

Abstract
Influenza viruses cause seasonal epidemics and pandemic outbreaks associated with significant morbidity and mortality, and a huge cost. Since resistance to the existing anti‐influenza drugs is rising, innovative inhibitors with a different mode of action are urgently needed. The influenza polymerase complex is widely recognized as a key drug target, given its critical role in virus replication and high degree of conservation among influenza A (of human or zoonotic origin) and B viruses. We here review the major progress that has been made in recent years in unravelling the structure and functions of this protein complex, enabling structure‐aided drug design toward the core regions of the PA endonuclease, PB1 polymerase, or cap‐binding PB2 subunit. Alternatively, inhibitors may target a protein–protein interaction site, a cellular factor involved in viral RNA synthesis, the viral RNA itself, or the nucleoprotein component of the viral ribonucleoprotein. The latest advances made for these diverse pharmacological targets have yielded agents in advanced (i.e., favipiravir and VX‐787) or early clinical testing, besides several experimental inhibitors in various stages of development, which are all covered here.
Keywords: influenza virus, antiviral, polymerase, cap‐snatching, nucleoside
1. INTRODUCTION
The influenza A, B, C, and D viruses belong to the Orthomyxoviridae, a family of enveloped viruses with a single‐stranded negative‐sense, and segmented RNA genome. Seasonal influenza A and B viruses affect each year approximately 5–10% of the adult and 20–30% of the pediatric population. In addition, there is permanent concern for sudden influenza pandemics, such as that of 2009 or the notorious “Spanish flu” of 1918.
A universal influenza vaccine that confers broad and long‐term protection remains the “Holy Grail” in influenza research,1 as vaccination is considered as the most effective way to prevent influenza virus infections. The existing tri‐ or quadrivalent vaccines contain the circulating A/H1N1, A/H3N2, and (one or two) B strains. They fail to provide broadly protective and long‐lasting immunity, require annual updating, and are only partially protective in some target populations.2, 3
For prevention or treatment of influenza virus infections, two classes of antiviral drugs are currently available. The M2‐blockers, amantadine and rimantadine, inhibit influenza A virus replication by occluding the M2 proton channel,4 but lack activity against influenza B virus.5 In addition, central nervous system side effects6 and the global spread of adamantane‐resistant influenza viruses7 undermine their clinical usefulness. The second group, the neuraminidase inhibitors (NAIs), prevents virus release from infected cells. Zanamivir requires administration by powder inhalation, while oseltamivir is administered orally as its prodrug oseltamivir phosphate. In the period 2007–2009, oseltamivir‐resistant influenza H1N1 viruses were disseminated worldwide.8 In contrast, resistance to zanamivir seems to be rare.9, 10 The search for novel M2‐inhibitors and NAIs with improved resistance profile and higher potency is still ongoing, as is further optimization of original lead compounds.11
In conclusion, the armamentarium to prevent and treat influenza infections is currently limited, and new influenza virus inhibitors with an entirely different mode of action are urgently required. In this review, we focus on the influenza virus polymerase that is widely recognized as a superior antiviral drug target,12 given its critical role in virus replication and high degree of sequence conservation in influenza A and B, particularly in the active sites for RNA binding, cleavage, or elongation. Before explaining the different antiviral strategies that are being explored, we provide relevant background on structure and functions of the viral polymerase. During recent years, major breakthroughs have been made in revealing the structure of this protein complex, its subunits, or the associated viral nucleoprotein (NP), creating unique opportunities for structure‐aided drug design.
2. STRUCTURE AND FUNCTIONS OF THE INFLUENZA VIRUS POLYMERASE COMPLEX
To avoid overlap with other recent reviews on this topic,13, 14, 15, 16 this section is limited to provide an update on new findings with direct or indirect relevance for inhibitor design. The influenza virus polymerase complex (schematically depicted in Fig. 1) is composed of three subunits: PB1, PB2 (polymerase basic protein‐1 and ‐2, respectively) and PA (polymerase acidic protein; referred to as P3 in the case of influenza C). The influenza A and B virions contain eight viral ribonucleoprotein (vRNP) segments, which have a double‐helical hairpin structure and each carry one polymerase heterotrimer. These vRNPs contain vRNA that is coated by viral NP molecules. Viral RNA synthesis occurs in the cell nucleus in two stages (Fig. 1). (i) For primary transcription of vRNA to mRNA, primers are generated by the PA‐dependent “cap‐snatching” reaction on cellular capped RNAs. The PB2 subunit initially binds the capped RNAs, and actual RNA synthesis is performed by PB1. (ii) Replication of vRNA proceeds via complementary RNA (cRNA) intermediates and occurs in a primer‐independent (de novo) manner. The nascent vRNA and cRNA replicates are immediately packaged into new vRNPs or cRNPs, respectively. The newly synthesized vRNP complexes are exported from the nucleus to the cytoplasm, for which different pathways have been proposed (reviewed in Eisfeld et al.14).
Figure 1.
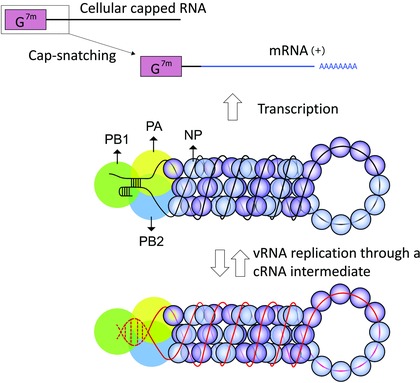
Role of the influenza virus polymerase complex in vRNA transcription and replication.
The influenza polymerase is a complex containing the PA, PB1, and PB2 proteins. One polymerase heterotrimer is attached to each vRNP segment inside the virion. These vRNPs have a double‐helical hairpin structure consisting of two antiparallel vRNA strands that are coated by NP molecules.17, 18
Transcription of vRNA to mRNA starts with the “cap‐snatching” reaction, in which cellular capped RNAs are bound by PB2 and cleaved, by PA, at 10–15 nucleotides from the cap, to yield primers for viral mRNA synthesis.19, 20 Termination and polyadenylation occur at a stretch of five to seven U residues near the 5ʹ end of the vRNA.21, 22 Replication of vRNA proceeds through full‐length complementary cRNAs, which are assembled as cRNP complexes. In both transcription and replication, RNA elongation is carried out by the PB1 subunit. The vRNA promoter is depicted as observed in the promoter‐bound polymerase crystal structures, in which its 5ʹ end forms a “hook” structure.23, 24 The cRNA promoter is drawn in dashed lines, since its conformation is yet to be determined. (Adapted from Portela et al.25)
The influenza virus polymerase is a major determinant of virus pathogenicity and host adaptation.26, 27, 28 One notable key factor is the PB2‐627 signature residue, which is glutamic acid in avian viruses and lysine in human viruses, and was recently linked to species differences in the host nuclear protein ANP32A.29
A. The PB2 Cap‐Binding Domain
The discovery that the cap‐binding activity resides in the PB2 subunit was made in 1981.30, 31 In 2008, Guilligay et al.32 reported the X‐ray structure of the PB2 cap‐binding domain (CBD; i.e., residues 318–483) of influenza A virus, complexed with the minimal cap analogue 7‐methylguanosine 5′‐triphosphate (m7GTP; Fig. 2A). A similar approach for the influenza B PB2‐CBD (Fig. 2B)33 revealed that the binding mode of m7GTP is similar in influenza A and B, indicating a conserved methylated cap recognition mechanism. Similar as in other cap‐binding proteins (such as the eukaryotic initiation factor eIF4E) in which the 7‐methylguanine moiety is sandwiched between two aromatic residues, a cation‐π packing interaction occurs between Phe404 and His357 in the influenza A PB2‐CBD, and Phe406 and Trp359 in the influenza B counterpart. However, the PB2‐CBD has a completely different protein fold compared to other cap‐binding proteins.32 It also contains a remarkable cluster of phenylalanine residues with one noticeable difference (Phe323 vs. Gln325; compare Fig. 2A and B) between the influenza A and B proteins. In two other crystal structures, that is, the influenza B PB2‐CBD in complex with GTP,33 and a Q325F mutant form of this protein in complex with m7GDP,34 an inverted conformation for the guanine and ribose moieties was seen. These data indicate structural flexibility of the influenza B PB2‐CBD, which explains its promiscuous cap recognition. In enzymatic assays with purified vRNPs35 and binding assays with isolated PB2‐CBD,33 influenza A displays strict specificity for m7G‐capped RNA, while the influenza B protein recognizes various cap structures, including unmethylated GpppG‐RNA.
Figure 2.
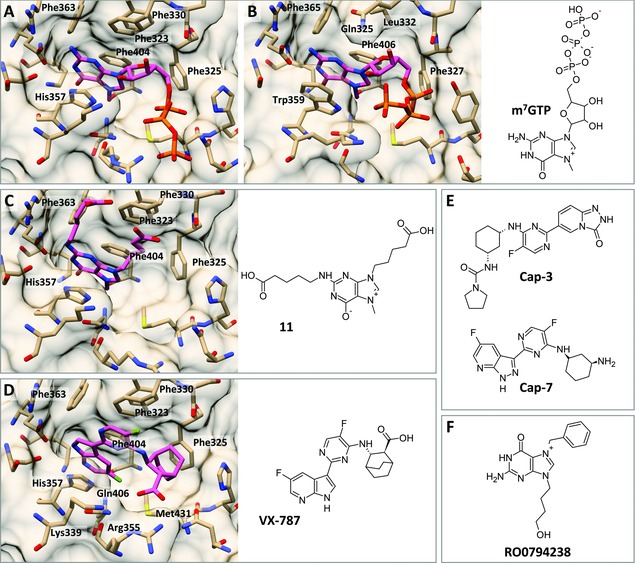
Chemical structures of the minimal cap analogue m7GTP and reported inhibitors of cap‐binding and X‐ray structures of the PB2‐CBD in complex with the corresponding ligands. (A–D) Comparison of the crystal structures of the PB2‐CBD of influenza A32 (A; PDB: 2VQZ) or influenza B33 (B; PDB: 5EFA) PB2‐CBD in complex with m7GTP; and influenza A PB2‐CBD with bound compound “11”36 (C; PDB: 4CB6) or VX‐78737 (D; PDB: 4P1U). (E) Cap‐binding inhibitors Cap‐3 and Cap‐7.38 (F) For RO0794238,39 direct binding to the PB2‐CBD could not be demonstrated.
B. The PA Endonuclease Domain
Until 2009, the endonuclease activity was thought to reside in PB140 or PB2.41 A major leap forward was made when two groups independently revealed42, 43 that the endonuclease catalytic site resides in the N‐terminal domain of PA (PA‐Nter; residues 1 to ∼195) and published the first crystal structures of the holo form of PA‐Nter (Fig. 3A and B). Its structural core contains a characteristic (P)DXN(D/E)XK motif (Fig. 3) conserved in all influenza viruses, which coordinates one, two, or three manganese or magnesium ions. It is still unresolved which and how many divalent metal ions are present in the native enzyme,42, 43, 44, 45, 46, 47, 48 although this information is critical for efficient inhibitor design. The discrepancies in the crystal structures with regard to the identity of these metal ions (Fig. 3A–F) are possibly related to the crystallization conditions (i.e., soaking vs. cocrystallization or different buffer composition, for instance related to pH or which metal ion was added). More conclusive evidence requires crystal structures obtained in the presence of an RNA substrate. As of today, only cocrystal structures with mononucleotides have been determined.44, 46, 49
Figure 3.
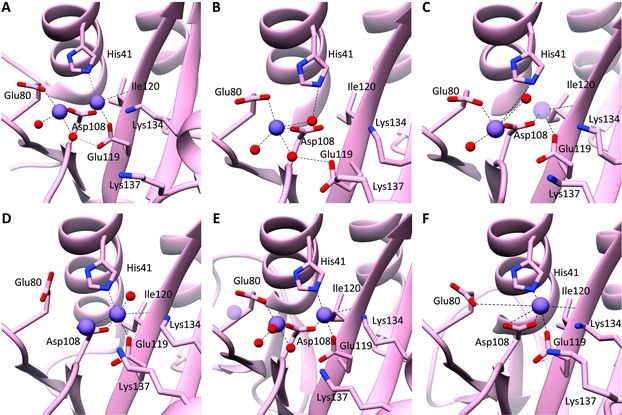
Comparison of PA‐Nter active site structures as determined by crystallography. The metal coordinating and/or catalytic residues are labeled. The metal ions and water molecules are shown as purple and red spheres, respectively. (A) The PA‐Nter crystal structure determined by Dias et al.42 (PDB: 2W69) contains two metal ions (two Mn2+ ions or one Mn2+ and one Mg2+). (B) Yuan et al.43 (PDB: 3EBJ) obtained a PA‐Nter crystal with one Mg2+ ion. (C) Zhao et al.44 (PDB: 3HW6) discerned one (Mg2+) metal (dark purple) or two (Mn2+) metal ions, depending on the crystallization conditions. (D) Dubois et al.45 (PDB: 4E5E) identified two Mn2+ ions. (E) Bauman et al.50 (PDB: 4M5Q) described two Mn2+ ions (in dark purple) in the PA‐Nter apo protein, and a third one (light purple) in the presence of a bound inhibitor (not shown; PDB: 4M5U). (F) In the crystal structure published by Tefsen et al.48 (PDB: 4NFZ), one Mn2+ ion is accommodated in the PA‐Nter active site.
In biochemical assays with isolated recombinant PA‐Nter51 or full‐length PA,52 the endonucleolytic activity is higher in the presence of manganese compared to magnesium. Experiments using isothermal titration calorimetry (ITC) showed a 500‐fold higher affinity for Mn2+ ions than for Mg2+ ions.51 This observation is at least partially explainable by the presence of a histidine residue in the PA‐Nter active site, as Mn2+ ions have higher affinity for histidine than Mg2+. However, these ITC measurements were performed in the absence of RNA substrate, which may be a different situation compared to the substrate‐bound enzyme state. Since the intracellular concentration of free Mg2+ is at least 1000‐fold higher than that of Mn2+, magnesium may be more biologically relevant.53, 54 Also, in the context of the heterotrimeric polymerase, efficient cleavage is seen in the presence of the less reactive Mg2+,55, 56 which can be attributed to tight RNA binding by the nearby PB2‐CBD.
In order to clarify this issue, Xiao and co‐workers performed molecular dynamics (MD) simulations to construct structural models of PA‐Nter in the presence of one or two magnesium ions and in complex with the RNA substrate.57 In Figure 4, both active site conformations and possible one‐ and two‐metal‐ion reaction mechanisms are compared and explained in full detail in the figure legend.
Figure 4.
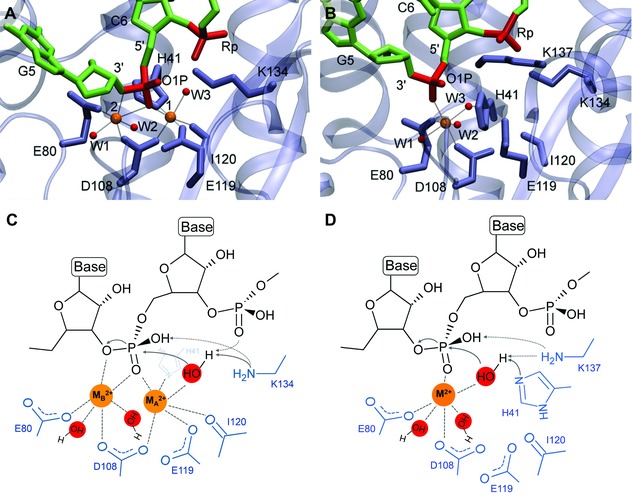
Comparison of the two‐ versus one‐metal‐ion models for the PA‐Nter active site. The metal ions are depicted as orange spheres, while the red spheres represent the water molecules coordinated to the metal ions. (A and B) Structural models obtained by molecular dynamics (MD) simulations of the PA‐Nter active site in complex with its RNA substrate, in the presence of two (A) or one Mg2+ ion(s) (B) [reprinted with permission from Xiao et al.57 Copyright 2014 American Chemical Society]. (C and D) Possible reaction mechanism of the PA‐Nter cleavage reaction, in the presence of two (C) or one (D) metal ion(s), based on the models of Xiao et al.57 In each case, the metal ion has six coordinated ligands (indicated by dotted lines). (C) In the two‐metal mechanism, both metal ions are coordinated to the scissile phosphodiester of the nucleic acid substrate, and the ribose 3ʹ‐O is coordinated apically to MB 2+. MA 2+ would lower the pKa of the attacking water molecule, together with the catalytic Lys134 and the adjacent 3ʹ phosphodiester, thus activating this water molecule as a nucleophile. MA 2+ might also assist the nucleophilic attack by moving toward MB 2+, bringing the activated water molecule closer to the scissile phosphodiester. MB 2+ can stabilize the pentacovalent intermediate formed during the SN2‐type phosphodiester bond cleavage. (D) When the one‐metal mechanism is operative, this metal ion is coordinated to the scissile phosphodiester. Its role would be to stabilize the transition state of the nucleophilic attack, like MB 2+ in the two‐metal mechanism. Additionally, the single metal ion could decrease the pKa of the nucleophilic water, together with the catalytic Lys137, while His41 would act as the general base that deprotonates the water molecule.
The two‐metal‐ion model is favored by the ITC data for Mn2+ binding to PA‐Nter.51 Doan et al.55 found that the endonuclease activity in vRNP complexes isolated from virions depends on metal ion concentration in a cooperative manner, with Hill coefficients close to or larger than 2. Also, synergistic activation of the cleavage activity was observed with combinations of different metal ions. This suggests that PA‐Nter requires two metal ions to perform RNA cleavage. The possibility was raised that binding of the second ion is stabilized when an RNA substrate or inhibitor is present.51, 57
C. The Heterotrimeric Influenza Polymerase Complex
The crystal structure of the large (∼260 kDa) polymerase complex was first resolved in 2014 by Cusack and co‐workers,23, 24 who succeeded to achieve high resolution (2.7 Å) structures of the polymerase heterotrimer, in complex with the vRNA promoter and originating from bat influenza A (FluA; Fig. 5A) or human influenza B (FluB; Fig. 5B) virus. The crystal structures of the apo influenza C polymerase complex (FluC; without bound promoter; Fig. 5C),58 and FluB polymerase in complex with a 5ʹ cRNA fragment (Fig. 5D),56 were revealed more recently.
Figure 5.
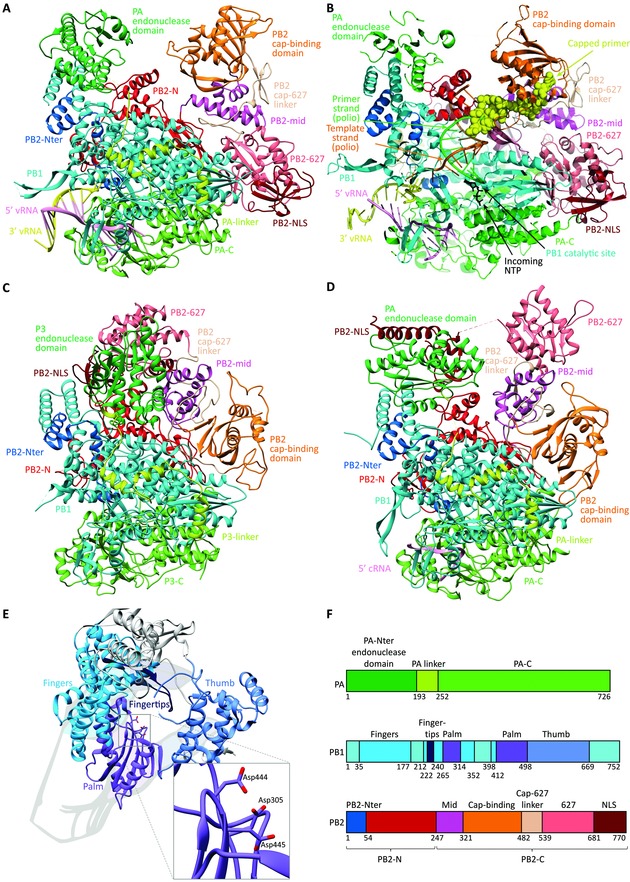
Comparison of the crystal structures of the heterotrimeric influenza polymerase complex containing full‐length PA, PB1, and PB2. The models are shown in the same orientation, and the same coloring was applied for the different subdomains. (A) Bat FluA polymerase with bound vRNA promoter [PDB: 4WSB].23 (B) Superposition model of the FluB polymerase crystal structure with a template–primer (orange–green) duplex and incoming NTP (black) (taken from a poliovirus polymerase crystal structure). The yellow spheres represent the capped primer bound to PB2, after cleavage by the PA endonuclease domain. This primer is now directed toward the PB1 catalytic cavity, where primer elongation occurs. (Adapted by permission from Macmillan Publishers Ltd: Nature, Reich et al.,24 copyright 2014.) (C) Influenza C polymerase (PDB: 5D98) in apo form.58 (D) FluB polymerase structure with bound cRNA 5′ end56 (PDB: 5EPI). (E) Domain arrangement of FluB PB1, illustrating the right‐hand‐like polymerase fold. The inset shows a closeup of the PB1 catalytic residues, which coordinate two divalent metal ions (not shown). (F) Subdomain names and color scheme as applied in panels A–E, based on the FluB polymerase numbering. For clarity, the PB1 subunit is colored uniformly in cyan in panels A–D, while its different subdomains are differentiated in panel E.
A first striking observation is the complex intertwining of the three subunits, which results in vastly more extensive intersubunit interactions than previously assumed (see other review articles13, 59 for the literature until 2013). Equally intriguing are some prominent conformational differences among the different crystal structures, which most likely represent functionally different states of the enzyme complex. Third, these crystallization studies are the first to reveal the architecture of the PB1 active site. These three insights undoubtedly create several new avenues for structure‐aided drug design.
The polymerase body is formed by PB1, enclosed by the PA linker (which connects the N‐ and C‐terminal domains of PA) on one side, and the N‐terminal domain of PB2 (PB2‐N) at the other side. Akin to other RNA‐dependent RNA‐polymerase (RdRp) enzymes, the overall structure of the catalytic core of PB1 resembles a right hand (Fig. 5E). It contains fingers, fingertips, palm, and thumb domains, formed mainly by conserved RdRp motifs designated “pre‐A”/F, A, B, C, D, and E. By comparison with other polymerase structures, three conserved aspartic acids (Asp305, Asp444, and Asp445 in FluB) within the active site of PB1 were identified, which have a crucial role in catalysis60 and together coordinate two divalent metal ions (Fig. 5, inset panel E).
The promoter‐bound FluA and FluB polymerase complexes have a similar “open” conformation, a U‐shaped structure (Fig. 5A and B), in which one arm is formed by PA‐Nter and the other by the PB2‐CBD. These domains face each other in the FluA structure, suggesting that this crystal represents the cap‐snatching mode. In the FluB structure, the PB2‐CBD is rotated by 70° toward the PB1 active site where primer elongation takes place (compare Fig. 5A and B, in orange). Hence, this FluB structure is thought to represent the cap‐dependent priming step. In sharp contrast, in the crystal structures of the apo FluC polymerase (i.e., without bound promoter58; Fig. 5C) and the 5ʹ cRNA‐bound FluB polymerase (Fig. 5D),56 the polymerase adopts a “closed” conformation in which cap‐snatching is disfavored; it is not yet clear whether these structures represent an inactive or “only replication‐competent” state. Several interdomain interactions immobilize the PB2‐CBD (Fig. 5C and D, in orange) and PA‐Nter (which is named P3 in the case of influenza C) is repositioned and packed against the PB2‐NLS‐domain (Fig. 5C and D, in maroon). This pronounced difference between the open and closed forms of the polymerase complex is related to a radical in situ rearrangement of a large part of PB2. Hence, the regulatory role of the vRNA promoter, as evidenced by enzymatic studies,61, 62 may be related to triggering the transition from the closed preactivation state to the active transcription state. The crystallization studies further indicated that the influenza polymerase complex contains several hinge regions that provide high flexibility to enable its multiple roles in viral RNA synthesis.56 Stabilization of one of its conformations by an inhibitor, such that a vital rearrangement cannot take place, would be an excellent approach to inhibit the polymerase complex.
D. Recent Structural Insights: A First Step Toward Solving Some Unknowns
The recent crystallographic analyses also help to interpret some issues that are already debated since many years.13 A first issue with relevance for inhibitor design is related to the potential substrate or sequence specificity of the cap‐snatching reaction. The observation that the capped host RNAs is cleaved at a distance of 10–15 nucleotides from the 5ʹ‐cap can be explained by the 50 Å distance between the cap‐binding and endonuclease domains in the “open” FluA structure.23 Regarding the type of RNA substrate, the previous assumption that only host (pre‐)mRNAs are targeted, is contradicted by recent data that noncoding RNAs are the primary source of capped primers.63, 64, 65 The evidence for sequence preference is inconclusive,19, 20, 42, 52, 62, 66, 67, 68 but recent deep sequencing studies63, 69 indicate a preference for capped primers ending with a purine residue. The selection could be determined20, 70, 71, 72 (i) at the level of RNA cleavage and/or (ii) transcription initiation when requiring base‐complementarity between the 3ʹ end of the vRNA template and a given cellular RNA. To gain insight into the first selection mechanism, a cocrystal of PA‐Nter (or the entire polymerase complex) with an RNA substrate is required. This could elucidate substrate‐dependent changes in the active site of PA‐Nter, with relevance for design of optimized PA inhibitors. One indication in favor of such base preference comes from crystallization studies of PA‐Nter in complex with a ribomononucleotide, which proved successful for uridine 5ʹ‐monophosphate (UMP)44, 46, 49 (Fig. 6H–J) and adenosine 5ʹ‐monophosphate (AMP),44 but not cytidine 5ʹ‐monophosphate (CMP) and guanosine 5ʹ‐monophosphate (GMP).44, 46
Figure 6.
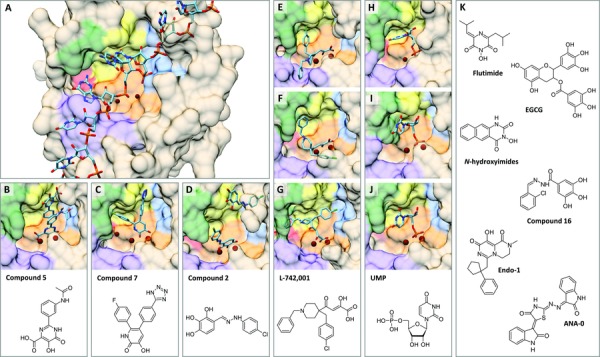
Comparison of the predicted orientation of RNA versus that of different ligands within the PA‐Nter active site; and chemical structures of representative PA inhibitors. (A) Hypothetical disposition of the RNA substrate in PA‐Nter with two bound Mg2+ ions, based on the molecular dynamics model (kindly provided by Dr. M. Alfonso‐Prieto), as performed by Xiao et al.57 The 3ʹ end of the mRNA is at the right‐hand side, while its 5ʹ end is located at the left. (B–D) Crystal structures of PA‐Nter in complex with (B) “compound 5”45 (PDB: 4E5J); (C) “compound 7”50 (PDB: 4M5U); or (D) “compound 2”73 (PDB: 5I13).
(E–G) Position of L‐742,001, as (E–F) determined by cocrystallization (E, PDB: 4E5H45 and F, PDB: 5CGV49) or (G) predicted by docking.74 (H–J) X‐ray structures of PA‐Nter with bound UMP, as determined by (H) Zhao et al.44 (PDB: 3HW3), (I) Kowalinski et al.46 (PDB: 4AWH), and (J) Song et al.49 (PDB: 5CL0). In the latter two crystal structures, the UMP‐binding mode fully overlaps. The binding regions in PA‐Nter are colored—in orange: metal binding and catalytic residues (His41, Glu80, Asp108, Glu119, Ile120, and Lys134); in yellow: pocket P1 (Ala37, Ile38, Leu42, and Lys34); in blue: pocket P2 (Thr40, Val122, Arg124, Tyr130, and Phe150); in purple: pocket P3 (Arg84, Trp88, Phe105, and Leu106); in red: pocket P4 (Leu16 and Gly81); in green: pocket P5 (Ala20, Tyr24, and Glu26). The bound molecules (i.e., RNA‐substrate or ligand) are shown in cyan, while the metal ions are colored dark red. (K) Chemical structures of flutimide,75 EGCG,76 N‐hydroxyimides,77 compound 16,78 Endo‐1,38 and ANA‐0.79
Substrate selection at the level of initiation is corroborated by the polymerase crystal structures, more specifically the model that was proposed24 (Fig. 5B) by superposition of the influenza B PB1 structure on the poliovirus polymerase primer–template complex.80 This model suggests that the 3ʹ end of the vRNA would pass the PB1 active site by three nucleotides, creating a template overhang that could form three base pairs with the capped primer.
A second intriguing issue is how the different conformations captured in the crystal structures of the heterotrimeric polymerase complex, relate to the cis/trans RNA synthesis model81 for which experimental support was provided by the cryo‐electron microscopic analyses of vRNPs.17, 18 Namely, the branched arrangement of the RNP complexes in which a smaller nascent RNP seems to bud from a larger full‐length RNP, indicate that genome replication is performed in trans by a second polymerase heterotrimer. On the other hand, vRNA transcription would be performed by the cis‐acting polymerase that resides in the vRNP complexes. Given the high flexibility of the polymerase heterotrimer (see above), it is plausible that structural rearrangements occur in each of its functional states which, furthermore, might also be regulated by specific host cell factors.
3. STRATEGIES TO INTERFERE WITH THE INFLUENZA VIRUS POLYMERASE, THE NP, OR AN ASSOCIATED HOST CELL FACTOR
A. General Reflections
In the following section, we describe the diverse strategies that have been explored to block influenza virus RNA synthesis, from the stage of rational design or serendipitous screening, to unravelling the presumed mechanism of action (which includes analysis of the resistance profile), and evaluation in cell culture or mouse models, or, if appropriate, in clinical trials (see Table I for a concise overview). We added in this table some published antiviral activity data, in the awareness that these values may show large variations depending on assay conditions, for instance related to virus input, incubation time, or the parameters used to monitor inhibition of virus replication or cytotoxicity of the test compounds. In our own experience, in vitro evaluation based on cytopathic effect (CPE) reduction is more stringent compared to plaque reduction or virus yield assays. In the latter approach, a reduction in virus titer of at least two‐log10 is required to conclude that a given compound has meaningful activity. For this reason, we marked with an asterisk the published compounds for which the anti‐influenza effect in cell culture has been confirmed in at least two independent studies. We also mentioned the published resistance mutations. Resistance studies help to reveal the precise interaction with the viral target and represent the gold standard to validate the proposed mechanism of action of any new class of antiviral molecules.
Table I.
Overview of Published Approaches to Interfere with the Influenza Virus Polymerase, the Nucleoprotein or a Host Factor Involved in Viral RNA Synthesis
| Compound name or code | Proposed mechanism of actiona | Antiviral activity in cell cultureb | Known spectrum of influenza virus activityc | Published resistance mutations | References |
|---|---|---|---|---|---|
| Inhibitors targeting PB1 (directly or indirectly) | |||||
| Nucleoside and nucleobase analogues | |||||
| T‐705 (in Phase 3/approved in Japan) | Alternative substrate for viral polymerase; chain termination and lethal mutagenesis | 0.1–10 μM82, 83, 84[*] | A, B, and C | PB1: V43I85 | 82, 83, 84, 85 |
| Ribavirin | Alternative substrate for viral polymerase; lethal mutagenesis; IMPDH inhibitor | 2.5–37 μM86, 87, 88; 2–3 log10 reductions in virus titer at 10 μM89 [*] | A, B, and C | PB1: V43I85 and D27N90 | 85, 86, 87, 88, 89, 90 |
| Prodrug 2 | Ribavirin prodrug | 0.5 μM87 | A | 87 | |
| Viramidine | Ribavirin prodrug | 8.6–132 μM88 | A and B | 88 | |
| 5‐Azacytidine | Alternative substrate for viral polymerase; lethal mutagenesis | 2–3 log10 reductions in virus titer at 10 μM89 | A | 89 | |
| 5‐Fluorouracil | Alternative substrate for viral polymerase; lethal mutagenesis | 2–3 log10 reductions in virus titer at 80 μM89 | A | 89 | |
| EICAR | IMPDH inhibitor | 1.5–8.6 μM91 | A and B | 91 | |
| ETCAR | IMPDH inhibitor | 1.2–19 μM92 | A and B | 92 | |
| Selenazofurin | IMPDH inhibitor | 0.7–1.2 μM93 | A and B | 93 | |
| Pyrazofurin | IMPDH inhibitor | 0.24–0.88 μM94 | A, B, and C | 94 | |
| LY217896 | IMPDH inhibitor | 2.9–12 μM95 | A and B | 95 | |
| Compound 3c (analogue of T‐705) | Alternative substrate for viral polymerase | 1.9 μM96 | A | 96 | |
| Compound 8a (analogue of T‐705) | ? | 5.6–7.4 μM97 | A | 97 | |
| 2ʹ‐Deoxy‐2ʹ‐fluoroguanosine | Alternative substrate for viral polymerase; nonobligate chain termination | 2–22 μM98, 99 [*] | A and B | ?d | 98, 99 |
| 2ʹ‐Substituted carba‐nucleoside analogues “6” and “10” | ? | 0.9–51 μM100 | A and B | 100 | |
| C‐3ʹ‐modified analogues | ? | 3.7–65 μg/ml102 | A and B | 102 | |
| 6‐Methyl‐7‐substituted‐7‐deaza purine nucleoside analogues “5x” and “5z” | ? | 3.6–7.0 μM103 | A | 103 | |
| Protide 9j | 2ʹ‐Deoxy‐2ʹ‐fluoroguanosine prodrug | EC99 = 12 μM104 | A | 104 | |
| 2′‐Deoxy‐2′‐fluorocytidine | Alternative substrate for RNA synthesis?; immunomodulator? | 0.05–8 μM105 | A and B | 105 | |
| 2ʹ‐2ʹ‐Difluorodeoxycytidine | Interferes with pyrimidine nucleotide synthesis | <0.032–1.2 μM105 | A and B | 105 | |
| Protide 23a | 2′‐Deoxy‐2′‐fluorouridine prodrug | EC99 = 49 μM106 | A | 106 | |
| Compound A3 | Interferes with pyrimidine nucleotide synthesis | 0.04–1 μM107 | A and B | 107 | |
| N10169 | Interferes with pyrimidine nucleotide synthesis | 3 μM108 | B | 108 | |
| Carbodine | Interferes with pyrimidine nucleotide synthesis | 0.6–36 μM94, 109, 110 [*] | A, B, and C | 94, 109, 110 | |
| Cyclopentenyl cytosine | Interferes with pyrimidine nucleotide synthesis | 9.6–26 μM94 | A, B, and C | 94 | |
| 3‐Deazaguanine | IMPDH inhibitor | 14–49 μM94 | A, B, and C | 94 | |
| Nonnucleoside analogues | |||||
| “367” | ? | 0.5 μM111 | A | PB1: H456P111 | 111 |
| ASN2 | ? | 3‐14 μM112 | A and B | PB1: Y499H112 | 111 |
| Inhibitors targeting PB2 | |||||
| VX‐787 (in Phase 2) | Inhibits cap‐binding | 0.32–2.8 nM37 | A | PB2; cell culture: Q306H, S324I/N/R; F404Y and N510T113; patients: M431I11 | 11, 37, 113 |
| Cap‐3 and Cap‐7 | Inhibit cap‐binding | 1.17–9 μM114 | A | 114 | |
| Inhibitors targeting PA | |||||
| AL‐794 (in Phase 1) | Endonuclease inhibitor | Undisclosed | |||
| S‐033188 (in Phase 2) | Endonuclease inhibitor | Undisclosed | |||
| L‐735,882 and analogues | Metal‐chelating endonuclease inhibitor | 0.18–1.6 μM115, 116 | A and B | 115, 116 | |
| L‐742,001 | Metal‐chelating endonuclease inhibitor | 0.35116 to 11117 μM [*] | A and B | PA: T20A,49, 74, 117 H41A,74 L42T,74 I79L,49 G81F/T/V,74 F105S,49 E119D,49 I120T,74 V122T74 | 49, 74, 116, 117 |
| Flutimide | Metal‐chelating endonuclease inhibitor | 5.9 μM75 | A and B | 75 | |
| Tetramic acid “36” | Metal‐chelating endonuclease inhibitor | 21 μM118 | A | 118 | |
| Phenethylphenylphthalimide analogs PPT‐65 and PPT‐66 | Metal‐chelating endonuclease inhibitor | 26–48 μM119 | A | 119 | |
| Carboxamides “42,” “44,” “45” | Metal‐chelating endonuclease inhibitor | 13–19 μM101 | A | 101 | |
| Pyrimidinol “26” | Metal‐chelating endonuclease inhibitor | 13 μM45 | A | 45 | |
| Hydroxypyridinone “7” | Metal‐chelating endonuclease inhibitor | 11 μM50 | A | 50 | |
| Hexanetetrone “4” and benzohydrazide “16” | Metal‐chelating endonuclease inhibitor | 18–23 μM78 | A | 78 | |
| (Tri)hydroxyphenyls “1,” “2,” and “3” | Metal‐chelating endonuclease inhibitor | 11–14 μM120 | A | 120 | |
| Dihydroxyindoles “10” and “15” | Metal‐chelating endonuclease inhibitor | EC99 = 5.7–12 μM121 | A | 121 | |
| THC19 | ? | 31–45 μM122 | A | PA: V44I and E165D122 | 122 |
| Aptamer PAN‐2 | ? | About 10 nM123 | A | 123 | |
| Pyridopiperazinediones Endo‐1, Endo‐8, and Endo‐9 | Metal‐chelating endonuclease inhibitor | 0.39–2 μM114 | A | 114 | |
| ANA‐0 | Endonuclease inhibitor | 0.8–4 μM79 | A | 79 | |
| PPI inhibitors designed against the PA‐PB1 interface | |||||
| Benzofuran derivatives | Inhibit PA‐PB1 assembly | 1–60 μM124, 125 | A | 124, 125 | |
| Benzbromarone and diclazuril | Inhibit PA‐PB1 assembly | 31–39 μM126 | A | 126 | |
| “1” and AL18 | Inhibit PA‐PB1 assembly | 8.3–23 μM127, 128 | A and B | 127, 128 | |
| Cycloheptathiophene‐3‐carboxamides “6” and “19” | Inhibit PA‐PB1 assembly | 15–28 μM129 | A and B | 129 | |
| Thiophene‐3‐carboxamides “7,” “10,” “’18,” and “19” | Inhibit PA‐PB1 assembly | 11–43 μM130 | A and B | 130 | |
| Triazolopyrimidines “16,” “31,” “’36,” and “37” | Inhibit PA‐PB1 assembly | 5–51 μM131 | A and B | 131 | |
| 4,6‐Diphenyl pyridines “1,” “11,” “15” | Inhibit PA‐PB1 assembly | 7.3–26 μM132 | A | 132 | |
| ANA‐1 | Inhibit PA‐PB1 assembly | 0.09–1.2 μM133 | A | 133 | |
| Inhibitors targeting NP | |||||
| Nucleozin | Inhibits cytoplasmic trafficking of vRNPs | 0.069–0.33 μM134 [*] | A | NP: Y52H/C,111, 135 Y289H,134, 135 N309K/T,134, 135, 136 | 111, 134, 135, 136 |
| PPQ‐581 | Inhibits cytoplasmic trafficking of vRNPs | 1 μM137 | A | NP: S377G137 | 137 |
| “3,” “7,” “’12,” and “23” | Disrupts NP dimerization by targeting the E339…R416‐salt bridge | 1.7–118 μM138 | A | 138 | |
| Naproxen | Blocks the RNA‐binding groove of NP | 11–25 μM139 | A | 139 | |
| F66 | Blocks the RNA‐binding groove of NP | ∼1–5 μM140 | A | 140 | |
| RK424 | Inhibits nuclear export of NP; inhibits NP–RNA, and NP–NP interactions | 0.40–0.63 μM141 | A | 141 | |
| Mycalamide analog “4” | Inhibits nuclear transport of NP? | 59–97% reduction of plaque formation at 32 μM142 | A | 142 | |
| Ingavirin (approved in Russia) | Diverse effects on NP functioning? | Inconsistent143, 144, 145 | A and B | 143, 144, 145 | |
| Other | |||||
| AVI‐7100 (in Phase 1) | Inhibition of M‐gene translation by gene silencing | 10–20 μM146 | A | 146 | |
| KPT‐335 (in Phase 1) | CRM1 inhibition | 0.01–0.42 μM147 | A and B | 147 | |
| BPR3P0128 | Inhibitor of cap‐binding | 0.05–0.19 μM148 | A and B | 148 | |
| DPQ | Inhibition of RNA synthesis by binding to 5′ vRNA promoter | 72–276 μM149 | A and B | 149 | |
| Geldanamycin, 17‐AAG, and 3beta‐acetoxydeoxodihydrogedunin | Hsp90 inhibition | 1–2 log10 reductions in virus titer150, 151 [*] | A | 150, 151 | |
| U0126 | MEK inhibition | 1.2–141 μM152, 153 [*] | A | 152, 153 | |
| PD‐0325901, AZD‐6244, AZD‐8330, and RDEA‐119 | MEK inhibition | 0.005–0.75 μM154 | A | 154 | |
| WV970 | Kinase inhibition | 0.015–0.24 μM155 | A and B | 155 | |
| Acetylsalicylic acid, pyrrolidine dithiocarbamate, SC75741 | NF‐κB inhibition | 1–4 log10 reductions in virus titer156, 157, 158 | A | 156, 157, 158 | |
| Bay 11–7082 | NF‐κB inhibition | 40–85% reduction in virus titer at 15 μM159 | A | 159 | |
| NSC23766 | Rac1 GTPase inhibition | 22 μM160 | A and B | 160 | |
For compounds with broader antiviral activity, only the mechanism of action proposed for influenza virus is given. Note that for many of the experimental compounds, the antiviral mode of action in cell culture is still unproven (e.g., because no resistance data are thus far available).
Unless stated otherwise, the EC50 value is given. The [*] annotation indicates that the antiviral activity was reported in two or more independent publications.
The information in this column indicates only the clearly defined activity spectrum. For most compounds, only a limited number of influenza viruses was tested, and hence, the activity spectrum was not (yet) specified.
During compound testing, influenza B virus is sometimes ignored, although it accounts for about 25% of seasonal influenza cases. The protein sequences of PA, PB1, and PB2 are, overall, highly conserved among influenza A and B viruses, but even subtle amino acid differences in a binding pocket may render an inhibitor inactive against either of the two virus types. Several inhibitor classes described below were designed to interact with critical functional residues (for instance crucial for binding the capped RNA or NTP substrates). Still, high inhibitor binding affinity often requires involvement of adjacent and less conserved sites, which can give influenza A or B specific activity, or a reduced barrier for the virus to acquire resistance. In other words, although the viral polymerase complex is, generally speaking, an excellent target for developing broad influenza A and B inhibitors that do not readily select for resistance, this theoretical assumption requires biological verification for every single class of new inhibitors.
Besides application in seasonal influenza infections, antiviral drugs are essential to combat serious zoonotic infections (in particular, by highly pathogenic avian influenza A viruses). This seems less of a concern for inhibitors that directly interact with active domains in PA, PB1, or PB2, since these regions are highly conserved among human and avian influenza A viruses. On the other hand, there are species‐dependent amino acid differences in some parts of the protein complex, supposedly related to a regulatory role for host cell factors such as ANP32A,29, 162 RNA polymerase II,163 or nuclear import or export proteins.14 The insights into the influenza virus host interactome are rapidly growing164 and will hopefully rationalize the concept of host cell based antiviral therapeutics. As of today, some approaches (described in Section 3.,H) have already been proposed, although their mechanistic details remain to be established.
B. Nucleoside and Nucleobase Analogue Inhibitors
1. Ribavirin and Structurally Related Carboxamide Analogues
Ribavirin [1‐(β‐d‐ribofuranosyl)‐1,2,4‐triazole‐3‐carboxamide; Fig. 7A] was already discovered in 1972.86 This nucleoside analogue inhibits many diverse RNA viruses including influenza virus. Ribavirin has been a first‐line therapeutic for hepatitis C virus infections, but its clinical utility in the management of influenza infections seems limited. In clinical trials performed in the 1970s and 1980s, ribavirin was found to be ineffective against experimentally induced influenza,165 though some benefit was provided at higher drug doses,166 and in vivo data support its potential usefulness in combination therapy.167 Although it has severe side effects (e.g., hemolytic anemia) and teratogenic properties,168 ribavirin may be a last resort for clinicians encountering rare cases of multidrug‐resistant influenza viruses.169
Figure 7.
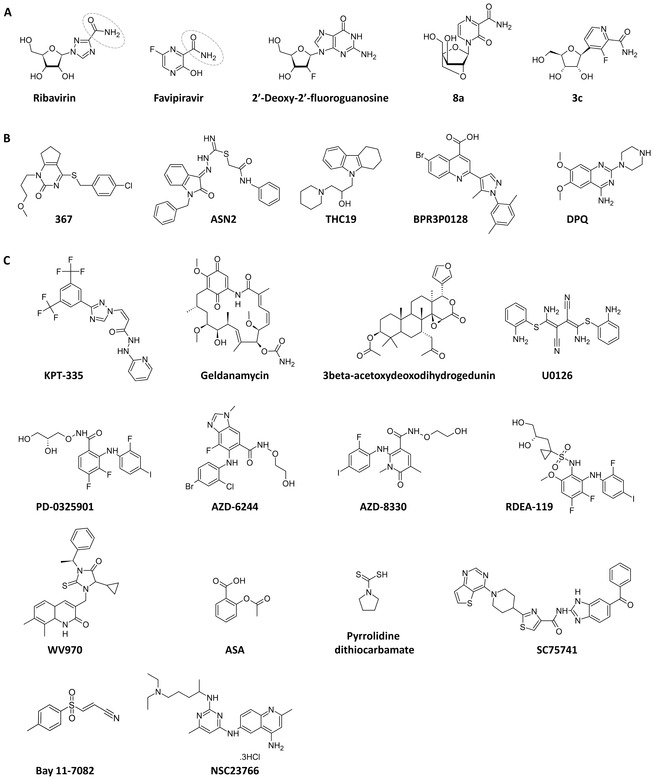
Chemical structures of proven or tentatively proposed polymerase inhibitors. (A) Nucleoside or nucleobase analogue inhibitors ribavirin,86 favipiravir82 (T‐705), 2ʹ‐deoxy‐2ʹ‐fluoroguanosine98 (2ʹ‐FdG), “8a,”97 and “3c.”96 The rotating carboxamide moiety (encircled) in ribavirin and favipiravir explains their ambiguous base pairing, since their base part mimics guanine as well as adenine. (B) Compounds for which the detailed mechanism of action at the level of the polymerase has not yet been revealed. (C) Compounds targeting a host cell factor that has been linked to viral polymerase function. See Table I for references on the individual compounds.
The mode of action of ribavirin is rather complex, since it appears to inhibit virus replication through a combination of different mechanisms. After phosphorylation by adenosine kinase170 or cytosolic 5ʹ‐nucleotidase II,171 ribavirin 5ʹ‐monophosphate inhibits the cellular enzyme inosine monophosphate dehydrogenase (IMPDH), resulting in a decrease in the intracellular GTP pool and, hence, indirect inhibition of viral RNA synthesis.172, 173, 174 Other proposed mechanisms include direct inhibition of the viral polymerase by ribavirin 5ʹ‐triphosphate,175, 176 immunosuppression,177 and lethal virus mutagenesis.85 It was recently demonstrated that ribavirin induces mutagenesis in the influenza virus genome by acting as an ambiguous purine analogue and increasing G‐to‐A and C‐to‐U mutations.85 Serial virus passaging in the presence of the compound gave rise to a mutant virus with twofold resistance to ribavirin and a lower degree of resistance to favipiravir (described below). This mutant virus carried mutation V43I in PB1, which was associated with increased polymerase fidelity and reduced pathogenicity in mice.85 This Val43 residue lies in close proximity of the putative NTP entrance channel, as predicted in the polymerase crystal structure of Pflug et al.,23 which could explain how the V43I mutation may affect the activity of ribavirin, for instance by modifying the interaction of the polymerase with GTP or ribavirin 5ʹ‐triphosphate. The last analysis is difficult to reconcile with the observation that ribavirin 5ʹ‐triphosphate is a rather weak inhibitor of the viral polymerase in enzymatic assays, with reported IC50 values of 70 μM173 or ∼100 μM.176 By using random mutagenesis, Binh et al.90 identified PB1 mutation D27N, which conferred a 1.8‐fold resistance to ribavirin. This mutation was also picked up by Pauly et al. after serial passaging of influenza virus under ribavirin.89 By inspecting the published crystal structure,23 we noticed that Asp27 is located outside the catalytic site, in the vicinity of the 5ʹ vRNA promoter binding site. Pauly et al. further confirmed that ribavirin and two other nucleoside analogues, 5‐azacytidine and 5‐fluorouracil, are lethal mutagens for influenza virus in vitro.89
With the aim to improve the efficacy or safety, Dong et al. obtained an alkoxyalkylphosphodiester prodrug of ribavirin, prodrug “2,” which still requires in vivo validation.87 Viramidine, the 3‐carboxamidine prodrug of ribavirin,178 was found to be slightly less active against influenza virus in vitro and in vivo, but also showed less toxicity.88 In addition to ribavirin, the following carboxamide‐containing nucleoside analogues exhibit broad antiviral activity, presumably by inhibition of IMPDH: 5‐fluoro‐1‐β‐d‐ribofuranosylimidazole‐4‐carboxamide (FICAR),179 5‐ethynyl‐1‐β‐d‐ribofuranosylimidazole‐4‐carboxamide (EICAR),91 5‐ethynyl‐1‐β‐d‐ribofuranosyl‐1H‐[1–3]triazole‐4‐carboxylic acid amide (ETCAR),92 selenazofurin,93 and pyrazofurin.94 All these are cytostatic molecules mainly explored as anticancer agents.180 The substituted thiadiazole compound LY217896 was shown to display broad anti‐influenza virus activity95 via IMPDH inhibition,181 but proved ineffective in a placebo‐controlled clinical trial.182
2. Favipiravir
Favipiravir (6‐fluoro‐3‐hydroxy‐2‐pyrazinecarboxamide; Fig. 7A), also known as T‐705, was approved in Japan in March 2014 for restricted use in uncomplicated influenza virus infections, and is currently in Phase 3 clinical trials in the USA and Europe. Unpublished clinical data suggest that the antiviral effects of favipiravir are similar to those of oseltamivir.183 In one reported Phase 2 study,11 favipiravir significantly reduced the time to resolution of symptoms. No signs of drug resistance were seen in more than 700 samples tested.
Favipiravir has broad activity against influenza A, B, and C viruses,82 including the 2009 pandemic A/H1N1 virus,184 highly pathogenic avian influenza H5N1185 and H7N9186 viruses, and virus strains with resistance to M2 blockers or NAIs.187 Among all nucleobase/nucleoside inhibitors of influenza virus reported thus far, favipiravir is absolutely unique in having a clearly superior therapeutic window (i.e., antiviral EC50 value in the range of 0.1–10 μM82, 83, 84 and no cytotoxicity at 6400 μM82).
Mechanistically, favipiravir is a nucleobase mimetic that undergoes intracellular conversion to its ribofuranosyl 5ʹ‐triphosphate metabolite. The cellular hypoxanthine guanine phosphoribosyltransferase (HGPRT) converts favipiravir into its ribose‐5′‐monophosphate (RMP), which is further metabolized, by cellular kinases, to favipiravir‐ribosyl‐5′‐triphosphate (favipiravir‐RTP).83 Its requirement for very high dosing (in the order of 1600–2400 mg per day in some clinical trials) may, at least partially, be related to the low efficiency of its metabolic activation.83 This limitation could be solved by designing prodrug forms of favipiravir that bypass one or more of its activation steps.
Favipiravir‐RTP is recognized by influenza virus RNA polymerase as an alternative for the natural substrates GTP and, to a lesser degree, ATP.188, 189, 190 The precise mechanism of action of favipiravir remains to be fully explained, and two nonmutually exclusive hypotheses have been proposed. Similarly to what is described above for ribavirin, favipiravir can cause lethal virus mutagenesis by inducing a high rate of mutations and generating a nonviable viral phenotype. In virus grown under favipiravir, Baranovich et al.84 observed a reduction in virus infectivity without a corresponding decrease in the number of viral RNA copies, together with a dose‐dependent increase in mutation frequency in the influenza virus genome (primarily G‐to‐A and C‐to‐U). The biochemical basis for this mutagenic effect was revealed in the enzymatic studies by Jin et al.,189 since the influenza polymerase was shown to efficiently incorporate favipiravir‐RTP both opposite to C and U, meaning that favipiravir‐RTP mimics GTP as well as ATP. This ambiguous base‐pairing behavior is related to the rotating carboxamide moiety (encircled in Fig. 7A). The second hypothesis, “non‐obligate chain termination,” is supported by the biochemical observation that incorporation of a single molecule of favipiravir‐RMP (which carries a normal 3ʹ‐hydroxyl group) into a nascent influenza RNA strand causes inhibition of viral RNA extension.190 However, another study did not confirm these results and concluded that at least two consecutive incorporation events of favipiravir‐RMP are needed to terminate viral RNA elongation.189 Hence, the RNA chain‐terminating effect may prevail at higher favipiravir concentrations, while at lower compound concentrations, the mutagenic effect may become apparent. Interestingly, the compound appears to have an exceptionally high barrier for selecting resistance, since no favipiravir‐resistant influenza virus was obtained in cell culture after up to 30 serial virus passages in the presence of favipiravir.84, 191 From a drug development perspective, this quality is of course clearly advantageous. On the other hand, it obstructs experiments aimed at providing conclusive evidence on favipiravir's mechanism of action.
Next to its activity against influenza virus,82 favipiravir inhibits the replication of various other RNA viruses.192 Until now, few structural analogues of favipiravir have been reported. The 6‐fluoro substituent is not required for antiviral activity,83, 191 whereas the 3‐hydroxyl group is indispensable.83 An example of a base‐modified analogue is “3c” (Fig. 7A), which displayed comparable antiviral activity as T‐705 in cell culture and influenza polymerase enzymatic assays.96 A recent example of a sugar‐modified analogue is the 2ʹ,4ʹ‐bridged analogue “8a” (Fig. 7A), which has anti‐influenza virus activity comparable to that of favipiravir.97 This compound has the 3ʹ‐hydroxyl of the pentose in the inverted xylo position. Evaluation of “8a” (or, rather, its 5ʹ‐triphosphate) at the level of the viral polymerase was not yet reported.
3. 2ʹ‐Deoxy‐2ʹ‐Fluoroguanosine and Other Nucleoside Analogues
2ʹ‐Deoxy‐2ʹ‐fluoroguanosine (2ʹ‐FdG; Fig. 7A) was reported years ago as a broad inhibitor of influenza A and B viruses in cell culture.98 Its analogues 2ʹ‐deoxy‐2ʹ‐fluoroadenosine and 2ʹ‐deoxy‐2ʹ‐fluoroinosine were significantly less active.99 In vivo, 2ʹ‐FdG was shown to reduce influenza virus titers in the respiratory tract of mice and ferrets.193 Mechanistically, 2ʹ‐FdG‐triphosphate was found to inhibit the influenza polymerase complex by nonobligate chain termination. This agrees with the observation that a virus selected for resistance to 2ʹ‐FdG, contained a polymerase with tenfold lower susceptibility to 2ʹ‐FdG‐triphosphate in an enzymatic assay.161 The identity of the amino acid changes in this mutant polymerase was not disclosed, which is unfortunate since this insight could be very helpful to explain the role of specific residues in the catalytic or other functional domain of PB1, as identified in the recent crystallographic studies.23, 24, 56, 58
During more recent years, 2ʹ‐substituted carba‐nucleoside analogues,100 C‐3ʹ‐modified analogues,102 and 6‐methyl‐7‐substituted‐7‐deaza purine nucleoside analogues 103 were reported to have anti‐influenza activity comparable to 2ʹ‐FdG. The antiviral activity of 2ʹ‐FdG and analogues is dependent on intracellular conversion to the active nucleoside 5ʹ‐triphosphate form. ProTide prodrugs were employed to overcome the first (rate‐limiting) phosphorylation step, and enable intracellular delivery of the nucleoside 5ʹ‐monophosphate species.194 This ProTide concept was successfully applied to 2ʹ‐FdG and its uridine analogue 2ʹ‐FdU.104, 106
The pyrimidine analogue 2ʹ‐deoxy‐2ʹ‐fluorocytidine (2ʹ‐FdC) seems more potent than 2ʹ‐FdG, with in vitro and in vivo activity against various strains of influenza A or B.105 It remains to be demonstrated whether the 5ʹ‐triphosphate of 2ʹ‐FdC acts as an alternative substrate for the influenza polymerase complex, as is the case for 2ʹ‐FdG. Alternatively, since 2ʹ‐FdC was shown to be cytostatic in cells,195 it could act as an immunomodulator in vivo, or inhibit influenza virus through inhibition of cellular enzymes which are involved in de novo pyrimidine biosynthesis. Similar to what is explained above for ribavirin and other inhibitors of IMPDH, nucleoside analogues that interfere with purine or pyrimidine nucleotide synthesis can give strong inhibition of influenza virus replication in cell culture. Unfortunately, the therapeutic window of this approach is too narrow to allow broad application in influenza virus therapy. A few examples are 2ʹ‐2ʹ‐difluorodeoxycytidine 196 (known as the anticancer drug gemcitabine), compound “A3,”107 and N10169,108 which lower the intracellular pyrimidine levels by inhibiting ribonucleotide reductase, dihydroorotate dehydrogenase, and orotidylate decarboxylase, respectively. Also, the anti‐influenza activity of the carbocyclic nucleoside analogues carbodine and cyclopentenyl cytosine has been linked to pyrimidine depletion through CTP‐synthetase inhibition.94, 197, 198, 199 For a few carbocyclic purine nucleoside analogues with borderline to modest activity against influenza virus, no mechanistic data are available.200 For the broad antiviral agent 3‐deazaguanine, a mechanism involving IMPDH inhibition was proposed.94, 201
4. Time to Revisit Nucleoside Inhibitors for Influenza?
That nucleoside analogues merit more attention in influenza drug development is nicely illustrated by the successful progression of T‐705. The unprecedented bonuses of this agent are: high resistance barrier, low cytotoxicity, and broad coverage of diverse RNA viruses. As for 2ʹ‐FdG, the relevance of the 2ʹ‐fluoro modification to achieve nonobligate chain terminators of RNA virus polymerases202 is underscored by the fact that this substitution is present in successful drugs for hepatitis C203 or drug candidates for respiratory syncytial virus therapy.204 An important aspect is the avoidance of inhibitory effects on cellular polymerases or enzymes of the purine or pyrimidine pathways, since this will de facto reduce the therapeutic index. Besides this relevance for drug development, nucleoside analogues (or their 5ʹ‐triphosphate forms) are unique tools to study the kinetics or fidelity of the influenza polymerase, and the role of specific PB1 residues.
C. Inhibitors of the Cap‐Snatching Reaction
1. Inhibitors of Cap‐Binding by PB2
As explained in Section 2,A, the first part of the cap‐snatching reaction by the influenza virus polymerase involves cap‐binding by PB2. Initially, the fact that PB2 recognizes capped RNA alike eukaryotic cap‐binding proteins such as eIF4E,205 raised doubts about the target drugability of the PB2‐CBD and the possibility to design influenza virus‐selective cap‐binding inhibitors. However, Hooker et al.39 used quantitative UV crosslinking to analyze the elements in cap analogues that contribute to their recognition by isolated vRNPs and eIF4E, and identified some notable differences. Based on this, compound RO0794238 (Fig. 2F) was designed as a cap analogue lacking a negative charge and containing an acyclic moiety instead of a ribose, with the aim to achieve superior potency and selectivity for PB2. This molecule was indeed able to inhibit cap‐binding by influenza virus vRNPs in a dose‐dependent manner, with no effect on eIF4E.39
The revelation of the cocrystal structure of the PB2‐CBD in complex with m7GTP32 (Fig. 2A and Section 2,A) finally enabled the rational design of selective inhibitors, given its unique protein fold and cluster of hydrophobic residues. Four compounds (e.g., compound “11”; Fig. 2C) displayed potent activity in a binding assay with the isolated PB2‐CBD but, unfortunately, were devoid of antiviral activity in influenza virus‐infected cells (possibly due to the presence of multiple negative charges, resulting in poor cellular uptake).36 The previously identified compound RO079423839 however appeared inactive in this binding assay. The authors speculated that RO0794238 might inhibit cap‐binding by PB2 in an indirect manner through its interaction with another part of the viral RNP complex. In addition, given its long substitution at the N‐7 position, it seems implausible that RO0794238 would be able to bind in the m7GTP pocket of the PB2‐CBD.
The validity of the PB2‐CBD as a drug target is best demonstrated by the clinical candidate VX‐787 (JNJ‐63623872 or JNJ‐872; Fig. 2D). In a Phase 2a challenge study, VX‐787 yielded a dose‐dependent decrease in symptom scores and duration of symptoms.11 Additional clinical trials with this promising compound are ongoing. Its discovery process was reported in 2014 by Clark et al.37 VX‐787 is an azaindole derivative that resulted from extensive iterative synthesis to develop PB2 inhibitors, which optimally occupy the m7GTP binding pocket, as demonstrated by cocrystallizing VX‐787 with the PB2‐CBD of influenza A (Fig. 2D). In particular, optimal interactions with the hydrophobic as well as basic residues were achieved by introducing the bicyclooctane‐carboxylate moiety. A further structure‐activity relationship (SAR) exploration with isosteric replacements of the carboxylic group of VX‐787 showed that the pKa value and orientation of the negative charge significantly affect both anti‐influenza potency and selectivity for unwanted protein kinase targets (the latter being related to compound binding to the ATP site of cellular kinases).206 VX‐787 possesses strong antiviral activity (i.e., EC50 values in the nanomolar range) in cellular assays with a broad range of influenza A virus strains, including NAI‐ and amantadine‐resistant isolates, 2009 pandemic H1N1, and circulating avian H5N1 strains.37, 207 Its activity against influenza B virus is negligible, which can be attributed to amino acid differences in the PB2‐CBD.208 In particular, the π‐stacking interaction between VX‐787 and Phe323 in the influenza A PB2‐CBD cannot be formed in the influenza B protein, which contains a glutamine (Q325) at the corresponding position (Fig. 2A and B). The apparent flexibility in the PB2‐CBD of influenza B (see Section 2,A) may be another factor complicating the development of cap‐binding inhibitors that cover both influenza A and B virus.
Resistance studies with VX‐787 in cell culture delivered six resistance mutations in PB2 (Q306H, S324I, S324N, S324R, F404Y, and N510T), which yielded at least 60‐fold reduction in VX‐787 sensitivity.207 In the Phase 2a clinical study, an M431I mutant virus was detected in a minority of the patients, which, in cell culture, displayed 57‐fold lower sensitivity to VX‐787, yet reduced viral fitness.11 In mice, prophylactic use of VX‐787 (which is orally bioavailable) provided 100% protection against the APR8 laboratory strain, a pandemic 2009 strain, or a highly pathogenic avian H5N1 virus. In addition, this inhibitor provided 100% survival when treatment was initiated up to 4 days after challenge with influenza virus.207
Recently, Roch et al.38 reported two compounds, Cap‐3 and Cap‐7 (Fig. 2E), which bind to the PB2‐CBD and inhibit transcription in an enzymatic assay with the polymerase complex. In addition, they inhibit virus replication in cell culture, albeit considerably less potently than VX‐787 (i.e., EC50 values in the range of 1–9 μM). Hitherto, mechanistic validation was not yet reported.
2. Metal‐Chelating Inhibitors of PA‐Nter Endonuclease Activity
The conserved nature of several amino acid residues inside the catalytic site of PA‐Nter implies that this domain is highly relevant to achieve inhibitors with broad activity against influenza A and B. The strategy explored thus far consists of metal‐chelating scaffolds containing coplanar oxygens to bind the divalent metal ion(s), which resembles the action principle of approved HIV integrase inhibitors.209, 210 The challenge is to achieve inhibitors, which optimally occupy the PA‐Nter catalytic site and surrounding regions, yet do not interact with metal‐dependent proteins of the host cell. The concept seems now validated since the recent introduction of two PA inhibitors (AL‐794 and S‐033188) into clinical trials.211
The literature contains diverse PA inhibitors with inhibitory activity in enzymatic assays with the viral polymerase or isolated PA‐Nter. It is important to note that for many of these agents, cell culture data on antiviral activity and, in particular, mechanism of action are not elaborated. Some compounds have a high anionic charge that impedes their cellular uptake. For example, we demonstrated that for EGCG (Fig. 6K), the activity in virus‐infected cells is related to inhibition of virus entry.74 To definitely prove that an assumed PA inhibitor acts upon PA in a virus/cell context, resistance studies should be carried out. As of today, this decisive mechanistic evidence is only available for L‐742,001 (Fig. 6E–G), since three laboratories including ours49, 74, 117 demonstrated that specific mutations in PA confer moderate (3‐) to high (>10‐fold) viral resistance to this agent. The substitutions were present in the catalytic core of PA or surrounding hydrophobic pockets (colored purple, green, red, and orange in Fig. 6), confirming that these PA regions are critical for the antiviral mode of action of L‐742,001. This molecule is the prototype of the first class of influenza endonuclease inhibitors, already discovered at Merck about two decades ago.115, 116 Among this series of 4‐substituted‐2,4‐dioxobutanoic acids with a characteristic β‐diketo acid motif, compound L‐742,001 was identified as a particularly potent inhibitor of the influenza virus endonuclease reaction in an enzymatic assay, and of virus replication in cells.49, 74, 116, 117 In a mouse model, the compound provided up to 4‐log10 reduction in virus lung titers.116 Strong in vivo activity was also reported for the recently discovered inhibitor ANA‐0 proposed to act upon PA (Fig. 6K).79 Other classes of reported endonuclease inhibitors (awaiting mechanistic validation in cell culture) include: flutimide 75 (Fig. 6K) and a series of more potent aromatic analogues,212 N‐hydroxamic acid and N‐hydroxyimide (Fig. 6K) compounds,77 tetramic acid derivatives,118 polyphenolic catechins,76 trihydroxyphenyl‐bearing compounds73, 78, 120 (Fig. 6D and K, compound 273 and compound 1678), phenethylphenylphthalimide analogues derived from thalidomide,119 macrocyclic bisbibenzyls,213 fullerenes,214 hydroxyquinolinones,215 hydroxypyridinones,47, 50 hydroxypyridazinones,216 hydroxypyrimidinones,216 β‐diketo acid (DKA), and DKA‐bioisosteric compounds,68 bis‐dihydroxyindolecarboxamides,121 2‐hydroxybenzamides,217 thiosemicarbazones,218 pyrimidinoles,45 pyridopiperazinediones 38 (Fig. 6K, Endo‐1), and miscellaneous compounds bearing distinct pharmacophoric fragments.45, 78, 101 All these different chemotypes have in common that they bear chelating motifs able to bind the bivalent metal ion(s) in the catalytic core of PA‐Nter.219
As explained in Section 2,B, the different crystal structures for PA‐Nter that have become available since 2009 converge on the overall structure of the active site, yet do not agree on whether the enzyme contains one or two (or possibly three) divalent metal ions in its catalytic site, nor on which metal ions (i.e., Mn2+ or Mg2+) are present in the native enzyme.42, 43, 44, 45, 46, 47 Knowledge of the precise number and identity of these metal ions is critical to design optimized metal‐chelating PA inhibitors. Similar to what was observed for HIV integrase,220 we found that the potency of metal‐chelating inhibitors in enzymatic assays with PA‐Nter can show large variations depending on which bivalent metal ion (Mg2+ or Mn2+) is used.68 This phenomenon can be explained by the different ligand preferences of these two metal ions. For example, magnesium generally binds oxygen atoms rather than nitrogen, while manganese has slightly greater affinity for nitrogen.221 Moreover, two tautomeric forms of an inhibitor can display a difference in metal preference. For DKA compounds, it was shown that Mn2+ preferentially binds to the diketo form, while Mg2+ predominantly recognizes the β‐keto‐enol form.222 Dependent on which metal ion is physiologically relevant, the compounds’ activity may increase by stabilization of the preferred tautomeric form, for instance by substituting the flexible β‐diketo acid (DKA) moiety by a “locked” motif having optimal geometry for bidentate metal coordination.
Crystallographic analyses of PA‐Nter in complex with diverse PA inhibitors have revealed that the catalytic center is surrounded by different hydrophobic pockets, which are amenable to inhibitor design.45, 46, 47, 49, 50, 120, 215, 216 Overall, the active site of PA‐Nter is quite spacious and flexible, and in silico design of optimized PA inhibitors is further complicated by the fact that inhibitors bind via an induced‐fit mechanism. Assuming that metal‐chelating PA inhibitors act as substrate or product mimics, precise insight into the binding mode of the RNA substrate or cleavage product is crucial to identify which of the hydrophobic pockets have the highest relevance for PA inhibitor design. A cocrystal structure for PA‐Nter in complex with an RNA substrate is highly needed. Three X‐ray structures are available of PA‐Nter in complex with UMP (Fig. 6H–J), which can be regarded as a mimic of one of the two cleavage products.44, 46, 49 Two distinct UMP‐binding poses were observed, particularly related to the position of the ribose and base, which underlines the impact of crystallization procedures (i.e., cocrystallization vs. soaking; addition of metal ions or not). In the context of PA, cocrystallization seems more reliable since it enables the binding pocket to adapt to the ligand. One particularly flexible region is the helix containing residue Tyr24, which, in two cocrystal structures, stacks with the uracil base46, 49 of UMP (Fig. 6I and J, in green) and also shows hydrophobic interactions with PA inhibitors like those shown in Figures 6C and E.45, 46, 49, 50 To accommodate these ligands, this Tyr24 moves or rotates compared to its pose in the apo form. Since, upon crystal soaking, this conformational change is unlikely to occur, the ligand may be forced into an alternative and biologically less relevant binding pose.
Another interaction site that seems of high relevance is Arg124. A role for this residue in RNA substrate binding was suggested by Yuan et al.43 This was corroborated by Xiao et al.,57 whose MD simulations (Fig. 6A) pointed to Arg124, Arg125, and Arg192 as the residues responsible for interaction with the 3ʹ end of the RNA substrate. Likewise, the relevance of this Arg124 pocket (in blue in Fig. 6) was suggested by our cell‐based antiviral studies in which we introduced mutations at different sites within or around the PA‐Nter active center, and analyzed the resistance of the mutant viruses to the prototype PA inhibitor L‐742,001.74 The data agreed to a docking model74 for L‐742,001 in PA‐Nter (Fig. 6G), but only partially agreed to the cocrystallization results45, 49 (Fig. 6E and F). In addition, an analogous orientation as for the docked L‐742,001 molecule was seen in the X‐ray structures of PA‐Nter in complex with the dihydropyrimidine‐based “compound 5”45 (Fig. 6B), and the particularly potent hydroxypyridinone‐based PA inhibitor “compound 7”50 (Fig. 6C). For both these compounds, the authors reported a role for Arg124 in binding of the inhibitor.
Consequently, we speculate that a PA inhibitor with a binding orientation that allows interactions with these RNA‐binding residues, mimics the 3ʹ end of the capped RNA substrate and is able to efficiently compete with the substrate. Presumably, the interactions with the 3ʹ end are essential for correct positioning and cleavage of the substrate. In contrast, the interactions between the 5ʹ end of the capped RNA and PA‐Nter (i.e., at the pocket colored in purple in Fig. 6) may be weaker, since the cap‐binding site in the PB2 subunit already ensures strong binding of the 5ʹ end. As a result, the pocket in PA‐Nter, which binds the 5ʹ end could possibly be more flexible. An additional argument is the fact that, once cleavage has occurred, the PA active site has to release the capped 5ʹ end in order to allow its relocation into the PB1 catalytic site for primer elongation and transcription of the viral mRNA. Hence, tight binding affinity of PA to the 5ʹ end of the capped RNA substrate would substantially decrease the rate of product release, which would be detrimental to the overall process of transcription.
3. PA Targeting Aptamers
Recently, a DNA aptamer library screening performed by Yuan et al.123 yielded a number of aptamers with binding affinity to either entire PA or PA‐Nter. Four aptamers were able to inhibit the endonuclease reaction and one of them showed broad influenza A inhibition when transfected into virus‐infected cells. At present, aptamers still require significant improvements in terms of pharmacokinetics and—dynamics before having broad clinical applicability. Nevertheless, this study demonstrates that aptamers can be excellent tools to identify new relevant binding pockets for inhibitors of the PA endonuclease or other viral target proteins.
D. Protein–Protein Interaction Inhibitors of the Influenza Polymerase Complex
The assembly of the three subunits (PB1, PB2, and PA) into a functional viral polymerase complex is essential for influenza RNA synthesis and virus replication. Thus, interference with its correct assembly through inhibition of a crucial protein–protein interaction (PPI) is currently explored as an innovative antiviral strategy. Until 2014, tailored design of assembly inhibitors was based on at that time available crystal structures for two specific interfaces in the polymerase heterotrimer (Fig. 8). The revelation of the full polymerase structure now creates the possibility to explore other inter‐ or intrasubunit interfaces, yet two important issues are: (i) the size of a specific interface, to allow fitting of small‐molecule inhibitors; and (ii) the conservation of its residues when envisaging both influenza A and B.
Figure 8.
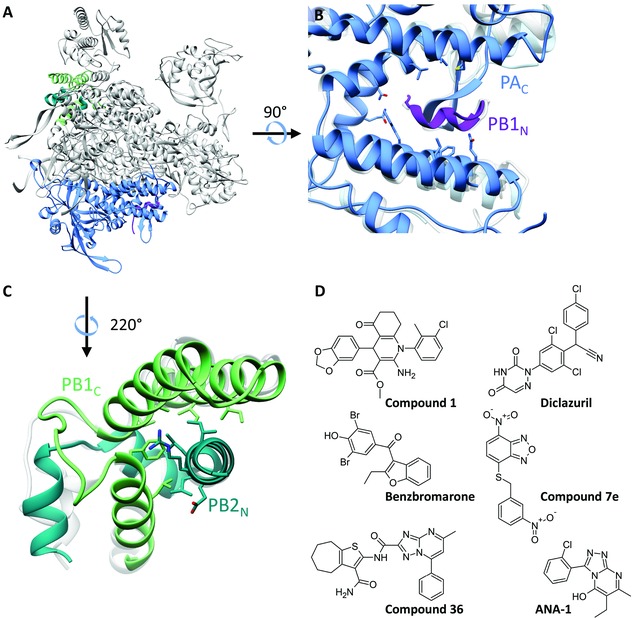
The two thus far explored protein–protein interaction (PPI) domains and chemical structures of PAC‐PB1N PPI inhibitors. (A) Location in the FluA polymerase crystal structure23 (PDB: 4WSB) of the two PPI domains, which have been targeted by peptides or small molecules. (B) Closeup showing a superposition of the crystal structures of the PAC‐PB1N interface223 (PDB: 3CM8) on that of the FluA polymerase (light gray) and the apo form of PAC 224 (light blue; PDB: 4IUJ). (C) Closeup showing a superposition of the crystal structure of the PB1C‐PB2N interface225 (PDB: 3A1G) on the FluA polymerase complex, in light gray. (D) Chemical structures of representative PAC‐PB1N‐PPI inhibitors “compound 1,”127 diclazuril,126 benzbromarone,126 “compound 7e,”124 “compound 36,”131 and ANA‐1.133
The structural details for one important PA‐PB1 interaction domain were revealed in 2008, based on cocrystallization of the PA C‐terminal domain (PAC, residues 239 or 256–716) with a short N‐terminal PB1 peptide (PB1N).223, 226 This PAC domain resembles a dragon's head that clamps the PB1N peptide. As shown in Figure 8B, the base of this pocket overlaps between the crystal structures of the PB1N‐bound PAC domain223 (in dark blue), the full polymerase heterotrimer23 (in gray), and the apo PAC domain224 (in light blue). In contrast, there is less overlap at the periphery, meaning that the binding groove is slightly more narrow in the structure of the PB1N‐bound PAC compared to the two other crystal structures. The less mobile base of the pocket appears relevant as the initial interaction point for PPI inhibitors; to these scaffolds, structural elements should be added to target also the more flexible protein parts on the periphery of the interface.
Three features explain the drugability of this PAC‐PB1N interface: several residues are conserved (among influenza A, B, and C), and the interface is hydrophobic and relatively small, implying that it can be targeted by small molecules.227 Initially, the concept to disrupt the PAC‐PB1N interface was explored with PB1‐derived peptides.228, 229, 230 Among a first series of small‐molecule inhibitors identified by in silico screening followed by a PAC‐PB1N biochemical interaction assay, “compound 1” emerged as particularly relevant given its broad anti‐influenza A and B virus activity in cell culture (Fig. 8D).127 Other early lead molecules with anti‐influenza A activity in cell culture are benzbromarone and diclazuril 126 and “compound 7e”124 (Fig. 8D). This was followed by rational development of diverse lead compounds,125, 129, 130, 131, 132, 231 some of which were used to generate a pharmacophore model for PAC‐PB1N interaction inhibitors.130 For the recently identified inhibitor ANA‐1 (Fig. 8D), the anti‐influenza activity was confirmed in a mouse model.133 On the basis of docking, the binding site of ANA‐1 in PAC was predicted to lie in an allosteric site adjacent to the PB1 interacting domain. Yet, for all the PAC‐PB1N interaction inhibitors reported thus far, mechanistic studies using cocrystallization or resistance selection remain to be performed to verify their antiviral mode of action and precise binding mode. This could also aid to design PAC‐PB1N assembly inhibitors, which establish hydrophilic besides hydrophobic interactions, thereby leading to better solubility and potentially higher antiviral potency than the current lead compounds.232
Another protein–protein interface that was validated with peptide inhibitors233, 234 but is as yet unexplored with small molecules, is the interaction domain between PB1C (residues 678–757; Fig. 8C, in light green) and PB2N (residues 1–37; in dark green). The crystal structure of this isolated domain was published in 2009225 and nicely overlaps with that in the full polymerase complex23 (in gray). Despite the fact that this PB1C‐PB2N interface carries conserved amino acids, the inhibition by a PB2N‐derived peptide seemed prone to strain dependency.235 One proposed hypothesis relates this observation to a conformational change in PB1, when it is bound in the PB1‐PA dimer intermediate, thereby rendering the PB2N‐binding part in PB1 inaccessible. Hence, this PB1‐PB2 interface appears a more challenging target for development of broad influenza virus inhibitors.234, 235
E. Gene Silencing Approaches
Pharmaceutical development of oligonucleotide inhibitors faces some common obstacles, particularly related to their low in vivo stability and inefficient cellular delivery. Antisense oligonucleotides function as a single strand and block mRNA processing or translation by binding to the mRNA to which they are complementary. One influenza virus inhibitor currently in Phase 1 clinical trials,236 is the antisense oligonucleotide AVI‐7100 (Radavirsen), a phosphorodiamidate morpholino oligomer (PMO). AVI‐7100 contains nonionic morpholino rings (instead of ribose rings) and three phosphorodiamidate intersubunit linkages, and was designed to interfere with translation and splicing of mRNA derived from the M‐gene. In preclinical studies, AVI‐7100 proved effective against influenza A virus infection in mice and ferrets, even when administered after virus challenge.146
The possible application of RNA interference (RNAi) for influenza therapy has been the subject of a series of studies.237 This includes siRNAs, small RNA duplexes, which trigger the destruction of specific mRNAs by associating with the RNA‐induced silencing complex. This concept can be valuable for PA, PB1, PB2, and NP since some regions in their nucleotide sequences are conserved among influenza A subtypes. The siRNAs for NP and PA were shown to prevent accumulation of their mRNAs, and reduce the mRNA, vRNA, and cRNA levels for other viral genes.238 siRNAs against NP, PA, and PB1 were shown to have antiviral activity in cell culture and mice.238, 239 5ʹ‐Triphosphate modification of siRNA (for NP) provided a dual effect by combining gene silencing with RIG‐I activation.240 Alternatively, to circumvent the poor delivery of siRNA, a lentiviral241 or Escherichia coli vector242 was explored to express NP‐ and PB1‐targeting, or NP‐ and PA‐targeting RNA interfering sequences, respectively.
F. Small‐Molecules Targeting the Viral NP
The viral NP directly interacts with the viral polymerase complex to support viral RNA synthesis.243, 244, 245 However, it primarily has a structural function since it forms the protein scaffold of the vRNP complexes through its homo‐oligomerizing246, 247, 248 and RNA‐binding properties.246, 249 The NP protein is crescent shaped with head, body, and tail domains246, 247 and a putative RNA‐binding groove (Fig. 9A and B).246 NP–NP oligomerization is the result of one tail loop inserting into the body of a neighboring monomer. This insertion is stabilized by intermolecular β‐sheets, hydrophobic interactions, and salt bridges.246
Figure 9.
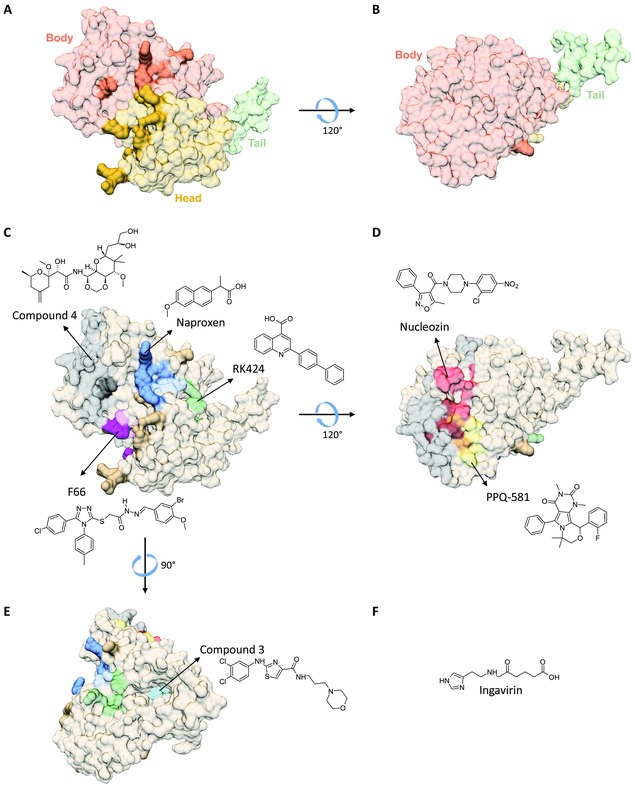
Structure of the viral nucleoprotein and chemical structures of proposed or proven NP inhibitors. The NP structures are based on the H1N1 NP X‐ray structure246 (PDB: 2IQH). The residues lining the RNA‐binding groove are shown in darker colors. (A) Head, body, and tail domains in NP. (B) As in panel A, but rotated 120° around the x‐axis. (C) Predicted binding sites for compound 4,142 naproxen139 and naproxen C0,250 RK424,141 and F66.140 (D) Binding site for nucleozin111, 134, 135 (proven by cocrystallization) and that predicted for PPQ‐581.137 The overlap of the nucleozin‐binding site with that of compound 4 or PPQ‐581 is colored in maroon and orange, respectively. (E) The model in (C) is rotated 90° around the y‐axis to show the internal tail loop binding cavity (in cyan), which is the predicted binding pocket for compound 3.138 (F) Chemical structure of Ingavirin®, which was reported to impair biogenesis and oligomerization of NP in vitro.144
The NP protein contains different sites that are amenable to inhibitor design and are briefly described below (Fig. 9C–E).136 A first series are NP‐aggregating agents such as the aryl piperazine amide compound called nucleozin (Fig. 9D).111, 134, 135, 251 This molecule forms bridges between NP subunits to give higher order NP oligomers.135 Depending on the assay, nucleozin exerts its activity prior to nuclear import of the vRNPs; at the stage of viral RNA synthesis; or during cytoplasmic trafficking of progeny vRNPs.252 It exhibits robust activity against influenza A virus in vitro and in vivo, yet is inactive against influenza B. Viral resistance to nucleozin emerged after only five cell culture passages.111, 134, 135 This agent is inherently inactive against some influenza virus strains (such as the 2009 pandemic H1N1 virus) that already carry one of its resistance mutations.111, 134 Another NP‐aggregating compound, PPQ‐581, shows no cross‐resistance to nucleozin and selects for a resistance mutation adjacent to the nucleozin‐binding site (Fig. 9D).137
A second type of NP inhibitors target the highly conserved Glu339‐Arg416 salt bridge among NP neighbor monomers.246 Shen et al.138 identified four small‐molecule influenza inhibitors, which are able to disrupt NP oligomerization, with compound “3” (Fig. 9E) being the most potent one in cell culture.
A third possibility is to target the RNA‐binding groove in NP. In silico screening139 led to the identification of naproxen (Fig. 9C), which was found to inhibit RNA binding by NP in biochemical assays, and display anti‐influenza virus properties in cell culture and mouse models. The latter activity is likely potentiated by naproxen's well‐known anti‐inflammatory effect. The analogue naproxen C0,250 which carries a 1,3‐dicarboxylated phenyl group instead of the propionic acid in naproxen, has higher NP binding affinity in vitro, but its activity in cell culture was not yet reported. A different virtual screening hit encoded F66 showed antiviral activity in cell culture and mouse models,140 yet its RNA‐NP disrupting activity and binding mode in NP remain to be established.
Compound RK424 141 was reported to inhibit influenza A virus in cell culture and mouse models, and interfere with RNA binding to NP, the NP–NP interaction, and nuclear export of NP. Although this complex mode of action remains to be verified, it seems to agree with the docking analysis for RK424, which predicted its binding in an NP pocket surrounded by the RNA‐binding groove, NP dimer interface, and nuclear export signal.141 Other proposed NP inhibitors are compound “4” (Fig. 9C) and Ingavirin® (Fig. 9F), a controversial143 influenza inhibitor that is commercially available in Russia. This agent was reported to impair biogenesis and oligomerization of NP in vitro144 and suppress influenza A and B infections in animal models.145, 253 However, since its broad antiviral activity encompasses unrelated respiratory viruses,145 Ingavirin® most likely targets a cellular rather than a virus‐specific component.
Taken together, besides nucleozin, several lead molecules to target NP have been proposed during recent years. Among these, nucleozin is the only inhibitor for which the mechanism has been firmly proven by cocrystallization and resistance analysis.
G. Less Defined Approaches to Inhibit the Influenza Polymerase Complex
For a number of compounds suggested to act on influenza virus polymerase activity in cell culture, the detailed mechanism of action has not yet been revealed. Compound “367”111 (Fig. 7B) was proposed to target PB1 since mutation H456P in PB1 rendered the virus resistant to “367” and its close analogue “715.” Compound “367” inhibited viral RNA synthesis in the vRNP reconstitution (minigenome) assay, an effect that possibly contributes to its antiviral activity in influenza virus‐infected cell cultures. Another small molecule that may target PB1 is “ASN2” (Fig. 7B). This compound inhibits influenza A virus polymerase function while simultaneously activating the innate immune system by inducing type I interferon expression.112 ASN2 inhibited viral transcription but not all genes were equally affected. It induced preferential downregulation of mRNAs encoded by the smallest genome segments, that is M1, M2, NS1, and nuclear‐export protein (NEP). Loss of NS1 expression resulted in induction of interferon‐I, which may prove beneficial in an in vivo setting. Resistance selection revealed that the antiviral activity of ASN2 is linked to the serine or tyrosine residue at position 499 in PB1.
Before the three‐dimensional structure of PB1 was revealed, it was impossible to analyze whether the identified resistance mutations for ASN2 and “367” (at position 499 and 456 of PB1, respectively) are located close to each other. Using the crystal structures published by Pflug et al.,23 we were now able to establish that both residues are located at the outside of PB1, close to the PB2‐627 domain (Fig. 5, in maroon) and at a distance of 15–20 Å from each other. Both molecules appear relevant not only in terms of drug development, but also as tools to unravel as yet unknown functions of particular regions in PB1.
Compound THC19 (Fig. 7B) was reported to inhibit influenza RNA synthesis and virus replication by targeting the PA subunit, yet its mode of action remains to be established.122 The quinoline‐based compound BPR3P0128 (Fig. 7B) was shown to strongly inhibit viral mRNA synthesis.148 This molecule inhibited binding and cleavage of capped RNA fragments by influenza polymerase complexes in nuclear extracts, yet did not compete with cap structures in an in vitro cap‐binding assay using isolated PB2. Based on this, it was hypothesized148 that BPR3P0128 might target the interaction between PB2 and a host factor, such as the heat shock proteins Hsp70 or Hsp90. Based on NMR‐based fragment screening, Lee et al.149 identified the small molecule DPQ (Fig. 7B) that appears to interact with the influenza vRNA promoter. This molecule was found to inhibit virus replication in a plaque reduction assay.149
H. Strategies Directed Toward Associated Host Cell Factors
The concept of targeting a host factor to indirectly inhibit influenza virus RNA synthesis is still in its infancy. Like all other viral pathogens, influenza viruses strictly rely on a variety of host factors to support their replication.254 Thus, targeting such a cellular component represents an alternative to traditional drugs that are directed toward a viral factor.255, 256, 257 This approach could diminish the emergence of resistance since the mutation rate of the host cells is intrinsically lower compared to that of the virus, and the virus most probably does not readily adapt itself to use an alternative cellular factor. Obviously, the inherent challenge of all host‐directed drug discovery campaigns is to ensure that inhibition of the cellular protein is not detrimental to the host. Hence, the pharmaceutical industry has been rather hesitant when considering host cell targets to treat virus infections.
As explained below, miscellaneous host targeting approaches for influenza have been published, which, at least conceptually, may be linked to indirect inhibition of the viral polymerase complex. The interpretation is complicated by the fact that many of these molecules have multiple pharmacological (such as anti‐inflammatory) effects. Hence, their therapeutic activity in mouse influenza models may be related to immune‐related besides antiviral effects. Although this deviates from the traditional view on antiviral therapy, it fits with an ongoing call for broader acting or immunomodulatory antiviral agents.258
One explored host‐targeting strategy is to interfere with nuclear transport. All details on the mechanisms for vRNP nuclear import and export can be found in a recent review.14 Inhibition of the cellular CRM1 protein by the cytotoxic agent leptomycin B was already shown several years ago to give nuclear retention of the vRNPs, presumably by disrupting the interaction between NP and the CRM1 nuclear export receptor.259 In 2014, the more selective CRM1‐antagonist KPT‐335 (Verdinexor; Fig. 7C), which is currently in Phase 1 clinical trials,260 was proven to increase survival in influenza virus‐infected mice.147
An alternative approach relates to the heat shock protein Hsp90, which is involved in assembly and nuclear transport of the viral RNA polymerase, possibly as a molecular chaperone for its protein subunits prior to the formation of a mature ternary polymerase complex.261 It was demonstrated that Hsp90 inhibitors (Fig. 7C) such as geldanamycin,150 its analogue 17‐allylamino‐17‐demethoxygeldanamycin150 (17‐AAG), and 3beta‐acetoxydeoxodihydrogedunin,151 impair influenza viral growth by reducing the levels of ribonucleoprotein complexes. Recently, Swale et al.262 revealed how the PA‐PB1 heterodimer assembles with host RanBP5 into a complex, probably being the PA‐PB1 import complex. Targeting the assembly or disintegration of this complex with inhibitors, could be a valuable antiviral strategy.
Another pharmacological class is protein kinase inhibitors to inhibit phosphorylation of a viral protein or crucial cellular factor. The MEK (mitogen‐activated protein kinase kinase) inhibitor U0126 (Fig. 7C) was shown to cause nuclear retention of viral RNPs, impaired function of the NEP, and concomitant inhibition of virus production.263 U0126 proved able to suppress propagation of influenza viruses, including highly pathogenic avian influenza viruses, and was reported to have low cytotoxicity in vitro and in vivo.152, 153 Four other MEK inhibitors (Fig. 7C), PD‐0325901, AZD‐6244, AZD‐8330, and RDEA‐119 (which are all orally available and at least in Phase 1 clinical trials for cancer therapy) demonstrated antiviral activity in vitro as single agents or in combination with oseltamivir.154 Also, the multiple kinase inhibitor WV970 (Fig. 7C) was reported to suppress influenza virus in cell culture through vRNP inhibition.155
The NF‐κB signaling pathway is critical for efficient replication of influenza virus, and its inhibition results in reduced virus titers. Mazur et al.156 reported that acetylsalicylic acid (aspirin; Fig. 7C) can efficiently diminish influenza virus replication in vitro and in vivo through NF‐κB inhibition, which blocks caspase‐mediated nuclear export of VRNPs. Three other NF‐κB inhibitors, pyrrolidine dithiocarbamate, SC75741, and Bay 11–7082 (Fig. 7C), were reported to significantly decrease influenza virus‐induced disease in mice.157, 158, 159, 264
Another host cell factor with diverse regulatory functions, the Rac1 GTPase, was demonstrated to have both virus‐supportive as antiviral effects. The Rac1 GTPase inhibitor NSC23766 (Fig. 7C) was found to possess anti‐influenza virus properties by affecting viral polymerase activity in a cell‐based (mini‐genome) assay. NSC23766 was able to reduce virus replication in mice and prolong the survival rate of infected mice.160
4. CONCLUSION AND PERSPECTIVES
For many viruses such as HIV, hepatitis B virus, hepatitis C virus, and herpesviruses, inhibitors of viral DNA or RNA polymerases or hereto related activities are the cornerstone of current antiviral interventions. For influenza virus, the development of viral polymerase inhibitors has been lagging behind, but presently seems to be at a turning point. During the past years, major progress was made in the structural elucidation of different influenza virus polymerase subdomains, and very recently of the entire heterotrimeric complex. In parallel, significant advances were achieved in unravelling the reaction mechanisms and functioning of the influenza replication machinery. These accomplishments fuel the ongoing search for unique influenza polymerase inhibitors, which has evolved from serendipitous discovery to rational structure‐aided drug design. It can be anticipated that, in the near future, some of these inhibitors will enter the clinic to fundamentally reshape the field of influenza therapy and prevention.
ACKNOWLEDGMENTS
The authors acknowledge financial support by the Geconcerteerde Onderzoeksacties (GOA/15/019/TBA) from the KU Leuven. They wish to thank Talitha Boogaerts, Leentje Persoons, Stijn Stevens, Ria Van Berwaer, and Wim van Dam for their dedicated contribution to their research work. Molecular graphics and analyses in Figures 2, 3, 5, 6, 8, 9 were performed with the UCSF Chimera package. Chimera is developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIGMS P41‐GM103311).265
Biographies
Annelies Stevaert obtained her Master of Science in Drug Development (2009) at the KU Leuven, Belgium and her Ph.D. as Doctor in Pharmaceutical Sciences (2015) at the Rega Institute for Medical Research, KU Leuven. She was holder of a Ph.D. grant from the Agency for Innovation by Science and Technology (IWT). Her current research is focused on the development of novel inhibitors of the influenza PA endonuclease.
Lieve Naesens obtained her degree of Master in Pharmaceutical Sciences (1987) at the KU Leuven, Belgium, and her Ph.D. as Doctor in Pharmaceutical Sciences (1993) at the Rega Institute for Medical Research, KU Leuven. In 2003, she was appointed as assistant and later associate professor at the Faculty of Medicine of the KU Leuven to teach courses in virology and immunology. She is currently a senior investigator and project leader in influenza virus research at the Laboratory of Virology and Chemotherapy, Rega Institute, KU Leuven. Her research is focused on the design and mechanism of action of novel antiviral compounds, with a particular interest in inhibitors targeting the processes of viral entry or genome replication of influenza virus.
REFERENCES
- 1. Krammer F, Palese P. Advances in the development of influenza virus vaccines. Nat Rev Drug Discov 2015;14:167–182. [DOI] [PubMed] [Google Scholar]
- 2. Simonsen L, Taylor RJ, Viboud C, Miller MA, Jackson LA. Mortality benefits of influenza vaccination in elderly people: An ongoing controversy. Lancet Infect Dis 2007;7:658–666. [DOI] [PubMed] [Google Scholar]
- 3. Madhi SA, Cutland CL, Kuwanda L, Weinberg A, Hugo A, Jones S, Adrian PV, van Niekerk N, Treurnicht F, Ortiz JR, Venter M, Violari A, Neuzil KM, Simões EA, Klugman KP, Nunes MC. Influenza vaccination of pregnant women and protection of their infants. N Engl J Med 2014;371:918–931. [DOI] [PubMed] [Google Scholar]
- 4. Gu RX, Liu LA, Wei DQ. Structural and energetic analysis of drug inhibition of the influenza A M2 proton channel. Trends Pharmacol Sci 2013;34:571–580. [DOI] [PubMed] [Google Scholar]
- 5. Pielak RM, Oxenoid K, Chou JJ. Structural investigation of rimantadine inhibition of the AM2‐BM2 chimera channel of influenza viruses. Structure 2011;19:1655–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jefferson T, Demicheli V, Di Pietrantonj C, Rivetti D. Amantadine and rimantadine for influenza A in adults. Cochrane Database Syst Rev 2006:CD001169. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD001169.pub3/pdf/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deyde VM, Xu X, Bright RA, Shaw M, Smith CB, Zhang Y, Shu Y, Gubareva LV, Cox NJ, Klimov AI. Surveillance of resistance to adamantanes among influenza A(H3N2) and A(H1N1) viruses isolated worldwide. J Infect Dis 2007;196:249–257. [DOI] [PubMed] [Google Scholar]
- 8. Moscona A. Global transmission of oseltamivir‐resistant influenza. N Engl J Med 2009;360:953–956. [DOI] [PubMed] [Google Scholar]
- 9. Thorlund K, Awad T, Boivin G, Thabane L. Systematic review of influenza resistance to the neuraminidase inhibitors. BMC Infect Dis 2011;11:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Samson M, Pizzorno A, Abed Y, Boivin G. Influenza virus resistance to neuraminidase inhibitors. Antiviral Res 2013;98:174–185. [DOI] [PubMed] [Google Scholar]
- 11. McKimm‐Breschkin JL, Fry AM. Meeting report: 4th ISIRV antiviral group conference: Novel antiviral therapies for influenza and other respiratory viruses. Antiviral Res 2016;129:21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Das K, Aramini JM, Ma LC, Krug RM, Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nat Struct Mol Biol 2010;17:530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fodor E. The RNA polymerase of influenza A virus: Mechanisms of viral transcription and replication. Acta Virol 2013;57:113–122. [DOI] [PubMed] [Google Scholar]
- 14. Eisfeld AJ, Neumann G, Kawaoka Y. At the centre: Influenza A virus ribonucleoproteins. Nat Rev Microbiol 2015;13:28–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ortín J, Martín‐Benito J. The RNA synthesis machinery of negative‐stranded RNA viruses. Virology 2015;479‐480:532–544. [DOI] [PubMed] [Google Scholar]
- 16. Reguera J, Gerlach P, Cusack S. Towards a structural understanding of RNA synthesis by negative strand RNA viral polymerases. Curr Opin Struct Biol 2016;36:75–84. [DOI] [PubMed] [Google Scholar]
- 17. Moeller A, Kirchdoerfer RN, Potter CS, Carragher B, Wilson IA. Organization of the influenza virus replication machinery. Science 2012;338:1631–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arranz R, Coloma R, Chichón FJ, Conesa JJ, Carrascosa JL, Valpuesta JM, Ortín J, Martín‐Benito J. The structure of native influenza virion ribonucleoproteins. Science 2012;338:1634–1637. [DOI] [PubMed] [Google Scholar]
- 19. Bouloy M, Plotch SJ, Krug RM. Globin mRNAs are primers for the transcription of influenza viral RNA in vitro . Proc Natl Acad Sci USA 1978;75:4886–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)‐dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981;23:847–858. [DOI] [PubMed] [Google Scholar]
- 21. Robertson JS, Schubert M, Lazzarini RA. Polyadenylation sites for influenza virus mRNA. J Virol 1981;38:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Poon LL, Pritlove DC, Fodor E, Brownlee GG. Direct evidence that the poly(A) tail of influenza A virus mRNA is synthesized by reiterative copying of a U track in the virion RNA template. J Virol 1999;73:3473–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pflug A, Guilligay D, Reich S, Cusack S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014;516:355–360. [DOI] [PubMed] [Google Scholar]
- 24. Reich S, Guilligay D, Pflug A, Malet H, Berger I, Crépin T, Hart D, Lunardi T, Nanao M, Ruigrok RW, Cusack S. Structural insight into cap‐snatching and RNA synthesis by influenza polymerase. Nature 2014;516:361–366. [DOI] [PubMed] [Google Scholar]
- 25. Portela A, Digard P. The influenza virus nucleoprotein: A multifunctional RNA‐binding protein pivotal to virus replication. J Gen Virol 2002;83:723–734. [DOI] [PubMed] [Google Scholar]
- 26. Boivin S, Cusack S, Ruigrok RW, Hart DJ. Influenza A virus polymerase: Structural insights into replication and host adaptation mechanisms. J Biol Chem 2010;285:28411–28417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gabriel G, Fodor E. Molecular determinants of pathogenicity in the polymerase complex. Curr Top Microbiol Immunol 2014;385:35–60. [DOI] [PubMed] [Google Scholar]
- 28. Rodriguez‐Frandsen A, Alfonso R, Nieto A. Influenza virus polymerase: Functions on host range, inhibition of cellular response to infection and pathogenicity. Virus Res 2015;209:23–38. [DOI] [PubMed] [Google Scholar]
- 29. Long JS, Giotis ES, O Moncorgé, Frise R, Mistry B, James J, Morisson M, Iqbal M, Vignal A, Skinner MA, Barclay WS. Species difference in ANP32A underlies influenza A virus polymerase host restriction. Nature 2016;529:101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ulmanen I, Broni BA, Krug RM. Role of two of the influenza virus core P proteins in recognizing cap 1 structures (m7GpppNm) on RNAs and in initiating viral RNA transcription. Proc Natl Acad Sci USA 1981;78:7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blaas D, Patzelt E, Kuechler E. Identification of the cap binding protein of influenza virus. Nucleic Acids Res 1982;10:4803–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guilligay D, Tarendeau F, Resa‐Infante P, Coloma R, Crépin T, Sehr P, Lewis J, Ruigrok RW, Ortín J, Hart DJ, Cusack S. The structural basis for cap binding by influenza virus polymerase subunit PB2. Nat Struct Mol Biol 2008;15:500–506. [DOI] [PubMed] [Google Scholar]
- 33. Xie L, Wartchow C, Shia S, Uehara K, Steffek M, Warne R, Sutton J, Muiru GT, Leonard VH, Bussiere DE, Ma X. Molecular basis of mRNA cap recognition by influenza B polymerase PB2 subunit. J Biol Chem 2016;291:363–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Y, Yang Y, Fan J, He R, Luo M, Zheng X. The crystal structure of the PB2 cap‐binding domain of influenza B virus reveals a novel cap recognition mechanism. J Biol Chem 2015;290:9141–9149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wakai C, Iwama M, Mizumoto K, Nagata K. Recognition of cap structure by influenza B virus RNA polymerase is less dependent on the methyl residue than recognition by influenza A virus polymerase. J Virol 2011;85:7504–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pautus S, Sehr P, Lewis J, Fortuné A, Wolkerstorfer A, Szolar O, Guilligay D, Lunardi T, Décout JL, Cusack S. New 7‐methylguanine derivatives targeting the influenza polymerase PB2 cap‐binding domain. J Med Chem 2013;56:8915–8930. [DOI] [PubMed] [Google Scholar]
- 37. Clark MP, Ledeboer MW, Davies I, Byrn RA, Jones SM, Perola E, Tsai A, Jacobs M, Nti‐Addae K, Bandarage UK, Boyd MJ, Bethiel RS, Court JJ, Deng H, Duffy JP, Dorsch WA, Farmer LJ, Gao H, Gu W, Jackson K, Jacobs DH, Kennedy JM, Ledford B, Liang J, Maltais F, Murcko M, Wang T, Wannamaker MW, Bennett HB, Leeman JR, McNeil C, Taylor WP, Memmott C, Jiang M, Rijnbrand R, Bral C, Germann U, Nezami A, Zhang Y, Salituro FG, Bennani YL, Charifson PS. Discovery of a novel, first‐in‐class, orally bioavailable azaindole inhibitor (VX‐787) of influenza PB2. J Med Chem 2014;57:6668–6678. [DOI] [PubMed] [Google Scholar]
- 38. Roch FF, Hinterkörner G, Menke J, Tang GQ, Cusack S, Butzendobler B, Buschmann H, Datta K, Wolkerstorfer A. An RNA hybridization assay for screening influenza A virus polymerase inhibitors using the entire ribonucleoprotein complex. Assay Drug Dev Technol 2015;13:488–506. [DOI] [PubMed] [Google Scholar]
- 39. Hooker L, Sully R, Handa B, Ono N, Koyano H, Klumpp K. Quantitative analysis of influenza virus RNP interaction with RNA cap structures and comparison to human cap binding protein eIF4E. Biochemistry 2003;42:6234–6240. [DOI] [PubMed] [Google Scholar]
- 40. Li ML, Rao P, Krug RM. The active sites of the influenza cap‐dependent endonuclease are on different polymerase subunits. EMBO J 2001;20:2078–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shi L, Summers DF, Peng Q, Galarz JM. Influenza A virus RNA polymerase subunit PB2 is the endonuclease which cleaves host cell mRNA and functions only as the trimeric enzyme. Virology 1995;208:38–47. [DOI] [PubMed] [Google Scholar]
- 42. Dias A, Bouvier D, Crépin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. The cap‐snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009;458:914–918. [DOI] [PubMed] [Google Scholar]
- 43. Yuan P, Bartlam M, Lou Z, Chen S, Zhou J, He X, Lv Z, Ge R, Li X, Deng T, Fodor E, Rao Z, Liu Y. Crystal structure of an avian influenza polymerase PA(N) reveals an endonuclease active site. Nature 2009;458:909–913. [DOI] [PubMed] [Google Scholar]
- 44. Zhao C, Lou Z, Guo Y, Ma M, Chen Y, Liang S, Zhang L, Chen S, Li X, Liu Y, Bartlam M, Rao Z. Nucleoside monophosphate complex structures of the endonuclease domain from the influenza virus polymerase PA subunit reveal the substrate binding site inside the catalytic center. J Virol 2009;83:9024–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. DuBois RM, Slavish PJ, Baughman BM, Yun MK, Bao J, Webby RJ, Webb TR, White SW. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog 2012;8:e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kowalinski E, Zubieta C, Wolkerstorfer A, Szolar OH, Ruigrok RW, Cusack S. Structural analysis of specific metal chelating inhibitor binding to the endonuclease domain of influenza pH1N1 (2009) polymerase. PLoS Pathog 2012;8:e1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Parhi AK, Xiang A, Bauman JD, Patel D, Vijayan RS, Das K, Arnold E, Lavoie EJ. Phenyl substituted 3‐hydroxypyridin‐2(1H)‐ones: Inhibitors of influenza A endonuclease. Bioorg Med Chem 2013;21:6435–6446. [DOI] [PubMed] [Google Scholar]
- 48. Tefsen B, Lu G, Zhu Y, Haywood J, Zhao L, Deng T, Qi J, Gao GF. The N‐terminal domain of PA from bat‐derived influenza‐like virus H17N10 has endonuclease activity. J Virol 2014;88:1935–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Song MS, Kumar G, Shadrick WR, Zhou W, Jeevan T, Li Z, Slavish PJ, Fabrizio TP, Yoon SW, Webb TR, Webby RJ, White SW. Identification and characterization of influenza variants resistant to a viral endonuclease inhibitor. Proc Natl Acad Sci USA 2016;113:3669–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bauman JD, Patel D, Baker SF, Vijayan RS, Xiang A, Parhi AK, Martínez‐Sobrido L, Lavoie EJ, Das K, Arnold E. Crystallographic fragment screening and structure‐based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem Biol 2013;8:2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Crépin T, Dias A, Palencia A, Swale C, Cusack S, Ruigrok RW. Mutational and metal binding analysis of the endonuclease domain of the influenza virus polymerase PA subunit. J Virol 2010;84:9096–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Noble E, Cox A, Deval J, Kim B. Endonuclease substrate selectivity characterized with full‐length PA of influenza A virus polymerase. Virology 2012;433:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Maret W. Metalloproteomics, metalloproteomes, and the annotation of metalloproteins. Metallomics 2010;2:117–125. [DOI] [PubMed] [Google Scholar]
- 54. Jahnen‐Dechent W, Ketteler M. Magnesium basics. Clin Kidney J 2012;5:i3–i14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Doan L, Handa B, Roberts NA, Klumpp K. Metal ion catalysis of RNA cleavage by the influenza virus endonuclease. Biochemistry 1999;38:5612–5619. [DOI] [PubMed] [Google Scholar]
- 56. Thierry E, Guilligay D, Kosinski J, Bock T, Gaudon S, Round A, Pflug A, Hengrung N, El Omari K, Baudin F, Hart DJ, Beck M, Cusack S. Influenza polymerase can adopt an alternative configuration involving a radical repacking of PB2 domains. Mol Cell 2016;61:125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Xiao S, Klein ML, LeBard DN, Levine BG, Liang H, MacDermaid CM, Alfonso‐Prieto M. Magnesium‐dependent RNA binding to the PA endonuclease domain of the avian influenza polymerase. J Phys Chem B 2014;118:873–889. [DOI] [PubMed] [Google Scholar]
- 58. Hengrung N, El Omari K, Serna M, I, Vreede FT , Cusack S, Rambo RP, Vonrhein C, Bricogne G, Stuart DI, Grimes JM, Fodor E. Crystal structure of the RNA‐dependent RNA polymerase from influenza C virus. Nature 2015;527:114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Resa‐Infante P, Jorba N, Coloma R, Ortín J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol 2011;8:207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Biswas SK, Nayak DP. Mutational analysis of the conserved motifs of influenza A virus polymerase basic protein 1. J Virol 1994;68:1819–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cianci C, Tiley L, Krystal M. Differential activation of the influenza virus polymerase via template RNA binding. J Virol 1995;69:3995–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rao P, Yuan W, Krug RM. Crucial role of CA cleavage sites in the cap‐snatching mechanism for initiating viral mRNA synthesis. EMBO J 2003;22:1188–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Koppstein D, Ashour J, Bartel DP. Sequencing the cap‐snatching repertoire of H1N1 influenza provides insight into the mechanism of viral transcription initiation. Nucleic Acids Res 2015;43:5052–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gu W, Gallagher GR, Dai W, Liu P, Li R, Trombly MI, Gammon DB, Mello CC, Wang JP, Finberg RW. Influenza A virus preferentially snatches noncoding RNA caps. RNA 2015;21:2067–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Clohisey S, Bertin N, Wise H, Smith N, Hussain S, Tomiou A, Hu J, Consortium Fantom5, Carninci P, Forrest A, Hayashizaki Y, Hume D, Digard P, Baillie K. Terminal dept single‐molecule sequencing of capped transcripts reveals cap‐snatching preferences of influenza A virus in human macrophages. Negative Strand Viruses Meeting; 2015. [Google Scholar]
- 66. Shaw MW, Lamb RA. A specific sub‐set of host‐cell mRNAs prime influenza virus mRNA synthesis. Virus Res 1984;1:455–467. [DOI] [PubMed] [Google Scholar]
- 67. Datta K, Wolkerstorfer A, Szolar OH, Cusack S, Klumpp K. Characterization of PA‐N terminal domain of influenza A polymerase reveals sequence specific RNA cleavage. Nucleic Acids Res 2013;41:8289–8299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Stevaert A, Nurra S, Pala N, Carcelli M, Rogolino D, Shepard C, Domaoal RA, Kim B, Alfonso‐Prieto M, Marras SAE, Sechi M, Naesens L. An integrated biological approach to guide the development of metal‐chelating inhibitors of influenza virus PA endonuclease. Mol Pharmacol 2015;87:323–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sikora D, Rocheleau L, Brown EG, Pelchat M. Deep sequencing reveals the eight facets of the influenza A/HongKong/1/1968 (H3N2) virus cap‐snatching process. Sci Rep 2014;4:6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hagen M, Chung TD, Butcher JA, Krystal M. Recombinant influenza virus polymerase: Requirement of both 5′ and 3′ viral ends for endonuclease activity. J Virol 1994;68:1509–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chung TD, Cianci C, Hagen M, Terry B, Matthews JT, Krystal M, Colonno RJ. Biochemical studies on capped RNA primers identify a class of oligonucleotide inhibitors of the influenza virus RNA polymerase. Proc Natl Acad Sci USA 1994;91:2372–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Geerts‐Dimitriadou C, Zwart MP, Goldbach R, Kormelink R. Base‐pairing promotes leader selection to prime in vitro influenza genome transcription. Virology 2011;409:17–26. [DOI] [PubMed] [Google Scholar]
- 73. Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Hoshino T. Two distinctive binding modes of endonuclease inhibitors to the N‐terminal region of influenza virus polymerase acidic subunit. Biochemistry 2016;55:2646–2660. [DOI] [PubMed] [Google Scholar]
- 74. Stevaert A, Dallocchio R, Dessì A, Pala N, Rogolino D, Sechi M, Naesens L. Mutational analysis of the binding pockets of the diketo acid inhibitor L‐742,001 in the influenza virus PA endonuclease. J Virol 2013;87:10524–10538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Tomassini JE, Davies ME, Hastings JC, Lingham R, Mojena M, Raghoobar SL, Singh SB, Tkacz JS, Goetz MA. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob Agents Chemother 1996;40:1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kuzuhara T, Iwai Y, Takahashi H, Hatakeyama D, Echigo N. Green tea catechins inhibit the endonuclease activity of influenza A virus RNA polymerase. PLoS Curr 2009;1:RRN1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cianci C, Chung TDY, Meanwell N, Putz H, Hagen M, Colonno RJ, Krystal M. Identification of N‐hydroxamic acid and N‐hydroxy‐imide compounds that inhibit the influenza virus polymerase. Antivir Chem Chemother 1996;7:353–360. [Google Scholar]
- 78. Chen E, Swift RV, Alderson N, Feher VA, Feng GS, Amaro RE. Computation‐guided discovery of influenza endonuclease inhibitors. ACS Med Chem Lett 2014;5:61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Yuan S, Chu H, Singh K, Zhao H, Zhang K, Kao RY, Chow BK, Zhou J, Zheng BJ. A novel small‐molecule inhibitor of influenza A virus acts by suppressing PA endonuclease activity of the viral polymerase. Sci Rep 2016;6:22880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Gong P, Peersen OB. Structural basis for active site closure by the poliovirus RNA‐dependent RNA polymerase. Proc Natl Acad Sci USA 2010;107:22505–22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jorba N, Coloma R, Ortín J. Genetic trans‐complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog 2009;5:e1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Furuta Y, Takahashi K, Fukuda Y, Kuno‐Maekawa M, Kamiyama T, Kozaki K, Nomura N, Egawa H, Minami S, Watanabe Y, Narita H, Shiraki K. In vitro and in vivo activities of anti‐influenza virus compound T‐705. Antimicrob Agents Chemother 2002;46:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Naesens L, Guddat LW, Keough DT, van Kuilenburg AB, Meijer J, Vande Voorde J, Balzarini J. Role of human hypoxanthine guanine phosphoribosyltransferase in activation of the antiviral agent T‐705 (favipiravir). Mol Pharmacol 2013;84:615–629. [DOI] [PubMed] [Google Scholar]
- 84. Baranovich T, Wong SS, Armstrong J, Marjuki H, Webby RJ, Webster RG, Govorkova EA. T‐705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro . J Virol 2013;87:3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cheung PP, Watson SJ, Choy KT, Fun Sia S, Wong DD, Poon LL, Kellam P, Guan Y, Malik Peiris JS, Yen HL. Generation and characterization of influenza A viruses with altered polymerase fidelity. Nat Commun 2014;5:4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Sidwell RW, Huffman JH, Khare GP, Allen LB, Witkowski JT, Robins RK. Broad‐spectrum antiviral activity of Virazole: 1‐beta‐D‐ribofuranosyl‐1,2,4‐triazole‐3‐carboxamide. Science 1972;177:705–706. [DOI] [PubMed] [Google Scholar]
- 87. Dong SD, Lin CC, Schroeder M. Synthesis and evaluation of a new phosphorylated ribavirin prodrug. Antiviral Res 2013;99:18–26. [DOI] [PubMed] [Google Scholar]
- 88. Sidwell RW, Bailey KW, Wong MH, Barnard DL, Smee DF. In vitro and in vivo influenza virus‐inhibitory effects of viramidine. Antiviral Res 2005;68:10–17. [DOI] [PubMed] [Google Scholar]
- 89. Pauly MD, Lauring AS. Effective lethal mutagenesis of influenza virus by three nucleoside analogs. J Virol 2015;89:3584–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Binh NT, Wakai C, Kawaguchi A, Nagata K. Involvement of the N‐terminal portion of influenza virus RNA polymerase subunit PB1 in nucleotide recognition. Biochem Biophys Res Commun 2014;443:975–979. [DOI] [PubMed] [Google Scholar]
- 91. De Clercq E, Cools M, Balzarini J, Snoeck R, Andrei G, Hosoya M, Shigeta S, Ueda T, Minakawa N, Matsuda A. Antiviral activities of 5‐ethynyl‐1‐beta‐D‐ribofuranosylimidazole‐4‐carboxamide and related compounds. Antimicrob Agents Chemother 1991;35:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Krajczyk A, Kulinska K, Kulinski T, Hurst BL, Day CW, Smee DF, Ostrowski T, Januszczyk P, Zeidler J. Antivirally active ribavirin analogues–4,5‐disubstituted 1,2,3‐triazole nucleosides: Biological evaluation against certain respiratory viruses and computational modelling. Antivir Chem Chemother 2014;23:161–171. [DOI] [PubMed] [Google Scholar]
- 93. Sidwell RW, Huffman JH, Call EW, Alaghamandan H, Cook PD, Robins RK. Activity of selenazofurin against influenza A and B viruses in vitro . Antimicrob Agents Chemother 1985;28:375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Shigeta S, Konno K, Yokota T, Nakamura K, De Clercq E. Comparative activities of several nucleoside analogs against influenza A, B, and C viruses in vitro . Antimicrob Agents Chemother 1988;32:906–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Colacino JM, DeLong DC, Nelson JR, Spitzer WA, Tang J, Victor F, Wu CY. Evaluation of the anti‐influenza virus activities of 1,3,4‐thiadiazol‐2‐ylcyanamide (LY217896) and its sodium salt. Antimicrob Agents Chemother 1990;34:2156–2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Wang G, Wan J, Hu Y, Wu X, Prhavc M, Dyatkina N, Rajwanshi VK, Smith DB, Jekle A, Kinkade A, Symons JA, Jin Z, Deval J, Zhang Q, Tam Y, Chanda S, Blatt L, Beigelman L. Synthesis and anti‐influenza activity of pyridine, pyridazine, and pyrimidine C‐nucleosides as favipiravir (T‐705) analogues. J Med Chem 2016;59:4611–4624. [DOI] [PubMed] [Google Scholar]
- 97. Gao J, Luo X, Li Y, Gao R, Chen H, Ji D. Synthesis and biological evaluation of 2‐oxo‐pyrazine‐3‐carboxamide‐yl nucleoside analogues and their epimers as inhibitors of influenza A viruses. Chem Biol Drug Des 2015;85:245–252. [DOI] [PubMed] [Google Scholar]
- 98. Tisdale M, Appleyard G, Tuttle JV, Nelson DJ, Nusinoff‐Lehrman S, Al Nakib W, Stables JN, Purifoy DJM, Powell KL, Darby G. Inhibition of influenza A and B viruses by 2′‐deoxy‐2′‐fluororibosides. Antivir Chem Chemother 1993;4:281–287. [Google Scholar]
- 99. Rollins BS, Elkhatieb AH, Hayden FG. Comparative anti‐influenza virus activity of 2′‐deoxy‐2′‐fluororibosides in vitro . Antiviral Res 1993;21:357–368. [DOI] [PubMed] [Google Scholar]
- 100. Abdel‐Magid AF. Influenza RNA‐dependent RNA polymerase (RdRp) inhibitors: potential new therapy for influenza treatment. ACS Med Chem Lett 2013;4:1133–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Baughman BM, Jake Slavish P, DuBois RM, Boyd VA, White SW, Webb TR. Identification of influenza endonuclease inhibitors using a novel fluorescence polarization assay. ACS Chem Biol 2012;7:526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Vedula MS, Jennepalli S, Aryasomayajula R, Rondla SR, Musku MR, Kura RR, Bandi PR. Novel nucleosides as potent influenza viral inhibitors. Bioorg Med Chem 2010;18:6329–6339. [DOI] [PubMed] [Google Scholar]
- 103. Lin C, Sun C, Liu X, Zhou Y, Hussain M, Wan J, Li M, Li X, Jin R, Tu Z, Zhang J. Design, synthesis, and in vitro biological evaluation of novel 6‐methyl‐7‐substituted‐7‐deaza purine nucleoside analogs as anti‐influenza A agents. Antiviral Res 2016;129:13–20. [DOI] [PubMed] [Google Scholar]
- 104. Meneghesso S, Vanderlinden E, Brancale A, Balzarini J, Naesens L, McGuigan C. Synthesis and biological evaluation of purine 2′‐fluoro‐2′‐deoxyriboside ProTides as anti‐influenza virus agents. ChemMedChem 2013;8:415–425. [DOI] [PubMed] [Google Scholar]
- 105. Kumaki Y, Day CW, Smee DF, Morrey JD, Barnard DL. In vitro and in vivo efficacy of fluorodeoxycytidine analogs against highly pathogenic avian influenza H5N1, seasonal, and pandemic H1N1 virus infections. Antiviral Res 2011;92:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Meneghesso S, Vanderlinden E, Stevaert A, McGuigan C, Balzarini J, Naesens L. Synthesis and biological evaluation of pyrimidine nucleoside monophosphate prodrugs targeted against influenza virus. Antiviral Res 2012;94:35–43. [DOI] [PubMed] [Google Scholar]
- 107. Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad‐spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci USA 2011;108:5777–5782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Smee DF, McKernan PA, Nord LD, Willis RC, Petrie CR, Riley TM, Revankar GR, Robins RK, Smith RA. Novel pyrazolo[3,4‐d]pyrimidine nucleoside analog with broad‐spectrum antiviral activity. Antimicrob Agents Chemother 1987;31:1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Shannon WM, Arnett G, Westbrook L, Shealy YF, O'Dell CA, Brockman RW. Evaluation of carbodine, the carbocyclic analog of cytidine, and related carbocyclic analogs of pyrimidine nucleosides for antiviral activity against human influenza type A viruses. Antimicrob Agents Chemother 1981;20:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rao JR, Jha AK, Rawal RK, Sharon A, Day CW, Barnard DL, Smee DF, Chu CK. (‐)‐Carbodine: Enantiomeric synthesis and in vitro antiviral activity against various strains of influenza virus including H5N1 (avian influenza) and novel 2009 H1N1 (swine flu). Bioorg Med Chem Lett 2010;20:2601–2604. [DOI] [PubMed] [Google Scholar]
- 111. Su CY, Cheng TJ, Lin MI, Wang SY, Huang WI, Lin‐Chu SY, Chen YH, Wu CY, Lai MM, Cheng WC, Wu YT, Tsai MD, Cheng YS, Wong CH. High‐throughput identification of compounds targeting influenza RNA‐dependent RNA polymerase activity. Proc Natl Acad Sci USA 2010;107:19151–19156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Ortigoza MB, Dibben O, Maamary J, Martínez‐Gil L, Leyva‐Grado VH, Abreu P Jr, Ayllon J, Palese P, Shaw ML. A novel small molecule inhibitor of influenza A viruses that targets polymerase function and indirectly induces interferon. PLoS Pathog 2012;8:e1002668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Byrn RA, Jones SM, Bennett HB, Bral C, Clark MP, Jacobs MD, Kwong AD, Ledeboer MW, Leeman JR, McNeil CF, Murcko MA, Nezami A, Perola E, Rijnbrand R, Saxena K, Tsai AW, Zhou Y, Charifson PS. Preclinical activity of VX‐787, a first‐in‐class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob Agents Chemother 2015;59:1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Roch FF, Hinterkorner G, Menke J, Tang GQ, Cusack S, Butzendobler B, Buschmann H, Datta K, Wolkerstorfer A. An RNA hybridization assay for screening influenza A virus polymerase inhibitors using the entire ribonucleoprotein complex. Assay Drug Dev Technol 2015;13:488–506. [DOI] [PubMed] [Google Scholar]
- 115. Tomassini JE, Selnick H, Davies ME, Armstrong ME, Baldwin J, Bourgeois M, Hastings JC, Hazuda D, Lewis J, McClements W, Ponticello G, Radzilowski E, Smith G, Tebben A, Wolfe A. Inhibition of cap (m7GpppXm)‐dependent endonuclease of influenza virus by 4‐substituted 2,4‐dioxobutanoic acid compounds. Antimicrob Agents Chemother 1994;38:2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hastings JC, Selnick H, Wolanski B, Tomassini JE. Anti‐influenza virus activities of 4‐substituted 2,4‐dioxobutanoic acid inhibitors. Antimicrob Agents Chemother 1996;40:1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nakazawa M, Kadowaki SE, Watanabe I, Kadowaki Y, Takei M, Fukuda H. PA subunit of RNA polymerase as a promising target for anti‐influenza virus agents. Antiviral Res 2008;78:194–201. [DOI] [PubMed] [Google Scholar]
- 118. Parkes KE, Ermert P, Fässler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J Med Chem 2003;46:1153–1164. [DOI] [PubMed] [Google Scholar]
- 119. Iwai Y, Takahashi H, Hatakeyama D, Motoshima K, Ishikawa M, Sugita K, Hashimoto Y, Harada Y, Itamura S, Odagiri T, Tashiro M, Sei Y, Yamaguchi K, Kuzuhara T. Anti‐influenza activity of phenethylphenylphthalimide analogs derived from thalidomide. Bioorg Med Chem 2010;18:5379–5390. [DOI] [PubMed] [Google Scholar]
- 120. Fudo S, Yamamoto N, Nukaga M, Odagiri T, Tashiro M, Neya S, Hoshino T. Structural and computational study on inhibitory compounds for endonuclease activity of influenza virus polymerase. Bioorg Med Chem 2015;23:5466–5475. [DOI] [PubMed] [Google Scholar]
- 121. Pala N, Stevaert A, Dallocchio R, Dessì A, Rogolino D, Carcelli M, Sanna V, Sechi M, Naesens L. Virtual screening and biological validation of novel influenza virus PA endonuclease inhibitors. ACS Med Chem Lett 2015;6:866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Yamada K, Koyama H, Hagiwara K, Ueda A, Sasaki Y, Kanesashi SN, Ueno R, Nakamura HK, Kuwata K, Shimizu K, Suzuki M, Aida Y. Identification of a novel compound with antiviral activity against influenza A virus depending on PA subunit of viral RNA polymerase. Microbes Infect 2012;14:740–747. [DOI] [PubMed] [Google Scholar]
- 123. Yuan S, Zhang N, Singh K, Shuai H, Chu H, Zhou J, Chow BK, Zheng BJ. Cross‐protection of influenza A virus infection by a DNA aptamer targeting the PA endonuclease domain. Antimicrob Agents Chemother 2015;59:4082–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Kessler U, Castagnolo D, Pagano M, Deodato D, Bernardini M, Pilger B, Ranadheera C, Botta M. Discovery and synthesis of novel benzofurazan derivatives as inhibitors of influenza A virus. Bioorg Med Chem Lett 2013;23:5575–5577. [DOI] [PubMed] [Google Scholar]
- 125. Pagano M, Castagnolo D, Bernardini M, Fallacara AL, Laurenzana I, Deodato D, Kessler U, Pilger B, Stergiou L, Strunze S, Tintori C, Botta M. The fight against the influenza A virus H1N1: Synthesis, molecular modeling, and biological evaluation of benzofurazan derivatives as viral RNA polymerase inhibitors. ChemMedChem 2014;9:129–150. [DOI] [PubMed] [Google Scholar]
- 126. Fukuoka M, Minakuchi M, Kawaguchi A, Nagata K, Kamatari YO, Kuwata K. Structure‐based discovery of anti‐influenza virus A compounds among medicines. Biochim Biophys Acta 2012;1820:90–95. [DOI] [PubMed] [Google Scholar]
- 127. Muratore G, Goracci L, Mercorelli B, Foeglein Á, Digard P, Cruciani G, Palù G, Loregian A. Small molecule inhibitors of influenza A and B viruses that act by disrupting subunit interactions of the viral polymerase. Proc Natl Acad Sci USA 2012;109:6247–6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Muratore G, Mercorelli B, Goracci L, Cruciani G, Digard P, Palù G, Loregian A. Human cytomegalovirus inhibitor AL18 also possesses activity against influenza A and B viruses. Antimicrob Agents Chemother 2012;56:6009–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Massari S, Nannetti G, Goracci L, Sancineto L, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Facchini M, Palù G, Cruciani G, Loregian A, Tabarrini O. Structural investigation of cycloheptathiophene‐3‐carboxamide derivatives targeting influenza virus polymerase assembly. J Med Chem 2013;56:10118–10131. [DOI] [PubMed] [Google Scholar]
- 130. Lepri S, Nannetti G, Muratore G, Cruciani G, Ruzziconi R, Mercorelli B, Palù G, Loregian A, Goracci L. Optimization of small‐molecule inhibitors of influenza virus polymerase: from thiophene‐3‐carboxamide to polyamido scaffolds. J Med Chem 2014;57:4337–4350. [DOI] [PubMed] [Google Scholar]
- 131. Massari S, Nannetti G, Desantis J, Muratore G, Sabatini S, Manfroni G, Mercorelli B, Cecchetti V, Palù G, Cruciani G, Loregian A, Goracci L, Tabarrini O. A broad anti‐influenza hybrid small molecule that potently disrupts the interaction of Polymerase Acidic Protein‐Basic Protein 1 (PA‐PB1) subunits. J Med Chem 2015;58:3830–3842. [DOI] [PubMed] [Google Scholar]
- 132. Trist IM, Nannetti G, Tintori C, Fallacara AL, Deodato D, Mercorelli B, Palù G, Wijtmans M, Gospodova T, Edink E, Verheij M, de Esch I, Viteva L, Loregian A, Botta M. 4,6‐Diphenylpyridines as promising novel anti‐influenza agents targeting the PA‐PB1 protein‐protein interaction: Structure‐activity relationships exploration with the aid of molecular modeling. J Med Chem 2016;59:2688–2703. [DOI] [PubMed] [Google Scholar]
- 133. Yuan S, Chu H, Zhao H, Zhang K, Singh K, Chow BK, Kao RY, Zhou J, Zheng BJ. Identification of a small‐molecule inhibitor of influenza virus via disrupting the subunits interaction of the viral polymerase. Antiviral Res 2016;125:34–42. [DOI] [PubMed] [Google Scholar]
- 134. Kao RY, Yang D, Lau LS, Tsui WH, Hu L, Dai J, Chan MP, Chan CM, Wang P, Zheng BJ, Sun J, Huang JD, Madar J, Chen G, Chen H, Guan Y, Yuen KY. Identification of influenza A nucleoprotein as an antiviral target. Nat Biotechnol 2010;28:600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Gerritz SW, Cianci C, Kim S, Pearce BC, Deminie C, Discotto L, McAuliffe B, Minassian BF, Shi S, Zhu S, Zhai W, Pendri A, Li G, Poss MA, Edavettal S, McDonnell PA, Lewis HA, Maskos K, Mörtl M, Kiefersauer R, Steinbacher S, Baldwin ET, Metzler W, Bryson J, Healy MD, Philip T, Zoeckler M, Schartman R, Sinz M, Leyva‐Grado VH, Hoffmann HH, Langley DR, Meanwell NA, Krystal M. Inhibition of influenza virus replication via small molecules that induce the formation of higher‐order nucleoprotein oligomers. Proc Natl Acad Sci USA 2011;108:15366–15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Cianci C, Gerritz SW, Deminie C, Krystal M. Influenza nucleoprotein: Promising target for antiviral chemotherapy. Antivir Chem Chemother 2013;23:77–91. [DOI] [PubMed] [Google Scholar]
- 137. Lin MI, Su BH, Lee CH, Wang ST, Wu WC, Dangate P, Wang SY, Huang WI, Cheng TJ, Lin OA, Cheng YE, Tseng YJ, Sun CM. Synthesis and inhibitory effects of novel pyrimido‐pyrrolo‐quinoxalinedione analogues targeting nucleoproteins of influenza A virus H1N1. Eur J Med Chem 2015;102:477–486. [DOI] [PubMed] [Google Scholar]
- 138. Shen YF, Chen YH, Chu SY, Lin MI, Hsu HT, Wu PY, Wu CJ, Liu HW, Lin FY, Lin G, Hsu PH, Yang AS, Cheng YS, Wu YT, Wong CH, Tsai MD. E339…R416 salt bridge of nucleoprotein as a feasible target for influenza virus inhibitors. Proc Natl Acad Sci USA 2011;108:16515–16520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Lejal N, Tarus B, Bouguyon E, Chenavas S, Bertho N, Delmas B, Ruigrok RW, Di Primo C, Slama‐Schwok A. Structure‐based discovery of the novel antiviral properties of naproxen against the nucleoprotein of influenza A virus. Antimicrob Agents Chemother 2013;57:2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Fedichev P, Timakhov R, Pyrkov T, Getmantsev E, Vinnik A. Structure‐based drug design of a new chemical class of small molecules active against influenza A nucleoprotein in vitro and in vivo . PLoS Curr 2011;3:RRN1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Kakisaka M, Sasaki Y, Yamada K, Kondoh Y, Hikono H, Osada H, Tomii K, Saito T, Aida Y. A novel antiviral target structure involved in the RNA binding, dimerization, and nuclear export functions of the influenza A virus nucleoprotein. PLoS Pathog 2015;11:e1005062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Hagiwara K, Kondoh Y, Ueda A, Yamada K, Goto H, Watanabe T, Nakata T, Osada H, Aida Y. Discovery of novel antiviral agents directed against the influenza A virus nucleoprotein using photo‐cross‐linked chemical arrays. Biochem Biophys Res Commun 2010;394:721–727. [DOI] [PubMed] [Google Scholar]
- 143. Leneva IA, Fediakina IT, Eropkin MI, Gudova NV, Romanovskaia AA, Danilenko DM, Vinogradova SM, Lepeshkin AI, Shestopalov AM. [Study of the antiviral activity of Russian anti‐influenza agents in cell culture and animal models]. Vopr Virusol 2010;55:19–27. [PubMed] [Google Scholar]
- 144. Semenova NP, Prokudina EN, Livov DK, Nebol'sin VE. Effect of the antiviral drug Ingaviruin on intracellular transformations and import into the nucleus of influenza A virus nucleocapsid protein. Vopr Virusol 2010;55:17–20. [PubMed] [Google Scholar]
- 145. Zarubaev VV, Garshinina AV, Kalinina NA, Shtro AA, Belyaevskaya SV, Slita AV, Nebolsin VE, Kiselev OI. Activity of Ingavirin (6‐[2‐(1H‐imidazol‐4‐yl)ethylamino]‐5‐oxo‐hexanoic acid) against human respiratory viruses in in vivo experiments. Pharmaceuticals 2011;4:1518–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Iversen P, Schnell F, Crumley S, Mourich D. Post exposure efficacy of AVI‐7100 against influenza A in mouse and ferret infection models. 22nd European Congress of Clinical Microbiology and Infectious Diseases; 2012.
- 147. Perwitasari O, Johnson S, Yan X, Howerth E, Shacham S, Landesman Y, Baloglu E, McCauley D, Tamir S, Tompkins SM, Tripp RA. Verdinexor, a novel selective inhibitor of nuclear export, reduces influenza a virus replication in vitro and in vivo . J Virol 2014;88:10228–10243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Hsu JT, Yeh JY, Lin TJ, Li ML, Wu MS, Hsieh CF, Chou YC, Tang WF, Lau KS, Hung HC, Fang MY, Ko S, Hsieh HP, Horng JT. Identification of BPR3P0128 as an inhibitor of cap‐snatching activities of influenza virus. Antimicrob Agents Chemother 2012;56:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lee MK, Bottini A, Kim M, Bardaro MF, Jr , Zhang Z, Pellecchia M, Choi BS, Varani G. A novel small‐molecule binds to the influenza A virus RNA promoter and inhibits viral replication. Chem Commun (Camb) 2014;50:368–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee G. Hsp90 inhibitors reduce influenza virus replication in cell culture. Virology 2008;377:431–439. [DOI] [PubMed] [Google Scholar]
- 151. Ozawa M, Shimojima M, Goto H, Watanabe S, Hatta Y, Kiso M, Furuta Y, Horimoto T, Peters NR, Hoffmann FM, Kawaoka Y. A cell‐based screening system for influenza A viral RNA transcription/replication inhibitors. Sci Rep 2013;3:1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Pascua PN, Lee JH, Song MS, Park SJ, Baek YH, Ann BH, Shin EY, Kim EG, Choi YK. Role of the p21‐activated kinases (PAKs) in influenza A virus replication. Biochem Biophys Res Commun 2011;414:569–574. [DOI] [PubMed] [Google Scholar]
- 153. Droebner K, Pleschka S, Ludwig S, Planz O. Antiviral activity of the MEK‐inhibitor U0126 against pandemic H1N1v and highly pathogenic avian influenza virus in vitro and in vivo . Antiviral Res 2011;92:195–203. [DOI] [PubMed] [Google Scholar]
- 154. Haasbach E, Hartmayer C, Planz O. Combination of MEK inhibitors and oseltamivir leads to synergistic antiviral effects after influenza A virus infection in vitro . Antiviral Res 2013;98:319–324. [DOI] [PubMed] [Google Scholar]
- 155. Sasaki Y, Kakisaka M, Chutiwitoonchai N, Tajima S, Hikono H, Saito T, Aida Y. Identification of a novel multiple kinase inhibitor with potent antiviral activity against influenza virus by reducing viral polymerase activity. Biochem Biophys Res Commun 2014;450:49–54. [DOI] [PubMed] [Google Scholar]
- 156. Mazur I, Wurzer WJ, Ehrhardt C, Pleschka S, Puthavathana P, Silberzahn T, Wolff T, Planz O, Ludwig S. Acetylsalicylic acid (ASA) blocks influenza virus propagation via its NF‐kappaB‐inhibiting activity. Cell Microbiol 2007;9:1683–1694. [DOI] [PubMed] [Google Scholar]
- 157. Wiesener N, Zimmer C, Jarasch‐Althof N, Wutzler P, Henke A. Therapy of experimental influenza virus infection with pyrrolidine dithiocarbamate. Med Microbiol Immunol 2011;200:115–126. [DOI] [PubMed] [Google Scholar]
- 158. Ehrhardt C, Rückle A, Hrincius ER, Haasbach E, Anhlan D, Ahmann K, Banning C, Reiling SJ, Kühn J, Strobl S, Vitt D, Leban J, Planz O, Ludwig S. The NF‐kappaB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol 2013;15:1198–1211. [DOI] [PubMed] [Google Scholar]
- 159. Pinto R, Herold S, Cakarova L, Hoegner K, Lohmeyer J, Planz O, Pleschka S. Inhibition of influenza virus‐induced NF‐kappaB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo . Antiviral Res 2011;92:45–56. [DOI] [PubMed] [Google Scholar]
- 160. Dierkes R, Warnking K, Liedmann S, Seyer R, Ludwig S, Ehrhardt C. The Rac1 inhibitor NSC23766 exerts anti‐influenza virus properties by affecting the viral polymerase complex activity. PLoS One 2014;9:e88520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Tisdale M, Ellis M, Klumpp K, Court S, Ford M. Inhibition of influenza virus transcription by 2′‐deoxy‐2′‐fluoroguanosine. Antimicrob Agents Chemother 1995;39:2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Sugiyama K, Kawaguchi A, Okuwaki M, Nagata K. pp32 and APRIL are host cell‐derived regulators of influenza virus RNA synthesis from cRNA. Elife 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Vreede FT, Chan AY, Sharps J, Fodor E. Mechanisms and functional implications of the degradation of host RNA polymerase II in influenza virus infected cells. Virology 2010;396:125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Watanabe T, Kawaoka Y. Influenza virus‐host interactomes as a basis for antiviral drug development. Curr Opin Virol 2015;14:71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Cohen A, Togo Y, Khakoo R, Waldman R, Sigel M. Comparative clinical and laboratory evaluation of the prophylactic capacity of ribavirin, amantadine hydrochloride, and placebo in induced human influenza type A. J Infect Dis 1976;133(Suppl):A114–A120. [DOI] [PubMed] [Google Scholar]
- 166. Stein DS, Creticos CM, Jackson GG, Bernstein JM, Hayden FG, Schiff GM, Bernstein DI. Oral ribavirin treatment of influenza A and B. Antimicrob Agents Chemother 1987;31:1285–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ilyushina NA, Hay A, Yilmaz N, Boon AC, Webster RG, Govorkova EA. Oseltamivir‐ribavirin combination therapy for highly pathogenic H5N1 influenza virus infection in mice. Antimicrob Agents Chemother 2008;52:3889–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. NIH . National Library of Medicine (US). Hazardous Substances Data Bank. Ribavirin. Hazardous Substances Databank Number: 6513. http://toxnet.nlm.nih.gov/.
- 169. van der Vries E, Stelma FF, Boucher CA. Emergence of a multidrug‐resistant pandemic influenza A (H1N1) virus. N Engl J Med 2010;363:1381–1382. [DOI] [PubMed] [Google Scholar]
- 170. Willis RC, Carson DA, Seegmiller JE. Adenosine kinase initiates the major route of ribavirin activation in a cultured human cell line. Proc Natl Acad Sci USA 1978;75:3042–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Wu JZ, Larson G, Walker H, Shim JH, Hong Z. Phosphorylation of ribavirin and viramidine by adenosine kinase and cytosolic 5′‐nucleotidase II: Implications for ribavirin metabolism in erythrocytes. Antimicrob Agents Chemother 2005;49:2164–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Streeter DG, Witkowski JT, Khare GP, Sidwell RW, Bauer RJ, Robins RK, Simon LN. Mechanism of action of 1‐beta‐D‐ribofuranosyl‐1,2,4‐triazole‐3‐carboxamide (Virazole), a new broad‐spectrum antiviral agent. Proc Natl Acad Sci USA 1973;70:1174–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Wray SK, Gilbert BE, Noall MW, Knight V. Mode of action of ribavirin: Effect of nucleotide pool alterations on influenza virus ribonucleoprotein synthesis. Antiviral Res 1985;5:29–37. [DOI] [PubMed] [Google Scholar]
- 174. Robins RK, Revankar GR, McKernan PA, Murray BK, Kirsi JJ, North JA. The importance of IMP dehydrogenase inhibition in the broad spectrum antiviral activity of ribavirin and selenazofurin. Adv Enzyme Regul 1985;24:29–43. [DOI] [PubMed] [Google Scholar]
- 175. Scholtissek C. Inhibition of influenza RNA synthesis by virazole (ribavirin). Arch Virol 1976;50:349–352. [DOI] [PubMed] [Google Scholar]
- 176. Eriksson B, Helgstrand E, Johansson NG, Larsson A, Misiorny A, Norén JO, Philipson L, Stenberg K, Stening G, Stridh S, Oberg B. Inhibition of influenza virus ribonucleic acid polymerase by ribavirin triphosphate. Antimicrob Agents Chemother 1977;11:946–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Graci JD, Cameron CE. Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol 2006;16:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Witkowski JT, Robins RK, Khare GP, Sidwell RW. Synthesis and antiviral activity of 1,2,4‐triazole‐3‐thiocarboxamide and 1,2,4‐triazole‐3‐carboxamidine ribonucleosides. J Med Chem 1973;16:935–937. [DOI] [PubMed] [Google Scholar]
- 179. De Clercq E, Luczak M. Fluoroimidazoles as antiviral agents and inhibitors of polynucleotide biosynthesis. Life Sci 1975;17:187–194. [DOI] [PubMed] [Google Scholar]
- 180. Minakawa N, Matsuda A. Mechanism‐based design of inosine 5‐monophosphate dehydrogenase inhibitors: Synthesis and biological activities of 5‐ethynyl‐1‐beta‐D‐ribofuranosylimidazole‐4‐carboxamide (EICAR) and its derivatives. Curr Med Chem 1999;6:615–628. [PubMed] [Google Scholar]
- 181. Birch GM, Colacino JM, Ehlhardt WJ, Balzarini J. The intracellular formation of a mononucleotide of the anti‐influenza agent 1,3,4‐thiadiazol‐2‐ylcyanamide (LY217896). Antivir Chem Chemother 1995;6:127–137. [Google Scholar]
- 182. Hayden FG, Tunkel AR, Treanor JJ, Betts RF, Allerheiligen S, Harris J. Oral LY217896 for prevention of experimental influenza A virus infection and illness in humans. Antimicrob Agents Chemother 1994;38:1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Zumla A, Memish ZA, Maeurer M, Bates M, Mwaba P, Al‐Tawfiq JA, Denning DW, Hayden FG, Hui DS. Emerging novel and antimicrobial‐resistant respiratory tract infections: New drug development and therapeutic options. Lancet Infect Dis 2014;14:1136–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Itoh Y, Shinya K, Kiso M, Watanabe T, Sakoda Y, Hatta M, Muramoto Y, Tamura D, Sakai‐Tagawa Y, Noda T, Sakabe S, Imai M, Hatta Y, Watanabe S, Li C, Yamada S, Fujii K, Murakami S, Imai H, Kakugawa S, Ito M, Takano R, Iwatsuki‐Horimoto K, Shimojima M, Horimoto T, Goto H, Takahashi K, Makino A, Ishigaki H, Nakayama M, Okamatsu M, Takahashi K, Warshauer D, Shult PA, Saito R, Suzuki H, Furuta Y, Yamashita M, Mitamura K, Nakano K, Nakamura M, Brockman‐Schneider R, Mitamura H, Yamazaki M, Sugaya N, Suresh M, Ozawa M, Neumann G, Gern J, Kida H, Ogasawara K, Kawaoka Y. In vitro and in vivo characterization of new swine‐origin H1N1 influenza viruses. Nature 2009;460:1021–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Kiso M, Takahashi K, Sakai‐Tagawa Y, Shinya K, Sakabe S, Le QM, Ozawa M, Furuta Y, Kawaoka Y. T‐705 (favipiravir) activity against lethal H5N1 influenza A viruses. Proc Natl Acad Sci USA 2010;107:882–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Cao RY, Xiao JH, Cao B, Li S, Kumaki Y, Zhong W. Inhibition of novel reassortant avian influenza H7N9 virus infection in vitro with three antiviral drugs, oseltamivir, peramivir and favipiravir. Antivir Chem Chemother 2013. [DOI] [PubMed] [Google Scholar]
- 187. Sleeman K, Mishin VP, Deyde VM, Furuta Y, Klimov AI, Gubareva LV. In vitro antiviral activity of favipiravir (T‐705) against drug‐resistant influenza and 2009 A(H1N1) viruses. Antimicrob Agents Chemother 2010;54:2517–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Furuta Y, Takahashi K, Kuno‐Maekawa M, Sangawa H, Uehara S, Kozaki K, Nomura N, Egawa H, Shiraki K. Mechanism of action of T‐705 against influenza virus. Antimicrob Agents Chemother 2005;49:981–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Jin Z, Smith LK, Rajwanshi VK, Kim B, Deval J. The ambiguous base‐pairing and high substrate efficiency of T‐705 (favipiravir) ribofuranosyl 5′‐triphosphate towards influenza A virus polymerase. PLoS One 2013;8:e68347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Sangawa H, Komeno T, Nishikawa H, Yoshida A, Takahashi K, Nomura N, Furuta Y. Mechanism of action of T‐705 ribosyl triphosphate against influenza virus RNA polymerase. Antimicrob Agents Chemother 2013;57:5202–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Furuta Y, Takahashi K, Shiraki K, Sakamoto K, Smee DF, Barnard DL, Gowen BB, Julander JG, Morrey JD. T‐705 (favipiravir) and related compounds: Novel broad‐spectrum inhibitors of RNA viral infections. Antiviral Res 2009;82:95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Furuta Y, Gowen BB, Takahashi K, Shiraki K, Smee DF, Barnard DL. Favipiravir (T‐705), a novel viral RNA polymerase inhibitor. Antiviral Res 2013;100:446–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Jakeman KJ, Tisdale M, Russell S, Leone A, Sweet C. Efficacy of 2′‐deoxy‐2′‐fluororibosides against influenza A and B viruses in ferrets. Antimicrob Agents Chemother 1994;38:1864–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Mehellou Y, Balzarini J, McGuigan C. Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009;4:1779–1791. [DOI] [PubMed] [Google Scholar]
- 195. Stuyver LJ, McBrayer TR, Whitaker T, Tharnish PM, Ramesh M, Lostia S, Cartee L, Shi J, Hobbs A, Schinazi RF, Watanabe KA, Otto MJ. Inhibition of the subgenomic hepatitis C virus replicon in huh‐7 cells by 2′‐deoxy‐2′‐fluorocytidine. Antimicrob Agents Chemother 2004;48:651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J, Yadav B, Aittokallio T, Kaukinen P, Ahola T, Kuivanen S, Vapalahti O, Kantele A, Tynell J, Julkunen I, Kallio‐Kokko H, Paavilainen H, Hukkanen V, Elliott RM, De Brabander JK, Saelens X, Kainov DE. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza A virus infection. J Biol Chem 2012;287:35324–35332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Shannon WM, Arnett G, Westbrook L, Shealy YF, O'Dell CA, Brockman RW. Evaluation of carbodine, the carbocyclic analog of cytidine, and related carbocyclic analogs of pyrimidine nucleosides for antiviral activity against human influenza type A viruses. Antimicrob Agents Chemother 1981;20:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. De Clercq E, Bernaerts R, Shealy YF, Montgomery JA. Broad‐spectrum antiviral activity of carbodine, the carbocyclic analogue of cytidine. Biochem Pharmacol 1990;39:319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. De Clercq E, Murase J, Marquez VE. Broad‐spectrum antiviral and cytocidal activity of cyclopentenylcytosine, a carbocyclic nucleoside targeted at CTP synthetase. Biochem Pharmacol 1991;41:1821–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Shealy YF, O'Dell CA, Arnett G. Synthesis and antiviral evaluation of carbocyclic analogues of 2‐amino‐6‐substituted‐purine 3′‐deoxyribofuranosides. J Med Chem 1987;30:1090–1094. [DOI] [PubMed] [Google Scholar]
- 201. Streeter DG, Koyama HH. Inhibition of purine nucleotide biosynthesis by 3‐deazaguanine, its nucleoside and 5′‐nucleotide. Biochem Pharmacol 1976;25:2413–2415. [DOI] [PubMed] [Google Scholar]
- 202. Fung A, Jin Z, Dyatkina N, Wang G, Beigelman L, Deval J. Efficiency of incorporation and chain termination determines the inhibition potency of 2′‐modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob Agents Chemother 2014;58:3636–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Sofia MJ, Chang W, Furman PA, Mosley RT, Ross BS. Nucleoside, nucleotide, and non‐nucleoside inhibitors of hepatitis C virus NS5B RNA‐dependent RNA‐polymerase. J Med Chem 2012;55:2481–2531. [DOI] [PubMed] [Google Scholar]
- 204. Wang G, Deval J, Hong J, Dyatkina N, Prhavc M, Taylor J, Fung A, Jin Z, Stevens SK, Serebryany V, Liu J, Zhang Q, Tam Y, Chanda SM, Smith DB, Symons JA, Blatt LM, Beigelman L. Discovery of 4′‐chloromethyl‐2′‐deoxy‐3′,5′‐di‐O‐isobutyryl‐2′‐fluorocytidine (ALS‐8176), a first‐in‐class RSV polymerase inhibitor for treatment of human respiratory syncytial virus infection. J Med Chem 2015;58:1862–1878. [DOI] [PubMed] [Google Scholar]
- 205. de la Luna S, Martínez C, Ortín J. Molecular cloning and sequencing of influenza virus A/Victoria/3/75 polymerase genes: Sequence evolution and prediction of possible functional domains. Virus Res 1989;13:143–155. [DOI] [PubMed] [Google Scholar]
- 206. Boyd MJ, Bandarage UK, Bennett H, Byrn RR, Davies I, Gu W, Jacobs M, Ledeboer MW, Ledford B, Leeman JR, Perola E, Wang T, Bennani Y, Clark MP, Charifson PS. Isosteric replacements of the carboxylic acid of drug candidate VX‐787: Effect of charge on antiviral potency and kinase activity of azaindole‐based influenza PB2 inhibitors. Bioorg Med Chem Lett 2015;25:1990–1994. [DOI] [PubMed] [Google Scholar]
- 207. Byrn RA, Jones SM, Bennett HB, Bral C, Clark MP, Jacobs MD, Kwong AD, Ledeboer MW, Leeman JR, McNeil CF, Murcko MA, Nezami A, Perola E, Rijnbrand R, Saxena K, Tsai AW, Zhou Y, Charifson PS. Preclinical activity of VX‐787, a first‐in‐class, orally bioavailable inhibitor of the influenza virus polymerase PB2 subunit. Antimicrob Agents Chemother 2015;59:1569–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Severin C, Rocha de Moura T, Liu Y, Li K, Zheng X, Luo M. The cap‐binding site of influenza virus protein PB2 as a drug target. Acta Crystallogr D Struct Biol 2016;72:245–253. [DOI] [PubMed] [Google Scholar]
- 209. Metifiot M, Marchand C, Pommier Y. HIV integrase inhibitors: 20‐Year landmark and challenges. Adv Pharmacol 2013;67:75–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Kirschberg T, Parrish J. Metal chelators as antiviral agents. Curr Opin Drug Discov Dev 2007;10:460–472. [PubMed] [Google Scholar]
- 211. Blair W, Cox C. Current landscape of antiviral drug discovery. F1000Research 2016;5:F1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Singh SB, Tomassini JE. Synthesis of natural flutimide and analogous fully substituted pyrazine‐2,6‐diones, endonuclease inhibitors of influenza virus. J Org Chem 2001;66:5504–5516. [DOI] [PubMed] [Google Scholar]
- 213. Iwai Y, Murakami K, Gomi Y, Hashimoto T, Asakawa Y, Okuno Y, Ishikawa T, Hatakeyama D, Echigo N, Kuzuhara T. Anti‐influenza activity of marchantins, macrocyclic bisbibenzyls contained in liverworts. PLoS One 2011;6:e19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Shoji M, Takahashi E, Hatakeyama D, Iwai Y, Morita Y, Shirayama R, Echigo N, Kido H, Nakamura S, Mashino T, Okutani T, Kuzuhara T. Anti‐influenza activity of C60 fullerene derivatives. PLoS One 2013;8:e66337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Sagong HY, Parhi A, Bauman JD, Patel D, Vijayan RSK, Das K, Arnold E, LaVoie EJ. 3‐hydroxyquinolin‐2(1H)‐ones as inhibitors of influenza A endonuclease. ACS Med Chem Lett 2013;4:547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Sagong HY, Bauman JD, Patel D, Das K, Arnold E, Lavoie EJ. Phenyl substituted 4‐hydroxypyridazin‐3(2H)‐ones and 5‐hydroxypyrimidin‐4(3H)‐ones: Inhibitors of influenza A endonuclease. J Med Chem 2014;57:8086–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Carcelli M, Rogolino D, Bacchi A, Rispoli G, Fisicaro E, Compari C, Sechi M, Stevaert A, Naesens L. Metal‐chelating 2‐hydroxyphenyl amide pharmacophore for inhibition of influenza virus endonuclease. Mol Pharm 2014;11:304–316. [DOI] [PubMed] [Google Scholar]
- 218. Rogolino D, Bacchi A, De Luca L, Rispoli G, Sechi M, Stevaert A, Naesens L, Carcelli M. Investigation of the salicylaldehyde thiosemicarbazone scaffold for inhibition of influenza virus PA endonuclease. J Biol Inorg Chem 2015;20:1109–1121. [DOI] [PubMed] [Google Scholar]
- 219. Rogolino D, Carcelli M, Sechi M, Neamati N. Viral enzymes containing magnesium: Metal binding as a successful strategy in drug design. Coord Chem Rev 2012;256:3063–3086. [Google Scholar]
- 220. Marchand C, Johnson AA, Karki RG, Pais GC, Zhang X, Cowansage K, Patel TA, Nicklaus MC, Burke TR, Jr , Pommier Y. Metal‐dependent inhibition of HIV‐1 integrase by beta‐diketo acids and resistance of the soluble double‐mutant (F185K/C280S). Mol Pharmacol 2003;64:600–609. [DOI] [PubMed] [Google Scholar]
- 221. Bock CW, Kaufman Katz A, Markham GD, Glusker JP. Manganese as a replacement for magnesium and zinc: Functional comparison of the divalent ions. J Am Chem Soc 1999;121:7360–7372. [Google Scholar]
- 222. Tchertanov L, Mouscadet JF. Target recognition by catechols and beta‐ketoenols: Potential contribution of hydrogen bonding and Mn/Mg chelation to HIV‐1 integrase inhibition. J Med Chem 2007;50:1133–1145. [DOI] [PubMed] [Google Scholar]
- 223. He X, Zhou J, Bartlam M, Zhang R, Ma J, Lou Z, Li X, Li J, Joachimiak A, Zeng Z, Ge R, Rao Z, Liu Y. Crystal structure of the polymerase PA(C)‐PB1(N) complex from an avian influenza H5N1 virus. Nature 2008;454:1123–1126. [DOI] [PubMed] [Google Scholar]
- 224. Moen SO, Abendroth J, Fairman JW, Baydo RO, Bullen J, Kirkwood JL, Barnes SR, Raymond AC, Begley DW, Henkel G, McCormack K, Tam VC, Phan I, Staker BL, Stacy R, Myler PJ, Lorimer D, Edwards TE. Structural analysis of H1N1 and H7N9 influenza A virus PA in the absence of PB1. Sci Rep 2014;4:5944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Sugiyama K, Obayashi E, Kawaguchi A, Suzuki Y, Tame JR, Nagata K, Park SY. Structural insight into the essential PB1‐PB2 subunit contact of the influenza virus RNA polymerase. EMBO J 2009;28:1803–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Obayashi E, Yoshida H, Kawai F, Shibayama N, Kawaguchi A, Nagata K, Tame JR, Park SY. The structural basis for an essential subunit interaction in influenza virus RNA polymerase. Nature 2008;454:1127–1131. [DOI] [PubMed] [Google Scholar]
- 227. Rognan D. Rational design of protein‐protein interaction inhibitors. Med Chem Commun 2015;6:51–60. [Google Scholar]
- 228. Ghanem A, Mayer D, Chase G, Tegge W, Frank R, Kochs G, García‐Sastre A, Schwemmle M. Peptide‐mediated interference with influenza A virus polymerase. J Virol 2007;81:7801–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Wunderlich K, Mayer D, Ranadheera C, Holler AS, Mänz B, Martin A, Chase G, Tegge W, Frank R, Kessler U, Schwemmle M. Identification of a PA‐binding peptide with inhibitory activity against influenza A and B virus replication. PLoS One 2009;4:e7517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wunderlich K, Juozapaitis M, Ranadheera C, Kessler U, Martin A, Eisel J, Beutling U, Frank R, Schwemmle M. Identification of high‐affinity PB1‐derived peptides with enhanced affinity to the PA protein of influenza A virus polymerase. Antimicrob Agents Chemother 2011;55:696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Tintori C, Laurenzana I, Fallacara AL, Kessler U, Pilger B, Stergiou L, Botta M. High‐throughput docking for the identification of new influenza A virus polymerase inhibitors targeting the PA‐PB1 protein‐protein interaction. Bioorg Med Chem Lett 2014;24:280–282. [DOI] [PubMed] [Google Scholar]
- 232. Massari S, Goracci L, Desantis J, Tabarrini O. Polymerase acidic protein‐basic protein 1 (PA‐PB1) protein‐protein interaction as a target for next‐generation anti‐influenza therapeutics. J Med Chem 2016. In press. [DOI] [PubMed] [Google Scholar]
- 233. Chase G, Wunderlich K, Reuther P, Schwemmle M. Identification of influenza virus inhibitors which disrupt of viral polymerase protein‐protein interactions. Methods 2011;55:188–191. [DOI] [PubMed] [Google Scholar]
- 234. Li C, Ba Q, Wu A, Zhang H, Deng T, Jiang T. A peptide derived from the C‐terminus of PB1 inhibits influenza virus replication by interfering with viral polymerase assembly. FEBS J 2013;280:1139–1149. [DOI] [PubMed] [Google Scholar]
- 235. Reuther P, Mänz B, Brunotte L, Schwemmle M, Wunderlich K. Targeting of the influenza A virus polymerase PB1‐PB2 interface indicates strain‐specific assembly differences. J Virol 2011;85:13298–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.U.S. National Library of Medicine. Information on clinical trials and human research studies: Testing the AVI‐7100 flu drug in healthy volunteers. https://clinicaltrials.gov/ct2/show/NCT01747148.
- 237. Barik S. siRNA for influenza therapy. Viruses 2010;2:1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Ge Q, McManus MT, Nguyen T, Shen CH, Sharp PA, Eisen HN, Chen J. RNA interference of influenza virus production by directly targeting mRNA for degradation and indirectly inhibiting all viral RNA transcription. Proc Natl Acad Sci USA 2003;100:2718–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Ge Q, Filip L, Bai A, Nguyen T, Eisen HN, Chen J. Inhibition of influenza virus production in virus‐infected mice by RNA interference. Proc Natl Acad Sci USA 2004;101:8676–8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Lin L, Liu Q, Berube N, Detmer S, Zhou Y. 5′‐Triphosphate‐short interfering RNA: Potent inhibition of influenza A virus infection by gene silencing and RIG‐I activation. J Virol 2012;86:10359–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Xu F, Liu G, Liu Q, Zhou Y. RNA interference of influenza A virus replication by microRNA‐adapted lentiviral loop short hairpin RNA. J Gen Virol 2015;96:2971–2981. [DOI] [PubMed] [Google Scholar]
- 242. Linke LM, Wilusz J, Pabilonia KL, Fruehauf J, Magnuson R, Olea‐Popelka F, Triantis J, Landolt G, Salman M. Inhibiting avian influenza virus shedding using a novel RNAi antiviral vector technology: Proof of concept in an avian cell model. AMB Express 2016;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Biswas SK, Boutz PL, Nayak DP. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J Virol 1998;72:5493–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Poole E, Elton D, Medcalf L, Digard P. Functional domains of the influenza A virus PB2 protein: Identification of NP‐ and PB1‐binding sites. Virology 2004;321:120–133. [DOI] [PubMed] [Google Scholar]
- 245. Marklund JK, Ye Q, Dong J, Tao YJ, Krug RM. Sequence in the influenza A virus nucleoprotein required for viral polymerase binding and RNA synthesis. J Virol 2012;86:7292–7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Ye Q, Krug RM, Tao YJ. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature 2006;444:1078–1082. [DOI] [PubMed] [Google Scholar]
- 247. Ng AK, Zhang H, Tan K, Li Z, Liu JH, Chan PK, Li SM, Chan WY, Au SW, Joachimiak A, Walz T, Wang JH, Shaw PC. Structure of the influenza virus A H5N1 nucleoprotein: Implications for RNA binding, oligomerization, and vaccine design. FASEB J 2008;22:3638–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Chan WH, Ng AK, Robb NC, Lam MK, Chan PK, Au SW, Wang JH, Fodor E, Shaw PC. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomerization and ribonucleoprotein activities. J Virol 2010;84:7337–7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Baudin F, Bach C, Cusack S, Ruigrok RW. Structure of influenza virus RNP. I. Influenza virus nucleoprotein melts secondary structure in panhandle RNA and exposes the bases to the solvent. EMBO J 1994;13:3158–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Tarus B, Bertrand H, Zedda G, Di PC, Quideau S, Slama‐Schwok A. Structure‐based design of novel naproxen derivatives targeting monomeric nucleoprotein of influenza A virus. J Biomol Struct Dyn 2015;33:1899–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251. Cheng H, Wan J, Lin MI, Liu Y, Lu X, Liu J, Xu Y, Chen J, Tu Z, Cheng YS, Ding K. Design, synthesis, and in vitro biological evaluation of 1H‐1,2,3‐triazole‐4‐carboxamide derivatives as new anti‐influenza A agents targeting virus nucleoprotein. J Med Chem 2012;55:2144–2153. [DOI] [PubMed] [Google Scholar]
- 252. Amorim MJ, Kao RY, Digard P. Nucleozin targets cytoplasmic trafficking of viral ribonucleoprotein‐Rab11 complexes in influenza A virus infection. J Virol 2013;87:4694–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Zarubaev VV, Beliaevskaia SV, Sirotkin AK, Anfimov PM, Nebol'sin VE, Kiselev OI, Reikhart DV. In vitro and in vivo effects of Ingavirin on the ultrastructure and infectivity of influenza virus. Vopr Virusol 2011;56:21–25. [PubMed] [Google Scholar]
- 254. Watanabe T, Kawakami E, Shoemaker JE, Lopes TJ, Matsuoka Y, Tomita Y, Kozuka‐Hata H, Gorai T, Kuwahara T, Takeda E, Nagata A, Takano R, Kiso M, Yamashita M, Sakai‐Tagawa Y, Katsura H, Nonaka N, Fujii H, Fujii K, Sugita Y, Noda T, Goto H, Fukuyama S, Watanabe S, Neumann G, Oyama M, Kitano H, Kawaoka Y. Influenza virus‐host interactome screen as a platform for antiviral drug development. Cell Host Microbe 2014;16:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Ludwig S, Zell R, Schwemmle M, Herold S. Influenza, a One Health paradigm—Novel therapeutic strategies to fight a zoonotic pathogen with pandemic potential. Int J Med Microbiol 2014;304:894–901. [DOI] [PubMed] [Google Scholar]
- 256. Watanabe T, Watanabe S, Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host Microbe 2010;7:427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Stertz S, Shaw ML. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect 2011;13:516–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Liu Q, Zhou YH, Yang ZQ. The cytokine storm of severe influenza and development of immunomodulatory therapy. Cell Mol Immunol 2016;13:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259. Elton D, Simpson‐Holley M, Archer K, Medcalf L, Hallam R, McCauley J, Digard P. Interaction of the influenza virus nucleoprotein with the cellular CRM1‐mediated nuclear export pathway. J Virol 2001;75:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.U.S. National Library of Medicine. Information on clinical trials and human research studies: Trial of safety and tolerability of oral Verdinexor (KPT‐335) in healthy adults. https://clinicaltrials.gov/ct2/show/NCT02431364.
- 261. Naito T, Momose F, Kawaguchi A, Nagata K. Involvement of Hsp90 in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 2007;81:1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Swale C, Monod A, Tengo L, Labaronne A, Garzoni F, Bourhis JM, Cusack S, Schoehn G, Berger I, Ruigrok RW, Crepin T. Structural characterization of recombinant IAV polymerase reveals a stable complex between viral PA‐PB1 heterodimer and host RanBP5. Sci Rep 2016;6:24727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Pleschka S, Wolff T, Ehrhardt C, Hobom G, Planz O, Rapp UR, Ludwig S. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol 2001;3:301–305. [DOI] [PubMed] [Google Scholar]
- 264. Haasbach E, Reiling SJ, Ehrhardt C, Droebner K, Rückle A, Hrincius ER, Leban J, Strobl S, Vitt D, Ludwig S, Planz O. The NF‐kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza A virus. Antiviral Res 2013;99:336–344. [DOI] [PubMed] [Google Scholar]
- 265. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem 2004;25:1605‐1612. [DOI] [PubMed] [Google Scholar]