

Abstract
Cytochromes P450 represent a family of enzymes that are responsible for the oxidative metabolism of a wide variety of xenobiotics. Although the mammalian P450s require interactions with their redox partners in order to function, more recently, P450 system proteins have been shown to exist as multi-protein aggregates that include the formation of P450•P450 complexes. Evidence has shown that the metabolism of some substrates by a given P450 can be influenced by the specific interaction of the enzyme with other forms of P450. Detailed kinetic analysis of these reactions in vitro has shown that the P450-P450 interactions can alter metabolism by changing the ability of a P450 to bind to its cognate redox partner, NADPH-cytochrome P450 reductase; by altering substrate binding to the affected P450; and/or by changing the rate of a catalytic step of the reaction cycle. This review summarizes the known examples of P450-P450 interactions that have been shown in vitro to influence metabolism and categorizes them according to the mechanism(s) causing the effects. P450-P450 interactions have the potential to cause major changes in the metabolism and elimination of drugs in vivo. Current research on the topic of P450-P450 interactions is focused on elucidating the extent of the functional effects of these interactions in vivo. This review also summarizes the evidence that the P450-P450 interactions influence metabolism in vivo and discusses the studies that will provide further insight into the extent of these effects in the future.
Keywords: cytochrome P450, protein-protein interactions, drug metabolism, enzyme inhibition, enzyme activation
Introduction
The cytochromes P450 (P450 or CYP) constitute a superfamily of enzymes that are expressed across species and are involved in the metabolism of both endogenous and exogenous compounds. The P450s from families 1, 2, and 3 are extremely important in the phase I metabolism of most lipophilic xenobiotics including an estimated 75 to 90% of prescribed drugs (Guengerich 2006;Lynch and Price 2007). These same P450s can also activate many pro-toxins and pro-carcinogens to harmful, reactive products that initiate the deleterious effects of being exposed to these compounds (Guengerich et al. 1996). Because of these aspects of P450-mediated metabolism, there is intensive research to understand and predict the roles they play in xenobiotic metabolism and disposition.
Mammalian P450s from families 1, 2, and 3 are typically expressed on the cytoplasmic side of the smooth endoplasmic reticulum (ER). However, there have been reported instances of expression in mitochondria and other membranes as well (Anandatheerthavarada et al. 1997;Dong et al. 2013;Genter et al. 2006;Robin et al. 1996). P450 function is inherently dependent on protein-protein interactions. The mixed function oxygenation of substrates by P450s depends on a complicated reaction scheme which uses two separate electron-delivery steps that are mediated through protein interactions with specific membrane-bound redox partners. These electrons are used to activate molecular oxygen to a reactive form that is capable of substrate oxidation and form a molecule of water in the process (White and Coon 1980). In most cases, the primary electron needed for catalysis is delivered by the NADPH cytochrome P450 reductase (CPR). The second electron can be delivered by either CPR or cytochrome b5 (b5) (Hildebrandt and Estabrook 1971). There have been reported instances in which b5 was able to deliver the first electron of the catalytic cycle (Yamazaki et al. 1996b), but examples of this process are rare. Interestingly, b5 has been shown to dramatically increase the rate of some P450-mediated reactions presumably by stimulating the rate of second electron delivery (Gruenke et al. 1995;Jansson and Schenkman 1987). However, stimulation of these reactions has also been attributed to allosteric effects resulting from the interaction of P450s and b5 – effects that do not require b5 to P450 electron transfer (Yamazaki et al. 1996a).
The utilization of molecular oxygen and extraneous electrons unfortunately leads to some negative consequences of P450-mediated metabolism. The productive metabolism of substrate can be aborted by loss of oxygen and reducing equivalents as either superoxide or hydrogen peroxide depending on the stage that the reaction becomes “uncoupled”. In an additional nonproductive mechanism, the activated oxygen that catalyzes oxidation of substrate can be reduced with an extra pair of electrons and protonated to produce an extra molecule of water (Loida and Sligar 1993).
In most tissues, the total concentration of P450 enzymes far exceeds that of CPR (Gomes et al. 2009;Peterson et al. 1976;Reed et al. 2011) raising the interesting probability that P450-mediated metabolism is limited by the interactions of the enzymes with the requisite redox carriers in the membrane. Furthermore, it is now established that P450s carry out their various roles, at least in part, as heteromeric and/or homomeric aggregates in the ER (reviewed in (Davydov 2011;Kandel and Lampe 2014;Reed and Backes 2012)). In fact, the limiting levels of redox carriers in the membranes are conducive to the formation and stabilization of the P450-P450 aggregates (Gut et al. 1982). Thus, metabolism by the enzymes relies on a complex interplay involving P450-P450 aggregation and the interaction of aggregated P450s with the limiting supply of redox partners.
Given the dynamics of protein-protein interactions involving the P450 monooxygenase system in the ER, it would not be surprising for the catalytic activities of P450s to be influenced by their physical interactions with one another. This is indeed the case as research has now demonstrated that the catalytic activities of P450s are often stimulated or inhibited by form-specific, physical interactions with other P450s (Davydov 2011;Reed & Backes 2012). Interestingly, these changes have been shown to be mediated by several mechanisms including changes in the affinities of the interacting P450s for binding to CPR (Backes et al. 1998;Kelley et al. 2006) and changes in the catalytic potential of the enzymes, both with regard to enzyme turnover (Davydov et al. 2015;Subramanian et al. 2009) and substrate specificity (Kenaan et al. 2013;Subramanian, Low, Locuson, & Tracy 2009).
The study of P450 aggregation, in itself, would not necessarily be a topic of interest in pharmacology unless the interactions of specific P450s influenced the rate and extent of drug metabolism and disposition. Since our last review on this topic (Reed & Backes 2012), the study of the physical interaction of P450s has shifted from an emphasis on proving that they do affect P450 function to understanding the nature and extent of these functional interactions as they occur in vivo in biological membranes. This review will focus on aspects of P450-P450 interactions that are related to changes in P450 activities by describing in detail, the specific P450 interactions that have been shown to alter P450-mediated metabolism and the mechanisms attributed to these changes. The review will also summarize the evidence showing that the functional interactions also occur in biological membranes.
Specific P450-P450 interactions and the underlying mechanisms that influence metabolism
Most of the specifics about the effects of P450-P450 interactions on enzyme activities have been derived from studies using purified, reconstituted P450 systems (Backes, Batie, & Cawley 1998;Davydov et al. 2005;Hazai and Kupfer 2005;Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013;Reed et al. 2010;Subramanian, Low, Locuson, & Tracy 2009;Subramanian et al. 2010). These studies have been important in identifying which P450s specifically interact to influence function, and also in defining the specific changes in P450 activities that occur as a result of these interactions. Furthermore, because the relative concentrations of proteins, substrates, and lipid can be controlled and systematically varied in the preparation and use of the purified, reconstituted system, these types of studies have been instrumental in elucidating the mechanisms by which the P450-P450 interactions influence drug metabolism.
It is appropriate to begin the topic of the functional effects of P450-P450 interactions with those involving CYP2B4-CYP1A2 because it has been the most significant in terms of establishing that these types of interactions can influence metabolism. In addition to the aforementioned kinetic data suggesting that the two P450s interact, there have been at least three lines of direct physical evidence demonstrating complex formation between CYP1A2 and CYP2B4. These include studies that 1) monitored changes in the hyperbaric sensitivity of the P450s (Davydov et al. 1992;Davydov et al. 1995;Davydov et al. 2000b); 2) measured fluorescence resonance energy transfer (FRET) when cysteine residues of the two P450s were labeled with different fluorescent molecules (Davydov et al. 2001); and 3) used cross-linking/immunoprecipitation to show the P450s interacted when they were reconstituted in the same lipid membranes but not when reconstituted in separate lipid vesicles that were subsequently mixed (Reed, Eyer, & Backes 2010). The cross-linking/immunoprecipitation study also was significant in showing that the changes in metabolism associated with the co-reconstitution of the P450s were only associated with the physical interaction of the two P450s (Reed, Eyer, & Backes 2010). The study also showed the interaction stimulated the CYP1A2-mediated metabolism of a luciferin-methyl ester substrate used in a commercially available P450GLO™ assay in addition to the previously reported effect on metabolism of ethoxyresorufin (Backes, Batie, & Cawley 1998) and inhibited the CYP2B4-mediated metabolism of 7-ethoxy-4-trifluoromethylcoumarin, 7-ethoxycoumarin, benzphetamine, and p-nitroanisole in addition to the previously reported effect on metabolism of pentoxyresorufin (Backes, Batie, & Cawley 1998;Cawley et al. 1995).
P450-P450 interactions that cause changes in CPR-binding
The first instance in which the mechanism was defined for the alteration of metabolism by a P450-P450 interaction involved the interaction of rabbit CYP1A2 and CYP2B4 (Backes, Batie, & Cawley 1998). This was accomplished by performing a detailed kinetic analysis of the metabolism by each P450 as a function of the CPR concentration. The apparent affinities of CYP2B4 and CYP1A2 for binding to CPR were approximately equal when determined independently by measuring the P450-specific metabolism of pentoxyresorufin and ethoxyresorufin, respectively. However, when reconstituted together, metabolism of ethoxyresorufin by CYP1A2 was dramatically stimulated 100 to 400% and that of pentoxyresorufin by CYP2B4 was inhibited 30 to 50% when the CPR concentration was subsaturating. However, as the CPR concentration was increased to exceed the total P450 concentration, the rates of metabolism of the substrates by the mixed P450 system roughly equaled the sum of the rates by the individual P450 systems. The data could be interpreted as the CYP1A2-CYP2B4 interaction causing CYP1A2 to have a greater affinity for binding to CPR when it was bound to CYP2B4. The higher level of binding with CYP1A2 caused CPR to interact less with CYP2B4 resulting in inhibition of pentoxyresorufin O-dealkylation. Thus, the changes in the rates of metabolism caused by this interaction are manifest only when the CPR concentration limits overall metabolism (as it does in vivo) and are not readily apparent when present in excess of the P450s. As discussed below, a subsequent study also demonstrated the interaction caused the CYP2B4- and CYP1A2-mediated metabolism of toluene to be inhibited and activated, respectively (Reed et al. 2013). The study examining toluene metabolism also showed that the interaction of CYP1A2 and CYP2B4 made the flow of electrons from CPR to the P450s more efficient as metabolism by the mixed P450 system was characterized by a dramatic decrease in the proportion of electrons being diverted towards the production of excess water (discussed in detail below).
When purified P450s are reconstituted with CPR and lipid, the proteins are added from concentrated stock solutions. Thus, when a mixed reconstituted system is prepared with two P450s added at a 1:1 ratio, the P450s are not going to mix completely to form a homogeneous suspension of mixed complexes. Instead, there will be an indeterminable mixture of homomeric associations of P450s in addition to the desired heteromeric association that is being studied. Because of this limitation, only the trends of the effects of P450-P450 interactions can be assessed in these types of studies and not the overall magnitude of the changes. Recently, the effects of fine particle suspensions on P450-mediated activities were determined (Reed et al. 2014). Various types of particle were evaluated and all were found to inhibit CYP1A2- and CYP2B4-mediated activities by competitive (Reed et al. 2015b) and noncompetitive (Reed et al. 2015a) mechanisms, respectively. The different particle effects on each P450 enabled our lab to ascertain the relative contributions of CYP1A2 and CYP2B4 to overall metabolism by the mixed P450 system (Reed, Dela Cruz, Lomnicki, & Backes 2015b). More specifically, the mechanism of enzyme inhibition by the particles using the mixed CYP1A2-CYP2B4 system would be predicted to be competitive if CYP1A2 was primarily responsible for metabolism but noncompetitive or mixed if the CYP2B4 made a significant contribution to the reaction. The CYP2B4-specific substrate, 7-ethoxy-4-trifluoromethylcoumarin was used to assess the effects of particles on metabolism by the mixed P450 system. In order to favor the likelihood that CYP2B4 formed a mixed complex with CYP1A2, the CYP1A2 concentration was present in three-fold excess to CYP2B4. Under these conditions, the rate of 7-ethoxy-4-trifluoromethylcoumarin metabolism was much lower (≈1/10th) than that by the simple reconstituted system of CYP2B4, and the mechanism of inhibition by the particles was found to be purely competitive. As a result, the data suggest that CYP2B4 is actually catalytically silent when in a complex with CYP1A2 (Figure 1) and not just inhibited 30 to 50% as suggested by previous studies (Reed, Eyer, & Backes 2010). This finding has important implications about drug metabolism mediated by CYP2B enzymes when there is also high expression of CYP1A2.
Figure 1. Effects of a P450-P450 interaction on metabolism when the mechanism involves a change in CPR-binding affinities of the interacting P450s.
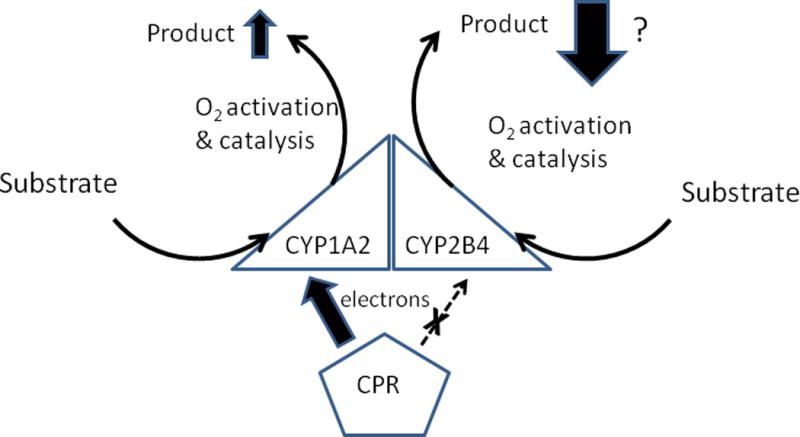
Metabolism of alkoxyresorufin substrates (Backes, Batie, & Cawley 1998;Reed, Eyer, & Backes 2010;Reed, Dela Cruz, Lomnicki, & Backes 2015b) by the mixed CYP1A2-CYP2B4 complex is represented by arrows for electron transfer from CPR; substrate binding; and oxygen activation/mono-oxygenation of substrate to form product. Larger arrows indicate an increase in the rates of a step relative to that catalyzed by the homomeric P450 complex. When the rate of a step is diminished by the P450-P450 interaction, a dashed arrow is used. The regular arrows indicate the step is unchanged. The ability of CYP2B4 to bind CPR is attenuated and possibly inhibited completely following interaction of the P450 with CYP1A2. Thus, an “X” is placed over the arrow for electron transfer, and a “?” is placed next to the large arrow indicating decreased product formation by this P450.
Having established that the CYP1A2-CYP2B4 interaction could influence metabolism by changing the relative levels of CPR-binding to the interacting P450s, the same mechanism was subsequently implicated in the stimulation of alkoxyresorufin metabolism caused by the interaction of CYP1A2 and CYP2E1 (Kelley, Cheng, & Backes 2006). Although the alkoxyresorufins tested (7-ethoxyresorufin and 7-pentoxyresorufin) were not selective probes for CYP1A2, the metabolism of the CYP2E1-specific substrate, N-nitrosodimethylamine, was not affected by the CYP1A2-CYP2E1 interaction. Thus, it may be presumed that the P450-P450 interaction, as in the case involving the CYP1A2-CYP2B4 interaction, caused stimulation of CYP1A2-mediated metabolism of the alkoxyresorufins by increasing its ability to bind CPR. The lack of an effect on metabolism of N-nitrosodimethylamine by CYP2E1 may be a substrate-specific effect, and it is possible that metabolism of other CYP2E1-specific substrates might be inhibited by the interaction of CYP2E1 with CYP1A2. It is also possible that other untested mechanisms may also influence the effects of the CYP1A2-CYP2E1 interaction as observed with those involving CYP1A2 and CYP2B4 (discussed below). At this point, the mechanism involving changes in the abilities of the P450s to bind CPR is the only established mechanism for the observed changes in metabolism that occurred as a result of the CYP2E1-CYP1A2 interaction.
P450-P450 interactions that affect specific steps of the P450 catalytic cycle
CYP2C9 has been shown to participate in several P450-P450 interactions that alter its catalytic behavior. In the interaction of CYP2C9 with CYP2D6 (Subramanian, Low, Locuson, & Tracy 2009) and CYP3A4 (Subramanian, Zhang, & Tracy 2010), the CYP2C9-mediated metabolism of S-flurbiprofen (in the presence of CYP2D6) and S-flurbiprofen and diclofenac (in the presence of CYP3A4) was dramatically inhibited. In both cases, the presence of CYP2C9 had no effect on metabolism by the other P450 (dextromethorphan by CYP2D6 and testosterone by CYP3A4). Unlike the interactions mentioned above, the functional effects of these other CYP2C9-P450 interactions also were observed at saturating reductase concentrations (CPR:P450 ≥ 3:1). Thus, the mechanism(s) by which these P450-P450 interactions modify enzymatic activities involved a change in specific catalytic steps of the P450 cycle and not necessarily in the affinities by which the P450s bind to CPR. A kinetic analysis of the metabolism of S-flurbiprofen by CYP2C9 with and without CYP2D6 indicated that the interaction with CYP2D6 lowered the Vmax of the reaction yet had no effect on the Km for S-flurbiprofen. However, when S-flurbiprofen binding to CYP2C9 was analyzed directly by spectral changes of the P450 heme group, the presence of CYP2D6 perturbed the binding so that the dissociation constant for S-flurbiprofen and CYP2C9 was increased 20-fold. The fact that Ksflurbiprofen was dramatically altered but not the Kmflurbiprofen suggests that the binding of CYP2D6 may destabilize the CYP2C9-flurbiprofen complex (increasing koff) in addition to lowering the kcat of the reaction (Figure 2).
Figure 2. Effects of a P450-P450 interaction on metabolism when the mechanism involves a change in the rate(s) of a catalytic step(s) of the P450 reaction cycles.
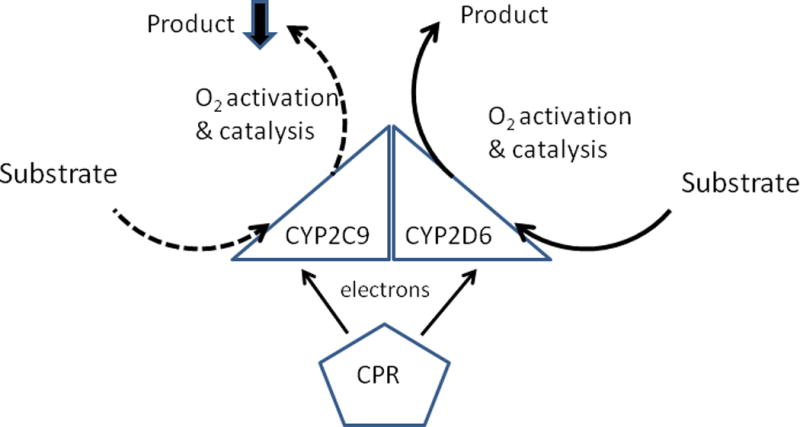
The diagram indicates the reported effects of the CYP2C9-CYP2D6 interaction on metabolism of S-flurbiprofen and dextromethorphan, respectively (Subramanian, Low, Locuson, & Tracy 2009). Arrows for the various steps of catalysis are described in Figure 1.
Although the effects of the CYP2C9-CYP3A4 interaction are very similar to those associated with the CYP2C9-CYP2D6 interaction (i.e. inhibition of CYP2C9-mediated catalysis at saturating CPR, a lower Vmax but unchanged Km for metabolism of both S-flurbiprofen and naproxen, and no effect on metabolism by the P450 paired with CYP2C9), the mechanism involving disrupted complex formation between CYP2C9 and its substrate was not assessed for this P450-P450 interaction. However, it seems likely that the effects of the CYP3A4-CYP2C9 interaction are driven by the same mechanism as that for CYP2C9 and CYP2D6. The CYP3A4-CYP2C9 study did go a step further to show a physical interaction between the P450s by co-immunoprecipitation using a specific CYP2C9 antibody.
In an elegant, recent study, both CYP3A4 and CYP3A5 were shown to participate in interactions with CYP2E1 that influenced the latter’s metabolism of 7-methoxy-4-trifluoromethylcoumarin (7MFC) (Davydov, Davydova, Sineva, & Halpert 2015). In other experiments, the study also used labeled enzymes that were capable of luminescence resonance energy transfer (LRET) upon interacting with one another. The LRET experiments demonstrated that all three P450s physically interacted with one another when incorporated in natural membranes. The interaction of either CYP3A4 or CYP3A5 with CYP2E1 resulted in the stimulation of 7MFC metabolism by CYP2E1. Metabolism by CYP2E1 was measured as a function of the 7MFC concentration in the presence and absence of CYP3A4 and CYP3A5. The interaction with either CYP3A4 or CYP3A5 increased the kcat of the CYP2E1-supported reaction but also unexpectedly resulted in a slight decrease in the binding affinity (increase in Km) for 7MFC. As metabolism of 7MFC by the interacting P450s was not rigorously examined as a function of the CPR concentration, the direct effect on the KD for the CPR•CYP2E1 complex was not determined. The activating effect of CYP3A proteins on metabolism by CYP2E1 was diminished at higher CPR:P450 ratios which may indicate that the CYP3A-CYP2E1 interaction also increased the binding affinity (or access) of CPR to the CYP2E1 moiety of the CYP3A-CYP2E1 complex in addition to the kcat of the reaction. Although CYP3A activated CYP2E1-mediated metabolism, there appeared to be no reciprocal effects of CYP2E1 on CYP3A-mediated 7-benzyloxyquinoline metabolism. With respect to the CYP3A4 and CYP3A5 interaction, CYP3A5 may have slightly activated metabolism by CYP3A4 by eliminating cooperativity of CYP3A4-mediated metabolism and in the process, the activating effect of α-naphthoflavone (Davydov et al. 2013;Davydov, Davydova, Sineva, & Halpert 2015).
P450-P450 interactions that affect both CPR binding affinity and catalytic steps
The bulk of studies on the mechanism of interaction of CYP1A2 and CYP2B4 focused on the increase in the apparent binding affinity of CPR and the CYP1A2 moiety of the P450-P450 complex. However, a subsequent study implicated an additional contributing mechanism when the effects of the CYP1A2-CYP2B4 interaction were assessed using toluene as a substrate (Reed, Cawley, & Backes 2013). This study was novel in that the effect of the interaction on reaction coupling was assessed by measuring the rates of NADPH oxidation and hydrogen peroxide generation. Toluene metabolism by these P450s results in the generation of three metabolites. Benzyl alcohol and p-cresol are formed specifically by CYP2B4, whereas o-cresol is formed exclusively by CYP1A2. Consistent with the previously discussed effects of the CYP1A2-CYP2B4 interaction, CYP1A2-mediated metabolism was dramatically activated and that by CYP2B4 was inhibited by the P450-P450 interaction.
Previous studies have shown that the rate-limiting step of metabolism in most CYP1A2 reactions involves hydrogen abstraction from the substrate in the final step of catalysis (Guengerich et al. 2004;Yun et al. 2000). As a result, the activated oxygen intermediate in the CYP1A2 active site is present for an extended amount of time allowing for the delivery of extra electrons by the CPR and in turn, the formation of excess water. This was indeed shown to be the case in the CYP1A2-mediated metabolism of toluene as 83% of the NADPH oxidized by a CYP1A2 reconstituted system was used in the formation of this excess water ((Reed, Cawley, & Backes 2013) and Table 1). To evaluate the effects of the P450-P450 interaction on reaction coupling by the enzymes, the rates of P450-specifc product formation and NADPH oxidation by the individual reconstituted systems were multiplied by the factors of change observed in the rates of substrate metabolism by the mixed reconstituted system containing both P450s. For instance, CYP1A2-specific o-cresol formation was stimulated 3.75-fold by interaction with CYP2B4. Therefore, if the coupling of NADPH oxidation to metabolism was not altered by the P450-P450 interaction, the rates of NADPH oxidation, hydrogen peroxide production, and excess water production would also be stimulated by 3.75-fold. Analysis of the reaction coupling of CYP1A2-mediated metabolism by the mixed system clearly indicated that the production of excess water by CYP1A2 was dramatically decreased by the P450-P450 interaction making the reaction much more efficient (only 52% of oxidized NADPH was used for excess water production by the mixed system). The ironic aspect of this finding is that the tighter binding of CPR to the CYP1A2-moiety of the mixed CYP1A2-CYP2B4 complex (as described above) might be expected to deliver excess electrons to the CYP1A2 active site and in turn, cause an increase in excess water production at the expense of substrate hydroxylation.
Table 1. Measured and predicted rates of P450-mediated NADPH oxidation, toluene metabolism, and the production of uncoupled reaction products catalyzed by simple and mixed reconstituted P450 systems.
The rates of NADPH consumption and the production of hydrogen peroxide, toluene metabolites, and excess water by P450 reconstituted systems were determined as described previously (Reed, Cawley, & Backes 2013). The rates are expressed as the average ± the standard error of five separate determinations. The table shows the rates of formation of the P450-specific toluene metabolites by the reconstituted systems containing lipid, CPR, and CYP2B4a, CYP1A2b, or a mixed system containing both P450sc. The theoretical rates of NADPH oxidation and the production of uncoupled reaction products by the mixed systemd were based on the relative rates of substrate oxidation in the simple and mixed systems. The theoretical rates are not merely the summation of rates measured using the simple systems containing either CYP1A2 or CYP2B4. Instead, the same proportionality factor relating the experimentally determined rates of CYP-specific substrate metabolism by the simple and mixed systems is also assumed to relate the rates of formation of uncoupled reaction products and NADPH oxidation by the simple and mixed systems. This assumption would be valid if only the rate of overall P450-mediated metabolism (and not the efficiency by which the reaction occurred) was altered by the P450-P450 interaction. Any significant deviation when comparing the “Experimental”c and “Theoretical”d groups points to a change in catalytic efficiency as compared to that observed with the simple systems. The results indicate that the mixed system consumes less NADPH and generates less “excess water” than predicted, and are consistent with electron flow being more efficiently coupled in the mixed reconstituted system. eExcess water refers specifically to that formed by excess reduction of the high valent, perferryl intermediate and not to the putative water molecule formed upon reduction, protonation, and heterolytic scission of molecular oxygen at an earlier stage of the catalytic cycle. As a result, the production of each molecule of excess water consumes two molecules of NADPH. This is why the rate of NADPH oxidation is not equal to the sum of the rates of formation of the reaction products.
| Product or Substrate | CYP2B4 systema | CYP1A2 systemb | Mixed system (Experimental results)c | Mixed system (Theoretical)d |
|---|---|---|---|---|
| NADPH oxidized | 12.7 ± 0.38 | 7.21 ±0 .23 | 14.14 ± 0.38 | 28.42 |
| Toluene metabolites | 2.61 ± 0.28 | 0.01 ± 0.002 | 1.51 ± 0.1 | 1.51 |
| H2O2 formed | 2.01 ± 0.63 | 1.20 ± 0.1 | 5.23 ± 0.86 | 4.68 |
| Excess H2O formede | 4.04 ± 1.29 | 3.00 ± 0.33 | 3.70 ± 1.34 | 11.16 |
The apparent contradiction could be rationalized if the changes in CYP1A2 reaction coupling were mediated specifically through an increase in the rate of hydrogen abstraction from the substrate by activated oxygen in the catalytic site of CYP1A2 (i.e kcat). Interestingly, peroxidative metabolism by CYP1A2 (which does not require electrons from NADPH) showed the P450-P450 interaction did not result in a significant change in the rate of P450-mediated metabolism relative to the individual P450 systems. Thus, the putative change in kcat resulting from the CYP1A2-CYP2B4 interaction seemed to be much smaller than the effect of the interaction on CPR-binding by CYP1A2. Interestingly, peroxidative metabolism by CYP1A2 was slightly activated by the physical presence of CPR in the absence of NADPH. This suggests CPR might play an effector role by binding to CYP1A2 in a way that causes electron flow to the CYP1A2 active site to be better regulated and coordinated with the formation of activated oxygen bound to the heme group. The easiest explanation, given the dramatic conformational changes that have been demonstrated for CPR (reviewed in (Laursen et al. 2011)), is that the binding of partially oxidized CPR (with interacting FAD and FMN groups) facilitates productive metabolism by CYP1A2, whereas binding of the extended, reduced form of CPR (with the FMN group rotated away from the FAD group) would negatively impact drug metabolism at the latter stage of the P450 catalytic cycle by favoring excess water production. An effector role for oxidized CPR has been proposed for other P450-mediated reactions (Davydov et al. 2010;Reed and Hollenberg 2003). This putative effector role for CPR would explain why increased binding between CPR and the CYP1A2 moiety of the mixed CYP2B4-CYP1A2 complex does not necessarily lead to an increase in excess water formation. In fact, by causing increased binding between CPR and CYP1A2, the CYP2B4-CYP1A2 interaction also contributes to the positive effector role of CPR.
A physical interaction between CYP2C9 and CYP2C19 has also been reported to cause both stimulation of CYP2C9-mediated metabolism of diclofenac and inhibition of CYP2C19-mediated metabolism of both methoxychlor and S-mephenytoin (Hazai & Kupfer 2005). The authors proposed that the P450-P450 interaction caused the CYP2C9 moiety of the complex to have a higher affinity to bind CPR resulting in stimulation of this enzyme in the same manner that CYP1A2 was affected by the interactions discussed above. By corollary, the inhibition of the CYP2C19 reactions was attributed to the CYP2C9 moiety sequestering CPR from binding to CYP2C19 moiety of the P450-P450 complex. Unfortunately, the data in this study did not conclusively validate this proposed mechanism. The binding affinity of CYP2C19 and CPR was very low in the absence of CYP2C9 (the enzyme was not saturated even at a 9:1 CPR:P450 ratio). Therefore, without additional experiments, the inhibition of CYP2C19 in the presence of CYP2C9 being the result of simple competition of independent P450s for limiting CPR could not be excluded. The most compelling data in the study to demonstrate a P450-P450 interaction showed that CYP2C9 activity was dramatically stimulated by increasing CYP2C19 concentrations even as the total CPR:P450 ratio decreased. However, in these experiments, the CPR concentration was saturating with respect to CYP2C9 (3:1 CPR:P450). Thus, under these conditions, the level of binding between CPR and the CYP2C9 would not be expected to increase by the addition of CYP2C19. The observed increase under the conditions of this experiment apparently demonstrated that the P450-P450 interaction resulted in an increase in the rate of an undetermined catalytic step of the CYP2C9 reaction. It is possible that a mechanism involving increased binding affinity between CYP2C9 and CPR also contributed to the observed effects. Therefore, this interaction is tentatively classified as one involving two mechanisms by which P450-mediated metabolism is altered.
Interactions between CYP2E1 and CYP2B4 have also been reported (Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013). CYP2E1 was shown to inhibit benzphetamine metabolism by CYP2B4 in a concentration dependent manner. When CYP2E1 was present at lower concentrations than CYP2B4, benzphetamine metabolism was inhibited competitively with respect to the CPR concentration when the reaction was initiated with NADPH; however, the tert-butyl hydroperoxide-supported reaction was only marginally affected. Interestingly, the Y422D CYP2E1 mutant that was shown to have an impaired ability to bind CPR was shown to be a less effective inhibitor of CYP2B4 activities (Lin et al. 2007). Conversely, the presence of CYP2B4 caused a decrease in the KD of the CYP2E1 and CPR complex when the rate of the CYP2E1-mediated metabolism of p-nitrophenol was measured as a function of the CPR concentration. These results are consistent with the P450-P450 interaction causing the binding affinity of CYP2E1 and CPR to be increased and that of CYP2B4 and CPR to be decreased. Thus, the effects of the interaction were similar to those described above for the interaction of CYP2B4 and CYP1A2.
However, when the CYP2E1 concentration greatly exceeded that of CYP2B4 (≥4-fold), the inhibition of CYP2B4-mediated metabolism was noncompetitive with respect to the CPR concentration, and it was shown that this inhibition was caused by a 30-fold increase in the Ks for CYP2B4 and benzphetamine. Thus, at higher concentrations, the CYP2E1 presumably formed a higher order complex with CYP2B4 that interfered with substrate binding and not CPR binding. In conjunction with the inhibition of CYP2B4, The CYP2E1 and CYP2B4 interaction resulted in dramatic stimulation in the kcat for the metabolism of p-nitrophenol by CYP2E1. Thus, the study showed that the interaction with CYP2B4 also increased an undefined catalytic step in the CYP2E1-mediated reaction. These findings show that the CYP2B4-CYP2E1 interaction has functional effects that are caused by both the mechanism involving altered CPR-binding affinities of the P450s and one involving alterations in the rates of catalytic steps of the P450 reaction cycle (Figure 3).
Figure 3. Effects of a P450-P450 interaction on metabolism when the mechanisms involve both a change in CPR-binding affinities and the rate(s) of a catalytic step(s) of the P450 reaction cycles.
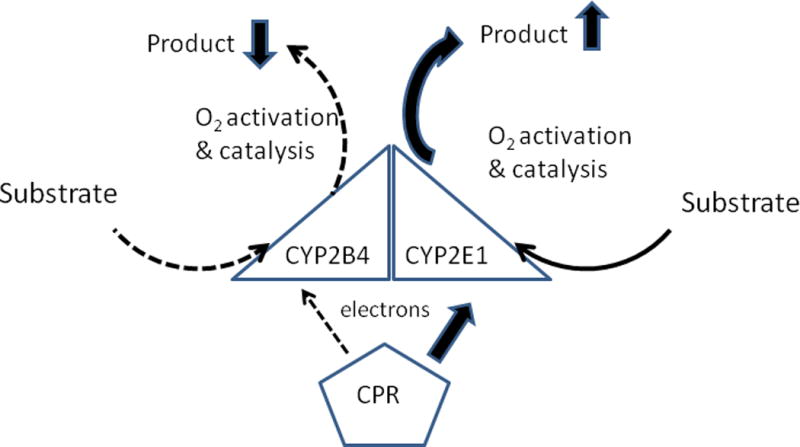
The diagram indicates the reported effects of the CYP2E1-CYP2B4 interaction on metabolism of p-nitrophenol and benzphetamine, respectively (Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013). Arrows for the various steps of catalysis are described in Figure 1.
P450-P450 interactions that influence metabolism by unknown mechanisms
Many of the earliest studies pertaining to P450-mediated metabolism simply reported evidence of functional interactions without specifying the mechanisms causing the effects. For instance, Kaminsky (Kaminsky et al. 1983;Kaminsky and Guengerich 1985) observed general effects of P450-P450 interactions on metabolism of warfarin when comparing the activity in rat liver microsomes and the sum of activities by eight different forms of purified rat P450s after the rates were adjusted to account for the limiting supply of CPR in the microsomes.
In one of the earliest studies to implicate that a specific P450-P450 interaction could modify P450-mediated activity, metabolism and P450-P450 interactions using six different forms of purified P450s (CYP1A1, CYP1A2, CYP2C9, CYP2E1, CYP2D6, and CYP3A4) were assessed (Yamazaki et al. 1997). The study demonstrated that the CYP3A4-mediated hydroxylation of testosterone was stimulated by both CYP1A1 and CYP1A2. Interestingly, an earlier study had confirmed a physical interaction between these P450s using chemical cross-linking and immunoprecipitation (Alston et al. 1991). Although the study did not attempt to determine the mechanism by which the CYP1A2 interaction caused stimulation of CYP3A4, the rates were determined at a relatively high 1:1 CPR:P450 ratio. Thus, the effect probably involved a catalytic step of CYP3A-mediated metabolism although an effect on the CPR-binding affinity of the CYP3A4 moiety could not be excluded. The interaction with CYP3A4 did not have any reciprocal effects on metabolism of theophylline by CYP1A2. Metabolism of specific probe substrates by most of the P450s examined in this study (bufuralol-CYP2D6, tolbutamide and S-warfarin-CYP2C9, chloroxazone-CYP2E1) was measured with all possible combinations of the other P450s to test for other P450-P450 interactions that had functional effects. No P450-P450 interactions were found to influence metabolism of these substrates. The study did not examine the effects of these other P450s on CYP1A-specific metabolism. Thus, it was not ascertained whether CYP2D6, CYP2E1, or CYP2C9 influenced the metabolism of theophylline by CYP1A2.
As one of the early studies to demonstrate the functional effects of P450-P450 interactions, the CYP3A4-CYP1A2 interaction was assessed with respect to two important aspects necessary to interpret many subsequent studies of P450-P450 interactions (Yamazaki, Gillam, Dong, Johnson, Guengerich, & Shimada 1997). First, the effects of the interaction with CYP3A4 was also examined using CYP1A2 from different species (rabbit and rat), and it was found that the forms from these other species also stimulated the metabolism of testosterone by CYP3A4. This is an important finding in extending the findings regarding interactions of P450s from animal species to those involving the orthologous forms in human. Secondly, the study examined many different CYP1A2 mutants containing modifications in the N-terminus. Many of the N-terminal mutants did not activate testosterone 6β-hydroxylation upon interaction with CYP3A4. One of the mutations that was effective involved a widely-used substitution of an E. coli expression sequence for the wild type N-terminal sequence of CYP1A2 (Dong et al. 1996a;Dong et al. 1996b). Addition of a thrombin site in proximity to the N-terminus of the protein in a region that was exposed to the cytosol stimulated CYP3A4-mediated metabolism after subsequent cleavage of this protein by thrombin. Interestingly, the uncleaved form of this mutant did not stimulate CYP3A4. When a thrombin site was inserted in a membrane-spanning region of the N-terminal sequence, the mutant did not activate metabolism by CYP3A4 with or without cleavage by thrombin. Thus, the results showed that some of these N-terminal mutants lose their native abilities to interact with and affect the function of other P450 forms, and caution must be used depending on the specific N-terminal mutation that is used. This part of the study was significant because the N-terminally modified proteins are almost universally used in current P450-P450 interaction studies (Davydov, Davydova, Sineva, & Halpert 2015;Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013;Subramanian, Low, Locuson, & Tracy 2009;Subramanian, Zhang, & Tracy 2010).
Homomeric P450-P450 interactions that affect function
Homomeric P450 interactions are also associated with affecting P450 function. This was reported for CYP1A2 where the metabolism of 7-pentoxyresorufin, 7-ethoxycoumarin, and 7-ethoxyresorufin by a simple reconstituted system with CYP1A2 as a function of the CPR concentration was hyperbolic at low CYP1A2 concentrations, but became sigmoidal under conditions where the proteins were more crowded in the membrane (Reed et al. 2012). Interestingly, the sigmoidal curves could be converted to hyperbolic curves by increasing the ionic strength of the buffer, conditions that would diminish the electrostatic CYP1A2•CYP1A2 complexes. These data are consistent with an equilibrium between CYP1A2•CYP1A2 complexes and CYP1A2 monomers where the monomers are more active. The study proceeded to use chemical cross-linking of protein amino groups to demonstrate that higher order aggregation of the enzyme was associated with conditions leading to inhibition of the catalytic activities. Finally, the aggregation of CYP1A2 was demonstrated in HEK 293T cells using BRET after co-expression of CYP1A2 cDNAs that were linked with the cDNA for either luciferase or green fluorescent protein (GFP) on the C-terminal end of the P450 sequence.
Interestingly, the study examining the homomeric interaction of CYP1A2 also showed the same general kinetic profiles for rabbit CYP2E1-mediated metabolism of N-dimethylnitrosamine as a function of the CPR concentration at different P450 concentrations (high P450 concentrations above 0.1 μM were sigmoidal, whereas those at lower concentrations were hyperbolic) (Reed, Connick, Cheng, Cawley, & Backes 2012). Thus, the metabolism of this substrate by CYP2E1 also appeared to be associated with a functional (inhibitory) homomeric interaction of the P450.
These findings were consistent with a comprehensive kinetic analysis of the metabolism of p-nitrophenol by reconstituted systems of rabbit CYP2E1 by Miller’s group (Jamakhandi et al. 2007). First, the study used a Job plot to show that metabolism by the P450 could not be explained by a simple 1:1 complex formation between CPR and CYP2E1. Instead, the data suggested higher order aggregates of the enzymes were in equilibrium with the 1:1 complex. The authors then compiled a comprehensive data set representing metabolism as a function of variations in both CYP2E1 and CPR concentrations. These data were used to mathematically discriminate between 32 possible mechanisms involving varying degrees of CYP2E1 and CPR aggregation. The best fit for the data was derived from models that included a P450 homodimer, a P4502:CPR2 quaternary complex, and possibly a P4502:CPR ternary complex in addition to the 1:1 P450:CPR complex. Interestingly, the best fit models indicated that the 1:1 CPR:CYP2E1 complex was the only functional complex, and the other two (or three depending on the model) complexes were catalytically silent (and thus inhibitory).
In contrast to the compelling kinetic data indicating CYP2E1 aggregation, studies using both BiFC (Ozalp et al. 2005) and FRET (Szczesna-Skorupa et al. 2003) to monitor aggregation failed to provide evidence of homomeric CYP2E1 interactions after co-expression of the modified cDNAs in either COS1 or HEK293 cells. Thus, unlike the case for homomeric functional interactions involving CYP1A2, the physical evidence supporting the case for homomeric interactions of CYP2E1 has remained elusive.
The presence of homomeric complexes affecting P450 function was not observed with all P450s tested. Despite both CPR-CYP1A2 and CPR-CYP2E1 exhibiting a more exaggerated sigmoidal responses in activity as a function of CPR as P450 crowding was increased, CYP2B4 showed simple hyperbolic behavior consistent with monomeric CYP2B4 interacting with CPR. This does not necessarily mean that CYP2B4 does not form homomeric complexes, as it is possible that CYP2B4•CYP2B4 are not kinetically distinct from the monomer (Reed, Connick, Cheng, Cawley, & Backes 2012). Taken together, the results show that P450s can form homomeric complexes; however, it is not certain that this is a universal characteristic of all P450s.
Studies conducted by Davydov and co-workers have provided evidence for the positional heterogeneity of homomeric P450 aggregates. Initially, the ferrous-CO complex of P450 2B4 was examined under hyperbaric conditions to find that 65 to 70% of the aggregated P450 was sensitive to a particular pressure range as indicated by a spectral transition to P420 (Davydov, Knyushko, & Hoa 1992). When the P450 was solubilized with a detergent, the entire population of CYP2B4 enzyme was subject to pressure-related inactivation indicating that the aggregation of the enzyme actually protected a segment of the CYP2B4 population from the hyperbaric conditions. These findings demonstrated the heterogeneous nature of the homomeric CYP2B4 complex. When the ferric P450 was analyzed, the authors also found that 65 to 70% of the aggregated ferric P450 was either not capable of or not responsive to the binding of substrate as indicated by an induced low to high spin state transition. Written another way, the ferrous-CO CYP2B4 moieties in the homomeric complex were arranged in a manner that protected 30 to 35% of the P450 from hyperbaric sensitivity, and in the ferric state, presumably the same 30 to 35% was responsive to substrate binding as indicated by a low to high spin state change of the ferric heme. Subsequently, it was shown that aggregated CYP1A2 was completely resistant to the pressure-induced inactivation over a comparable range of pressure, and in the presence of excess CYP1A2, CYP2B4 no longer exhibited this pressure-sensitive conversion of P420 (Davydov, Petushkova, Archakov, & Hoa 2000b). The study demonstrated a difference in the physical characteristics of the two P450s when present in homomeric aggregates and indicated the active site of a fraction of the total CYP2B4 population was much more subject to hydration that prevented a substrate-induced spin state change when in a homomeric aggregate than it was in an aggregate including CYP1A2.
Davydov and co-workers have also provided evidence for positional heterogeneity by examining the pre-steady state reduction of P450s by dithionite. Multi-phasic reduction (biphasic or triphasic) of aggregated CYP2B4 (Davydov et al. 1985) or CYP3A4 (Davydov, Fernando, Baas, Sligar, & Halpert 2005) was observed in solution or when the P450s were incorporated in proteoliposomes. Interestingly, the study with CYP3A4 indicated three phases of reduction of approximately equal amplitudes and showed that the low spin form of P450 was exclusively reduced in the fast phase, whereas the slow phase mainly represented reduction of the high spin form. This would indicate that the soluble reductant has better access to the low spin active site probably as a result of a higher degree of hydration in proximity to the heme group. Substrate binding increased the proportion of high spin enzyme and quantitatively decreased the proportion of enzyme reduced in the fast phase. The middle phase of reduction appeared to represent enzyme that was subject to a substrate-induced spin state change but unlike the high spin form reduced in the slow phase of reduction, the spin shift did not affect the rate at which this population of CYP3A4 was reduced by dithionite.
In a subsequent study, the kinetics of CYP3A4 reduction were measured through protein-protein interactions involving a redox partner (Davydov, Sineva, Sistla, Davydova, Frank, Sligar, & Halpert 2010). For these experiments, the soluble reductase domain of P450BM3 (Davydov et al. 2000a;Sevrioukova et al. 1996) was used instead of the membrane-bound CPR in order to simplify the interpretation of the kinetics. In contrast to the results observed using dithionite as a reductant, the high spin form was reduced in the fast phase and low spin form was reduced in the slow phase indicating that the high spin form within the P450 aggregate was better able to interact with the redox partner than the low spin form.
In both of the studies examining reduction of CYP3A4, physical evidence was provided to show the multi-phasic reduction was attributable to oligomerization of the CYP3A4. First, it was shown that the reduction process became mono-phasic upon solubilization with detergent. Next, the authors assessed reduction of monomeric CYP3A4 using nanodiscs™ (Baas et al. 2004;Davydov, Sineva, Sistla, Davydova, Frank, Sligar, & Halpert 2010). Nanodiscs utilize phospholipid and a membrane scaffolding protein that allows for the stabilization of small, discoidal lipid-bilayer particles that contain monomeric enzyme when mixed at appropriate relative concentrations. After incorporation of monomeric CYP3A4 into nanodiscs the reduction became biphasic (from triphasic) and the amplitudes of the phases correlated with the proportions of high and low spin forms of the P450. Upon addition of saturating concentrations of substrate, the enzyme totally converted to the high spin form, and the reduction of CYP3A4 in the nanodiscs became monophasic in the process indicating the functionality of the monomeric enzyme. Finally, the reduction kinetics also provided evidence that aggregated CYP3A4 dissociated into smaller aggregates (possibly monomeric) as the lipid:P450 ratios of the liposomes into which it was incorporated increased from 112:1 to 726:1. Because the lipid:protein ratio in microsomes is approximately 23:1 (Depierre and Dallner 1975;Wibo et al. 1971), P450 enzymes would probably be entirely aggregated in vivo.
Physical evidence of aggregation was also demonstrated in this study with a FRET-based assay (fluorescence energy resonance) using BODIPY iodoacetamide to label cysteine residues in CYP3A4. Originally, a quadruple mutant of CYP3A4 was engineered in which four cysteines were replaced with serine or alanine (Tsalkova et al. 2007). The selective re-introduction of Cys back into the quadruple mutant then allowed for labeling with Cys-reactive fluorescent probes and cross-linkers. These Cys mutants of CYP3A4 not only provided evidence for CYP3A4 homomeric interactions but also established approximate distances between cysteines in the interacting CYP3A4 units of homomeric oligomers (Davydov, Davydova, Sineva, Kufareva, & Halpert 2013;Davydov, Davydova, Sineva, & Halpert 2015;Tsalkova, Davydova, Halpert, & Davydov 2007).
In addition to correlating the kinetic changes with the P450 surface density in proteoliposomes, Davydov et al. also provided physical evidence of a binding equilibrium over the 100:1 to 726:1 range of lipid:P450 ratios by using time–resolved LRET (luminescence resonance energy transfer) of the CYS-labeled CYP3A4 mutants (discussed in previous paragraph) that were labeled with cysteine-reactive probes (Davydov, Davydova, Sineva, Kufareva, & Halpert 2013). One aspect of CYP3A4-mediated metabolism that has been well studied is cooperativity with respect to substrate resulting in atypical, often sigmoidal, enzyme kinetics (reviewed in (Davydov and Halpert 2008;Denisov and Sligar 2012)). These effects can be observed upon varying concentrations of a single substrate (homotropic) or by the presence of different compounds (heterotropic). One heterotropic activator of CYP3A4 is α-naphthoflavone (ANF). In the presence of this effector, sigmoidal enzyme kinetics related to metabolism of some substrates is eliminated, resulting in enzyme activation at limiting substrate concentrations. In large part, these effects are attributed to multiple substrate binding sites within the active site of the P450 (Houston and Galetin 2005). However, Davydov and others (Davydov, Davydova, Sineva, Kufareva, & Halpert 2013) provided evidence using lipid dilution and LRET between the aforementioned fluorescent-modified CYS mutants that ANF binding to CYP3A4 changed the aggregation state of the P450. The altered aggregation state was shown to be associated with the ANF-related changes in enzyme kinetics for the metabolism of 7-benzyloxy-4-trifluoromethylcoumarin. These findings indicate that some cases of homomeric and heteromeric cooperativity may be attributable to substrate/effector binding causing changes in P450 aggregation instead of changes in the way substrate binds within the active site. With respect to ANF at least, it appears that its binding site may actually be outside the active site of the enzyme (Davydov, Davydova, Sineva, Kufareva, & Halpert 2013).
Substrate specificity of the effects of P450-P450 interactions
In our description of the effects of P450-P450 interactions, we have been careful to state the substrates with which these effects are observed. This is because the metabolism of some substrates is not affected by the interaction of the same P450 pairs discussed above. For instance, it appeared that the functional interaction of CYP1A and CYP2B that has been so extensively characterized in the metabolism of a variety of substrates (discussed above) did not affect metabolism of 3,4-benzo[a]pyrene by CYP1A2 (West and Lu 1972). Similarly, there was no evidence of the interaction of CYP3A4 and CYP2C9 affecting metabolism of S-warfarin and tolbutamide by the latter (Yamazaki, Gillam, Dong, Johnson, Guengerich, & Shimada 1997) even though CYP2C9-mediated metabolism of naproxen was inhibited by the interaction (Subramanian, Zhang, & Tracy 2010). In addition, the mutual effects resulting from the interaction of CYP2B4 and CYP2E1 in the metabolism of benzphetamine and p-nitrophenol (Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013), respectively, were not observed for the metabolism of 7-ethoxyresorufin, 7-pentoxyresorufin, aniline, and N-nitrosodimethylamine by the P450 pair (Kelley, Cheng, & Backes 2006). Consistency is not even observed with respect to homomeric interactions as the CYP1A2-mediated metabolism of 7-ethoxy-4-trifluoromethylcoumarin was clearly hyperbolic as a function of CPR concentration even though metabolism of the structurally similar 7-ethoxycoumarin by the enzyme was highly sigmoidal at high concentrations of the P450 (Reed, Connick, Cheng, Cawley, & Backes 2012). Finally, homomeric interactions involving CYP2E1 did appear to have effects on metabolism of N-nitrosodimethylamine but not that of 7-ethoxy-4-trifluoromethylcoumarin (Reed, Connick, Cheng, Cawley, & Backes 2012). It is interesting that homomeric effects of the CYP2E1 interaction were observed for metabolism of N-nitrosodimethylamine. However, no effects of the CYP1A2-CYP2E1 interaction were observed on the CYP2E1-mediated metabolism of the same substrate (Kelley, Cheng, & Backes 2006). In some cases, these substrate differences were observed by the same investigator; however, in others the differences were between different laboratories. Consequently, some of the discrepancies may be the result of differences in reconstitution conditions as noted by several investigators (Causey et al. 1990;Reed et al. 2006;Reed et al. 2008;Subramanian, Low, Locuson, & Tracy 2009).
Many of the screens of potential new drugs measure their metabolism in systems containing single expressed human P450 enzymes. Although these systems provide information on metabolism by human P450 enzymes, the potential for P450s to form complexes that influence function can lead to discrepancies that make in vitro to in vivo extrapolation more difficult. An example of this problem was illustrated with a report by Hazai and Kupfer (Hazai & Kupfer 2005) in the study of the CYP2C9-CYP2C19 interaction. This is made more complex when considering that such effects may only be presented by some substrates. Consequently, extrapolation of in vitro metabolism of a new drug or foreign compound to better predict the in vivo condition may require measurement of substrate metabolism using systems containing multiple P450 enzymes to account for P450 supramolecular complex formation.
Studies to demonstrate that P450-P450 interactions influence metabolism in natural membranes
A preponderance of the evidence for the functional effects of P450-P450 interactions stem from work using purified, reconstituted systems, with most of these studies using the synthetic lipid, DLPC, to reconstitute the P450 activity; however, there is ample evidence that the P450-P450 interactions found with reconstituted systems also can be found in cellular systems and in vivo.
Some studies of P450-P450 interactions have better simulated the ER-membrane environment by using labor-intensive methods to physically incorporate P450s into monolamellar, bilayer vesicles (Davydov, Fernando, Baas, Sligar, & Halpert 2005;Reed, Eyer, & Backes 2010). These types of studies also have the added benefit of being able to use the physiologic lipid composition of the ER membrane. Recently, Davydov et al. have developed methods to physically incorporate P450s in preformed bilayer vesicles (Davydov, Sineva, Sistla, Davydova, Frank, Sligar, & Halpert 2010) or in the microsomal membranes of insect cells (Supersomes™) (Davydov, Davydova, Sineva, & Halpert 2015). To get CPR in the latter system, Supersomes from insect cells transfected with a CPR-expressing construct were used as this protein will not readily incorporate in a preformed membrane. Thus, the methods utilizing preformed vesicle incorporation are limited in the ability to modulate CPR levels which is imperative when testing mechanisms that involve changes in CPR-binding affinities. Although the method is relatively easy, it has only been used with truncated, N-modified P450s. It is not clear if the wild type, full length P450 enzymes would incorporate into the preformed membranes. Thus, the use of the method has the potential to overlook P450-P450 interactions mediated by the wild type, N-terminal regions.
Because of the limitations with purified, reconstituted P450 systems, it has been necessary to confirm that P450-P450 interactions affect enzyme function in vivo. One approach in this regard has been to demonstrate the functional effects in the microsomes from tissues. The use of microsomes circumvents the doubts involving the lipid composition, the relative concentrations of lipids and P450s, and the manner in which the P450s associate with the lipid. In order to validate the relevance of the effects of the CYP1A2-CYP2B4 interaction observed in purified, reconstituted systems (Backes, Batie, & Cawley 1998), rates of substrate metabolism were compared using microsomes from rabbits treated with phenobarbital, β-naphthoflavone, and both compounds in order to induce expression of CYP2B4, CYP1A2, and both P450s, respectively (Cawley et al. 2001). After quantifying the amounts of both P450s and CPR in the microsomes from the different treatment groups, it was shown that CYP1A-mediated metabolism was activated and that by CYP2B4 was inhibited in the microsomes from rabbits treated with both inducers relative to the activities observed in microsomes from animals treated with only one of the inducers. Thus, the effects predicted from the studies with the purified, reconstituted systems were confirmed in the experiments with microsomes.
A recent publication has used a hepatocyte model as a better simulation of in vivo conditions to provide evidence for the functional effects of the CYP3A4-CYP2C9 interaction (Ramsden et al. 2014). The study used the HepatoPac™ culture system which has been shown to retain the phenotypic enzymatic activities of fresh hepatocytes for several weeks. By virtue of the prolonged metabolic capacity of this culture system, the researchers were able to examine the effects of CYP3A4 on CYP2C9-mediated activity by modulating CYP3A4 expression using small interference RNA (siRNA) and a subsequent recovery phase after removal of the siRNA. Consistent with the effects of the P450-P450 interaction in reconstituted systems (Subramanian, Zhang, & Tracy 2010), higher CYP3A4 expression was associated with lower CYP2C9 activity. In fact, the percentage decrease in CYP3A4 expression upon treatment with the siRNA was approximately equal to the percentage increase in the rate of the CYP2C9-mediated metabolism of diclofenac by the hepatocytes. In addition, removal of the CYP3A4 siRNA resulted in a recovery of the pre-treatment expression levels and in turn, inhibition of CYP2C9-mediated metabolism.
A recent study using immunoprecipitation followed by LC-MS protein identification provided a sense of the extent and diversity of protein interactions with P450 in vivo (Li et al. 2011). Protein complexes involving rabbit CYP2C2 were identified in mouse liver after infection of the mice with an adenovirus containing a Flag- and His-tagged recombinant CYP2C2 cDNA. Immunoprecipitation of the Flag-tagged protein from liver microsomes followed by purification of the precipitates on a nickel-agarose affinity column (by virtue of the His-tag) resulted in co-isolation of 25 proteins. Interestingly, most of the proteins associated with CYP2C2 utilized NADH/NADPH directly or indirectly through interactions with redox partners. The sedimentation patterns of the complexes on a sucrose step gradient suggested that the P450 participated in a variety of heterogeneous protein complexes with masses ranging up to 600,000 Da. Many complexes of enzymes with the modified CYP2C2 were characteristic of a metabolon that could catalyze sequential phase I and phase II metabolism of drugs. Although the study did not assess the effects of protein interactions on CYP2C2-mediated activity, the interactions of CYP2C2 with both CYP3A and CYP2D forms were detected, consistent with the P450-P450 interactions that were shown to influence human CYP2C9-mediated activity in purified, reconstituted systems (Subramanian, Low, Locuson, & Tracy 2009;Subramanian, Zhang, & Tracy 2010).
Another approach to demonstrate the effects of P450-P450 interactions in biological membranes has been to express P450 chimeras that are capable of FRET or BRET upon physically interacting with one another in cell culture systems. The Kemper lab pioneered these types of studies with P450s by looking at various aspects of CYP2C2 interaction and localization in the ER. These studies have contributed greatly to demonstrating that P450-P450 interactions occur as a natural consequence of their expression in biological membranes.
Initially, the fluorescence emitted by different amino acid segments of CYP2C2 fused to green fluorescent protein (GFP) was measured after transient expression in COS1 cells (Szczesna-Skorupa et al. 1998). Fluorescence imaging showed that both the N-terminal sequence and the cytosolic domain of CYP2C2 conferred localization to the ER. Photobleaching studies indicated that 80% to 90% of the different CYP2C2 domains were laterally mobile within the membrane. Interestingly, the catalytic activity of CYP2C2-GFP was only slightly less than that by the wild type CYP2C2 indicating that the addition of GFP did not greatly alter protein function.
Later studies by Kemper’s group examined protein interactions by measuring FRET following interactions involving CFP (cyan fluorescent protein)- and YFP (yellow fluorescent protein)-labeled CYP2 family forms (CYP2C1, CYP2C2 and CYP2E1) and CPR (Szczesna-Skorupa, Mallah, & Kemper 2003). This study demonstrated oligomerization of CYP2C2 and the N-terminal domains of both CYP2C1 and CYP2C2 but not cytosolic CYP2C2 in which the N-terminal region was truncated. Interestingly, in direct contradiction to the conclusions of studies with purified CYP2E1 (Jamakhandi, Kuzmic, Sanders, & Miller 2007;Kelley, Cheng, & Backes 2006;Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013) (discussed above), FRET was not detected for the interaction of CFP- and YFP-labeled CYP2E1. Similarly, interaction of CYP2C2-CFP and CYP2E1-YFP was not detected by FRET, although both of the CFP-labeled CYP forms were shown to interact with YFP-labeled CPR (Jamakhandi, Kuzmic, Sanders, & Miller 2007;Kelley, Cheng, & Backes 2006;Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013). Furthermore, only the cytosolic domain of CYP2C2 was needed for complexation of the P450 with CPR.
A subsequent study used bimolecular fluorescence complementation (BiFC) to demonstrate interactions of CYP2C2 (and its individual amino acid domains), CYP2E1, and CPR after expression of chimeric forms in COS-1 cells (Ozalp, Szczesna-Skorupa, & Kemper 2005). For these experiments the interaction of chimeric forms of P450s and/or CPR were detected by transfecting a pair of chimeras in which the N-terminal ends were derived from one of the proteins in the P450 system and the C-terminal ends of the two chimeras were either the C-terminal or N-terminal ends of YFP. Interaction of the P450-related chimeras resulted in reconstitution (and in turn, the fluorescence) of full length YFP after excitation with 425 nm light.
In agreement with the FRET study (described above), the homomeric interactions of CYP2C2 and its N-terminal end sequence were demonstrated in addition to the interaction of the N-terminal end with the full length CYP2C2. Consistent with their FRET experiments, CYP2E1 did not appear to form homomeric oligomers. However, the heteromeric interaction of CYP2C2 and CYP2E1 was detected and was presumed to be mediated by the cytoplasmic domains of the P450s, unlike the homomeric interaction of CYP2C2. The BiFC study also showed that CPR formed homomeric complexes after expression in COS-1 cells, and suggested that both the N-terminal and cytosolic domains of CYP2C2 contributed to binding to CPR.
Taken together, the BiFC and FRET studies demonstrate the natural tendency of some P450 system proteins to interact after expression in the ER of cells, eliminating the doubts inherent when preparing reconstituted systems from stock solutions of purified enzymes. It was also shown that the catalytic activities of the expressed P450s could be measured using the chimeric fusion proteins. Finally, the studies offer the ability to modulate the expression levels of the proteins by varying the amount of cDNA transfected into the cells. These three characteristics of the fluorescent studies will allow for mechanistic questions to be addressed about the effects of the P450-P450 interactions.
Conclusion
The physical interaction of P450s can influence the rates of metabolism of specific substrates by various P450 isoforms. This review has summarized studies pertaining to this phenomenon and when possible has classified the P450-P450 interactions on the basis of the mechanisms by which they affect metabolism. P450-P450 interactions can alter P450-mediated metabolism by affecting the abilities of P450s in a complex to bind to CPR, to bind to substrate, or by both types of mechanisms. Mechanisms have also been proposed that involve changes in the rates of catalytic steps of the enzymatic cycle other than substrate binding.
An important aspect of pharmacology concerns understanding the metabolism and disposition of drugs in humans. Clearly, alterations in enzyme activities caused by the physical interaction of P450s will limit the accuracy of these methods and indeed, there are many examples in the literature where in vivo clearance of a drug is poorly predicted using parameters obtained in vitro (Houston and Kenworthy 2000;Stringer et al. 2009). The physical interactions of P450s can have profound effects on the metabolism of substrates and can influence metabolism and disposition of xenobiotics. This fact has been evidenced by the studies from the Kupfer lab showing that metabolism of the pesticide, methoxychlor, by human liver microsomes did not involve CYP2C19 even though it was the major P450 implicated when comparing the rates of metabolism by various expression systems containing only one P450 (Hu et al. 2004;Hu and Kupfer 2002). It is imperative to understand the specific changes that result from the physical interactions of different P450s so that we can better predict/anticipate outcomes resulting from exposure to drugs, toxins, and carcinogens that are P450 substrates.
Efforts are now focused on examining the extent to which P450-P450 interactions affect metabolism in vivo. Table 2 shows a summary of the studies pertaining to the effects of P450-P450 interactions. The table also shows the P450-P450 interactions that have not been tested for effects on P450 function. It is clear from the table that only a fraction of the full scope of P450-P450 interactions has been tested for functional effects. As a result, we know very little with regards to the extent to which this phenomenon affects drug metabolism in vivo. Studies utilizing BRET, FRET, or BiFC of appropriately labeled P450 chimeras have the potential to assess the relative tendencies of different P450s to interact and to determine the effects of specific P450 interactions on enzymatic function. This information will be useful in making predictions about the levels of drug metabolism in animals/humans.
Table 2.
Summary of P450-mediated reactions that are affected by P450-P450 interactions and the mechanism(s)a that cause these effects
| CYP1A2 | CYP2A6 | CYP2B | CYP2C8 | CYP2C9 | CYP2C19 | CYP2E1 | CYP2D6 | CYP3A4 | |
|---|---|---|---|---|---|---|---|---|---|
| CYP1A2 | −/−Rb*; NEb* | NT | +/−Bc–g*; NEh* | NT | NT | NT | +R/NEi* | NT | NT/+?j |
| CYP2A6 | NT | NT | NT | NT | NT | NT | NEk | NT | NT |
| CYP2B6 | −/+Bc–g* | NT | NEb*, −/−l* | NT | NT | NT | NEi*; −/+Bm* | NT | NT |
| CYP2C8 | NT | NT | NT | NT | NT | NT | NT | NT | NT |
| CYP2C9 | NEi | NT | NT | NT | NT | +/−Bn | NT | −C/NEo | −C/NEp; NEj |
| CYP2C19 | NT | NT | NT | NE | −/+Bn | NT | NT | NT | NT |
| CYP2E1 | NE/+Ri* | NEj | NEi*; +/−Bm* | NT | NT | NT | NEb −/−Rb*; −/−Cq* | NEi/NT | NEj; +C/NEr |
| CYP2D6 | NT | NT | NT | NT | NE/−Co | NT | NT/NEj | NT | NT/NE.j NEr |
| CYP3A4 | +?j/NT | NT | NT | NT | NE/−Cp | NT | NE/+Cr NTj | NE/NTj NEs | −/−Cr,t,u |
orthologous P450s tested from other species besides humans.
Mechanism(s) of interaction: R, alteration in CPR binding; C, alteration in catalytic step; B, both mechanisms involved. NT, interaction not tested; NE, no effect; ?, mechanism not determined. The symbol on the left side of the hash refers to the effect (+ or −) on the P450 on the left side of the table in the same row, and the one on the right of the hash refers to the effect on the P450 listed at the top of the table in the same column.
(Reed, Connick, Cheng, Cawley, & Backes 2012) (7-ethoxyresorufin; 7-ethoxy-4-trifluoromethylcoumarin; benzphetamine; p-nitrophenol; N-nitrosodimethylamine; 7-ethoxyresorufin);
(Backes, Batie, & Cawley 1998) (alkoxyresorufins)
Davydov, 2001(alkoxyresorufins)
(Reed, Eyer, & Backes 2010) (CYP1A2-P450Glo assay™;7-ethoxyresorufin; benzphetamine; 7-ethoxy-4-triflurormethylcoumarin; 7-ethoxycoumarin; p-nitroanisole);
(Reed, Cawley, & Backes 2013) (toluene);
(Kelley et al. 2005) (alkoxyresorufins; 7-ethoxy-4-trifluoromethylcoumarin);
(West & Lu 1972) (benzo-a-pyrene);
(Kelley, Cheng, & Backes 2006) (alkoxyresorufins; N-nitrosodimethylamine;aniline);
(Yamazaki, Gillam, Dong, Johnson, Guengerich, & Shimada 1997) (testosterone; S-warfarin; tolbutamide; chloroxazone);
(Tan et al. 1997) (coumarin; N-nitrosodimethylamine);
study by Davydov et al. (Davydov, Karyakin, Binas, Kurganov, & Archakov 1985) has shown functional heterogeneity with respect to P450 reduction, not substrate metabolism;
(Kenaan, Shea, Lin, Zhang, Pratt-Hyatt, & Hollenberg 2013) (benzphetamine; p-nitrophenol);
(Hazai & Kupfer 2005) (diclofenac; methoxychlor; S-mephenytoin);
(Subramanian, Low, Locuson, & Tracy 2009) (S-flurbiprofen; dextromethorphan);
(Subramanian, Zhang, & Tracy 2010) (S-flurbiprofen; naproxen; testosterone);
(Jamakhandi, Kuzmic, Sanders, & Miller 2007) (p-nitrophenol);
(Davydov, Davydova, Sineva, & Halpert 2015) (7-methoxy-4-trifluoromethylcoumarin; 7-benzyloxy-4-trifluoromethylcoumarin);
(Li et al. 1999) (testosterone;bufuralol);
(Fernando et al. 2008) – functional heterogeneity demonstrated by P450 reduction;
(Davydov, Davydova, Sineva, Kufareva, & Halpert 2013) (7-benzyloxy-4-trifluoromethylcoumarin, ANF).
Acknowledgments
This work was supported by US Public Health Service Research Grants from the National Institute of Environmental Health Sciences [ES013648 to WLB, JRR and ES004344 to WLB].
References
- Alston K, Robinson RC, Park SS, Gelboin HV, Friedman FK. Interactions among cytochromes P-450 in the endoplasmic reticulum. Detection of chemically cross-linked complexes with monoclonal antibodies. Journal of Biological Chemistry. 1991;266(2):735–739. [PubMed] [Google Scholar]
- Anandatheerthavarada HK, Addya S, Dwivedi RS, Biswas G, Mullick J, Avadhani NG. Localization of multiple forms of inducible cytochromes P450 in rat liver mitochondria: Immunological characteristics and patterns of xenobiotic substrate metabolism. Archives of Biochemistry and Biophysics. 1997;339:136–150. doi: 10.1006/abbi.1996.9855. [DOI] [PubMed] [Google Scholar]
- Baas BJ, Denisov IG, Sligar SG. Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Archives of Biochemistry and Biophysics. 2004;430(2):218–228. doi: 10.1016/j.abb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Backes WL, Batie CJ, Cawley GF. Interactions among P450 enzymes when combined in reconstituted systems: formation of a 2B4-1A2 complex with a high affinity for NADPH-cytochrome P450 reductase. Biochemistry. 1998;37(37):12852–12859. doi: 10.1021/bi980674a. [DOI] [PubMed] [Google Scholar]
- Causey KM, Eyer CS, Backes WL. Dual role of phospholipid in the reconstitution of cytochrome P- 450 LM2-dependent activities. Molecular Pharmacology. 1990;38:134–142. [PubMed] [Google Scholar]
- Cawley GF, Batie CJ, Backes WL. Substrate-dependent competition of different P450 isozymes for limiting NADPH-cytochrome P450 reductase. Biochemistry. 1995;34:1244–1247. doi: 10.1021/bi00004a018. [DOI] [PubMed] [Google Scholar]
- Cawley GF, Zhang S, Kelley RW, Backes WL. Evidence supporting the interaction of CYP2B4 and CYP1A2 in microsomal preparations. Drug Metab Dispos. 2001;29(12):1529–1534. [PubMed] [Google Scholar]
- Davydov DR. Microsomal monooxygenase as a multienzyme system: the role of P450-P450 interactions. Expert Opin Drug Metab Toxicol. 2011;7(5):543–558. doi: 10.1517/17425255.2011.562194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Davydova NY, Sineva EV, Halpert JR. Interactions among Cytochromes P450 in Microsomal Membranes: Oligomerization of cytochromes P450 2E1, 3A5, and 2E1 and its functional consequences. Journal of Biological Chemistry. 2015;290(6):3850–3864. doi: 10.1074/jbc.M114.615443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Davydova NY, Sineva EV, Kufareva I, Halpert JR. Pivotal role of P450-P450 interactions in CYP3A4 allostery: the case of alpha-naphthoflavone. Biochemical Journal. 2013;453(2):219–230. doi: 10.1042/BJ20130398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Deprez E, Hoa GH, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI. High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution: evidence for conformational inhomogeneity in the oligomers. Archives of Biochemistry and Biophysics. 1995;320:330–344. doi: 10.1016/0003-9861(95)90017-9. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Fernando H, Baas BJ, Sligar SG, Halpert JR. Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: heterogeneity of the enzyme caused by its oligomerization. Biochemistry. 2005;44(42):13902–13913. doi: 10.1021/bi0509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Halpert JR. Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin Drug Metab Toxicol. 2008;4(12):1523–1535. doi: 10.1517/17425250802500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davydov DR, Kariakin AA, Petushkova NA, Peterson JA. Association of cytochromes P450 with their reductases: opposite sign of the electrostatic interactions in P450BM-3 as compared with the microsomal 2B4 system. Biochemistry. 2000a;39(21):6489–6497. doi: 10.1021/bi992936u. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Karyakin AV, Binas B, Kurganov BI, Archakov AI. Kinetic studies on reduction of cytochromes P-450 and b5 by dithionite. European Journal of Biochemistry. 1985;150(1):155–159. doi: 10.1111/j.1432-1033.1985.tb09001.x. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Knyushko TV, Hoa GH. High pressure induced inactivation of ferrous cytochrome P-450 LM2 (IIB4) CO complex: evidence for the presence of two conformers in the oligomer. Biochemical and Biophysical Research Communications. 1992;188:216–221. doi: 10.1016/0006-291x(92)92372-5. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Petushkova NA, Archakov AI, Hoa GH. Stabilization of P450 2B4 by its association with P450 1A2 revealed by high-pressure spectroscopy. Biochemical and Biophysical Research Communications. 2000b;276(3):1005–1012. doi: 10.1006/bbrc.2000.3596. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Petushkova NA, Bobrovnikova EV, Knyushko TV, Dansette P. Association of cytochromes P450 1A2 and 2B4: are the interactions between different P450 species involved in the control of the monooxygenase activity and coupling? Adv Exp Med Biol. 2001;500:335–338. doi: 10.1007/978-1-4615-0667-6_53. [DOI] [PubMed] [Google Scholar]
- Davydov DR, Sineva EV, Sistla S, Davydova NY, Frank DJ, Sligar SG, Halpert JR. Electron transfer in the complex of membrane-bound human cytochrome P450 3A4 with the flavin domain of P450BM-3: the effect of oligomerization of the heme protein and intermittent modulation of the spin equilibrium. Biochimica et Biophysica Acta. 2010;1797(3):378–390. doi: 10.1016/j.bbabio.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Sligar SG. A novel type of allosteric regulation: functional cooperativity in monomeric proteins. Archives of Biochemistry and Biophysics. 2012;519(2):91–102. doi: 10.1016/j.abb.2011.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depierre JW, Dallner G. Structural aspects of the membrane of the endoplasmic reticulum. Biochimica et Biophysica Acta. 1975;415(4):411–472. doi: 10.1016/0304-4157(75)90006-4. [DOI] [PubMed] [Google Scholar]
- Dong H, Shertzer HG, Genter MB, Gonzalez FJ, Vasiliou V, Jefcoate C, Nebert DW. Mitochondrial targeting of mouse NQO1 and CYP1B1 proteins. Biochemical and Biophysical Research Communications. 2013;435(4):727–732. doi: 10.1016/j.bbrc.2013.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong MS, Bell LC, Guo Z, Phillips DR, Blair IA, Guengerich FP. Identification of retained N-formylmethionine in bacterial recombinant mammalian cytochrome P450 proteins with the N-terminal sequence MALLLAVFL…: roles of residues 3–5 in retention and membrane topology. Biochemistry. 1996a;35(31):10031–10040. doi: 10.1021/bi960873z. [DOI] [PubMed] [Google Scholar]
- Dong MS, Yamazaki H, Guo ZY, Guengerich FP. Recombinant human cytochrome P450 1A2 and an N-terminal- truncated form: Construction, purification, aggregation properties, and interactions with Flavodoxin, Ferredoxin, and NADPH-cytochrome P450 reductase. Archives of Biochemistry and Biophysics. 1996b;327:11–19. doi: 10.1006/abbi.1996.0086. [DOI] [PubMed] [Google Scholar]
- Fernando H, Halpert JR, Davydov DR. Kinetics of electron transfer in the complex of cytochrome P450 3A4 with the flavin domain of cytochrome P450BM-3 as evidence of functional heterogeneity of the heme protein. Archives of Biochemistry and Biophysics. 2008;471(1):20–31. doi: 10.1016/j.abb.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genter MB, Clay CD, Dalton TP, Dong H, Nebert DW, Shertzer HG. Comparison of mouse hepatic mitochondrial versus microsomal cytochromes P450 following TCDD treatment. Biochemical and Biophysical Research Communications. 2006;342(4):1375–1381. doi: 10.1016/j.bbrc.2006.02.121. [DOI] [PubMed] [Google Scholar]
- Gomes AM, Winter S, Klein K, Turpeinen M, Schaeffeler E, Schwab M, Zanger UM. Pharmacogenomics of human liver cytochrome P450 oxidoreductase: multifactorial analysis and impact on microsomal drug oxidation. Pharmacogenomics. 2009;10(4):579–599. doi: 10.2217/pgs.09.7. [DOI] [PubMed] [Google Scholar]
- Gruenke LD, Konopka K, Cadieu M, Waskell L. The Stoichiometry of the Cytochrome P-450-catalyzed Metabolism of Methoxyflurane and Benzphetamine in the Presence and Absence of Cytochrome b(5) Journal of Biological Chemistry. 1995;270(42):24707. doi: 10.1074/jbc.270.42.24707. available from: http://www.jbc.org/cgi/content/abstract/270/42/24707. [DOI] [PubMed] [Google Scholar]
- Guengerich FP. A malleable catalyst dominates the metabolism of drugs. Proc Natl Acad Sci USA. 2006;103(37):13565–13566. doi: 10.1073/pnas.0606333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP, Krauser JA, Johnson WW. Rate-limiting steps in oxidations catalyzed by rabbit cytochrome P450 1A2. Biochemistry. 2004;43(33):10775–10788. doi: 10.1021/bi0491393. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Ueng YF, Kim BR, Langouet S, Coles B, Iyer RS, Thier R, Harris TM, Shimada T, Yamazaki H, Ketterer B, Guillouzo A. Activation of toxic chemicals by cytochrome p450 enzymes – Regio- and stereoselective oxidation of aflatoxin B1. Advances in Experimental Medicine and Biology. 1996;387:7–15. doi: 10.1007/978-1-4757-9480-9_2. [DOI] [PubMed] [Google Scholar]
- Gut J, Richter C, Cherry RJ, Winterhalter KH, Kawato S. Rotation of cytochrome P-450. II. Specific interactions of cytochrome P-450 with NADPH-cytochrome P-450 reductase in phospholipid vesicles. Journal of Biological Chemistry. 1982;257(12):7030–7036. [PubMed] [Google Scholar]
- Hazai E, Kupfer D. Interactions between CYP2C9 and CYP2C19 in reconstituted binary systems influence their catalytic activity: possible rationale for the inability of CYP2C19 to catalyze methoxychlor demethylation in human liver microsomes. Drug Metab Dispos. 2005;33(1):157–164. doi: 10.1124/dmd.104.001578. [DOI] [PubMed] [Google Scholar]
- Hildebrandt A, Estabrook RW. Evidence for the participation of cytochrome b 5 in hepatic microsomal mixed-function oxidation reactions. Archives of Biochemistry and Biophysics. 1971;143(1):66–79. doi: 10.1016/0003-9861(71)90186-x. [DOI] [PubMed] [Google Scholar]
- Houston JB, Galetin A. Modelling atypical CYP3A4 kinetics: principles and pragmatism. Archives of Biochemistry and Biophysics. 2005;433(2):351–360. doi: 10.1016/j.abb.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Houston JB, Kenworthy KE. In vitro-in vivo scaling of CYP kinetic data not consistent with the classical Michaelis-Menten model. Drug Metab Dispos. 2000;28(3):246–254. [PubMed] [Google Scholar]
- Hu Y, Krausz K, Gelboin HV, Kupfer D. CYP2C subfamily, primarily CYP2C9, catalyses the enantioselective demethylation of the endocrine disruptor pesticide methoxychlor in human liver microsomes: use of inhibitory monoclonal antibodies in P450 identification. Xenobiotica. 2004;34(2):117–132. doi: 10.1080/00498250310001644535. [DOI] [PubMed] [Google Scholar]
- Hu Y, Kupfer D. Metabolism of the endocrine disruptor pesticide-methoxychlor by human P450s: pathways involving a novel catechol metabolite. Drug Metab Dispos. 2002;30(9):1035–1042. doi: 10.1124/dmd.30.9.1035. [DOI] [PubMed] [Google Scholar]
- Jamakhandi AP, Kuzmic P, Sanders DE, Miller GP. Global analysis of protein-protein interactions reveals multiple CYP2E1-reductase complexes. Biochemistry. 2007;46(35):10192–10201. doi: 10.1021/bi7003476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson I, Schenkman JB. Influence of cytochrome b5 on the stoichiometry of the different oxidative reactions catalyzed by liver microsomal cytochrome P-450. Drug Metab Dispos. 1987;15(3):344–348. [PubMed] [Google Scholar]
- Kaminsky LS, Guengerich FP. Cytochrome P-450 isozyme/isozyme functional interactions and NADPH-cytochrome P-450 reductase concentrations as factors in microsomal metabolism of warfarin. European Journal of Biochemistry. 1985;149:479–489. doi: 10.1111/j.1432-1033.1985.tb08950.x. [DOI] [PubMed] [Google Scholar]
- Kaminsky LS, Guengerich FP, Dannan GA, Aust SD. Comparisons of warfarin metabolism by liver microsomes of rats treated with a series of polybrominated biphenyl congeners and by the component-purified cytochrome P-450 isozymes. Archives of Biochemistry and Biophysics. 1983;225(1):398–404. doi: 10.1016/0003-9861(83)90045-0. [DOI] [PubMed] [Google Scholar]
- Kandel SE, Lampe J. Role of protein-protein interactions in cytochrome P450-mediated drug metabolism and toxicity. Chemical Reserarch in Toxicology. 2014;27(9):1474–1486. doi: 10.1021/tx500203s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RW, Cheng D, Backes WL. Heteromeric Complex Formation between CYP2E1 and CYP1A2: Evidence for the Involvement of Electrostatic Interactions. Biochemistry. 2006;45(51):15807–15816. doi: 10.1021/bi061803n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley RW, Reed JR, Backes WL. Effects of ionic strength on the functional interactions between CYP2B4 and CYP1A2. Biochemistry. 2005;44(7):2632–2641. doi: 10.1021/bi0477900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenaan C, Shea EV, Lin HL, Zhang H, Pratt-Hyatt MJ, Hollenberg PF. Interactions between CYP2E1 and CYP2B4: effects on affinity for NADPH-cytochrome P450 reductase and substrate metabolism. Drug Metab Dispos. 2013;41(1):101–110. doi: 10.1124/dmd.112.046094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen T, Jensen K, Moller BL. Conformational changes of the NADPH-dependent cytochrome P450 reductase in the course of electron transfer to cytochromes P450. Biochimica et Biophysica Acta. 2011;1814(1):132–138. doi: 10.1016/j.bbapap.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Li B, Yau P, Kemper B. Identification of cytochrome P450 2C2 protein complexes in mouse liver. Proteomics. 2011;11(16):3359–3368. doi: 10.1002/pmic.201100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DN, Pritchard MP, Hanlon SP, Burchell B, Wolf CR, Friedberg T. Competition between cytochrome P-450 isozymes for NADPH-cytochrome P-450 oxidoreductase affects drug metabolism. Journal of Pharmacology and Experimental Therapeutics. 1999;289(2):661–667. [PubMed] [Google Scholar]
- Lin HL, Myshkin E, Waskell L, Hollenberg PF. Peroxynitrite inactivation of human cytochrome P450s 2B6 and 2E1: heme modification and site-specific nitrotyrosine formation. Chemical Reserarch in Toxicology. 2007;20(11):1612–1622. doi: 10.1021/tx700220e. [DOI] [PubMed] [Google Scholar]
- Loida PJ, Sligar SG. Molecular recognition in cytochrome P-450: mechanism for the control of uncoupling reactions. Biochemistry. 1993;32(43):11530–11538. doi: 10.1021/bi00094a009. [DOI] [PubMed] [Google Scholar]
- Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76(3):391–396. [PubMed] [Google Scholar]
- Ozalp C, Szczesna-Skorupa E, Kemper B. Bimolecular fluorescence complementation analysis of cytochrome p450 2c2, 2e1, and NADPH-cytochrome p450 reductase molecular interactions in living cells. Drug Metab Dispos. 2005;33(9):1382–1390. doi: 10.1124/dmd.105.005538. [DOI] [PubMed] [Google Scholar]
- Peterson JA, Ebel RE, O’Keeffe DH, Matsubara T, Estabrook RW. Temperature dependence of cytochrome P-450 reduction. A model for NADPH-cytochrome P-450 reductase:cytochrome P-450 interaction. Journal of Biological Chemistry. 1976;251:4010–4016. [PubMed] [Google Scholar]
- Ramsden D, Tweedie DJ, Chan TS, Tracy TS. Altered CYP2C9 activity following modulation of CYP3A4 levels in human hepatocytes: an example of protein-protein interactions. Drug Metab Dispos. 2014;42(11):1940–1946. doi: 10.1124/dmd.114.057901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Backes WL. Formation of P450.P450 complexes and their effect on P450 function. Pharmacology and Therapeutics. 2012;133(3):299–310. doi: 10.1016/j.pharmthera.2011.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Brignac-Huber LM, Backes WL. Physical incorporation of NADPH-cytochrome P450 reductase and cytochrome P450 into phospholipid vesicles using glycocholate and Bio-Beads. Drug Metabolism and Disposition. 2008;36(3):582–588. doi: 10.1124/dmd.107.018473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Cawley GF, Ardoin TG, Dellinger B, Lomnicki SM, Hasan F, Kiruri LW, Backes WL. Environmentally persistent free radicals inhibit cytochrome P450 activity in rat liver microsomes. Toxicology and Applied Pharmacology. 2014;277(2):200–209. doi: 10.1016/j.taap.2014.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Cawley GF, Backes WL. Inhibition of cytochrome P450 1A2-mediated metabolism and production of reactive oxygen species by heme oxygenase-1 in rat liver microsomes. Drug Metab Lett. 2011;5(1):6–16. doi: 10.2174/187231211794455253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Cawley GF, Backes WL. Interactions between cytochromes P450 2B4 (CYP2B4) and 1A2 (CYP1A2) lead to alterations in toluene disposition and P450 uncoupling. Biochemistry. 2013;52(23):4003–4013. doi: 10.1021/bi400422a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Connick JP, Cheng D, Cawley GF, Backes WL. Effect of Homomeric P450•P450 Complexes on P450 Function. Biochemical Journal. 2012;446:489–497. doi: 10.1042/BJ20120636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Cruz AL, Lomnicki SM, Backes WL. Inhibition of cytochrome P450 2B4 by environmentally persistent free radical-containing particulate matter. Biochemical Pharmacology. 2015a;95(2):126–132. doi: 10.1016/j.bcp.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Dela Cruz AL, Lomnicki SM, Backes WL. Environmentally persistent free radical-containing particulate matter competitively inhibits metabolism by cytochrome P450 1A2. Toxicology and Applied Pharmacology. 2015b;289(2):223–230. doi: 10.1016/j.taap.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Eyer M, Backes WL. Functional interactions between cytochromes P450 1A2 and 2B4 require both enzymes to reside in the same phospholipid vesicle: evidence for physical complex formation. Journal of Biological Chemistry. 2010;285(12):8942–8952. doi: 10.1074/jbc.M109.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed JR, Hollenberg PF. Examining the mechanism of stimulation of cytochrome P450 by cytochrome b5: the effect of cytochrome b5 on the interaction between cytochrome P450 2B4 and P450 reductase. J Inorg Biochem. 2003;97(3):265–275. doi: 10.1016/s0162-0134(03)00275-7. [DOI] [PubMed] [Google Scholar]
- Reed JR, Kelley RW, Backes WL. An evaluation of methods for the reconstitution of cytochromes P450 and NADPH P450 reductase into lipid vesicles. Drug Metab Dispos. 2006;34(4):660–666. doi: 10.1124/dmd.105.006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin MA, Maratrat M, Le Roy M, Le Breton FP, Bonierbale E, Dansette P, Ballet F, Mansuy D, Pessayre D. Antigenic targets in tienilic acid hepatitis – Both cytochrome P450 2C11 and 2C11-tienilic acid adducts are transported to the plasma membrane of rat hepatocytes and recognized by human sera. Journal of Clinical Investigation. 1996;98:1471–1480. doi: 10.1172/JCI118936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevrioukova I, Truan G, Peterson JA. The Flavoprotein Domain of P450BM-3: Expression, Purification, and Properties of the Flavin Adenine Dinucleotide- and Flavin Mononucleotide-Binding Subdomains. Biochemistry. 1996;35(23):7528–7535. doi: 10.1021/bi960330p. [DOI] [PubMed] [Google Scholar]
- Stringer RA, Strain-Damerell C, Nicklin P, Houston JB. Evaluation of recombinant cytochrome P450 enzymes as an in vitro system for metabolic clearance predictions. Drug Metab Dispos. 2009;37(5):1025–1034. doi: 10.1124/dmd.108.024810. [DOI] [PubMed] [Google Scholar]
- Subramanian M, Low M, Locuson CW, Tracy TS. CYP2D6-CYP2C9 protein-protein interactions and isoform-selective effects on substrate binding and catalysis. Drug Metab Dispos. 2009;37(8):1682–1689. doi: 10.1124/dmd.109.026500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian M, Zhang H, Tracy TS. CYP2C9-CYP3A4 protein-protein interactions in a reconstituted expressed enzyme system. Drug Metabolism and Disposition. 2010;38(6):1003–1009. doi: 10.1124/dmd.109.030155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna-Skorupa E, Chen CD, Rogers S, Kemper B. Mobility of cytochrome P450 in the endoplasmic reticulum membrane. Proc Natl Acad Sci USA. 1998;95(25):14793–14798. doi: 10.1073/pnas.95.25.14793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczesna-Skorupa E, Mallah B, Kemper B. Fluorescence resonance energy transfer analysis of cytochromes P450 2C2 and 2E1 molecular interactions in living cells. Journal of Biological Chemistry. 2003;278(33):31269–31276. doi: 10.1074/jbc.M301489200. [DOI] [PubMed] [Google Scholar]
- Tan YZ, Patten CJ, Smith P, Yang CS. Competitive interactions between cytochromes P450 2A6 and 2E1 for NADPH-cytochrome P450 oxidoreductase in the microsomal membranes produced by a baculovirus expression system. Archives of Biochemistry and Biophysics. 1997;342(1):82–91. doi: 10.1006/abbi.1997.9995. [DOI] [PubMed] [Google Scholar]
- Tsalkova TN, Davydova NY, Halpert JR, Davydov DR. Mechanism of interactions of alpha-naphthoflavone with cytochrome P450 3A4 explored with an engineered enzyme bearing a fluorescent probe. Biochemistry. 2007;46(1):106–119. doi: 10.1021/bi061944p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SB, Lu AYH. Reconstituted liver microsomal enzyme system that hydroxylates drugs, other foreign compounds and endogenous substrates. V. Competition between cytochromes P-450 and P-448 for reductase in 3,4-benzpyrene hydroxylation. Archives of Biochemistry and Biophysics. 1972;153:298–303. doi: 10.1016/0003-9861(72)90448-1. [DOI] [PubMed] [Google Scholar]
- White RE, Coon MJ. Oxygen activation by cytochrome P-450. Annu Rev Biochem. 1980;49:315–356. doi: 10.1146/annurev.bi.49.070180.001531. [DOI] [PubMed] [Google Scholar]
- Wibo M, Amar-Costesec A, Berthet J, Beaufay H. Electron microscope examination of subcellular fractions. 3. Quantitative analysis of the microsomal fraction isolated from rat liver. The Journal of Cell Biology. 1971;51(1):52–71. doi: 10.1083/jcb.51.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Gillam EMJ, Dong MS, Johnson WW, Guengerich FP, Shimada T. Reconstitution of recombinant cytochrome P450 2C10(2C9) and comparison with cytochrome P450 3A4 and other forms: Effects of cytochrome P450-P450 and cytochrome P450-b5 interactions. Archives of Biochemistry and Biophysics. 1997;342(2):329–337. doi: 10.1006/abbi.1997.0125. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Johnson WW, Ueng YF, Shimada T, Guengerich FP. Lack of electron transfer from cytochrome b5 in stimulation of catalytic activities of cytochrome P450 3A4 – Characterization of a reconstituted cytochrome P450 3A4 NADPH-cytochrome P450 reductase system and studies with apo-cytochrome b5. Journal of Biological Chemistry. 1996a;271:27438–27444. doi: 10.1074/jbc.271.44.27438. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Nakano M, Gillam EMJ, Bell LC, Guengerich FP, Shimada T. Requirements for cytochrome b5 in the oxidation of 7- ethoxycoumarin, chlorzoxazone, aniline, and N-nitrosodimethylamine by recombinant cytochrome P450 2E1 and by human liver microsomes. Biochemical Pharmacology. 1996b;52:301–309. doi: 10.1016/0006-2952(96)00208-0. [DOI] [PubMed] [Google Scholar]
- Yun CH, Miller GP, Guengerich FP. Rate-determining steps in phenacetin oxidations by human cytochrome P450 1A2 and selected mutants. Biochemistry. 2000;39(37):11319–11329. doi: 10.1021/bi000869u. [DOI] [PubMed] [Google Scholar]