

Abstract
The homeodomain transcription factor Pdx-1 has important roles in pancreas and islet development as well as in β-cell function and survival. We previously reported that Pdx-1 overexpression stimulates islet cell proliferation, but the mechanism remains unclear. Here, we demonstrate that overexpression of Pdx-1 triggers proliferation largely by a non-cell-autonomous mechanism mediated by soluble factors. Consistent with this idea, overexpression of Pdx-1 under the control of a β-cell-specific promoter (rat insulin promoter [RIP]) stimulates proliferation of both α and β cells, and overexpression of Pdx-1 in islets separated by a Transwell membrane from islets lacking Pdx-1 overexpression activates proliferation in the untreated islets. Microarray and gene ontology (GO) analysis identified inhibin beta-B (Inhbb), an activin subunit and member of the transforming growth factor β (TGF-β) superfamily, as a Pdx-1-responsive gene. Overexpression of Inhbb or addition of activin B stimulates rat islet cell and β-cell proliferation, and the activin receptors RIIA and RIIB are required for the full proliferative effects of Pdx-1 in rat islets. In human islets, Inhbb overexpression stimulates total islet cell proliferation and potentiates Pdx-1-stimulated proliferation of total islet cells and β cells. In sum, this study identifies a mechanism by which Pdx-1 induces a soluble factor that is sufficient to stimulate both rat and human islet cell proliferation.
INTRODUCTION
Type 1 and type 2 diabetes ultimately arise from loss of functional islet β-cell mass (1–3). Islet transplantation and pharmaceutical activation of β-cell regeneration have been considered strategies for treatment of these diseases, but both approaches are hindered by the lack of druggable pathways that reliably induce human β-cell replication (4). Furthermore, factors that induce β-cell growth must do so without altering key β-cell functions, such as glucose-stimulated insulin secretion (GSIS), or causing cellular or DNA damage (5). For example, overexpression of cyclin D or cyclin-dependent kinase 6 (CDK6) induces human β-cell proliferation with retention of function (6) but leads to DNA damage, as measured by staining for γ-H2A member histone family member X (γ-H2AX) (5).
Our laboratory has demonstrated that Pdx-1 and Nkx6.1, two homeobox domain transcription factors well known for key roles in islet β-cell development, activate proliferation when overexpressed in rat or human islets while maintaining and enhancing GSIS, respectively (7–10). Importantly, the increase in proliferation induced by either factor has little to no impact on γ-H2AX expression, suggesting that these factors engage “safe” pathways of islet cell replication. In adult islets, Pdx-1 expression is restricted to β and δ cells, and Nkx6.1 expression is restricted to β cells. Importantly, expression of these transcription factors is low or absent in nonislet cells, except in the central nervous system, suggesting that deeper knowledge of the molecular pathways that they activate might be an entrée to development of islet cell-specific proliferative agents.
These positive features of Nkx6.1- and Pdx-1-mediated islet cell replication have led us to explore the pathways by which they engage the core cell cycle machinery (7–10). In our previous studies using adenovirus vectors containing the constitutive cytomegalovirus (CMV) promoter to drive transgene expression, we found that Nkx6.1 overexpression in rat islets causes almost exclusive proliferation of β cells, whereas Pdx-1 overexpression activates both β- and α-cell replication (7, 9, 10). Here, we sought to understand the molecular pathway by which Pdx-1 overexpression activates both α- and β-cell replication. We found that Pdx-1 overexpression exerts its effects on islet cell replication via a non-cell-autonomous mechanism involving induction of a secreted soluble factor or factors. The transforming growth factor β (TGF-β) family member inhibin beta-B (Inhbb) is identified as one factor mediating the proliferative effect of Pdx-1.
MATERIALS AND METHODS
Cell culture and reagents.
Pancreatic islets were isolated from male Wistar rats and cultured as previously described (7, 11, 12) under a protocol approved by the Duke University Institutional Animal Care and Use Committee. Human islets were obtained from the Integrated Islet Distribution Program (http://iidp.coh.org).
For activin ligand studies, rat or human islets were treated with recombinant activin B (R&D Systems) at 100 ng/ml for 72 h. Fresh medium and ligand were added each day. For activin neutralization studies, recombinant human or mouse activin receptor Fc chimeras (AcRIIA and AcRIIB; R&D Systems) or a tumor necrosis factor alpha (TNF-α) receptor Fc chimera (R&D Systems) was added to rat islet culture medium at 100 ng/ml. Fresh medium and recombinant chimera protein were added each day. In all experiments, ethynyl deoxyuridine (EdU) was added 18 h prior to cell harvest.
Use of recombinant adenoviruses.
For gene overexpression studies, cytomegalovirus (CMV) promoter-driven recombinant adenoviruses containing mouse Pdx-1 (AdCMV-mPdx-1), bacterial β-galactosidase (AdCMV-β-Gal), or green fluorescent protein (GFP; AdCMV-GFP) were constructed and used as described previously (8, 9, 13). We constructed CMV promoter-driven adenoviruses containing human Pdx-1 (hPdx-1), rat Noggin, rat Bmp3, rat Fstl5, and rat Inhbb by cloning the cDNA constructs into a pAdTrack shuttle vector and using the Ad-Easy system to generate the recombinant adenoviruses, as previously described (14). The use of the pAdTrack shuttle vector allowed us to track the transduced cells with GFP as this vector also contains a separate CMV promoter that drives the expression of GFP.
We also generated a new Tet-On inducible β-cell-specific adenoviral system. We cloned mouse Pdx-1 into the HindIII site of plasmid GO791, an inducible and β-cell-specific adenoviral shuttle vector. This pShuttle-based dual-cassette vector utilizes the rat insulin I promoter (RIP) to directly drive expression of the Tet-On activator (Clontech) (see Fig. 2D). Upon activation with doxycycline (Dox), expressed Tet-On activates the upstream inducible promoter, stimulating expression of Pdx-1 or other cDNA inserts.
FIG 2.

Tet-On inducible expression of RIP-mPdx-1 is specific to β cells. (A and B) Primary rat islets were treated with an adenovirus containing a Tet-On inducible rat insulin promoter upstream of mPdx-1 (RIP-mPdx-1) for 120 h. Doxycycline (Dox; 100 ng/ml) was added (Dox+) to one set of islets transduced with RIP-mPdx-1 to turn on gene expression (Dox was replenished each day). The islets that did not receive Dox (Dox−) served as a control. Rat islets were dispersed, fixed, and stained for Pdx-1 and insulin (A) or Pdx-1 and glucagon (B), followed by visualization using a fluorescence microscope (arrows indicate Pdx-1 and insulin costaining). (C) The percentage of total cells, β cells, and α cells overexpressing mPdx-1 was quantitated using ImageJ software. The data represent the means and SEM of three independent experiments. **, P < 0.001. (D) Overview of plasmid GO791, an inducible β-cell-specific adenoviral shuttle vector, comprised of a dual-expression cassette cloned between the adenoviral left-hand inverted terminal repeat (LITR) and right arm of pShuttle (shown in red) (Agilent Technologies). RIP controls expression of the Tet-On transcription factor. Upon activation with doxycycline, the Tet-On monomer activates the upstream inducible promoter, stimulating expression of Pdx-1. TRE, Tet response element; MIN, minimal promoter; PA, polyadenylation signal. (E) Primary rat islets were treated with AdCMV-β-Gal or AdCMV-mPdx-1 for 96 h or Tet-On inducible AdRIP-mPdx-1 for 120 h. Doxycycline (Dox; 100 ng/ml) was added daily to one set of islets transduced with RIP-mPdx-1 to turn on Pdx-1 expression. EdU was added during the last 18 h of incubation. Quantitative reverse transcription-PCR (qRT-PCR) was used to measure mRNA levels of mPdx-1 (RQ, relative quantity). The data represent the means and SEM of three independent experiments. **, P < 0.01 compared to the level in the β-Gal control. (F) Pdx-1 protein levels were measured by immunoblot (IB) analysis in the absence (−) or presence (+) of doxycycline, using γ-tubulin as a loading control. The immunoblot is representative of three independent experiments. INS, insulin; GCG, glucagon.
Pools of 200 islets were cultured in 2 ml of RPMI medium (10% fetal calf serum, [FCS] and 7.8 mM glucose), treated with viruses at a concentration of ∼2 × 109 particles/ml medium for 18 h, and then cultured in virus-free medium until the islets were collected for assays at 78 h postransduction unless otherwise indicated. To activate the inducible Tet-On system, islets were cultured in medium containing 100 ng/ml doxycycline for 102 h following the 18 h of incubation with adenovirus, and fresh doxycycline was added each day.
Gateway entry plasmids were obtained from the DNASU plasmid repository (15) for Artn (MmCD00081334), BDNF (HsCD00039652), Gdf11 (MmCD00083502), GFRA3 (HsCD00039690), IGSF1 (HsCD00082225), INHBB (HsCD00042725), NMB (HsCD00505263), RET (HsCD00506047), SCUBE1 (HsCD00297185), TNFRSF11B (HsCD00041216), and WNT4 (HsCD00512640) and were used to generate adenoviruses under the control of the CMV promoter as previously described (16).
All recombinant adenoviruses were shown to be E1A deficient using a reverse transcription-PCR (RT-PCR) screen as previously described (17).
Transwell assay.
Rat islets were left untreated or transduced with adenovirus in normal medium for 18 h as described above. After 18 h of incubation with adenovirus, islets were washed three times in serum-free (SF) medium (RPMI medium with 7.8 mM glucose) to remove serum and remaining viral particles. In the bottom of a 12-well plate, 50 untreated islets were placed in 1 ml of SF medium containing EdU. In the Transwell (3-μm pore size, polyethylene terephthalate [PET] membrane; Corning, MA), 200 islets transduced with either AdCMV-β-Gal control or AdCMV-mPdx-1 adenovirus were placed in 500 μl of SF medium containing EdU for a total of 1.5 ml of SF medium per Transwell setup. The medium was left unchanged for the remainder of the experiment. Islets from the top and bottom of the Transwell chambers were harvested for real-time PCR analysis and EdU incorporation at 78 h postransduction.
Glucose-stimulated insulin secretion.
Islets were cultured in the presence of activin B (100 ng/ml) or vehicle (0.3% bovine serum albumin [BSA] in phosphate-buffered saline [PBS]) for 3 days. Insulin secretion was measured at 2.5 and 16.7 mM glucose in triplicate groups of 25 islets as previously described (18).
EdU incorporation and immunofluorescence staining.
For EdU labeling, a 1:1,000 dilution of EdU-labeling reagent (Invitrogen) was added to islet culture medium during the last 18 h of cell culture unless otherwise specified. Islets were harvested and processed for immunofluorescence analyses as previously described (9). Briefly, islets were dispersed using trypsin-EDTA, plated on poly-d-lysine-coated coverslips (BD Biosciences), and fixed using neutral buffered formalin. EdU was detected using a Click-iT 555 kit (Invitrogen) according to the manufacturer's protocol. For predispersed islet cell assays, islets were isolated and treated with Accutase (Gibco) to achieve a single-cell suspension. Islet cells were then counted and plated on human bladder carcinoma (HTB9) cell matrix-coated (19) or poly-d-lysine-coated 96-well plates (Corning) for 16 h before virus or peptide treatment. For antibody staining, dispersed islet cells were incubated overnight with anti-guinea pig insulin (catalog number A0564; Dako), anti-mouse glucagon (251754; Abbiotec), anti-rabbit γ-H2AX (9718; Cell Signaling), anti-rabbit cleaved caspase-3 (9664; Cell Signaling), or anti-rabbit Pdx-1 (ab47467; Abcam) antibody, followed by detection with secondary antibodies. The cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured and analyzed using either OpenLab software and ImageJ software or a Cellomics ArrayScan VTI High-Content (HC) cell-based imaging system (Thermo).
Measurement of RNA levels.
RNA was isolated using an RNeasy kit (Qiagen), and cDNA was made using iScript (Bio-Rad). Real-time PCR assays were performed using a ViiA7 detection system and software (Applied Biosystems). The primers used for rat cyclin D1 (CCND1), rat CCND2, human peptidylprolyl isomerase A (PPIA), human Pdx-1 (hPdx-1), human CCND1, and human CCND2 were TaqMan-based Assays on Demand (Applied Biosystems). All other primers were designed and used with SYBR green (Bio-Rad). Primer sequences are available upon request. All rat islet mRNA levels were normalized to endogenous cyclophilin levels, and all human islet mRNA levels were normalized to the PPIA level.
Immunoblot analysis.
Cells or islets were harvested and lysed in ice-cold radioimmunoprecipitation assay (RIPA) buffer (Sigma) containing protease (BD Biosciences) and phosphatase (Sigma) inhibitors. Lysates were precleared by centrifugation (13,000 rpm at 4°C for 10 min), resolved on 4 to 12% NuPAGE gels (Invitrogen), and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% milk in Tris-buffered saline with 0.1% Tween (TBS-T) for 30 min, followed by overnight incubation at 4°C with the following diluted primary antibodies: Pdx-1 (catalog number 47267; Abcam) and γ-tubulin (catalog number T5326; Sigma). Goat anti-mouse IRDye 800CW (1:10,000) and Alexa Fluor 680–goat anti-rabbit IgG (1:10,000) antibodies were used to detect primary antibodies using an Odyssey CLx system (Li-Cor).
Statistical analysis.
Data are presented as means ± standard errors of the means (SEM). For statistical significance analysis, a two-tailed t test was used. For multiple group comparisons, analysis of variance (ANOVA) with a Bonferroni posttest was used. P values of less than 0.05 were considered significant.
RESULTS
Pdx-1-stimulated islet cell proliferation occurs in cells that do not overexpress Pdx-1.
We have previously demonstrated that adenovirus-mediated overexpression of Pdx-1 in rat pancreatic islets under the control of a constitutive (CMV) promoter stimulates both α- and β-cell proliferation (9). Here, we sought to determine if Pdx-1 activates proliferation via a cell-autonomous mechanism. We first treated rat islets with the adenovirus-containing mouse Pdx-1 cDNA under the control of the CMV promoter (AdCMV-mPdx-1) as used in our previous study (9). Following treatment with this virus or a control virus (AdCMV-β-Gal), we measured EdU incorporation and stained for Pdx-1 expression. Treatment with AdCMV-mPdx-1 resulted in an overall increase in EdU incorporation of 6-fold relative to that in AdCMV-β-Gal-treated cells (from 0.8% ± 0.3% EdU-positive [EdU+] cells to 5.5% ± 0.9% positive cells), similar to what we have reported previously (9). We found that most of the EdU-positive (proliferating) cells in the culture were not those with bright Pdx-1 staining due to overexpression but, rather, were the adjoining cells with low, presumably endogenous Pdx-1 expression (Fig. 1A). Quantification of signals in multiple images similar to the image shown in Fig. 1A revealed that the majority (approximately 75%) of the proliferating cells (EdU+) from islets treated with the AdCMV-mPdx-1 adenovirus did not overexpress Pdx-1 (Fig. 1B).
FIG 1.

Cell-extrinsic effect of Pdx-1 to promote islet cell proliferation. (A and B) Primary rat islets were treated with AdCMV-β-Gal or AdCMV-mPdx-1 adenovirus for 96 h, and EdU was added during the last 18 h of incubation. Rat islets were dispersed, fixed, and stained for EdU and Pdx-1 (EdU and Pdx-1 expression is indicated by red and green staining, respectively). EdU incorporation in islet cells following exposure to AdCMV-β-Gal (A, top panel) or AdCMV-mPdx-1 (A, bottom panel) was visualized using a fluorescence microscope (arrows indicate EdU+ cells that overexpress mPdx-1). The percentage of proliferating cells (EdU+) with endogenous or overexpressed Pdx-1 was quantitated using ImageJ software. The data represent the means and SEM of three independent experiments. ***, P < 0.001. (C and D) Primary rat islets were treated with AdCMV-GFP or AdCMV-Pdx-1 for 96 h, and EdU was added during the last 18 h of incubation. Rat islets were dispersed, fixed, and stained for DAPI, EdU, and GFP (EdU and GFP expression is indicated by red and green staining, respectively; DAPI staining is not shown). EdU incorporation for GFP control (C, top panel) and GFP hPdx-1 (C, bottom panel) treatments was visualized using a fluorescence microscope (arrows indicate cells coexpressing EdU and GFP). The percentage of proliferating cells (EdU+) that were positive (GFP+) or negative (GFP−) for GFP expression following treatment with GFP hPdx-1 adenovirus was quantitated using ImageJ software. The data represent the means and SEM of three independent experiments. **, P < 0.01.
We next constructed a new adenovirus vector containing both CMV-hPdx-1 (human Pdx-1 cDNA) and CMV-GFP expression cassettes (AdCMV-hPdx-1/CMV-GFP). Using this vector, GFP expression identifies adenovirus-transduced cells that coexpress GFP and hPdx-1, while EdU incorporation identifies proliferating cells. Figure 1C shows images of dispersed rat islets transduced with recombinant adenovirus either lacking the Pdx-1 cDNA (GFP control; top panels) or containing both the human Pdx-1 and GFP transgenes (GFP hPdx-1; bottom panels). Treatment with the AdCMV-hPdx-1/CMV-GFP virus resulted in an overall 6-fold increase of EdU incorporation compared to the level in the GFP control virus (from 1% ± 0.1% EdU-positive cells to 6.2% ± 0.4% EdU-positive cells), similar to results with the AdCMV-mPdx-1 adenovirus system. Consistent with the findings shown in Fig. 1B, we found that 80% of the EdU-positive (proliferating) cells in the culture were negative for GFP staining, showing that these cells were not transduced with the AdCMV-hPdx-1/CMV-GFP adenovirus (Fig. 1C and D). Thus, these two independent sets of experiments using two different adenovirus vectors strongly suggest that Pdx-1 induces expression of a soluble factor that stimulates proliferation of adjoining rat islet cells.
β-Cell-specific overexpression of mPdx-1 stimulates both α-cell and β-cell proliferation.
We next tested if β-cell-specific overexpression of Pdx-1 is sufficient to drive both α- and β-cell replication. To accomplish this, we developed an adenovirus vector containing a Tet-On inducible rat insulin promoter (RIP) to express Pdx-1 (AdRIP-mPdx-1) only in insulin-positive β cells. We first validated the cell type specificity of this new system using immunofluorescence to detect colocalization of Pdx-1 with insulin or glucagon (Fig. 2A and B). The data shown in Fig. 2C indicate that in the presence of doxycycline (Dox; 100 ng/ml), 10% of total cells and 15% of β cells overexpressed mPdx-1, whereas less than 0.5% of α cells overexpressed mPdx-1. The Tet-On inducible promoter was tightly regulated as no Pdx-1 was detected in the absence of Dox (Fig. 2C and 3A). We further characterized this new system by measuring Pdx-1 mRNA, Pdx-1 protein, and EdU incorporation in islets treated with the AdRIP-mPdx-1 or AdCMV-mPdx-1 system. AdRIP-mPdx-1 and Dox treatment of rat islets caused similar increases in Pdx-1 mRNA and protein levels as did treatment with AdCMV-mPdx-1 (Fig. 2E and F), and both AdRIP-mPdx-1 and AdCMV-mPdx-1 induced a robust increase in EdU incorporation compared to levels in their respective controls (Fig. 3).
FIG 3.

β-Cell-specific overexpression of mPdx-1 stimulates rat islet cell proliferation. Primary rat islets were treated with AdCMV-β-Gal or AdCMV-mPdx-1 for 96 h or Tet-On inducible AdRIP-mPdx-1 for 120 h. Doxycycline (Dox; 100 ng/ml) was added daily to one set of islets transduced with RIP-mPdx-1 to turn on Pdx-1 expression. EdU was added during the last 18 h of incubation. (A) Rat islets were dispersed, fixed, and stained for EdU and Pdx-1 (EdU and Pdx-1 are indicated by red and green staining, respectively). EdU incorporation for AdCMV-β-Gal control, AdCMV-mPdx-1, RIP-mPdx-1 without Dox (Dox−; control), and RIP-mPdx-1 with Dox (Dox+) treatments was visualized using a fluorescence microscope. (B) The percentage of EdU+ cells for each treatment was quantitated using ImageJ software. Data represent the means and SEM of three independent experiments. ns, not significant; **, P < 0.001 compared to the level for the β-Gal control.
After validation of the new inducible system, we tested if β-cell-specific overexpression of Pdx-1 stimulates proliferation of islet β and α cells as previously observed with a CMV-driven Pdx-1 expression system. The images in Fig. 4A show EdU incorporation after treatment of islets with AdRIP-mPdx-1 in the presence of Dox. The β and α cells were identified by insulin and glucagon staining, respectively (Fig. 4A, white arrows indicate EdU+ β cells, and yellow arrows indicate EdU+ α cells). In the presence of Dox, β cell-specific overexpression of mPdx-1 stimulated a clear increase in proliferation of both β cells and α cells (Fig. 4B and C). These data show that overexpression of Pdx-1 specifically in β cells induces proliferation of multiple islet cell types, consistent with the generation of a secreted, soluble factor.
FIG 4.

β-Cell-specific overexpression of mPdx-1 stimulates both β- and α-cell proliferation. Primary rat islets were treated with Tet-On inducible AdRIP-mPdx-1 for 120 h. Doxycycline (Dox; 100 ng/ml) was added daily to one set of islets transduced with RIP-mPdx-1 to turn on Pdx-1 expression. EdU was added during the last 18 h of incubation. Rat islets were dispersed, fixed, and stained for DAPI, EdU, insulin, and glucagon (indicated by blue, green, and red staining, respectively). (A) EdU incorporation for RIP-mPdx-1 Dox− (images not shown) and Dox+ treatments was visualized using a fluorescence microscope (white arrows indicate proliferating insulin-positive cells, and yellow arrows indicate proliferating glucagon-positive cells). (B and C) The percentages of β cells (B) and α cells (C) expressing EdU for each treatment were quantitated using ImageJ software. The data represent the means and SEM of three independent experiments. **, P < 0.01; ***, P < 0.001.
Pdx-1 overexpression induces a soluble factor that stimulates islet cell proliferation.
To further validate the soluble factor hypothesis, we used a transwell assay system in which untreated (acceptor) islets were cocultured with donor islets pretreated with AdCMV-β-Gal control or AdCMV-Pdx-1 virus, with the acceptor and donor islets separated by a membrane barrier (schematic shown in Fig. 5A). Importantly, the membrane (3-μm pore size) allows the free exchange of proteins and other factors while preventing cell-cell contact. Although virus-treated donor islets were washed repeatedly prior to an experiment (see Materials and Methods), it remained possible that remnant viral particles diffused from donor to acceptor islets across the barrier. To eliminate this possibility, we measured Pdx-1 mRNA levels in the donor and acceptor islets. As expected, Fig. 5B shows that only the donor islets transduced with CMV-Pdx-1 had increased Pdx-1 mRNA levels, whereas untreated acceptor islets maintained endogenous Pdx-1 levels (comparable to those of the AdCMV-β-Gal donor control). AdCMV-mPdx-1 treatment of the donor islets increased their total EdU incorporation by approximately 4-fold compared to the level in the AdCMV-β-Gal-treated donor islets (Fig. 5C). In the untreated acceptor islets, a significant 2-fold increase in EdU+ cells was observed in islets paired with AdCMV-Pdx-1 donor islets relative to islets paired with AdCMV-β-Gal donor islets (Fig. 5D). These findings are consistent with Pdx-1-mediated induction of a soluble factor(s) that traverses the Transwell membrane to activate islet cell replication.
FIG 5.

Pdx-1 produces a soluble factor(s) that stimulates rat islet cell proliferation. Primary rat islets were left untreated or treated with AdCMV-β-Gal or AdCMV-mPdx-1 adenovirus. After overnight adenoviral treatment, islets were washed three times with serum-free (SF) medium containing 0.1% BSA to remove serum and adenoviral particles. Untreated islets (50 islets; acceptor islets) were placed in the bottom of a 12-well plate containing 1 ml of SF medium and EdU, and islets treated with either AdCMV-β-Gal (200 islets; donor islets) or AdCMV-mPdx-1 (200 islets; donor islets) adenovirus were placed in the upper chamber of a Transwell (3-μm pore size) containing 500 μl of medium and EdU. Islets were cultured for 72 h without medium changes. (A) Schematic of experimental setup. (B) Quantitative reverse transcription-PCR (qRT-PCR) was used to measure Pdx-1 mRNA levels (RQ, relative quantity). The data represent the means and SEM of three independent experiments. **, P < 0.01 compared to the level for the AdCMV-β-Gal control. (C and D) Rat islets were dispersed, fixed, and stained for DAPI, EdU, and Pdx-1. The percentages of donor islet cells (C) and acceptor islet cells (D) expressing EdU for each treatment were quantitated using ImageJ software. The data represent the means and SEM of six independent experiments. *, P < 0.05; ***, P < 0.001.
Identification of candidate soluble factors by microarray/GO analysis and RT-PCR.
To identify potential soluble factors that are regulated by Pdx-1, we performed gene ontology (GO) analysis on our previously published microarray data collected from rat islets that were transduced with the AdCMV-mPdx-1 adenovirus for 48 h (9). We identified 21 candidate soluble factors and two transmembrane receptors induced by Pdx-1 overexpression (Table 1). We selected 12 of these genes for further study, including four TGF-β superfamily genes, namely, noggin (Nog), bone morphogenetic protein 3 (Bmp3), follistatin-like 5 (Fstl5), and inhibin beta-B (Inhbb). Also included were the interacting receptor subunits Gfra3 and Ret and the Gfra3 ligand artemin (Artn); the Artn/Gfra3/Ret ligand/receptor system has been implicated in regulation of cell proliferation in nonislet cells (20, 21). We also performed studies on one additional TGF-β superfamily gene that was downregulated in the microarray analysis, Tnfrsf11b/osteoprotegerin, based on a recent report of its proliferative activity in rodent and human islets (22). Measurement of mRNA levels for all 12 of these genes in rat islets transduced with AdCMV-mPdx-1 by quantitative RT-PCR (qRT-PCR) confirmed significant upregulation of Nog, Bmp3, Fstl5, Inhbb, Artn, Bdnf, Scube1, Gfra3, and Nmb compared to levels in rat islets transduced with AdCMV-β-Gal (Fig. 6A). In contrast, Igsf1 and Ret levels tended to increase without achieving statistical significance, and Tnfrsf11b/osteoprotegerin mRNA levels were unchanged in response to Pdx-1 overexpression.
TABLE 1.
Secreted factors and transmembrane receptors upregulated by Pdx-1 overexpressiona
| Symbol | Description | Fold change |
|---|---|---|
| Nog | Noggin, Nog | 11.72 |
| Cbln2 | Cerebellin 2 precursor protein, Cbln2 | 6.66 |
| Igfbp3 | Insulin-like growth factor binding protein 3, Igfbp3 | 6.56 |
| Adamts4 | ADAM metallopeptidase with thrombospondin type 1 motif, 4 | 6.11 |
| Gfra3 | Glial cell line-derived neurotrophic factor family receptor alpha 3, Gfra3 | 5.34 |
| Bmp3 | Bone morphogenetic protein 3, Bmp3 | 5.07 |
| Fstl5 | Follistatin-like 5, Fstl5 | 4.97 |
| Thsd7a | Thrombospondin, type I, domain containing 7A | 4.16 |
| Mmp16 | Matrix metallopeptidase 16, Mmp16 | 4.15 |
| Nmb | Neuromedin B precursor—rat, RGD1562710 | 4.12 |
| Scube1 | Signal peptide, CUB domain, EGF-like 1, Scube1b | 3.49 |
| Nid2 | Nidogen 2, Nid2 | 3.4 |
| Fam19a5 | Family with sequence similarity 19 (chemokine [C-C motif]-like), member A5, Fam19a5 | 2.88 |
| Serpini1 | Serine (or cysteine) peptidase inhibitor, clade I, member 1, Serpini1 | 2.88 |
| Col4a5 | Collagen, type IV, alpha 5, Col4a5 | 2.63 |
| Spata6 | Spermatogenesis-associated 6, Spata6 | 2.6 |
| Ret | Proto-oncogene | 2.31 |
| F13a1 | Coagulation factor xiii, a1 subunit, F13a1 | 2.3 |
| Bdnf | Brain-derived neurotrophic factor, Bdnf | 2.23 |
| Igsf1 | Immunoglobulin superfamily, member 1, Igsf1 (isoform 3) | 2.17 |
| Vwa5b1 | von Willebrand factor A domain containing 5B1 | 2.16 |
| Inhbb | Inhibin beta-B, Inhbb | 2.16 |
| Cp | Ceruloplasmin, Cp | 2.12 |
Rat islets were treated with AdCMV-β-Gal or AdCMV-Pdx-1 for 48 h. RNA from the treated islets was harvested for microarray analysis (11). GO analysis was then performed on the microarray data. This analysis identified 21 secreted factors and two transmembrane receptors that are upregulated by Pdx-1 overexpression.
CUB, complement, Uegf, Bmp1; EGF, epidermal growth factor.
FIG 6.

Identification and screening of candidate soluble factors induced by Pdx-1 overexpression. A subset of the genes identified by microarray and GO analysis that encode candidate soluble factors induced by Pdx-1 (Table 1) was studied further by RT-PCR and overexpression. (A) Primary rat islets were treated with AdCMV-mPdx-1 or Ad-CMV-β-Gal or AdCMV-GFP control adenovirus for 48 h. Quantitative reverse transcription-PCR (qRT-PCR) was used to measure mRNA levels of the indicated genes (RQ, relative quantity). Data represent the means and SEM of two to three independent experiments. (B and C) Primary rat islets were dispersed into a single-cell suspension and seeded onto an HTB9 matrix-coated 96-well plate for 16 h before treatment with adenoviruses encoding the indicated cDNAs, the Ad-CMV-β-Gal control virus, or the AdCMV-GFP control adenovirus for 96 h, and EdU was added during the last 18 h of incubation. Cells were then fixed and stained for DAPI, EdU, and insulin. EdU incorporation into total islet cells (B) or insulin-positive β cells (C) were visualized and quantitated using the Cellomics ArrayScan VTI high-content imaging system. *, P < 0.05; **, P < 0.01; ***, P < 0.001, compared to the level for β-Gal or the GFP control.
Screening of candidate soluble factors by adenovirus-mediated overexpression.
We next constructed recombinant adenoviruses containing cDNAs encoding all 12 of the genes characterized in Fig. 6A, under the control of the CMV promoter, and confirmed transgene expression from all of these vectors (data not shown). We used dispersed rat islet cells for this screen rather than intact rat islets as a more efficient method for testing multiple vectors. Dispersed rat islet cells were treated with each of the recombinant adenoviruses as well as the AdCMV-Pdx-1 or AdCMV-β-Gal control virus, followed by measurement of EdU incorporation into total islet cells (Fig. 6B) or into islet β cells identified by insulin coexpression (Fig. 6C). Overexpression of noggin, Bmp3, Fstl5, Artn, Bdnf, Scube1, Gfra3, Nmb, Igsf1, or Ret did not enhance total islet cell or β-cell EdU incorporation, and these factors were not studied further. In addition to the expected increase in total islet cell and β-cell proliferation in response to Pdx-1 overexpression, two of the selected target genes, osteoprotegerin (Tnfrsf11b) and the activin/inhibin subunit inhibin-beta B (Inhbb), were sufficient to activate rat β-cell proliferation when overexpressed. Because the proliferative effect of osteoprotegerin has recently been reported by another group (22) and given that this gene was not upregulated by Pdx-1 overexpression in our microarray and qRT-PCR experiments, we focused our further studies on Inhbb.
The impact of overexpressed Inhbb was studied in more detail by comparing effects in intact rat islets to those in predispersed rat islet cells, both in the presence and absence of co-overexpression of Pdx-1. For experiments using two adenoviruses per condition, we controlled for similar adenovirus titers among groups by measuring the percentage of GFP-positive (GFP+) cells and also confirmed that levels of Pdx-1 expression were the same across the islet groups (data not shown). We confirmed that overexpression of Inhbb was sufficient to activate total islet cell and β-cell proliferation in dispersed islet cell preparations, and we demonstrated that Inhbb overexpression potentiated the proliferative effect of Pdx-1 overexpression (Fig. 7B). In intact rat islets, the ability of Inhbb to potentiate Pdx-1-mediated total islet cell and β-cell proliferation was again evident, but Inhbb alone did not activate proliferation in the intact islet setting (Fig. 7A). We do not know why Inhbb overexpression is sufficient to activate islet cell proliferation in dispersed islet cells but not intact islets. One possibility is that dispersed islet cells are plated on HTB9 matrix, whereas no matrix is provided for intact islets. The matrix may have provided accessory factors that enhance/enable proliferative effects of overexpressed Inhbb. It is also possible that viral transduction efficiency was higher in dispersed islet cells than in intact islets or that dispersal of islet cells by Accutase treatment disrupted proteins at the cell surface needed for mediating the effects of overexpressed Inhbb. Further studies will be required to resolve this issue.
FIG 7.

Effects of Inhbb overexpression on intact rat islets and dispersed rat islet cells. (A and B) Intact rat islets (A) or predispersed rat islet cells (B) were treated with AdCMV-β-Gal, AdCMV-Inhbb, AdCMV-Pdx-1, or AdCMV-Pdx-1 plus AdCMV-Inhbb (see Materials and Methods for details). Intact rat islet cells were dispersed, fixed, and stained for DAPI, EdU, and insulin and analyzed with a high-content imager. Predispersed rat islet cells were fixed, stained, and analyzed in the same way. Data are representative of three independent experiments. *, P < 0.05; **, P < 0.01, ***, P < 0.001, compared to the value for the β-Gal control. (C and D) Intact rat islets were treated with AdCMV-GFP, AdCMV-hPdx-1 plus AdCMV-GFP, or AdCMV-Pdx-1 plus AdCMV-Inhbb adenoviruses for 96 h. qRT-PCR was used to measure mRNA levels of cyclin D1 or cyclin D2, as indicated (RQ, relative quantity). The data represent the means and SEM of three independent experiments. *, P < 0.01, for a comparison of results between hPdx-1 plus GFP and hPdx-1 plus Inhbb.
We have previously demonstrated that Pdx-1 upregulates cyclins D1 and D2, and we have also shown that the cyclin D/cdk4 complex is necessary for Pdx-1-stimulated proliferation (9). Here, we investigated if coexpression of Inhbb and Pdx-1 further alters the expression of cyclins D1 and D2. Cooverexpression of Inhbb and Pdx-1 in intact rat islets increased cyclin D1 mRNA levels above the already elevated levels in islets expressing Pdx-1 alone (Fig. 7C) but did not affect the level of cyclin D2 (Fig. 7D). Thus, Inhbb-mediated enhancement of the proliferative effect of Pdx-1 may be mediated by increased cyclin D1 expression.
Activin B and activin receptors mediate Pdx-1-stimulated islet cell proliferation.
The inhibin genes (i.e., the Inha, Inhba, and Inhbb genes) can produce several soluble homodimeric and heterodimeric ligands, including activin A, activin B, and activin AB, but only activin B is a homodimer comprised exclusively of Inhbb-encoded subunits (23, 24). We therefore investigated the effect of activin B alone or in combination with Pdx-1 overexpression on rat islet cell proliferation. Recombinant activin B protein caused a clear increase in EdU incorporation into total islet cells and islet β cells, both in intact (Fig. 8A) and dispersed (Fig. 8B) islets. Activin B also potentiated the proliferative effect of Pdx-1 overexpression in both intact and dispersed rat islet preparations (Fig. 8A and B).
FIG 8.
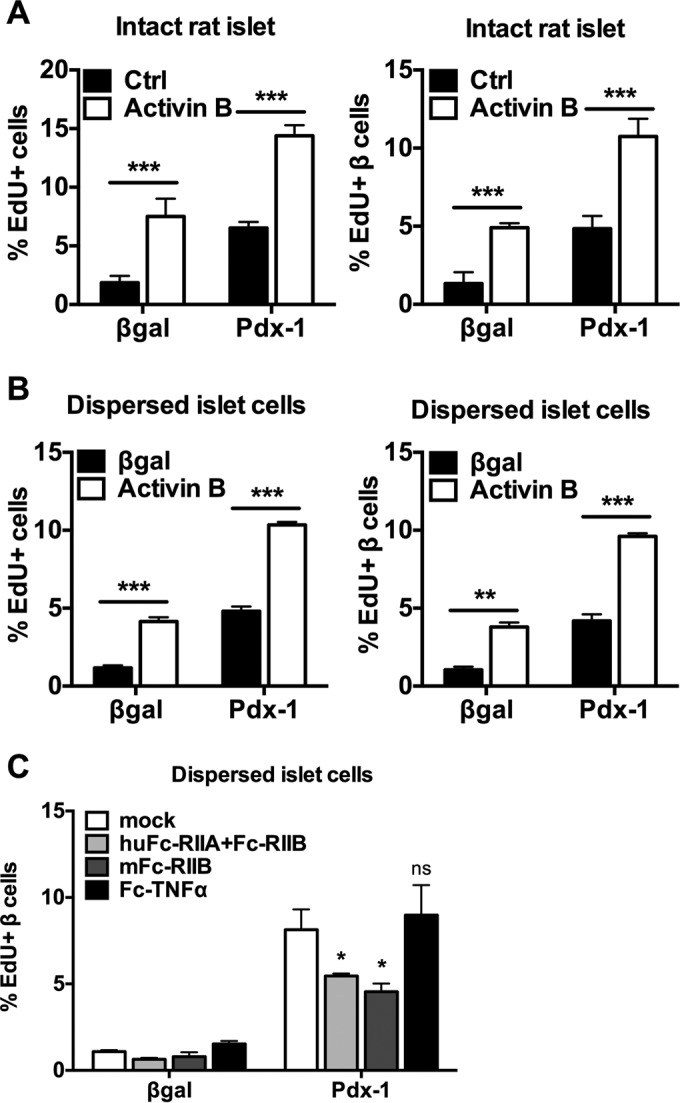
Activin B and activin receptors mediate Pdx-1-stimulated islet cell proliferation. (A and B) Intact rat islets or dispersed rat islet cells were treated with AdCMV-β-Gal or AdCMV-mPdx-1 for 18 h and then cultured for an additional 78 h in the absence (Ctrl) or presence of 100 ng/ml activin B. EdU was added during the last 18 h of incubation. Cells were stained for DAPI, EdU, and insulin and were visualized and quantitated with a high-content imager. The data are representative of three independent experiments. (C) Dispersed rat islet cells were treated with AdCMV-mPdx-1 or AdCMV-β-Gal adenovirus. Recombinant activin receptor Fc chimeras (Fc-RIIA and Fc-RIIB; 100 μg/ml) or the control TNF-α receptor Fc chimera (Fc-TNF-α; 100 μg/ml) was added each day (mock, culture medium alone). EdU was added during the last 18 h of culture, and the islets were harvested 96 h postransduction. Rat islet cells were fixed and stained for DAPI, EdU, and insulin. EdU incorporation was visualized and quantitated using a high-content imager. Data are representative of three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To investigate if activin signaling is important for Pdx-1-stimulated β-cell proliferation, we used recombinant activin receptor Fc chimeras (AcRIIA and AcRIIB). These recombinant chimeras consist of the activin receptor ligand-binding domains fused to Fc and serve as dominant negative reagents by neutralizing activin ligands in the culture medium. The Fc-RIIB chimera blocked the β-cell-proliferative effect of AdCMV-hPdx-1 treatment to an extent similar to that obtained by coincubation with mouse Fc-RIIB (mFc-RIIB) and human Fc-RIIA (huFc-RIIA), whereas a control Fc chimera against TNF-α had no effect (Fig. 8C). These data suggest that activin signaling is necessary for the full proliferative effect of overexpressed Pdx-1.
Activin B has no damaging effects on glucose-stimulated insulin secretion, insulin content, or markers of cell stress/survival.
Some agents that stimulate islet cell proliferation can simultaneously damage islets by causing impairment of glucose-stimulated insulin secretion (GSIS), decreased insulin content, or activation of DNA damage and programmed cell death (5, 25, 26). These effects have not been observed when Pdx-1 or Nkx6.1 overexpression is used to activate islet cell replication (9–12), but it remains possible that Inhbb or activin B treatment could have such effects. Treatment of dispersed rat islet cells with activin B for 72 h had no effects on GSIS (Fig. 9A), insulin content (Fig. 9B), expression of the DNA damage marker γ-H2AX (Fig. 9C), or levels of cleaved caspase-3 (Fig. 9D). Thus, activin B is an agent that stimulates islet cell proliferation with no overt damaging side effects.
FIG 9.
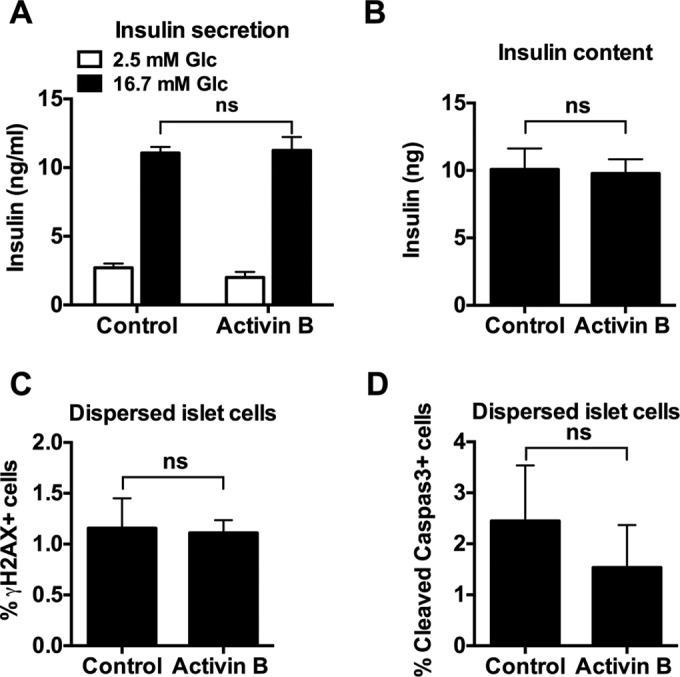
Activin B has no damaging effects on β-cell viability and does not interfere with β-cell function. (A and B) Primary rat islets were treated with activin B (100 ng/ml) or vehicle (0.3% BSA in PBS) for 72 h. Insulin secretion was measured in the presence of 3 or 16.7 mM glucose, and insulin content was measured by enzyme-linked immunosorbent assay. (C and D) Primary rat islets were dispersed into a single-cell suspension and seeded on HTB9 matrix-coated 96-well plates. Cells were treated with activin B (100 ng/ml) or vehicle (0.3% BSA in PBS) for 72 h, followed by fixation and staining with γ-H2AX and cleaved caspase-3 antibodies. Cells were visualized and quantified using a high-content imager. ns, not significant.
Overexpression of Inhbb stimulates human islet cell proliferation.
We have previously shown that Pdx-1 overexpression in intact human islets causes a modest but statistically significant increase in total islet cell proliferation as measured by [3H]thymidine uptake (9). Here, we investigated the effects of Inhbb overexpression in dispersed human islet cells using EdU incorporation as the measure of proliferation. Inhbb overexpression caused a significant increase in total islet cell proliferation (P < 0.001) but not β-cell proliferation (P = 0.06) (Fig. 10). Inhbb overexpression also significantly potentiated Pdx-1-mediated total islet cell and β-cell proliferation (Fig. 10).
FIG 10.
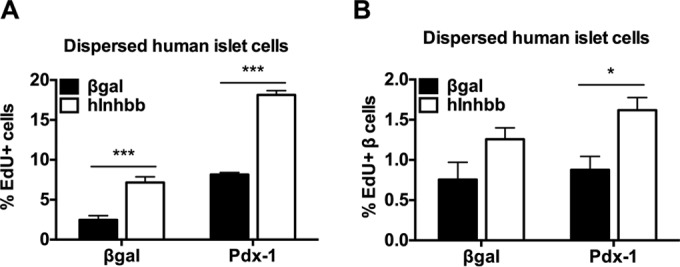
Overexpression of Inhbb stimulates human islet proliferation. Human islets were dispersed and treated with AdCMV-Pdx-1 (Pdx-1) or AdCMV-β-Gal in the absence (black bars) or presence (white bars) of AdCMV-Inhbb. EdU was added during the last 18 h of incubation, and EdU incorporation into total islet cells (A) or islet β cells (B) was measured. Human islet cells were fixed and stained for DAPI, EdU, and insulin and visualized and quantitated with a high-content imager. *, P < 0.05; ***, P < 0.001, compared to the value for the AdCMV-β-Gal control.
DISCUSSION
Controlled expansion of functional β-cell mass could represent a new therapeutic strategy for both major forms of diabetes, but methods for stimulating human islet cell replication without damage to islet function or health have been elusive. Recently, two groups reported identification of small molecules that stimulate human β-cell replication via inhibition of dual-specificity tyrosine kinase-1a (DYRK1A) (27, 28). Because the utility of these molecules for clinical intervention remains unknown, development of alternative strategies should continue. In recent years, our group has demonstrated that Nkx6.1 and Pdx-1 activate islet cell proliferation pathways that are distinct and additive, such that the combined overexpression of the two factors stimulates rodent islet cell replication by more than 15-fold (7, 9, 10). Consistent with these additive effects, there are several fundamental differences between the mechanisms of Nkx6.1- and Pdx-1-mediated islet cell proliferation. First, overexpression of Nkx6.1 from an adenovirus vector under the control of the constitutive CMV promoter (causing expression in all islet cells) stimulates proliferation of only β cells, whereas overexpression of Pdx-1 via the same vector stimulates proliferation of both α and β cells (9). Second, Nkx6.1 stimulates β-cell proliferation via activation of the Nr4a1 and Nr4a3 orphan nuclear receptors, which in turn induce expression of the E2F1 and cyclin E cell cycle activators and suppress expression of the p21cip cell cycle inhibitor (10). In contrast, Pdx-1-mediated proliferation is induced at least in part by activation of transient receptor potential channel 3 (TRPC3) and TRPC6 calcium channels leading to extracellular signal-regulated kinase 1 and 2 (ERK1/2) and cyclin D activation (9). Consistent with these results, the cyclin D/cdk4 inhibitor PD0332991 blocks Pdx-1- but not Nkx6.1-mediated islet cell proliferation (11).
Left unclear from our previous studies was the question of whether Pdx-1 works in a cell-autonomous fashion to stimulate proliferation only in islet cells in which it is overexpressed. Using three separate adenovirus constructs, we show here that Pdx-1 stimulates islet cell proliferation at least in part through production of a cell-extrinsic soluble factor(s). Using an inducible β-cell-specific (RIP) virus, we found that expression of Pdx-1 in β cells activates both α- and β-cell proliferation, suggesting that the putative soluble factor can originate in the β cell. We also used a transwell assay to show that overexpression of Pdx-1 in islets separated by a membrane from islets lacking Pdx-1 expression activates proliferation in the untreated islets. Together, these results demonstrate a novel non-cell-autonomous mechanism of Pdx-1-stimulated islet cell proliferation.
We then used microarray and bioinformatics analyses to identify genes that are upregulated by Pdx-1 overexpression in rat islets and that are predicted to encode soluble factors or transmembrane receptors with a potential role in cell replication. We found two genes that are sufficient to induce proliferation in dispersed rat islet cells, including osteoprotegerin (confirming a report by another group [22]) and Inhbb.
Inhbb is a member of the TGF-β superfamily, consisting of molecules that signal via the SMAD network (29). In the current study, overexpression of Pdx-1 induced expression of four TGF-β superfamily members, but among these, only Inhbb enhanced islet cell proliferation. Inha, Inhba, and Inhbb are three distinct genes that produce monomeric peptides (α, β-A, and β-B) that form inhibin and activin dimers. The specific dimers formed depend on the pairing of subunits and include inhibin A (α- and β-A), inhibin B (α- and β-B), activin A (β-A and β-Α), activin B (β-B and β-B), and activin AB (β-A and β-B) (23, 24).
Various effects of activins have been reported in pancreatic islets. Activin A potentiates glucose-stimulated insulin secretion in rat (30, 31) and human (32) islets at various glucose concentrations. Activin A has also been reported to increase BrdU incorporation in rat β cells, but the effect of activin A on α-cell replication was not studied (33). Activin A has also been used in combination with various other factors to induce partial conversion of the exocrine cell line, AR42J, to insulin-producing cells (34), and more recently, activins have emerged as important reagents in studies seeking to convert human embryonic stem cells to β cells (35, 36). In whole animals, one report has shown that treatment with activin A and betacellulin increases β-cell mass and insulin content while reducing plasma glucose levels in a streptozotocin (STZ)-treated neonatal rat β-cell regeneration model (37). β-Cell-specific overexpression of the inhibitory SMAD7 in mice inhibits SMAD2/3 signaling in the activin/TGF-β pathway, leading to decreased insulin content and serum insulin as well as impaired glucose tolerance, thereby suggesting a role of the TGF-β pathway in regulating functional β-cell mass (38). Furthermore, conventional knockout of follistatin-like 3 (Fstl3), a natural activin antagonist, results in β-cell hyperplasia and increased pancreatic cell number and size (39). Finally, conventional knockout of Inhbb in mice results in a decrease in islet mass accompanied by a decrease in the percentage of α cells in islets (40). In this previous study, by Bonomi et al., no changes in glucose homeostasis were noted in young knockout mice, but insulin levels decreased in older animals. Taken together, these data suggest important roles of activin/inhibin family members in control of functional β-cell mass.
Several new findings emerged from the current study, as follows. (i) Pdx-1 overexpression in rat islet cells, including β cells, generates a soluble factor or factors that stimulate islet cell proliferation. (ii) There have been no previous reports of specific roles of the Inhbb gene or the activin B protein (comprised of two Inhbb subunits) in regulation of β-cell proliferation. This includes the prior studies on global Inhbb knockout mice, in which islet cell proliferation was not measured (40). Here, we show that overexpression of Inhbb or treatment with recombinant activin B is sufficient to stimulate islet cell and β-cell proliferation in dispersed rat islet cell cultures while also potentiating the proliferative effects of Pdx-1 overexpression. In dispersed human islet cell cultures, Inhbb overexpression enhanced total islet cell proliferation and potentiated effects of Pdx-1 overexpression. (iii) We demonstrate that overexpression of Pdx-1 stimulates Inhbb expression and activin signaling, and we show that neutralization of this ligand with Fc-activin receptor chimeric antibodies partially interferes with Pdx-1-mediated β-cell proliferation. These data demonstrate that Pdx-1-stimulated islet cell proliferation is mediated in part by induction of Inhbb but also suggest that other, yet to be identified, factors may be required for the full proliferative response.
Other soluble factors and hormones have been reported to stimulate β-cell proliferation, including glucose, insulin, placental lactogen, betacellulin, Wnt3A, platelet-derived growth factor AA (PDGF-AA), and betatrophin (33, 41–45), among others. However, none of these factors have been reported to stimulate β-cell proliferation in adult human islets. PDGF-AA has a potent effect to stimulate juvenile human islet β-cell proliferation but is inactive in adult human islets apparently due to loss of PDGF signaling as a function of islet age (45). The current study joins another recent report showing human islet cell proliferative effects of serpin B1 (46) as evidence for the existence of peptidic factors capable of enhancing functional β-cell mass. Future efforts will be focused on identification of other Pdx-1-induced factors that may complement the human islet cell proliferative effects of Inhbb overexpression or activin B addition.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health β-Cell Biology Consortium (BCBC) (U01 DK-089538) to C.B.N., grants from the Juvenile Diabetes Research Foundation (JDRF) to C.B.N. (17-2011-15) and H.E.H. (17-2011-614), postdoctoral fellowships from the American Diabetes Association to L.Z. (1-16-PDF-136) and the JDRF to H.L.H. (3-2009-561) and a sponsored research agreement from Janssen (to H.E.H.).
We thank Danhong Lu, Helena Winfield, Lisa Poppe, Taylor Rosa, and Paul Anderson for expert technical assistance. We are also very grateful to Robert Mercer and family for generous support over many years for our diabetes and islet research.
REFERENCES
- 1.Weir GC, Bonner-Weir S. 2004. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes 53(Suppl 3):S16–S21. doi: 10.2337/diabetes.53.suppl_3.S16. [DOI] [PubMed] [Google Scholar]
- 2.Bouwens L, Rooman I. 2005. Regulation of pancreatic beta-cell mass. Physiol Rev 85:1255–1270. doi: 10.1152/physrev.00025.2004. [DOI] [PubMed] [Google Scholar]
- 3.Muoio DM, Newgard CB. 2008. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 4.Hohmeier HE, Tran VV, Chen G, Gasa R, Newgard CB. 2003. Inflammatory mechanisms in diabetes: lessons from the beta-cell. Int J Obes Relat Metab Disord 27(Suppl 3):S12–S6. doi: 10.1038/sj.ijo.0802493. [DOI] [PubMed] [Google Scholar]
- 5.Rieck S, Zhang J, Li Z, Liu C, Naji A, Takane KK, Fiaschi-Taesch NM, Stewart AF, Kushner JA, Kaestner KH. 2012. Overexpression of hepatocyte nuclear factor-4α initiates cell cycle entry, but is not sufficient to promote beta-cell expansion in human islets. Mol Endocrinol 26:1590–1602. doi: 10.1210/me.2012-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, Cherok E, Takane KK, Scott DK, Stewart AF. 2010. Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 59:1926–1936. doi: 10.2337/db09-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schisler JC, Fueger PT, Babu DA, Hohmeier HE, Tessem JS, Lu D, Becker TC, Naziruddin B, Levy M, Mirmira RG, Newgard CB. 2008. Stimulation of human and rat islet β-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol Cell Biol 28:3465–3476. doi: 10.1128/MCB.01791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stephens SB, Schisler JC, Hohmeier HE, An J, Sun AY, Pitt GS, Newgard CB. 2012. A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta-cell survival and function. Cell Metab 16:33–43. doi: 10.1016/j.cmet.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, Newgard CB, Hohmeier HE. 2013. Pdx-1 activates islet α- and β-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol Cell Biol 33:4017–4029. doi: 10.1128/MCB.00469-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tessem JS, Moss LG, Chao LC, Arlotto M, Lu D, Jensen MV, Stephens SB, Tontonoz P, Hohmeier HE, Newgard CB. 2014. Nkx6.1 regulates islet β-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc Natl Acad Sci U S A 111:5242–5247. doi: 10.1073/pnas.1320953111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Milburn JL Jr, Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M, BeltrandelRio H, Newgard CB, Johnson JH, Unger RH. 1995. Pancreatic β-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 270:1295–1299. [DOI] [PubMed] [Google Scholar]
- 12.Naber SP, McDonald JM, Jarett L, McDaniel ML, Ludvigsen CW, Lacy PE. 1980. Preliminary characterization of calcium binding in islet-cell plasma membranes. Diabetologia 19:439–444. doi: 10.1007/BF00281823. [DOI] [PubMed] [Google Scholar]
- 13.Schisler JC, Jensen PB, Taylor DG, Becker TC, Knop FK, Takekawa S, German M, Weir GC, Lu D, Mirmira RG, Newgard CB. 2005. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc Natl Acad Sci U S A 102:7297–7302. doi: 10.1073/pnas.0502168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. 1998. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A 95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seiler CY, Park JG, Sharma A, Hunter P, Surapaneni P, Sedillo C, Field J, Algar R, Price A, Steel J, Throop A, Fiacco M, LaBaer J. 2014. DNASU plasmid and PSI:Biology-Materials repositories: resources to accelerate biological research. Nucleic Acids Res 42:D1253–D1260. doi: 10.1093/nar/gkt1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jensen MV, Haldeman JM, Zhang H, Lu D, Huising MO, Vale WW, Hohmeier HE, Rosenberg P, Newgard CB. 2013. Control of voltage-gated potassium channel Kv2.2 expression by pyruvate-isocitrate cycling regulates glucose-stimulated insulin secretion. J Biol Chem 288:23128–23140. doi: 10.1074/jbc.M113.491654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavine JA, Raess PW, Davis DB, Rabaglia ME, Presley BK, Keller MP, Beinfeld MC, Kopin AS, Newgard CB, Attie AD. 2010. Contamination with E1A-positive wild-type adenovirus accounts for species-specific stimulation of islet cell proliferation by CCK: a cautionary note. Mol Endocrinol 24:464–467. doi: 10.1210/me.2009-0384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen MV, Joseph JW, Ilkayeva O, Burgess S, Lu D, Ronnebaum SM, Odegaard M, Becker TC, Sherry AD, Newgard CB. 2006. Compensatory responses to pyruvate carboxylase suppression in islet beta-cells. Preservation of glucose-stimulated insulin secretion. J Biol Chem 281:22342–22351. [DOI] [PubMed] [Google Scholar]
- 19.Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, Fiaschi-Taesch N, Shamji A, Clemons PA, Stewart AF, Schreiber SL, Wagner BK. 2012. A human islet cell culture system for high-throughput screening. J Biomol Screen 17:509–518. doi: 10.1177/1087057111430253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Andres R, Forgie A, Wyatt S, Chen Q, de Sauvage FJ, Davies AM. 2001. Multiple effects of artemin on sympathetic neurone generation, survival and growth. Development 128:3685–3695. [DOI] [PubMed] [Google Scholar]
- 21.Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS, Leitner ML, Araki T, Johnson EM Jr, Milbrandt J. 1998. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRα3-RET receptor complex. Neuron 21:1291–1302. doi: 10.1016/S0896-6273(00)80649-2. [DOI] [PubMed] [Google Scholar]
- 22.Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, Vasavada RC. 2015. Osteoprotegerin and Denosumab stimulate human beta cell proliferation through inhibition of the receptor activator of NF-κB ligand pathway. Cell Metab 22:77–85. doi: 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burger HG, Igarashi M. 1988. Inhibin: definition and nomenclature, including related substances. Mol Endocrinol 2:391–392. doi: 10.1210/mend-2-4-391. [DOI] [PubMed] [Google Scholar]
- 24.Ying SY, Czvik J, Becker A, Ling N, Ueno N, Guillemin R. 1987. Secretion of follicle-stimulating hormone and production of inhibin are reciprocally related. Proc Natl Acad Sci U S A 84:4631–4635. doi: 10.1073/pnas.84.13.4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee SH, Hao E, Levine F, Itkin-Ansari P. 2011. Id3 upregulates BrdU incorporation associated with a DNA damage response, not replication, in human pancreatic β-cells. Islets 3:358–366. doi: 10.4161/isl.3.6.17923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harb G, Vasavada RC, Cobrinik D, Stewart AF. 2009. The retinoblastoma protein and its homolog p130 regulate the G1/S transition in pancreatic β-cells. Diabetes 58:1852–1862. doi: 10.2337/db08-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, Stewart AF. 2015. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat Med 21:383–388. doi: 10.1038/nm.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang YQ, Kamireddy A, Swafford A, Powers AF, Walker J, Lamb J, Bursalaya B, DiDonato M, Harb G, Qiu M, Filippi CM, Deaton L, Turk CN, Suarez-Pinzon WL, Liu Y, Hao X, Mo T, Yan S, Li J, Herman AE, Hering BJ, Wu T, Martin Seidel H, McNamara P, Glynne R, Laffitte B. 2015. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat Commun 6:8372. doi: 10.1038/ncomms9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wrana JL. 2013. Signaling by the TGFβ superfamily. Cold Spring Harb Perspect Biol 5:a011197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Totsuka Y, Tabuchi M, Kojima I, Shibai H, Ogata E. 1988. A novel action of activin A: stimulation of insulin secretion in rat pancreatic islets. Biochem Biophys Res Commun 156:335–339. doi: 10.1016/S0006-291X(88)80845-3. [DOI] [PubMed] [Google Scholar]
- 31.Verspohl EJ, Ammon HP, Wahl MA. 1993. Activin A: its effects on rat pancreatic islets and the mechanism of action involved. Life Sci 53:1069–1078. doi: 10.1016/0024-3205(93)90260-A. [DOI] [PubMed] [Google Scholar]
- 32.Florio P, Luisi S, Marchetti P, Lupi R, Cobellis L, Falaschi C, Sugino H, Navalesi R, Genazzani AR, Petraglia F. 2000. Activin A stimulates insulin secretion in cultured human pancreatic islets. J Endocrinol Invest 23:231–234. doi: 10.1007/BF03343713. [DOI] [PubMed] [Google Scholar]
- 33.Brun T, Franklin I, St-Onge L, Biason-Lauber A, Schoenle EJ, Wollheim CB, Gauthier BR. 2004. The diabetes-linked transcription factor PAX4 promotes β-cell proliferation and survival in rat and human islets. J Cell Biol 167:1123–1135. doi: 10.1083/jcb.200405148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mashima H, Yamada S, Tajima T, Seno M, Yamada H, Takeda J, Kojima I. 1999. Genes expressed during the differentiation of pancreatic AR42J cells into insulin-secreting cells. Diabetes 48:304–309. doi: 10.2337/diabetes.48.2.304. [DOI] [PubMed] [Google Scholar]
- 35.Wiater E, Vale W. 2012. Roles of activin family in pancreatic development and homeostasis. Mol Cell Endocrinol 359:23–29. doi: 10.1016/j.mce.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 36.Sulzbacher S, Schroeder IS, Truong TT, Wobus AM. 2009. Activin A-induced differentiation of embryonic stem cells into endoderm and pancreatic progenitors—the influence of differentiation factors and culture conditions. Stem Cell Rev 5:159–173. doi: 10.1007/s12015-009-9061-5. [DOI] [PubMed] [Google Scholar]
- 37.Li L, Yi Z, Seno M, Kojima I. 2004. Activin A and betacellulin: effect on regeneration of pancreatic beta-cells in neonatal streptozotocin-treated rats. Diabetes 53:608–615. doi: 10.2337/diabetes.53.3.608. [DOI] [PubMed] [Google Scholar]
- 38.Smart NG, Apelqvist AA, Gu X, Harmon EB, Topper JN, MacDonald RJ, Kim SK. 2006. Conditional expression of Smad7 in pancreatic beta cells disrupts TGF-β signaling and induces reversible diabetes mellitus. PLoS Biol 4:e39. doi: 10.1371/journal.pbio.0040039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mukherjee A, Sidis Y, Mahan A, Raher MJ, Xia Y, Rosen ED, Bloch KD, Thomas MK, Schneyer AL. 2007. FSTL3 deletion reveals roles for TGF-β family ligands in glucose and fat homeostasis in adults. Proc Natl Acad Sci U S A 104:1348–1353. doi: 10.1073/pnas.0607966104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonomi L, Brown M, Ungerleider N, Muse M, Matzuk MM, Schneyer A. 2012. Activin B regulates islet composition and islet mass but not whole body glucose homeostasis or insulin sensitivity. Am J Physiol Endocrinol Metab 303:E587–E596. doi: 10.1152/ajpendo.00177.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paris M, Bernard-Kargar C, Berthault MF, Bouwens L, Ktorza A. 2003. Specific and combined effects of insulin and glucose on functional pancreatic beta-cell mass in vivo in adult rats. Endocrinology 144:2717–2727. doi: 10.1210/en.2002-221112. [DOI] [PubMed] [Google Scholar]
- 42.Parsons JA, Brelje TC, Sorenson RL. 1992. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 130:1459–1466. doi: 10.1210/endo.130.3.1537300. [DOI] [PubMed] [Google Scholar]
- 43.Rulifson IC, Karnik SK, Heiser PW, ten Berge D, Chen H, Gu X, Taketo MM, Nusse R, Hebrok M, Kim SK. 2007. Wnt signaling regulates pancreatic beta cell proliferation. Proc Natl Acad Sci U S A 104:6247–6252. doi: 10.1073/pnas.0701509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi P, Park JS, Melton DA. 2013. Betatrophin: a hormone that controls pancreatic beta cell proliferation. Cell 153:747–758. doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Chen H, Gu X, Liu Y, Wang J, Wirt SE, Bottino R, Schorle H, Sage J, Kim SK. 2011. PDGF signalling controls age-dependent proliferation in pancreatic beta-cells. Nature 478:349–355. doi: 10.1038/nature10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Ouaamari A, Dirice E, Gedeon N, Hu J, Zhou JY, Shirakawa J, Hou L, Goodman J, Karampelias C, Qiang G, Boucher J, Martinez R, Gritsenko MA, De Jesus DF, Kahraman S, Bhatt S, Smith RD, Beer HD, Jungtrakoon P, Gong Y, Goldfine AB, Liew CW, Doria A, Andersson O, Qian WJ, Remold-O'Donnell E, Kulkarni RN. 2016. SerpinB1 promotes pancreatic β cell proliferation. Cell Metab 23:194–205. doi: 10.1016/j.cmet.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]