

Abstract
Background
MiR-9 is reportedly involved with many diseases, such as acute myeloid leukemia and liver oncogenesis. In the present study we investigated the molecular mechanism, including the potential regulator and signaling pathways, of MYOCD, which is the gene that in humans encodes the protein myocardin.
Material/Methods
We searched the online miRNA database (www.mirdb.org) with the “seed sequence” located within the 3′-UTR of the target gene, and then validated MYOCD to be the direct gene via luciferase reporter assay system, and further confirmed it in cultured cells by using Western blot analysis and realtime PCR.
Results
We established the negative regulatory relationship between miR-9 and MYOCD via studying the relative luciferase activity. We also conducted realtime PCR and Western blot analysis to study the mRNA and protein expression level of MYOCD between different groups (intracranial aneurysm vs. normal control) or cells treated with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors, indicating the negative regulatory relationship between miR-9 and MYOCD. We also investigated the relative viability of smooth muscle cells when transfected with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors to validate that miR-9 t negatively interferes with the viability of smooth muscle cells. We then investigated the relative contractility of smooth muscle cells when transfected with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors, and the results showed that miR-9 weakened contractility.
Conclusions
Our findings show that dysregulation of miR-9 is responsible for the development of IA via targeting MYOCD. miR-9 and its direct target, MYOCD, might novel therapeutic targets in the treatment of IA.
MeSH Keywords: Intracranial Aneurysm; MicroRNAs; Myocytes, Smooth Muscle
Background
As a common vascular disease, rupture of intracranial aneurysm (IA) usually leads to subarachnoid hemorrhage (SAH). Even though numerous acquired hazards related to IAs have been confirmed, heritable conditions are involved in the formation of IAs; however, these syndromes consist of less than 1% of all intracranial aneurysms in humans [1]. IA rupture results in SAH, which account for approximately 50% of deaths in these cases. IAs are a common vascular disease, with an approximate frequency of unruptured IAs of 2% to 6% among adults, and is recognized as a complex disorder associated with environmental and genetic risk factors. Known risk factors include positive family history, increasing age, hypertension, and smoking [2]. It is difficult to determine how the molecular mechanisms evolved. Genomewide genetic association studies, DNA linkage studies, and mRNA expression studies based on microarray provide new ways to explore the molecular mechanisms underlying the pathobiology of IAs and identify the genetic risk factors [3].
More and more unruptured aneurysms are being identified because of advances in diagnostic modalities, but it is unclear if they should be treated. When it comes to the necessity of receiving a surgery for unruptured aneurysms, the surgical morbidity and mortality and natural history should be considered. Therefore, it is essential to identify the mechanism underlying aneurysmal rupture. In the beginning, hemodynamic stress might play an important role in aneurysm rupture. A tiny locus in the wall where the normal structure of vasculum vanished is commonly found in the unruptured aneurysms during surgery. The fragile site might be the precise location for rupture, but the mechanism by which the fragility of the aneurysmal wall takes place remains unknown. Recent studies demonstrated that apoptosis participates in the natural developmental phase and pathological conditions [4].
As a group of non-coding, small RNAs, miRNA negatively regulates expression of genes by inducing degradation of mRNAs, inhibition of translation, and sequenced repression of protein synthesis. About 30% of the encoding genes in the human genome are regulated by miRNA in a posttranscriptional manner, which bind to the target mRNAs’ 3′untranslated region (3′UTR) preferentially and incorporate into the RNA-induced silencing complex (RISC). Then, RISC suppress expression of genes either by inhibiting translation or inducing degradation of mRNAs [5]. Among the numerous miRNA-regulated target genes, many are involved in biological pathways, such as apoptosis and immune response [6]. Phase-specific expression and tissue-specific expression are critical features of miRNA expression, which might imply that miRNAs exert different physiological functions in different tissues [7,8]. Some miRNAs have physiological functions in the cardiovascular system. For instance, there is an increased level of miR-1 when smooth muscle cells (SMC) differentiate, leading to accumulating expression level of SMC-specific contractile proteins. miR-133 serves as the main regulator in the phenotypic switch of vascular smooth muscle cells in vivo and in vitro [9]. Notably, miR-145 is involved in vessel wall thickness, and lack of miR-145 leads to reduction of the vessel wall and subsequent SMC hypotrophy [10]. There is a downregulation of miR-145 in experimentally induced aneurysms, atherosclerosis, and vascular injury [11,12].
Zhang et al. identified many miRNAs that were differentially expressed in the smooth muscle cells collected from the patients with IA compared to those without IA; miR-9 is one of those differentially expressed miRNAs [13]. Using the online miRNA database, we found that MYOCD is a virtual target of miR-9, and MYOCD has been reported to be involved in the pathogenesis of IA by regulating the contractility of smooth muscle cells [14]. In the present study we explored the relationship between miR-9 and MYOCD, finding that downregulation of MYOCD caused by upregulation of miR-9 leads to the development of IA.
Material and Methods
Patients and samples
This study comprised 13 IA patients diagnosed through digital subtraction angiography (DSA). Full-thickness vessel wall tissue samples were collected from the ruptured IA patients who underwent microsurgical clipping. Ten middle meningeal artery segments (with controls matched for sex, age, and blood pressure) were obtained during standard neurosurgical procedures (traumatic hematoma and tumor resection). Tissue samples were snap-frozen in liquid nitrogen and sent directly to the laboratory for the following experiments. The study protocol was approved by the Ethics Committee of Capital Medical University, and written informed consent was obtained from each patient before the study started.
Real-time polymerase chain reaction
Realtime polymerase chain reaction (PCR) was performed for quantification of RNA. In brief, we extracted total RNA from culture cells or sample tissues with Trizol reagent column-purified with RNeasy kit (QIAGEN AG, Basel, Switzerland). Complementary DNA (cDNA) was synthesized using the High-Capacity cDNA Archive kit in accordance with the manufacturer’s instructions (Applied Biosystems, Life Technologies, Carlsbad, CA). Subsequently, real-time PCR was performed using the LightCycler 480 System (Roche Applied Science, Mannheim, Germany). The cycler protocol was 5 min at 95°C, 40 cycles of 15 s at 95°C, 60 s at 60°C, and 5 min at 72°C. Gene of interest expression was normalized to the reference genes U6 and β-actin. Fold expression was calculated using the 2−ΔΔCt method.
Cell proliferation assay
Cell proliferation was measured by the Cell Counting Kit-8 assay (CCK-8) (Dojindo, Kumamoto, Japan). After the same number of SMCs was seeded for 24 h, cells were transfected with miR-9 mimics or MYOCD siRNA or negative control, and incubated in DMEM supplemented with different serum for the indicated time. After the treatment, 10 ll CCK-8 solution was added to each well, and cells were cultured for 2 h more at 37°C. After that, the absorbance was read at 450 nm using a microplate absorbance reader (Bio-Rad Laboratories, Hercules, CA). Cells in the control group were considered 100% viable.
SMC contractile assay
SMC contractile assay was used to determine the contractility of SMCs with different treatments, as previously described [13].
Cell culture and transfection
SMCs grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, penicillin/streptomycin, and glutamine at 37°C in a humidified atmosphere with 5% CO2. SMCs were transfected with small interfering RNAs (siRNAs) in 12-well plates at a density of 100 000 or 130 000 cells per well. Transfections were performed with Dharmafect1 (GE Dharmacon, Lafayette, CO) according to the manufacturer’s recommendations. For plasmid transfection, SMCs were seeded in 12-well plates at a density of 120 000 cells per well. After 24 h, transfections were performed with Fugene HD (Promega, Madison, WI), according to the manufacturer’s instructions.
Luciferase assays
3′UTR of MYOCD was amplified and inserted into psiCHECK-2, and the predicted binding site sequence was mutated using a mutagenesis kit (Stratagene). SMCs were seeded in 12-well plates at a density of 120 000 cells per well 24 h prior to transfection. Cells were cotransfected with 100 ng of psiCHECK-2 + MYOCD3′UTR (wild-type or mutant) and 100 nmol/L miR-9 mimics with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. In all cases, expression levels were normalized to a constitutively expressed firefly luciferase gene in psiCHECK-2. At 48 h after transfection, firefly luciferase and Renilla luciferase activities were measured consecutively with a dual-luciferase reporter system and a luminometer (Promega). Each experiment was performed in triplicate.
Western blot analysis
In brief, protein levels of sample tissues or culture cell were determined using Bradford dye (Bio-Rad Laboratories, Hercules, CA, USA). For Western blot analysis, 10-mg aliquots of total protein were isolated by SDS-PAGE (12%) and transferred to nitrocellulose membranes (GE Healthcare Bio-Sciences, Pittsburgh, PA) in NuPAGE transfer buffer (Invitrogen Life Technologies) containing 10% methanol. The membranes were blocked then probed with specific primary antibodies for 2 h and secondary antibody for another 2 h.Antibodies used in this study were: anti-MYOCD antibody (1:2000, SCBT, Santa Cruz, CA) and anti-β-actin antibody (1:10000, SCBT, Santa Cruz, CA). Blots were visualized with the ECL Chemiluminescence Detection System (GE Healthcare Bio-Sciences).
Statistical analyses
SPSS version 19.0 software (IBM, Chicago, IL) was used for data processing. In vitro data were analyzed using the unpaired Student t-test or one-way ANOVA. P<0.05 were considered statistically significant.
Results
MYOCD was virtual target of miR-9
MiR-9 has been reported to be involved with many diseases, such as acute myeloid leukemia and liver oncogenesis. In order to understand the role of MiR-9 in intracranial aneurysm, we used online miRNA target prediction tools to search for the regulatory gene of miR-9, and consequently identified MYOCD as the candidate target gene of miR-9 in smooth muscle cells with the ‘seed sequence’ in the 3′UTR (Figure 1).To validate the regulatory relationship between miR-9 and MYOCD, we also conducted luciferase activity reporter assay in smooth muscle cells, and found that only the luciferase activity from the cells cotransfected with miR-9 and wild-type MYOCD 3′UTR decreased significantly Figure 2), while cells cotransfected with miR-9 and mutant MYOCD 3′UTR were comparable compared with scramble control (Figure 2). The results confirmed that MYOCD was a validated target of miR-9 in smooth muscle cells. To further determine the modulatory relationship between miR-9 and MYOCD, we analyzed the correlation between the expression level of miR-9 and MYOCD mRNA among the tissues (n=24); as shown in Figure 3, they had a negative regulatory relationship (r=−05148, p<0.05).
Figure 1.

MYOCD as the candidate target gene of miR-9 in smooth muscle cells with the ‘seed sequence’ in the 3′UTR.
Figure 2.
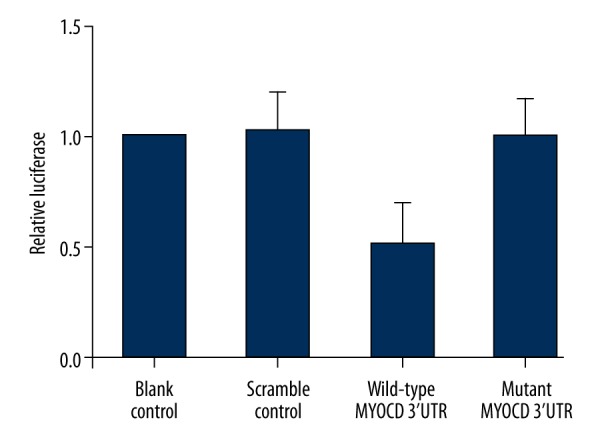
Luciferase activity reporter assay was conducted to verify MYOCD as the direct target gene of miR-9.
Figure 3.
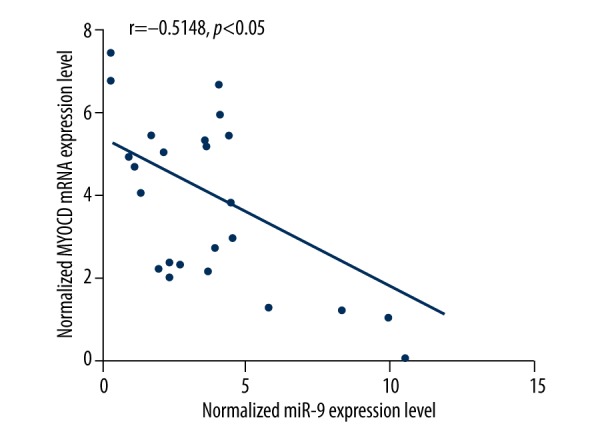
Correlation between expression level of miR-9 and MYOCD mRNA between IA and normal control (IA, n=13; normal groups, n=11).
Determination of expression patterns of miR-9 and MYOCD in tissues with different groups
The tissues of 2 different groups (IA, n=13; normal groups, n=11) were used to further explore the impacts on the interaction between miR-9 and MYOCD 3′UTR. Using realtime PCR, we found the expression of miR-9 was increased in IA group (Figure 4A) compared with the normal group, while the expression of MYOCD mRNA (Figure 4B) decreased in the IA group compared with the normal group; the expression of MYOCD protein (Figure 4C) was measured by densitometry analysis; we found it decreased in the IA group compared with the normal group. To further validate the hypothesis of the negative regulatory relationship between miR-9 and MYOCD, we investigated the mRNA/protein expression level of MYOCD of smooth muscle cells. We transfected the smooth muscle cells with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors. As shown in Figure 5, the MYOCD protein (upper panel) and mRNA expression level (lower panel) of smooth muscle cells treated with miR-9 mimics and MYOC siRNA were apparently lower than in the scramble control, while cells treated miR-9 inhibitors were apparently higher than in the scramble control, validating the negative regulatory relationship between miR-9 and MYOCD.
Figure 4.

The expression of miR-9 increased in the IA group (A) compared with the normal control group, while the expression of MYOCD mRNA (B) and protein (C) decreased in the IA group compared with the normal control group.
Figure 5.

When we transfected the smooth muscle cells with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors, the expression level of MYOCD protein (A) and mRNA (B) treated with miR-9 mimics and MYOCD siRNA decreased, while cells treated miR-9 inhibitors increased.
MiR-9 and MYOCD interfered with viability of smooth muscle cells
We also investigated the relative viability of smooth muscle cells when transfected with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors. Cells transfected with miR-9 inhibitors showed evidently upregulated viability (Figure 6) compared with the scramble controls, while cells transfected with miR-9 mimics and MYOCD siRNA showed comparably lower viability, indicating miR-9 negatively interfered with the viability of smooth muscle cells, while MYOCD positively interfered with the viability of smooth muscle cells.
Figure 6.

Cells transfected with miR-9 inhibitors showed evidently higher viability (A), while cells transfected with miR-9 mimics and MYOCD siRNA showed comparably lower viability. Cells transfected with miR-9 inhibitors showed evidently enhanced contractility (B), while cells transfected with miR-9 mimics and MYOCD siRNA showed comparably attenuated contractility.
MiR-9 and MYOCD interfered with the contractility of smooth muscle cells
We then investigated the relative contractility of smooth muscle cells when transfected with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors. When transfected with miR-9 mimics and MYOCD siRNA, the contractility of smooth muscle cells was lower, while cells transfected with miR-9 inhibitors showed comparably higher contractility. The results indicated miR-9 weakened contractility and MYOCD enhanced contractility.
Discussion
MiRNAs have been reported to be functionally involved in many cellular activities, including contractility of SMCs. For example, miR-1 expression could be induced by SRF in cardiomyocytes and miR-1 has a significant influence on skeletal and cardiac muscle cells differentiation and proliferation [15,16]. MiR-1 targets histone deacetylase 4 to promote differentiation of cardiomyocytes [15]. The overexpression of miR-1 targets Hand2 to reduce the ventricular cardiomyocytes’ proliferation in the development of heart [16]. Notably, miR-133 together with miR-1 participate in the regulation of sarcomeric actin organization in skeletal muscle of zebrafish by targeting actin-binding and actin-related proteins of muscle [17]. The arrhythmogenesis might be promoted by miR-1’s binding to connexin 43, inwardly correcting B56a, a unit of protein phosphatase 2A regulatory and Kir2.1, a subunit of Kþ channel [18,19]. In this study, we used online miRNA target prediction tools to search for the regulatory gene of miR-9, and consequently identified MYOCD as the candidate target gene of miR-9 in smooth muscle cells with the ‘seed sequence’ in the 3′UTR. Furthermore, we conducted luciferase reporter assay, and found that only the luciferase activity from the cells cotransfected with miR-9 and wild-type MYOCD 3′UTR decreased significantly, while cells cotransfected with miR-9 and mutant MYOCD 3′UTR were comparable compared with scramble control. The results confirmed that MYOCD was a validated target of miR-9 in smooth muscle cells.
It is believed that MYOCD represses SMC proliferation by independent mechanisms. First and foremost, MYOCD interferes with binding of p65 DNA to reduce the activity of target genes, which are mediated by p65 [20]. Secondly, Has-miR-1 reduces the expression of kinase PIM-1, which can promote proliferation of SMCs in vivo and in vitro, leading to inhibition of SMC proliferation [21,22]. The low Has-miR-1 expression level in tissues and vascular SMCs and MYOCD can induce expression of has-miR-1 in cultured bladders or SMCs of rat bladder in vivo to reduce the expression of connexin 43 (GJA1) [21,23,24]. Other mechanisms also participate in the cell cycle inhibition induced by MYOCD. For instance, MYOCD induces calponin expression, which can inhibit SMC proliferation [25,26]. In the present study, we collected the tissue samples of 2 different groups (IA, n=13; normal group, n=11), and found the expression of miR-9 increased in the IA group (Figure 4A) compared with the normal group, while the expression of MYOCD mRNA (Figure 4B) decreased in the IA group compared with the normal group.
We chose MYOCD and miR-9 as a model system because: 1) miR-9 was reported to be differentially expressed in IA SMCs in comparison to the normal control [27]; 2) it has been confirmed that MYOCD is essential in the process of enhancing contractility of vessels by expressing a unique group of signaling molecules, contractile agonist receptors, and contractile proteins to fulfill the task [28,29]; 3) delicate expression level change of MYOCD was critical in determining percentage of survival and IA was demonstrated by the IA mouse model [30]; and 4) partial or complete loss of MYOCD can lead to dramatic deceleration of IA in the IA mouse model [13].
It’s reported that myocardin plays a key role in differentiation of SMC lineage and vasculogenesis. Elevated expression level of MYOCD increases contractile proteins of smooth muscle, leading to development of contractility of smooth muscle [31,32]. When vascular injury occurs, differentiated SMCs experience a unique phenotype transformation known as transforming from a quiescent contractile state to a proliferative synthetic state [33,34]. The progress of IA initiation is accelerated by proliferative phenotyping; the size of new layers of intramural thrombi mediated by apposition remarkably increased within the vessel wall. In aneurysm pathogenesis, frequent subadventitial hemorrhages from the new vasa vasorum might be a risk factor; consequently, it is believed that IA is a proliferative disorder of the vessel wall resulting from extravascular activity [35,36]. In the present study, we investigated the relative viability and contractility of smooth muscle cells when transfected with scramble control, miR-9 mimics, MYOCD siRNA, and miR-9 inhibitors. Cells transfected with miR-9 inhibitors showed evidently upregulated viability and contractility when compared with the scramble controls, while cells transfected with miR-9 mimics and MYOCD siRNA showed comparably lower viability and contractility, indicating miR-9 negatively interfered with the viability and contractility of smooth muscle cells.
Conclusions
These findings show that dysregulation of miR-9 is responsible for the development of IA via targeting MYOCD. miR-9 and its direct target, MYOCD, might be a novel therapeutic targets in the treatment of IA.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Bourcier R, Redon R, Desal H. Genetic investigations on intracranial aneurysm: Update and perspectives. J Neuroradiol. 2015;42(2):67–71. doi: 10.1016/j.neurad.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 2.Francis SE, Tu J, Qian Y, Avolio AP, et al. A combination of genetic, molecular and haemodynamic risk factors contributes to the formation, enlargement and rupture of brain aneurysms. J Clin Neurosci. 2013;20(7):912–18. doi: 10.1016/j.jocn.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 3.Tromp G, Weinsheimer S, Ronkainen A, Kuivaniemi H. Molecular basis and genetic predisposition to intracranial aneurysm. Ann Med. 2014;46(8):597–606. doi: 10.3109/07853890.2014.949299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Savill J. Apoptosis in disease. Eur J Clin Invest. 1994;24(11):715–23. doi: 10.1111/j.1365-2362.1994.tb01067.x. [DOI] [PubMed] [Google Scholar]
- 5.Angaji SA, Hedayati SS, Poor RH, et al. Application of RNA interference in treating human diseases. J Genet. 2010;89(4):527–37. doi: 10.1007/s12041-010-0073-3. [DOI] [PubMed] [Google Scholar]
- 6.Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009;84(1):55–71. doi: 10.1111/j.1469-185X.2008.00061.x. [DOI] [PubMed] [Google Scholar]
- 7.Ji R, Cheng Y, Yue J, et al. MicroRNA expression signature and antisense-mediated depletion reveal an essential role of MicroRNA in vascular neointimal lesion formation. Circ Res. 2007;100(11):1579–88. doi: 10.1161/CIRCRESAHA.106.141986. [DOI] [PubMed] [Google Scholar]
- 8.Lagos-Quintana M, Rauhut R, Yalcin A, et al. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12(9):735–39. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 9.Torella D, Iaconetti C, Catalucci D, et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011;109(8):880–93. doi: 10.1161/CIRCRESAHA.111.240150. [DOI] [PubMed] [Google Scholar]
- 10.Xin M, Small EM, Sutherland LB, et al. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009;23(18):2166–78. doi: 10.1101/gad.1842409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng Y, Liu X, Yang J, et al. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009;105(2):158–66. doi: 10.1161/CIRCRESAHA.109.197517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elia L, Quintavalle M, Zhang J, et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009;16(12):1590–98. doi: 10.1038/cdd.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang M, Ren Y, Wang Y, et al. Regulation of smooth muscle contractility by competing endogenous mRNAs in intracranial aneurysms. J Neuropathol Exp Neurol. 2015;74(5):411–24. doi: 10.1097/NEN.0000000000000185. [DOI] [PubMed] [Google Scholar]
- 14.Xu D, Gu JT, Yi B, et al. Requirement of miR-9-dependent regulation of Myocd in PASMCs phenotypic modulation and proliferation induced by hepatopulmonary syndrome rat serum. J Cell Mol Med. 2015;19(10):2453–61. doi: 10.1111/jcmm.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen JF, Mandel EM, Thomson JM, et al. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat Genet. 2006;38(2):228–33. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436(7048):214–20. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 17.Mishima Y, Abreu-Goodger C, Staton AA, et al. Zebrafish miR-1 and miR-133 shape muscle gene expression and regulate sarcomeric actin organization. Genes Dev. 2009;23(5):619–32. doi: 10.1101/gad.1760209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Terentyev D, Belevych AE, Terentyeva R, et al. miR-1 overexpression enhances Ca(2+) release and promotes cardiac arrhythmogenesis by targeting PP2A regulatory subunit B56alpha and causing CaMKII-dependent hyperphosphorylation of RyR2. Circ Res. 2009;104(4):514–21. doi: 10.1161/CIRCRESAHA.108.181651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang B, Lin H, Xiao J, et al. The muscle-specific microRNA miR-1 regulates cardiac arrhythmogenic potential by targeting GJA1 and KCNJ2. Nat Med. 2007;13(4):486–91. doi: 10.1038/nm1569. [DOI] [PubMed] [Google Scholar]
- 20.Tang RH, Zheng XL, Callis TE, et al. Myocardin inhibits cellular proliferation by inhibiting NF-kappaB(p65)-dependent cell cycle progression. Proc Natl Acad Sci USA. 2008;105(9):3362–67. doi: 10.1073/pnas.0705842105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Yin H, Jiang Y, et al. Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011;31(2):368–75. doi: 10.1161/ATVBAHA.110.218149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katakami N, Kaneto H, Hao H, et al. Role of pim-1 in smooth muscle cell proliferation. J Biol Chem. 2004;279(52):54742–49. doi: 10.1074/jbc.M409140200. [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Hu G, Betts C, et al. Transforming growth factor-beta1-induced transcript 1 protein, a novel marker for smooth muscle contractile phenotype, is regulated by serum response factor/myocardin protein. J Biol Chem. 2011;286(48):41589–99. doi: 10.1074/jbc.M111.250878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imamura M, Sugino Y, Long X, et al. Myocardin and microRNA-1 modulate bladder activity through connexin 43 expression during post-natal development. J Cell Physiol. 2013;228(9):1819–26. doi: 10.1002/jcp.24333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Z, Grange RW, Walsh MP, Kamm KE. Adenovirus-mediated transfer of the smooth muscle cell calponin gene inhibits proliferation of smooth muscle cells and fibroblasts. FEBS Lett. 1997;413(3):441–45. doi: 10.1016/s0014-5793(97)00944-7. [DOI] [PubMed] [Google Scholar]
- 26.Hossain MM, Hwang DY, Huang QQ, et al. Developmentally regulated expression of calponin isoforms and the effect of h2-calponin on cell proliferation. Am J Physiol Cell Physiol. 2003;284(1):C156–67. doi: 10.1152/ajpcell.00233.2002. [DOI] [PubMed] [Google Scholar]
- 27.Boettger T, Beetz N, Kostin S, et al. Acquisition of the contractile phenotype by murine arterial smooth muscle cells depends on the Mir143/145 gene cluster. J Clin Invest. 2009;119(9):2634–47. doi: 10.1172/JCI38864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84(3):767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 29.Owens GK. Molecular control of vascular smooth muscle cell differentiation. Acta Physiol Scand. 1998;164(4):623–35. doi: 10.1111/j.1365-201x.1998.tb10706.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen J, Kitchen CM, Streb JW, Miano JM. Myocardin: A component of a molecular switch for smooth muscle differentiation. J Mol Cell Cardiol. 2002;34(10):1345–56. doi: 10.1006/jmcc.2002.2086. [DOI] [PubMed] [Google Scholar]
- 31.Zheng XL. Myocardin and smooth muscle differentiation. Arch Biochem Biophys. 2014;543:48–56. doi: 10.1016/j.abb.2013.12.015. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida T, Kawai-Kowase K, Owens GK. Forced expression of myocardin is not sufficient for induction of smooth muscle differentiation in multipotential embryonic cells. Arterioscler Thromb Vasc Biol. 2004;24(9):1596–601. doi: 10.1161/01.ATV.0000137190.63214.c5. [DOI] [PubMed] [Google Scholar]
- 33.Liu ZP, Wang Z, Yanagisawa H, Olson EN. Phenotypic modulation of smooth muscle cells through interaction of Foxo4 and myocardin. Dev Cell. 2005;9(2):261–70. doi: 10.1016/j.devcel.2005.05.017. [DOI] [PubMed] [Google Scholar]
- 34.Xie WB, Li Z, Miano JM, et al. Smad3-mediated myocardin silencing: A novel mechanism governing the initiation of smooth muscle differentiation. J Biol Chem. 2011;286(17):15050–57. doi: 10.1074/jbc.M110.202747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huttunen T, von und zu Fraunberg M, Frösen J, et al. Saccular intracranial aneurysm disease: distribution of site, size, and age suggests different etiologies for aneurysm formation and rupture in 316 familial and 1454 sporadic eastern Finnish patients. Neurosurgery. 2010;66(4):631–38. doi: 10.1227/01.NEU.0000367634.89384.4B. discussion 638. [DOI] [PubMed] [Google Scholar]
- 36.Krings T, Mandell DM, Kiehl TR, et al. Intracranial aneurysms: From vessel wall pathology to therapeutic approach. Nat Rev Neurol. 2011;7(10):547–59. doi: 10.1038/nrneurol.2011.136. [DOI] [PubMed] [Google Scholar]