Wang H et al APOE genotype and an ApoE‐mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem 2003; 278:48529–33. [DOI] [PubMed] [Google Scholar]
- 30. Lee SI, Lee SY, Yoo WH. Association of apolipoprotein E polymorphism with bone mineral density in postmenopausal women with rheumatoid arthritis. Rheumatology 2005; 44:1067–8. [DOI] [PubMed] [Google Scholar]
- 31. Maehlen MT, Provan SA, de Rooy DP et al Associations between APOE genotypes and disease susceptibility, joint damage and lipid levels in patients with rheumatoid arthritis. PLoS One 2013; 8:e60970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jonas A. Lecithin cholesterol acyltransferase. Biochim Biophys Acta 2000; 1529:245–56. [DOI] [PubMed] [Google Scholar]
- 33. Fessler MB. Regulation of adaptive immunity in health and disease by cholesterol metabolism. Curr Allergy Asthma Rep 2015; 15:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid‐activated nuclear receptors. Nature 2008; 454:470–7. [DOI] [PubMed] [Google Scholar]
- 35. Duewell P, Kono H, Rayner KJ et al NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010; 464:1357–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis 2014; 72:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. May P, Bock HH, Nofer JR. Low density receptor‐related protein 1 (LRP1) promotes anti‐inflammatory phenotype in murine macrophages. Cell Tissue Res 2013; 354:887–9. [DOI] [PubMed] [Google Scholar]