

Abstract
Multidrug-resistant Enterobacteriaceae, notably Escherichia coli and Klebsiella pneumoniae, have become major health concerns worldwide. Resistance to effective therapeutics is often carried by class I and II integrons that can confer insensitivity to carbapenems, extended spectrum beta-lactamases, the antifolate trimethoprim, fluoroquinolones and aminoglycosides. Specifically of interest to the study here, a prevalent gene (dfrA1) coding for an insensitive dihydrofolate reductase (DHFR) confers 190- or 1000-fold resistance to trimethoprim for K. pneumoniae and E. coli, respectively. Attaining inhibition of both the wild-type and resistant forms of the enzyme is critical for new antifolates. For several years, we have been developing the propargyl-linked antifolates (PLAs) as effective inhibitors against trimethoprim-resistant DHFR enzymes. Here, we show that the PLAs are active against both the wild-type and DfrA1 DHFR proteins. We report two high resolution crystal structures of DfrA1 bound to potent PLAs. The structure-activity relationships and crystal structures will be critical in driving the design of broadly active inhibitors against wild-type and resistant DHFR.
Keywords: Enterobacteriaceae, antifolate, resistance, trimethoprim, DfrA1
Introduction
Infections caused by Gram-negative bacteria are a significant global healthcare threat owing primarily to the increasing number of multidrug-resistant strains. Escherichia coli and Klebsiella pneumoniae, members of the Enterobacteriaceae family, are often specifically noted for their drug resistance, mortality rates and burden on the healthcare system. While there are many mechanisms by which these bacteria have become resistant to antibiotics, horizontal gene transfer of plasmid-encoded resistance elements is among the most common. The emergence of carbapenem-resistant Enterobacteriaceae (CRE) has become a particular concern as these bacteria harbor plasmids encoding resistance genes to almost all antibiotics. With the mortality rate of patients who contract a CRE bloodstream infection approaching 50 % (http://www.cdc.gov/drugresistance/threat-report-2013/), there is a great need for antibiotics targeting these bacteria. CRE are defined by the presence of β-lactamases that inactivate carbapenems and cephalosporins, which are considered some of the most effective treatments for infections caused by Gram-negative bacteria. However, the threat caused by CRE infections is increased as the genes encoding carbapenemases and extended spectrum β-lactamases are often found on type I and II integrons in conjunction with genes responsible for resistance to fluoroquinolones, folate pathway inhibitors and aminoglycosides. As a recent study (1) showed, class I integrons are present in 51.1 % of strains and carry recurring resistance genes for a wide spectrum of antibiotics including β-lactams, chloramphenicol, trimethoprim, erythromycin, aminoglycosides and rifampicin. In fact, it is the multidrug-resistant phenotype associated with class I and II integrons that truly defines the threat associated with CRE infections.
Inhibitors of the folate pathway, specifically trimethoprim and sulfamethoxazole (TMP/SMX, administered as Bactrim®), are used clinically in the treatment of infections caused by E. coli and K. pneumoniae. Trimethoprim, an inhibitor of the essential enzyme dihydrofolate reductase (DHFR) and sulfamethoxazole, an inhibitor of dihydropteroate synthase (DHPS), have been used for decades to effectively treat uncomplicated urinary tract infections and skin and soft tissue infections. TMP/SMX has also been successfully used to treat CRE infections (2); however, increasing resistance to both agents have limited its use, relegating DHFR and DHPS inhibition as an underutilized treatment strategy. In Gram-negative bacteria, TMP resistance has increased over the years (3) and now occurs in approximately 30 % of all strains (4-8). The primary mechanism of this resistance has been the acquisition of TMP-resistant dfr genes encoded within integrons. Two families of extrachromosomal DHFR enzymes, DfrA and DfrB, mediate high-level TMP resistance. The DfrA encompasses a family of over 30 homologous enzymes that maintain 64-88% sequence identity. Within this family of extrachromosomal dfr genes, dfrA1 is the most prevalent, accounting for 38 % of dfr-possessing strains (9-11). Not only is dfrA1 carried on type I and II integrons, making it a critical mechanism of antifolate resistance, it is frequently found in combination with extended spectrum β-lactamases, metallo-β-lactamases and Klebsiella pneumoniae carbapenemases (12-14). As the protein product of dfrA1 is highly resistant to TMP, identification of antifolates that are potent inhibitors of DfrA1 would be a significant advancement in overcoming antifolate resistance and provide a much needed treatment option for CRE infections.
For several years, we have been developing the class of propargyl-linked antifolates (PLAs) to be effective inhibitors of TMP-resistant species of DHFR (15-18). Recently, we reported the activity of several of these compounds against K. pneumoniae and we reported the crystal structure of K. pneumoniae DHFR (KpDHFR) bound to two PLA compounds (19). Here, we expand on that work to report that several of the PLAs not only inhibit the wild-type DHFR enzymes from Enterobacteriaceae but also significantly inhibit the DfrA1 enzyme as well. We also report, for the first time, two high resolution crystal structures of the DfrA1 enzyme. These structures of the protein DfrA1 bound to two PLAs are critical for the development of inhibitors that are effective against wild-type K. pneumoniae and E. coli as well as DfrA1.
Results and Discussion
Propargyl-linked Antifolates are Effective Antibacterial Agents for Enterobacteriaceae
As described above, we have been developing the PLAs to be potent antibacterial agents targeting wild-type and trimethoprim-resistant Enterobacteriaceae. To examine a number of chemotypes that differ in the B and C rings as potential agents that could exhibit broad inhibition, we evaluated the antibacterial activity of 11 compounds from our collection against K. pneumoniae. Most have good antibacterial activity, with several possessing MIC values at 1 μg/mL or lower (Table 1). We then evaluated several of the compounds, including TMP, in time-kill assays with E. coli (ATCC 25922) intended to determine whether they are bacteriostatic or bactericidal (Figure 1). TMP, as has been shown previously, is bacteriostatic against E. coli. Importantly, compounds 8 and 9 show bactericidal properties. In a third experiment, we determined that the antibacterial activity results from the inhibition of DHFR by performing the MIC evaluation in the presence of 20 μg/mL thymidine, one of the end-products of the depleted folate pathway. For the tested compounds (3, 6, and TMP), MIC values in the presence of thymidine were all greater than 20 μg/mL, showing that growth inhibition is on-target. Overall, these experiments show that the compounds penetrate and kill Gram-negative bacteria and exhibit on-target inhibition; notably, at least two are bactericidal. In summary, these appear to be promising antibacterial agents.
Table 1.
Inhibition of DHFR Enzymes
| Cmpd # |
Compound | MIC Kp (μg/mL) |
DfrA1 IC50 (μM) |
Ec IC50 (μM) |
Kp IC50 (μM) |
|---|---|---|---|---|---|
| TMP |
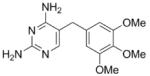
|
0.0781 | 20.166 ± 1.690 |
0.02 ± 0.002 |
0.106 ± 0.006 |
| 1 |
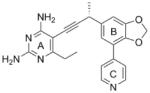
|
5 | 0.438 ± 0.035 |
0.075 ± 0.005 |
0.158 ± 0.026 |
| 2 |
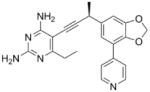
|
0.625 | 1.716 ± 0.274 |
0.019 ± 0.003 |
0.046 ± 0.0044 |
| 3 |
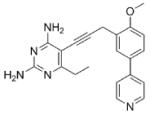
|
1.25 | 0.182 ± 0.005 |
0.083 ± 0.007 |
0.108 ± 0.008 |
| 4 |
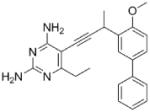
|
>20 | 1.934 ± 0.023 |
0.053 ± 0.004 |
0.053 ± 0.0035 |
| 5 |
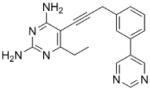
|
5 | 1.875 ± 0.099 |
0.097 ± 0.01 |
0.261 ± 0.0126 |
| 6 |
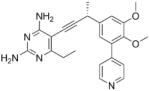
|
0.625 | 1.948 ± 0.231 |
0.171 ± 0.005 |
0.175 ± 0.0221 |
| 7 |
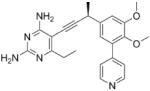
|
1.25 | 4.476 ± 0.446 |
0.076 ± 0.0048 |
0.127 ± 0.0249 |
| 8 |
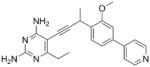
|
1 | 0.967 ± 0.031 |
0.101 ± 0.005 |
0.093 ± 0.0002 |
| 9 |
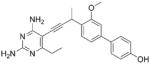
|
1.25 | 17.98 ± 0.504 |
0.033 ± 0.002 |
0.079 ± 0.0149 |
| 10 |
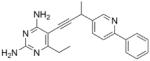
|
5 | 1.713 ± 0.304 |
N/A* | 0.019 ± 0.0013 |
| 11 |
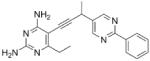
|
>20 | 18.649 ± 0.229 |
0.183 ± 0.045 |
0.333 ± 0.0095 |
| 12 |
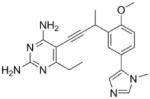
|
0.3125 | 2.558 ± 0.206 |
0.018 ± 0.002 |
0.059 ± 0.016 |
| 13 |
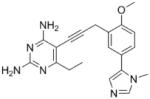
|
0.625 | 1.124 ± 0.047 |
0.046 ± 0.004 |
0.068 ± 0.004 |
| 14 |
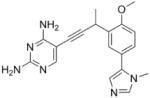
|
2.5 | 10.18 ± 0.277 |
0.023 ± 0.002 |
0.319 ± 0.021 |
| 15 |
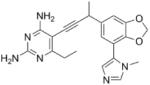
|
0.3125 | 0.826 ± 0.056 |
0.037 ± 0.001 |
0.283 ± 0.0004 |
| 16 |
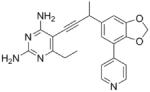
|
0.625 | 0.783 ± 0.020 |
0.047 ± 0.011 |
0.016 ± 0.001 |
| 17 |
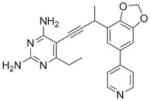
|
2.5 | 0.366 ± 0.034 |
0.039 ± 0.004 |
0.211 ± 0.011 |
N/A: stock was depleted before this evaluation could be completed
Figure 1.

Time-kill curves for TMP and compounds 8, 9, and 16 against E. coli.
Analysis of the DfrA1 enzyme
A sequence alignment of DHFR enzymes from the chromosomal Gram-negative bacteria E. coli and K. pneumoniae, DfrA1 from these bacteria as well as Staphylococcus aureus, a representative TMP-sensitive Gram-positive bacterium, (Figure 1) shows that the DfrA1 enzyme shares many conserved domains with other species of DHFR, suggesting that its structure is highly homologous. However, it also possesses several residue substitutions that are frequently found in other species of TMP-resistant DHFRs. For example, Ile 94 in wild-type KpDHFR is replaced with a smaller serine residue in DfrA1, which would be predicted to have diminished interactions with the methylene bridge of TMP. Additionally, Leu 28 that forms van der Waals interactions with the pyrimidine and trimethoxyphenyl rings and is common to TMP-sensitive enzymes, is replaced with Gln in the resistant enzymes. Again, this substitution is predicted to reduce van der Waals interactions with TMP. Finally, in the wild-type enzymes, there is an extra proline residue in the loop comprised of residues 50-54. The loss of this proline in the resistant enzymes is expected to reshape the interactions this loop typically has with the antifolates.
Propargyl-linked Antifolates are Active against the A1 Enzyme
As the PLAs are more highly functionalized than TMP and have been designed to have interactions with TMP-resistant enzymes, we expected that several PLAs would be potent inhibitors of the DfrA1 enzyme in addition to the wild-type enzymes. Overall, we began by screening a number of compounds in our existing library (Table 1: compounds 1-11) that are characterized by a diaminopyrimidine substituted at the C6 position, a propargyl linker that may be functionalized by a methyl group, a substituted B-ring and a C-ring heterocycle. The C-ring is either attached in a meta position relative to the B-ring (compounds 1-7) or a para-position (compounds 8-11). Methods for the synthesis of compounds 1, 2 and 6, 7 (17), 3-5 (18) as well as 8-11 (21) have been previously published.
Using purified K. pneumoniae DHFR, E. coli DHFR and DfrA1 DHFR in a spectroscopic assay that monitors the oxidation of the cofactor, NADPH, we determined 50 % inhibition concentrations (IC50 values) for TMP and the PLAs in Table 1. Remarkably, the inhibition data show that TMP is ~200- or 1000-fold less potent against the DfrA1 enzyme relative to its value against K. pneumoniae or E. coli wild-type DHFR, respectively. We also found that compounds with a benzodioxalane at the B-ring (1 and 2) along with compounds with a simple 2′-methoxy phenyl B-ring (specifically 3-6) exhibited relatively potent IC50 values less than 2 μM. Compounds with a C-ring in the para-position were generally poor inhibitors of the DfrA1 enzyme. While compound 8 has an IC50 value of ~1 μM, compounds 9-11 have IC50 values above 1 μM. In order to explore the active series with additional compounds, we synthesized compounds 12-17, extending methods that we have previously published (17, 18, 21) (purity characteristics are shown in Supporting Information).
Excitingly, several of the PLAs are active against both the wild-type and DfrA1 enzymes. For example, compound 12 is observed to be 10-fold more potent than TMP against the DfrA1 enzyme. Replacing the propargylic methyl with a hydrogen (compound 13) increases affinity by 2-fold. An ethyl substitution at the C6 position on the diaminopyrimidine ring is responsible for a 4-fold increase in activity as compared to a hydrogen (compare 14 to 12). Replacing the phenyl ring with benzodioxalane (15) and/or replacing the imidazole ring with a pyridyl ring (16) increases affinity to the sub-micromolar range. Overall, among this group of inhibitors, compounds 3 and 17 are the most potent inhibitors with IC50 values of 0.182 and 0.366 μM against the DfrA1 enzyme. Strikingly, these compounds do not lose significant activity relative to the K. pneumoniae wild-type DHFR and exhibit only minor losses relative to the E. coli DHFR.
We also verified that the new compounds have antibacterial activity (Table 1), showing that many exhibit MIC values less than 1 μg/mL. Compound 16 was selected for evaluation in a time-kill assay; we confirmed that this inhibitor also shows bactericidal activity (Figure 1). In a final evaluation, compounds 16 and 17 were shown to be rescued by the presence of 20 μg/mL thymidine, demonstrating on-target growth inhibition.
Crystal structure of DfrA1
In order to further understand the structure-activity relationships of the PLAs and to promote the design of highly potent inhibitors of wild-type and resistant enzymes, we determined high resolution crystal structures of DfrA1 bound to NADPH and the two most potent compounds, 3 and 17. Diffraction data for the crystals were collected at the University of Connecticut and Brookhaven National Synchrotron Light Source and extended to 1.95 and 1.87 Å, respectively (crystallography details are found in Table 2; omit electron density of the ligands is shown in Supporting Information, Figure S1). The crystals belong to space group P3121 and have two molecules in the asymmetric unit. Overall, the DfrA1 enzyme adopts the classical DHFR fold with an eight-stranded beta-sheet and four flanking alpha-helices. Similar to other ternary structures of DHFR, the 2-amino group and N1 of the pyrimidine ring of compound 17 form conserved hydrogen bonds with residue Glu 27 and the 4-amino group forms a hydrogen bond with the backbone carbonyl oxygen of Met 5 (Figure 2a). The nicotinamide group of NADPH forms π-π interactions with the propargyl linker, stabilizing the complex. The B- and C-rings (see letters in Table 1) form van der Waals interactions with Phe 31, Met 50 and Leu 53. In the complex with compound 3 (Figure 2b), the B- and C-rings adopt two different conformations, exploiting two possible pockets in the enzyme. Both pockets are defined by the residues Phe 31, Met 50 and Leu 53; however, two alternate conformations of Met 50 and Leu 53 are observed, corresponding to the two different conformations of the ligand. Additionally, Gln 28 forms a hydrogen bond with the pyridyl nitrogen, regardless of the conformation of the biaryl ring system.
Table 2.
Data collection and structure determination statistics
| Kp DfrA1:NADPH:3 | Kp DfrA1:NADPH:17 | |
|---|---|---|
| PDB ID | 5ECC | 5ECX |
| Space group | P3121 | P3121 |
| No. monomers in asymmetric unit |
2 | 2 |
| Unit cell (a, b, c in Å; α,β, γ in degrees) |
76.199 76.199 113.784 90.00 90.00 120.00 |
76.070 76.070 113.929 90.00 90.00 120.00 |
| Resolution (Å) | 28.54-1.87 | 38.04-1.95 |
| Completeness % (last shell, %) |
90.12 (50) | 99.50 (100.00) |
| Unique reflections | 29,029 | 159,164 |
| Redundancy (last shell) | 5.1 (1.8) | 5.60 (5.45) |
| Rsym, (last shell) | 0.060 (0.526) | 0.053 (0.56) |
| <I/σ> (last shell) | 31.8 (0.8) | 10.8 (1.9) |
| R-factor/Rfree | 0.1729/0.2088 | 0.2154/0.2656 |
| No. of atoms (protein, ligands, solvent) |
3,003 | 2,720 |
| Rms deviation bond lengths (Å), angles (deg) |
0.008. 1.198 | 0.008, 1.251 |
| Average B factor for protein (Å2) |
39.54 | 49.16 |
| Average B factor for ligand (Å2) |
NADPH: 37.15 Ligand: 40.20 |
NADPH: 47.20 Ligand: 59.95 |
| Average B factor for solvent molecules (Å2) |
49.99 | 53.47 |
| Residues in most favored regions (%)a |
98.77 | 98.11 |
| Residues in additional allowed regions (%)a |
1.23 | 1.89 |
| Residues in disallowed regions (%)a |
0.00 | 0.00 |
| Collection Location | BNL X4 | UConn |
Figure 2.

Structural alignment of the sequences of the wild-type DHFR from E. coli (PDB 1DRE) (20), K. pneumoniae (PDB ID 4OR7) (19), and Staphylococcus aureus (PDB ID 3SGY) (18). The sequences of DfrA1 from E. coli (GenBank ADH82150.1) and DfrA1 from K. pneumoniae (GenBank ADH82140.1) are aligned to the DHFR enzymes with structures. Active site residues are shown in red.
The structures of DfrA1 also reveal the basis of resistance to TMP (Figure 2c). The substitution of Ser 96 in DfrA1 for a bulkier residue (Ile, Leu or Phe) in TMP-sensitive enzymes such as wild-type E. coli, K. pneumoniae or S. aureus removes significant van der Waals contacts with the methylene bridge. Importantly, Gln 28 in DfrA1 DHFR is usually a hydrophobic residue in TMP-sensitive enzymes (eg. Leu); the substitution removes van der Waals interactions with both the pyrimidine and trimethoxyphenyl rings and introduces a polar residue in a hydrophobic region of the inhibitor. The substitution of Met 50 for the Ile found in TMP-sensitive enzymes also reduces van der Waals interactions. In one conformation observed in the DfrA1 structure, Met 50 would appear to sterically interfere with the trimethoxyphenyl ring, in the other conformation it makes very limited interactions.
A number of positive interactions that are unique to the PLAs explain much of the potency observed against the DfrA1 enzyme relative to TMP (Figures 2a and b). For example, Phe 31 forms key hydrophobic interactions with the propargyl linker and the C6-ethyl substitution on the pyrimidine ring explaining the 4-fold loss in activity upon replacement with a C6-hydrogen. While the substitution of Gln 28 is detrimental to TMP binding, the carboxamide forms a hydrogen bond with the pyridyl nitrogen of both compounds 3 and 17. A hydrophobic pocket consisting of Met 50, Leu 53, Ile 20, and Thr 46 accommodates the dioxalane or phenyl (B-ring) as well as the pyridyl or imidazole (C-rings) and their hydrophobic substituents, forming extensive van der Waals interactions.
A comparison of the DfrA1 enzyme to wild-type K. pneumoniae (Fig. 2d) or E. coli DHFR reveals the effects of the residue substitutions on binding PLAs. Conserved interactions between the inhibitors and wild-type DHFR or DfrA1 include Phe 31, Thr 46 as well as Ec/Kp Asp 27::A1 Glu 27 as this does not have a significant effect on binding the pyrimidine ring. The additional hydrogen bond formed by Gln 28 in the DfrA1 enzyme explains the greater potency for pyridyl-containing compounds. A loop at the active site (Ser 49-Pro 55) is one residue longer in the wild-type enzyme relative to the DfrA1 enzyme. This insertion alters the interaction of residues 52-53 from the wild-type or 53 from DfrA1. The residue substitution Ec/Kp Ile 94::A1 Ser 96 removes interactions with the propargyl linker, possibly explaining why some of the PLAs lose affinity for the DfrA1 enzyme relative to the wild-type.
Conclusions
Integrons carrying antibiotic resistance-conferring genes are a major threat for the increased spread of resistant Gram-negative bacteria. The most common extrachromosomal DHFR variant is DfrA1, which renders trimethoprim ineffective. Here, we present data showing that the propargyl-linked antifolates are effective inhibitors of the wild-type and DfrA1 proteins. Crystal structures of two of the most potent PLAs with the DfrA1 protein reveal the basis of the affinity of the PLAs and the structural origins of the enzyme’s resistance to trimethoprim. The PLAs represent an excellent lead series to develop inhibitors that are potent against wild-type and resistant DHFR from Enterobacteriaceae, which may be an important route for overcoming antibiotic resistance in these pathogens.
Methods
Antibacterial activity
Minimum inhibitory concentrations were determined using K. pneumoniae (ATCC 10031) and the microdilution broth assay with an inoculum of 1 × 105 CFU/mL in Isosensitest Broth (Oxoid). Growth was monitored at A600 using the Alamar Blue assay; the MIC is defined as the lowest concentration of inhibitor to completely inhibit growth. MIC values in the presence of 20 μg/mL thymidine were also determined for compounds 3, 6, 16 and 17.
Time-kill curves
Inocula were prepared from an overnight culture of E. coli 25922 grown at 37°C in LB media. The overnight culture was then adjusted to a 0.5 McFarland standard and diluted to a final concentration of ~5 × 105 cfu/mL in IsoSensitest media. Five flasks were prepared: 1 growth control (no inhibitor) and 4 inhibitor flasks (trimethoprim and Compounds 8, 9 and 16). Final concentrations of inhibitors were made to 4x their respective MIC values. Aliquots from each sample were taken at 0, 2, 4, 6, 8, and 12 hours to determine colony counts which were performed by making appropriate dilutions in physiological saline and plating 60 μL on pre-warmed LB agar.
Chemistry
Trimethoprim was purchased from Sigma. Compounds 1-11 have been previously synthesized, characterized and published as noted. Compounds 12-17 were synthesized and evaluated for this work. Synthetic procedures followed published practices (17, 18, 21); characterization and purity data using an HPLC method with two solvent systems are presented in Supporting Information.
Cloning, Expression and Enzyme Purification
The Klebsiella pneumoniae dfrA1 and E. coli DHFR genes were synthesized and cloned in the pET41a(+) expression vector containing a C-terminus histidine tag by GenScript. Recombinant protein was expressed in E. coli BL21(DE3) cells, purified via nickel affinity chromatography and subsequently desalted into 20 mM Tris, 20% glycerol, 0.1 mM EDTA, and 2 mM DTT (pH 8.0) using a PD-10 column (GE Healthcare). Protein was concentrated to ~10 mg/mL, flash frozen with liquid nitrogen and stored at −80 °C.
Enzyme Inhibition Assays
Enzyme inhibition assays were performed as previously described by monitoring the rate of NADPH oxidation by DHFR at an absorbance of 340 nm(19). Assays were carried out in a buffer containing 20 mM TES pH 7.0, 50 mM KCl, 10 mM 2-mercaptoethanol, 0.5 mM EDTA and 1 mg/mL BSA. Protein (1 mg/mL) was incubated with NADPH (0.1 mM) and inhibitor at concentrations near the estimated IC50 for five minutes at 25 °C before the reaction was initiated with dihydrofolate (0.1 mM). Enzyme inhibition was measured in triplicate and the average IC50 is reported with standard deviations.
Crystallization
Klebsiella pneumoniae DfrA1 DHFR was crystallized using the hanging-drop vapor diffusion method. Purified protein (20 mg/mL) was incubated with compound 3 or 17 (1 mM) and NADPH (2 mM) for 2 hours on ice. An equal volume of protein:ligand:NADPH complex was mixed with the optimized crystallization solution consisting of 20 mM imidazole pH 8.5, 300 mM calcium chloride and 15 % PEG 6,000. Crystals were observed within 3-5 days at 4 °C. Crystals were incubated in cryo-protectant buffer containing 15 % glycerol prior to flash-cooling with liquid nitrogen.
Data Collection and Refinement
Diffraction data for DfrA1 bound to compound 3 were collected at Brookhaven NSLS on beamline X25A and the data indexed and scaled using HKL 2000. Diffraction data for DfrA1 bound to compound 17 were collected at the University of Connecticut Protein X-Ray Crystallography Facility on a Rigaku HighFlux HomeLab system with d*TREK used for indexing and scaling. Structures were refined and validated using non-crystallographic symmetry and structure restraints with the PHENIX suite (22) while COOT (23) was used throughout the model building process. A model of E. coli DHFR (PDB ID: 1RX2) (20) was initially used for the molecular replacement to solve the structure of DfrA1 bound to 17 and NADPH, which was subsequently used to probe for the structure of DfrA1 bound to 3 and NADPH. The inhibitor PDB and CIF files were generated with PRODRG (24). Data collection and refinement statistics are reported in Table 2.
Supplementary Material
Figure 3.

Crystal structures of DfrA1 bound to potent inhibitors: a) Crystal structure of DfrA1 (dark green) bound to compound 17 (light green) with NADPH (salmon) b) Crystal structure of DfrA1 (dark blue) bound to compound 3 (magenta) with both conformations shown c) DfrA1 (dark green) bound to compound 17 (light green) and a model of TMP (gold) d) DfrA1 (dark green) bound to compound 17 (light green) superimposed with a crystal structure of KpDHFR (cyan). Residue differences between the wild-type and resistant enzyme are noted.
Acknowledgements
We gratefully acknowledge the support of NIH AI104841 to ACA and DLW.
Abbreviations
- CRE
carbapenem-resistant Enterobacteriaceae
- TMP
trimethoprim
- DHFR
dihydrofolate reductase
- PLA
propargyl-linked antifolate
- NADPH
β-nicotinamide adenine dinucleotide phosphate
Footnotes
Author Information
MNL performed enzyme assays, determined and interpreted crystal structures, wrote manuscript; NGD synthesized compounds; DLW supervised synthetic chemistry and assisted with manuscript preparation; ACA supervised biochemistry and crystallography, assisted with manuscript preparation.
HPLC traces showing purity analysis of compounds 12-17, 1H and 13C spectra for compounds 12-17 and omit density for the crystal structures of DfrA1 with compounds 3 and 17 are provided in Supporting Information. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Li B, Hu Y, Wang Q, Yi Y, Woo P, Jing H, Zhu B, Liu C. Structural diversity of class I integrons and their associated gene cassettes in Klebsiella pneumoniae isolates from a hospital in China. PLoS ONE. 2013;8:e75805. doi: 10.1371/journal.pone.0075805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lee G, Burgess D. Treatment of Klebsiella pneumoniae carbapenemase (KPC) infections: a review of published case series and case reports. Ann Clin Microbiol Antimicrob. 2012;11:32. doi: 10.1186/1476-0711-11-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Brown P, Freeman A, Foxman B. Prevalence and predictors of trimethoprim-sulfamethoxazole resistance among uropathogenic Escherichia coli isolates in Michigan. Clin. Infect. Dis. 2002;34:1061–1066. doi: 10.1086/339491. [DOI] [PubMed] [Google Scholar]
- (4).Garcia M, Bellido J, Rodriguez J. In vitro susceptibility of community-acquired urinary tract pathogens to commonly used antimicrobial agents in Spain: a comparative multicenter study (2002-2004) J. Chemother. 2007;19:263–270. doi: 10.1179/joc.2007.19.3.263. [DOI] [PubMed] [Google Scholar]
- (5).Tsay R, Siu L, Fung C, Chang F. Characteristics of bacteremia between community-acquired and nosocomial Klebsiella pneumoniae infection: risk factor for mortality and the impact of capsular serotypes as a herald for community-acquired infection. Arch. Intern. Med. 2002;162:1021–1027. doi: 10.1001/archinte.162.9.1021. [DOI] [PubMed] [Google Scholar]
- (6).Zhanel G, Karlowsky J, Harding G, Carrie A, Mazzulli T, Low D, Group, T. C. U. I. S. Hoban D. A Canadian National Surveillance Study of Urinary Tract Isolates from Outpatients: Comparison of the Activities of Trimethoprim-sulfamethoxazole, ampicillin, mecillinam, nitrofurantoin and ciprofloxacin. Antimicrob. Agents Chemother. 2000;44:1089–1092. doi: 10.1128/aac.44.4.1089-1092.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Talan D, Krishnadasan A, Abrahamian F, Stamm W, Moran G, group, E. I. N. S. Prevalence and risk factor analysis of trimethoprim-sulfamethoxazole—and fluoroquinolone-resistant Escherichia coli infection among emergency department patients with pyelonephritis. Clin. Infect. Dis. 2008;47:1150–1158. doi: 10.1086/592250. [DOI] [PubMed] [Google Scholar]
- (8).Lee J, Oh J, Cho J, Park J, Kim J, Seol S, Cho D. The prevalence of trimethoprim-resistance-conferring dihydrofolate reductase genes in urinary isolates of Escherichia coli in Korea. J. Antimicrob. Chemother. 2001;47:599–604. doi: 10.1093/jac/47.5.599. [DOI] [PubMed] [Google Scholar]
- (9).Blahna M, Zalewski C, Reuer J, Kahlmeter G, Foxman B, Marrs C. The role of horizontal gene transfer in the spread of trimethoprim-sulfamethoxazole resistance among uropathogenic Escherichia coli in Europe and Canada. J. Antimicrob. Chemother. 2006;57:666–672. doi: 10.1093/jac/dkl020. [DOI] [PubMed] [Google Scholar]
- (10).Brolund A, Sundqvist M, Kahlmeter G, Grape M. Molecular characterisation of trimethoprim resistance in Escherichia coli and Klebsiella pneumoniae during a two-year intervention on trimethoprim use. PLoS ONE. 2010;5:e9233. doi: 10.1371/journal.pone.0009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Seputiene V, Povilonis J, Ruzauskas M, Pavilonis A, Suziedeliene E. Prevalence of trimethoprim resistance genes in Escherichia coli isolates of human and animal origin in Lithuania. J. Med. Microbiol. 2010;59:315–322. doi: 10.1099/jmm.0.015008-0. [DOI] [PubMed] [Google Scholar]
- (12).Chen L, Chavda K, Al Laham N, Melano R, Jacobs M, Bonomo R, Krieiswirth B. Complete nucleotide sequence of a blaKPC-harboring IncI2 plasmid and its dissemination in New Jersey and New York hospitals. Antimicrob. Agents Chemother. 2013;57:5019–5025. doi: 10.1128/AAC.01397-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Pournaras S, Poulou A, Voulgari E, Vrioni G, Kristo I, Tsakris A. Detection of the new metallo-β-lactamase VIM-19 along with KPC-2, CMY-2 and CTX-M-15 in Klebsiella pneumoniae. J Antimicrob Chemother. 2010;65:1604–1607. doi: 10.1093/jac/dkq190. [DOI] [PubMed] [Google Scholar]
- (14).van Essen-Zandbergen A, Smith H, Veldman K, Mevius D. Occurrence and characteristics of class 1, 2 and 3 integrons in Escherichia coli, Salmonella and Campylobacter spp. in the Netherlands. J Antimicrob Chemother. 2007;59:746–750. doi: 10.1093/jac/dkl549. [DOI] [PubMed] [Google Scholar]
- (15).Frey K, Lombardo M, Wright D, Anderson A. Towards the Understanding of Resistance Mechanisms in Clinically Isolated Trimethoprim-resistant, Methicillin-resistant Staphylococcus aureus Dihydrofolate Reductase. J. Struc. Biol. 2010;170:93–97. doi: 10.1016/j.jsb.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Frey K, Viswanathan K, Wright D, Anderson A. Prospectively screening novel antibacterial inhibitors of dihydrofolate reductase for mutational resistance. Antimicrob. Agents and Chemother. 2012;56:3556–3562. doi: 10.1128/AAC.06263-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Keshipeddy S, Reeve S, Anderson A, Wright D. Nonracemic antifolates stereoselectively recruit alternate cofactors and overcome resistance in S. aureus. J. Am. Chem. Soc. 2015;137:8983–8990. doi: 10.1021/jacs.5b01442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Viswanathan K, Frey K, Scocchera E, Martin B, Swain P, Alverson J, Priestley N, Anderson A, Wright D. Toward new Therapeutics for Skin and Soft Tissue Infections: Propargyl-linked Antifolates Are Potent Inhibitors of MRSA and Streptococcus pyogenes. PLoS ONE. 2012;7:e29434. doi: 10.1371/journal.pone.0029434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lamb K, Lombardo M, Alverson J, Priestley N, Wright D, Anderson A. Crystal structures of Klebsiella pneumoniae dihydrofolate reductase bound to propargyl-linked antifolates reveal features for potency and selectivity. Antimicrob. Agents and Chem. 2014;58:7484–7491. doi: 10.1128/AAC.03555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sawaya, Kraut J. Loop and Subdomain Movements in the Mechanism of Escherichia coli Dihydrofolate Reductase: Crystallographic Evidence. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- (21).G-Dayanandan N, Paulsen J, Viswanathan K, Keshipeddy S, Lombardo M, Zhou W, Lamb K, Sochia A, Alverson J, Priestley N, Wright D, Anderson A. Propargyl-linked Antifolates are Dual Inhibitors of Candida albicans and Candida glabrata. J. Med. Chem. 2014;57:2643–2656. doi: 10.1021/jm401916j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Adams P, Afonine P, Bunkóczi G, Chen V, Davis I, Echools N, Headd J, Hung L-W, Kapral G, Grosse-Kunstleve R, McCoy A, Moriarty N, Oeffner R, Read R, Richardson D, Richardson J, Terwilliger T, Zwart P. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (24).Schüttelkopf A, van Aalten D. PRODRG - a tool for high-throughput crystallography of protein-ligand complexes. Acta Cryst. 2004;D60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.