

SUMMARY
Remodeling of the tricarboxylic acid (TCA) cycle is a metabolic adaptation accompanying inflammatory macrophage activation. During this process, endogenous metabolites can adopt regulatory roles that govern specific aspects of inflammatory response, as recently shown for succinate, which regulates the pro-inflammatory IL-1β-HIF-1α axis. Itaconate is one of the most highly induced metabolites in activated macrophages, yet its functional significance remains unknown. Here, we show that itaconate modulates macrophage metabolism and effector functions by inhibiting succinate dehydrogenase-mediated oxidation of succinate. Through this action, itaconate exerts anti-inflammatory effects when administered in vitro and in vivo during macrophage activation and ischemia-reperfusion injury. Using newly generated Irg1−/− mice, which lack the ability to produce itaconate, we show that endogenous itaconate regulates succinate levels and function, mitochondrial respiration, and inflammatory cytokine production during macrophage activation. These studies highlight itaconate as a major physiological regulator of the global metabolic rewiring and effector functions of inflammatory macrophages.
In Brief
Macrophage activation is accompanied by TCA cycle remodeling, resulting in endogenous metabolites moonlighting as regulatory mediators of the inflammatory response. Lampropoulou et al. investigate the role of itaconate, one of the most highly induced metabolites in activated macrophages, and show that itaconate regulates succinate levels, mitochondrial respiration, and cytokine production.
INTRODUCTION
Macrophage activation by lipopolysaccharide (LPS) is accompanied by marked metabolic changes including upregulation of aerobic glycolysis (Everts et al., 2014), impaired mitochondrial respiration, disruption of the TCA cycle (Jha et al., 2015), and accumulation of succinate (Kelly and O’Neill, 2015). While a significant portion of this metabolic adaptation is transcriptionally controlled (e.g., via modulation of Nos2 and Cox2), succinate represents an example of a metabolite that affects major inflammatory pathways in both immune and non-immune cells by controlling IL-1β expression, HIF-1α activity, and ROS production (Kelly and O’Neill, 2015). However, the mechanism(s) regulating succinate levels in inflammatory macrophages have remained unknown.
Itaconate has been recently shown to be made by macrophages in response to LPS (Strelko et al., 2011) and certain infections (Michelucci et al., 2013) and is generated by the mitochondria-associated enzyme (Degrandi et al., 2009) immune responsive gene 1 (Irg1). Itaconate is believed to have anti-bacterial function due to its ability to inhibit isocitrate lyase, a bacterial glyoxylate shunt enzyme and due to its bactericidal effect when added at supraphysiological concentrations to macrophage-free S. enterica, M. tuberculosis, and L. pneumonophilia cultures (Michelucci et al., 2013) (Naujoks et al., 2016). An effect of itaconate on mammalian enzymes also was reported in the context of Sdh inhibition (Ackermann and Potter, 1949) (Németh et al., 2016). It is unknown, however, whether this biochemical effect of itaconate translates into macrophage metabolism and function. Here, using exogenous itaconate as well as newly generated Irg1−/− mice, we report that itaconate potently modulates macrophage activation and inflammatory responses by controlling TCA cycle remodeling.
RESULTS AND DISCUSSION
Itaconate Treatment Has Anti-inflammatory Effects during Macrophage Activation
Itaconate production is one of the hallmarks of macrophage activation on both the transcriptional and metabolic levels: Irg1 is one of the most highly induced enzymes in activated macrophages, and itaconate accumulates at high levels within cells (Figure 1A). Metabolomic profiling of culture supernatants from activated bone marrow-derived macrophages (BMDMs) revealed that itaconate was also secreted, as suggested previously (Strelko et al., 2011) (Figure 1B). The magnitude of itaconate production prompted us to investigate its potential regulatory role.
Figure 1. Itaconate Has Anti-inflammatory Effects on Macrophage Activation.

(A) Volcano plots showing transcripts (left) and metabolites (right) that are differentially expressed between resting and activated BMDM (LPS + IFN-γ, 24 hr).
(B) Relative expression of intracellular and secreted itaconate by BMDM at indicated time points after activation (LPS + IFN-γ).
(C) Histogram of intracellular iNOS expression determined by flow cytometry in BMDM untreated or DI-pretreated (0.25 mM, 12 hr) and then stimulated with LPS + IFN-γ (24 hr).
(D) IL-12 levels in BMDM culture supernatants from (C).
(E) IL-6 and TNF-α secreted by untreated or DI-pretreated BMDM and stimulated with LPS (24 hr).
(F) Heatmap of selected inflammatory marker genes unstimulated (Uns), LPS-stimulated (LPS), DI-pretreated (DI + Uns), DI-pretreated and then LPS-stimulated BMDM (4 hr).
(G) Mature IL-1β and IL-18 secreted by BMDM untreated or DI-pretreated, then stimulated with LPS (4 hr) and ATP (45 min).
(H) Mature IL-1β levels secreted by BMDM untreated or DI-pretreated and stimulated with LPS and nigericin (Nig.) or monosodium urate crystals (MSU).
(I) Western blot analysis of NLRP3, pro-IL-1β, and ASC in lysates of BMDM untreated or DI-pretreated and stimulated as in (G). α-tubulin was used as loading control. Blot shown is representative of two independent experiments.
For (B), (E), (G), and (H), data are shown as mean ± SEM (n = 3). p values were calculated using two-tailed Student’s t test (B and E) or one-way ANOVA compared to untreated stimulated cells (G and H). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. See also Figure S1.
We first tested the effect of exogenously added itaconate on the inflammatory response induced after LPS or LPS + IFN-γ stimulation. We treated mouse BMDM with physiologically relevant doses of dimethyl itaconate (DI) (Figure S1A), a membrane-permeable non-ionic form of itaconate. Pretreatment with DI suppressed iNOS protein expression (Figure 1C) and IL-12p70 and IL-6 secretion (Figures 1D and 1E), thereby interfering with activation of pro-inflammatory macrophages. In contrast, TNF-α levels remained unchanged (Figure 1E), indicating that the effects of DI-treatment were not due to global inhibition of NF-κB-dependent gene expression. To determine the specific pathways affected by itaconate, we performed global transcriptional profiling by RNA-seq on BMDM pretreated with DI or vehicle and then stimulated with LPS. Differential gene expression confirmed that DI-treatment led to downregulation of a spectrum of pro-inflammatory transcripts (Figures 1F and S1B), including Nos2, Il6, and Il12b.
The RNA-seq analysis also revealed that DI-pretreatment modulated the expression of several LPS-regulated genes involved in inflammasome activation and function (Figure 1F), including Il1b, Il18, P2rx7, Casp1, and Pycard (ASC). Indeed, DI potently inhibited production of mature IL-1β and IL-18 induced under prototypical NLRP3-activating conditions, namely LPS-driven priming (signal 1) followed by signal 2 inducers ATP, nigericin, and monosodium urate crystals (Figures 1G and 1H), whereas it affected inflammasome-induced cytotoxicity only moderately (Figure S1C). DI-treated BMDM also had impaired IL-1β production upon AIM-2-dependent inflammasome activation (Figure S1D), suggesting a broader regulatory effect on the inflammasome. The decreased protein expression of pro-IL-1β, ASC, and NLRP3 in DI-treated cells (Figure 1I) indicated that itaconate-mediated inhibition of inflammasome function was due primarily to a defective priming phase.
In view of this anti-inflammatory action of itaconate, and given its known cidal ability on extracellular bacteria, we next determined its effect on intracellular bacteria during macrophage infection. To this end, we infected BMDMs with Salmonella typhimurium, an intracellular Gram-negative bacterium that triggers TLR4 signaling and NLRP3 inflammasome activation (Broz et al., 2010) in the presence or absence of DI pretreatment. Consistent with our previous findings, infection-induced IL-1β, IL-6, and nitric oxide (NO), but not TNF-α, were abrogated in DI-treated cells (Figure S1E), whereas the number of intracellular bacteria was comparable between DI-treated and control BMDM (Figure S1F), indicating that the anti-inflammatory effects of itaconate at non-cytotoxic concentrations did not result from direct bactericidal activity. Rather, itaconate likely has a regulatory role, as supported by Irg1 induction in the viral infection context (Cho et al., 2013) (Figure S1G), indicating it has regulatory functions that are not specific to anti-bacterial response.
Exogenous Itaconate Inhibits Succinate Dehydrogenase In Vitro and In Vivo
As perturbations in cellular metabolism can lead to transcriptional defects in IL-1β production and inflammasome activation (Moon et al., 2015), we hypothesized that itaconate exerts its anti-inflammatory action, in part, by interfering with cellular metabolism. Using computational analysis of transcriptional data (Becker and Palsson, 2008), we investigated the possible rewiring of the metabolic flux triggered by itaconate in the absence of LPS. Using a flux balance analysis framework, we extended the metabolic model originally formulated for RAW 264.7 macrophage cell line (Bordbar et al., 2012) by including several reactions and enzymes that were absent in the original model (e.g., Irg1 and itaconate, see Experimental Procedures for details). We searched for fluxes in untreated and itaconate-treated conditions that were most consistent with the RNA-seq data (see Experimental Procedures). A comparative network highlights three types of metabolic flux change in response to itaconate treatment (Figures 2A and S2A): decreased metabolic flux (blue edges), increased metabolic flux (red), and reactions insensitive to itaconate addition (gray).
Figure 2. Itaconate Inhibits Sdh Activity In Vitro and In Vivo and Modulates ROS-Mediated Tissue Damage during Ischemia-Reperfusion Injury.

(A) Comparative network showing changes in the magnitude of predicted fluxes between unstimulated macrophages with and without itaconate treatment.
(B) Extracellular acidification rate (ECAR) measured in DI-treated or untreated BMDM. Data are shown as mean ± SEM of 10–15 replicates from one of two representative experiments.
(C) Chemical structure of succinate, malonate, and itaconate.
(D) Relative succinate levels in resting BMDM treated or not with DI. Data are shown as mean ± SEM (n = 3). p values were calculated using two-tailed Student’s t test.
(E) Lineweaver-Burk plot (left), Dixon plot (right), and calculated Km and Ki values. Data shown are mean ± SD (L-B plot, n = 3) and mean (Dixon plot, n = 3).
(F) Representative Evans Blue and TTC stained sections of hearts subjected to IR injury, after DI or saline treatment.
(G and H) Quantitation of area-at-risk (AAR) and infarct area (IA) as percent of AAR (G) and left ventricular (LV) myocardium (H) (saline, n = 8; DI, n = 7). p values were calculated using two-tailed Student’s t test.
(I) Percentage change in ROS (over respective normoxic controls) in neonatal rat cardiac myocytes (NRCMs) subjected to hypoxia for 24 hr in the presence of DI or diluent.
(J) Percentage of cell death in NRCMs treated as in (H). For (I) and (J), p values were calculated using post hoc test after one-way ANOVA (n = 8/condition).
(K) Histograms of mROS expression detected using MitoSox dye in BMDM pretreated or not with DI and activated with LPS (3 hr).
(L) Fold change in mROS mean fluorescence intensity (relative to medium) measured in (K).
Data are shown as mean ± SEM (n = 3, each in duplicates). p value was calculated using two-tailed Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001. See also Figure S2.
Two features of the computational analysis were apparent. First, itaconate addition was predicted to increase lactate dehydrogenase (Ldh) production. We confirmed this experimentally using Seahorse technology on unstimulated BMDM with and without DI treatment. We observed that extracellular acidification rate (ECAR), which occurs as a consequence of lactate accumulation in the medium, was increased in the presence of itaconate (Figure 2B). The second prediction was that itaconate addition should decrease the metabolic flux through Sdh. This suggested that itaconate might inhibit Sdh competitively, conceivably due the structural similarity between itaconate, succinate, and malonate, the latter a known Sdh inhibitor (Figure 2C). Indeed, 67 years ago, an inhibitory effect of itaconate on Sdh was postulated based on similar effects of malonate and itaconate on mitochondria function (Ackermann and Potter, 1949). To evaluate this hypothesis, we compared the activity of purified Sdh in the presence or absence of itaconate. Notably, itaconate dose-dependently blocked the activity of Sdh when succinate was used near physiological concentration 1 mM (Bennett et al., 2009) (Figure S2B), and itaconate treatment of resting BMDM led to increased succinate levels (Figure 2D). Kinetic analysis confirmed the competitive mode of inhibition of Sdh by itaconate (Figure 2E, left), and Dixon plot analysis showed Ki for itaconate 0.22 mM (Figure 2E, right). The calculated Km value for succinate was 0.29 ± 0.8 mM. These results are in line with the molar amounts of intracellular succinate and itaconate present in LPS-activated macrophages at 24 hr (Figure S2C). These data suggest that the anti-inflammatory effects of itaconate in BMDM were likely due to inhibition of Sdh. Supporting this notion, pretreatment with dimethyl malonate also inhibited IL-1β production after LPS + ATP stimulation (Figure S2D).
We next evaluated whether itaconate inhibits Sdh activity in vivo. A recent study (Chouchani et al., 2014) highlighted the proinflammatory role of succinate oxidation in the context of cardiac ischemia-reperfusion (IR) injury. mROS generation during IR injury was proposed to occur in the mitochondria as a result of reverse electron transport (RET), whereby Sdh was fueled by the succinate that accumulated during ischemia. Accordingly, inhibition of Sdh by dimethyl malonate limited IR injury and mROS levels (Chouchani et al., 2014). We used this model to test whether itaconate would inhibit Sdh-mediated oxidation in vivo. Intravenous infusion of DI during ischemia markedly reduced myocardial infarct size (Figures 2F and 2G). While the area-at-risk was similar between the two groups (Figure 2H), the 42% reduction in infarct size with DI treatment was comparable to that seen after dimethyl malonate administration (Chouchani et al., 2014), suggesting a common mechanism of action. To evaluate this hypothesis, we used an in vitro assay that mimics myocardial infarction injury by subjecting neonatal cardiomyocytes to hypoxic insult (Ma et al., 2012). Pretreatment with DI attenuated the hypoxia-induced increase in ROS generation (Figure 2I) and conferred dose-dependent protection from hypoxia-induced cell death (Figure 2J).
Therefore, we assessed whether itaconate-mediated Sdh inhibition influenced mROS generation. Pretreatment with DI impaired the ability of BMDM to upregulate mROS in response to LPS (Figures 2K and 2L). As interfering with mROS can affect inflammasome priming (Bauernfeind et al., 2011), the blunted mROS response mechanistically links Sdh inhibition and the anti-inflammatory effects of itaconate on IL-1β and IL-18 production (Figure 1G). Furthermore, it suggests that succinate processing rather than accumulation is important for the inflammatory rewiring of macrophages.
Endogenous Itaconate Regulates Metabolic Remodeling, Succinate Levels, and Mitochondrial Respiration in Inflammatory Macrophages
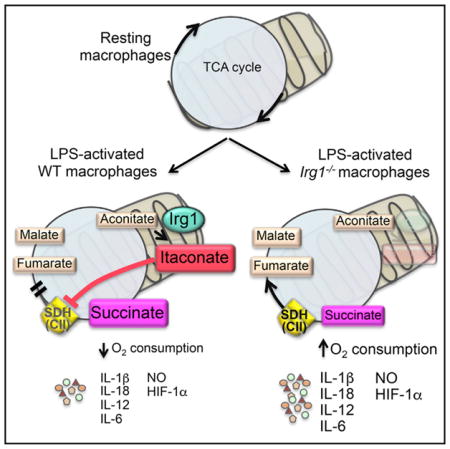
To test the physiological relevance of endogenous itaconate, we generated mice with a targeted disruption of the Irg1 gene (Figure S3). As BMDMs from Irg1−/− mice failed to produce or secrete itaconate (Figure 3A) after stimulation with LPS and IFN-γ, we conclude that Irg1 is the only enzyme carrying out itaconate synthesis under these conditions. LPS-activated WT macrophages show increased concentration of fumarate and malate due to an active aspartate-argininosuccinate shunt (Jha et al., 2015), as well as accumulation of succinate, presumably due to itaconate inhibition. To confirm this, we metabolically profiled Irg1−/− BMDMs and observed marked changes indicative of altered Sdh activity. Lack of Irg1 expression led to abrogation of succinate accumulation, whereas fumarate and malate concentrations were yet increased (Figure 3B). Thus, in the absence of endogenous itaconate, Sdh remained active and oxidized succinate to fumarate, which was rapidly converted to malate (Figure 3C).
Figure 3. Endogenous Itaconate Controls TCA Cycle Remodeling and Succinate Levels.

(A) Relative expression of secreted and intracellular itaconate by WT and Irg1−/− BMDM after activation with LPS + IFN-γ.
(B) Relative expression of succinate, fumarate, and malate in cell extracts from (A).
(C) Scheme showing how itaconate regulates TCA flow in LPS-activated macrophages.
(D) Basal oxygen consumption rate by resting (medium) and LPS-activated BMDM (24 hr) from WT and Irg1−/− mice.
Data are shown as mean ± SEM (A and B, n = 3; D, n = 2 total technical replicates 13–55). p values were calculated using two-tailed Student’s t test. *p < 0.05; ***p < 0.001; ****p < 0.0001. See also Figure S3.
Besides its role in the TCA cycle, Sdh is also part (as complex II) of the mitochondrial electron transport system. In LPS-activated macrophages, mitochondrial respiration decreases significantly (Kelly and O’Neill, 2015). Thus, we tested whether itaconate-mediated inhibition of Sdh influenced mitochondrial function by measuring oxygen consumption rates (OCR). Remarkably, contrary to WT cells, Irg1−/− BMDMs showed increased OCR at 24 hr post activation (Figure 3D), demonstrating that, by inhibiting Sdh, endogenous itaconate modulates mitochondrial respiration, as recently proposed for exogenous itaconate (Németh et al., 2016).
Endogenous Itaconate Regulates Macrophage Inflammatory Responses
We next assessed macrophage activation in the absence of endogenous itaconate. We used RNA-seq to profile differences in gene expression of LPS-activated wild-type (WT) and Irg1−/− BMDMs. Notably, the transcriptional signature in Irg1−/− cells was essentially inversely correlated with itaconate-treated WT cells: genes upregulated in Irg1−/− cells were downregulated in itaconate-treated WT BMDMs (Figure 4A), providing additional evidence that endogenous itaconate functions in a manner similar to exogenously added DI. Consistent with these observations, LPS-activated Irg1−/− BMDM produced more IL-12, NO, and IL-6, but similar amounts of TNF-α, compared to WT cells (Figures 4B and 4C). Irg1−/− BMDM also sustained higher expression of mature IL-1β and IL-18 under conditions that stimulate NLPR3 (Figure 4D). Consistent with the known IL-1β-promoting effect of HIF-1α (Tannahill et al., 2013), we observed increased HIF-1α mRNA and protein levels in Irg1−/− cells and, reciprocally, suppression of HIF-1α expression in DI-treated BMDM (Figures 4E–4G). Thus, changes in HIF-1α and IL-1β production correlate with efficiency of succinate oxidation by Sdh. These observations raise the possibility that the HIF-1α-IL-1β axis is linked to the efficiency and directionality of the electron transport chain in activated macrophages rather than direct signaling through succinate accumulation.
Figure 4. LPS-Activated Irg1−/− Macrophages, which Lack Itaconate, Show Augmented Inflammatory Responses.

(A) Transcriptional signatures of activated Irg1−/− and DI-treated activated WT BMDM are inversely related.
(B) IL-12 and NO levels in supernatants of WT and Irg1−/− BMDM stimulated with LPS + IFN-γ (24 hr).
(C) IL-6 and TNF-α levels in supernatants of LPS-activated (24 hr) WT and Irg1−/− BMDM.
(D) Mature IL-1β and IL-18 levels in supernatants of WT and Irg1−/− BMDM stimulated with LPS (4 hr) and ATP.
(E) Relative hif1a mRNA expression in LPS-activated WT and Irg1−/− BMDM (4 hr).
(F) Western blot analysis of HIF-1α in lysates of WT and Irg1−/− BMDM activated as in (E); α-tubulin was used as a loading control. Blot shown is representative of two independent experiments.
(G) Western blot analysis of HIF-1α in lysates of WT BMDM untreated or DI-treated and activated with LPS (24 hr). Blot shown is representative of two independent experiments.
Data in (B)–(E) are shown as mean ± SEM (n = 3/group, each in 2–3 replicates). p values were calculated with two-tailed Student’s t test. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In summary, our work identifies itaconate as part of a posttranscriptional mechanism that governs TCA cycle remodeling and macrophage activation via its inhibitory effect on Sdh and regulation of succinate levels. Our results expand the physiological roles of itaconate beyond a direct anti-bacterial action, to include regulation of TLR-mediated inflammatory cytokine production, and provide a physiological regulatory mechanism to control electron transport chain flow, succinate levels, ROS production, and tissue inflammation.
EXPERIMENTAL PROCEDURES
Mice
Irg1−/−(MGI:103206) mice were generated at Washington University after receiving embryonic stem cells (ESCs) (Irg1tm1a(KOMP)Wtsi) from the Knockout Mouse Project Repository (KOMP, University of California, Davis) containing an insertion cassette between exons 3 and 4. This cassette prevents transcription of downstream exons 4 and 5 and production of mature protein. Irg1−/− C57BL/6N ESCs were microinjected into (Cg)-Tyrc-2J/J albino recipient female C57BL/6 mouse blastocysts. Chimeric mice with black coat color were selected and bred to wild-type C57BL/6N mice. Homozygous Irg1−/− mice were generated by intercrossing the heterozygous animals and confirmed by PCR. Irg1−/− mice were fertile and exhibited normal Mendelian frequencies, and BMDM from both sexes were used.
C57BL/6N WT mice from Charles River Laboratories were used as age-matched controls. Mice were maintained at Washington University under specific pathogen-free conditions in accordance with Federal and University guidelines and protocols approved by the Animal Studies Committee of Washington University.
Differentiation and Activation of Macrophages
BMDM were prepared from 6- to 8-week-old mice as described (Jha et al., 2015) and seeded at 105 cells/well in 96-well tissue-culture plates in RMPI-1640 medium (ThermoFisher) supplemented with 10% FBS, 2 mM L-glutamine (Thermo Fisher Scientific), and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific). Cells were treated or not with DI (0.25 mM, 12 hr; Sigma) and activated as shown with LPS (E. coli 0111:B4; 100 ng/mL; Sigma) ± IFNγ (50 ng/mL; Peprotech). For inflammasome activation, cells were primed with LPS (100 ng/mL, 4 hr) before addition of ATP (3 mM, 45 min; Sigma), nigericin (5 mM; 1 hr; Sigma) or monosodium urate crystals (250 μg/mL; 4 hr; InvivoGen).
Flow Cytometry
Cells were incubated with anti-CD16/32 (clone 93, Biolegend), surface stained for F4/80 (clone BM8, eBioscience) and CD11b (clone M1/70; BD PharMingen) together with LIVE/DEAD dye (Invitrogen), followed by intracellular iNOS stain with anti-NOS2 (clone C-11; Santa Cruz Biotechnology) and FITC-conjugated IgG (Clone A85-1; BD PharMingen) using the BD cytofix/cytoperm Kit (BD Biosciences). Cells were acquired on Canto II flow cytometer (BD Biosciences), and data were analyzed with FlowJo v.9.5.2 software (Tree Star).
Quantification of Cytokines and Nitric Oxide
Cytokines were quantified using DuoSet kits for IL-1β/IL-1F2, TNF-α, and IL-6 (all R&D systems), IL-12 ELISA MAX Deluxe Set (BioLegend), and IL-18 ELISA kit (Medical & Biological Laboratories). Nitric oxide was detected with Griess Reagent System (Promega).
Western Blotting
Cells were lysed in RIPA Lysis Buffer (Santa Cruz) and heat-denatured in reducing sample buffer (Thermo Fisher Scientific). Proteins were separated on 4%–20% polyacrylamide gradient gels (BioRad) and transferred onto PVDF membranes. Non-specific binding was blocked with 5% skim milk, and membranes were probed with primary antibodies to IL-1β (1:1,000; 12507S, Cell Signaling), Nlrp3 (1:500; NBP2-12446, Novus), HIF-1α (1:500; NB100-449, Novus), α-tubulin (1:2,000; 2125S, Cell Signaling), and ASC (1:1,000; sc-22514-R, Santa Cruz), followed by incubation with anti-rabbit-HRP (1:10,000; sc-2030, Santa Cruz) and Clarity western ECL substrate (Bio-Rad).
Sdh Activity Assay
Sdh was purified from the BV2 macrophage cell line and its activity measured in the presence of itaconate using the Complex II Enzyme Activity Microplate Assay Kit (Abcam) as per manufacturer’s protocol. Substrates were diluted in activity buffer and added to the phospholipid mixture 5 min before adding the activity solution. For kinetic analysis, sodium succinate hexahydrate was used as indicated in activity buffer.
Extracellular Flux Analysis
Real-time extracellular acidification and oxygen consumption rates were measured using Seahorse technology as described (Huang et al., 2014).
Metabolite Profiling by GC-MS
Cellular metabolites were extracted from an equal number of cells per sample and analyzed by GC-MS as described (Vincent et al., 2015). Briefly, intracellular metabolism was quenched with 800 μl of 80% methanol. To analyze secreted metabolites, 20 μl supernatants were added to 800 μl of 80% methanol. D-myristic acid (750 ng/sample) was used as internal standard. Extracts were dried by vacuum centrifuge and pellets resuspended in 30 μl pyridine-containing 10 mg/mL methoxyamine hydrochloride, before being derivatized using N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide. Metabolite abundance was expressed relative to the internal standard.
RNA-Seq Analysis
mRNA was extracted with oligo-dT beads (Invitrogen), and libraries were prepared and quantified as described (Vincent et al., 2015).
Flux Balance Analysis
To investigate possible rewiring of the metabolic fluxes, we used flux balance analysis framework (FBA). Using the RAW 264.7 macrophage cell line metabolic model (Bordbar et al., 2012) and an algorithm similar to GIMME (Becker and Palsson, 2008) and MADE (Jensen and Papin, 2011), we simulated fluxes in untreated and DI-treated conditions based on their consistency with the RNA-seq data for each condition. See Supplemental Information for detailed analysis.
Myocardial Ischemia-Reperfusion Model
In vivo ischemia-reperfusion modeling was done as described (Ma et al., 2012). 8- to 10-week-old mice were anesthetized and subjected to open chest procedure of reversible left anterior descending artery ligation for 30 min and subsequent reperfusion for 2 hr. Saline or DI (4 mg/kg/min) was infused intravenously 10 min before and during ischemia. A cardioplegic solution followed by Evans Blue (after reocclusion of the LAD) was injected in a retrograde manner through the aorta in situ. The left ventricle was then sectioned into five slices, incubated in TTC (triphenyltetrazolium chloride) at 37°C (30 min), and images analyzed with ImageJ (NIH).
Hypoxia Modeling in Neonatal Rat Cardiac Myocytes
Neonatal rat cardiac myocytes were isolated and cultured as described (Ma et al., 2012). Cells were subjected to hypoxia in an oxygen control cabinet (Coy Laboratories) mounted within an incubator and equipped with oxygen controller and sensor for continuous oxygen level monitoring. A mixture of 95% nitrogen and 5% CO2 was utilized to create hypoxia, and oxygen levels in the chamber were monitored and maintained at <1%. Cell death was assessed with the Live-Dead Cytotoxicity Viability kit for Mammalian cells (Invitrogen) and ROS detected by flow cytometry with fluorescent carboxy-H2DCFDA (after incubation in 10 μmol/L for 30 min) (Ma et al., 2012).
mROS Measurement
Cells were treated with DI (0.25 mM, 12 hr) or vehicle, loaded with 5 μM MitoSOX (Invitrogen) at 37°C for 30 min in HBSS supplemented with 0.1% BSA, and rinsed with warm culture medium, before stimulation with LPS 1 hr later (for 3 hr) and analysis on Canto II flow cytometer (Becton Dickinson).
Statistical Analysis
Statistical analyses were performed in GraphPad Prism 6 software using statistical tests indicated for each experiment.
Supplementary Material
Highlights.
Exogenous itaconate exerts anti-inflammatory effects in vitro and in vivo
Itaconate inhibits Sdh and regulates succinate levels in LPS-activated macrophages
Itaconate controls mitochondrial respiration changes in inflammatory macrophages
Itaconate limits IL-1β, IL-18, IL-6, IL-12, NO, and HIF-1α, but not TNF-α, production
Acknowledgments
National Institutes of Health (NIH) grant R01 AI104972 (M.S.D.) supported this study. S.N. was supported by a DFG fellowship. The Irg1−/− targeting allele and ESCs were created by the Mouse Biology Program (https://www.mousebiology.org/) at the University of California Davis. The ESCs were generated by the trans-NIH Knock-Out Mouse Project (KOMP) and obtained from the KOMP Repository (https://www.komp.org/). NIH grants to Velocigene at Regeneron Inc. (U01HG004085) and the CSD Consortium (U01HG004080) funded the generation of gene-targeted vectors and ESCs for over 8,500 genes in the KOMP Program and archived and distributed by the KOMP Repository at UC Davis and CHORI (U42RR024244). We acknowledge technical assistance from the GCRC Metabolomics Core Facility at McGill University. E.E.V. and R.G.J. were supported by grants from the Canadian Institutes for Health Research (CIHR MOP-142259 to R.G.J.). A.D. was supported by grants from NIH (HL107594) and Department of Veterans Affairs (1I01BX001969). A.S. was supported by a grant from the Government of the Russian Federation (074-U01).
Footnotes
ACCESSION NUMBERS
The accession number for the RNA sequencing data reported in this paper is GEO: GSE82043.
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at http://dx.doi.org/10.1016/j.cmet.2016.06.004.
AUTHOR CONTRIBUTIONS
V.L. and M.N.A. conceived and designed the study. V.L. performed many of the immunological experiments, flow cytometry, Seahorse experiments, and data analysis. A.S. performed computational analysis of RNA-seq data and flux balance analysis. M.B. performed western blots, sdh activity assay, and mROS measurement in macrophages. E.L. performed genomics analysis and helped with experiments. S.N. and M.S.D. generated the Irg1−/− mice. E.E.V., T.G., and R.G.J. performed metabolomics profiling. L.C.-B provided Salmonella typhimurium cultures. C.J.W., X.M., and V.L. performed the ischemia-reperfusion model experiments and data analysis, under the supervision of A.D. S.C.-C.H. helped with Seahorse experiments. E.J.D, S.K., G.J.R., and M.S.D. contributed to study design and critical editing of the manuscript. V.L. and M.N.A. wrote the initial draft of the manuscript.
References
- Ackermann WW, Potter VR. Enzyme inhibition in relation to chemotherapy. Proc Soc Exp Biol Med. 1949;72:1–9. doi: 10.3181/00379727-72-17313. [DOI] [PubMed] [Google Scholar]
- Bauernfeind F, Bartok E, Rieger A, Franchi L, Núñez G, Hornung V. Cutting edge: reactive oxygen species inhibitors block priming, but not activation, of the NLRP3 inflammasome. J Immunol. 2011;187:613–617. doi: 10.4049/jimmunol.1100613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker SA, Palsson BO. Context-specific metabolic networks are consistent with experiments. PLoS Comput Biol. 2008;4:e1000082. doi: 10.1371/journal.pcbi.1000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat Chem Biol. 2009;5:593–599. doi: 10.1038/nchembio.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A, Mo ML, Nakayasu ES, Schrimpe-Rutledge AC, Kim YM, Metz TO, Jones MB, Frank BC, Smith RD, Peterson SN, et al. Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol. 2012;8:558. doi: 10.1038/msb.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P, Newton K, Lamkanfi M, Mariathasan S, Dixit VM, Monack DM. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J Exp Med. 2010;207:1745–1755. doi: 10.1084/jem.20100257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Proll SC, Szretter KJ, Katze MG, Gale M, Jr, Diamond MS. Differential innate immune response programs in neuronal subtypes determine susceptibility to infection in the brain by positive-stranded RNA viruses. Nat Med. 2013;19:458–464. doi: 10.1038/nm.3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515:431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degrandi D, Hoffmann R, Beuter-Gunia C, Pfeffer K. The proinflammatory cytokine-induced IRG1 protein associates with mitochondria. J Interferon Cytokine Res. 2009;29:55–67. doi: 10.1089/jir.2008.0013. [DOI] [PubMed] [Google Scholar]
- Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15:323–332. doi: 10.1038/ni.2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SC, Everts B, Ivanova Y, O’Sullivan D, Nascimento M, Smith AM, Beatty W, Love-Gregory L, Lam WY, O’Neill CM, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PA, Papin JA. Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics. 2011;27:541–547. doi: 10.1093/bioinformatics/btq702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42:419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci USA. 2013;110:7820–7825. doi: 10.1073/pnas.1218599110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, Choi AM. mTORC1-Induced HK1-Dependent Glycolysis Regulates NLRP3 Inflammasome Activation. Cell Rep. 2015;12:102–115. doi: 10.1016/j.celrep.2015.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, Kempa S, Peter A, Mollenkopf HJ, Dorhoi A, et al. IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathog. 2016;12:e1005408. doi: 10.1371/journal.ppat.1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Németh B, Doczi J, Csete D, Kacso G, Ravasz D, Adams D, Kiss G, Nagy AM, Horvath G, Tretter L, et al. Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J. 2016;30:286–300. doi: 10.1096/fj.15-279398. [DOI] [PubMed] [Google Scholar]
- Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, Roberts MF. Itaconic acid is a mammalian metabolite induced during macrophage activation. J Am Chem Soc. 2011;133:16386–16389. doi: 10.1021/ja2070889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent EE, Sergushichev A, Griss T, Gingras MC, Samborska B, Ntimbane T, Coelho PP, Blagih J, Raissi TC, Choinière L, et al. Mitochondrial Phosphoenolpyruvate Carboxykinase Regulates Metabolic Adaptation and Enables Glucose-Independent Tumor Growth. Mol Cell. 2015;60:195–207. doi: 10.1016/j.molcel.2015.08.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.