

Abstract
Key points
Absence seizures are accompanied by spike‐and‐wave discharges in cortical electroencephalograms. These complex paroxysmal activities, affecting the thalamocortical networks, profoundly alter cognitive performances and preclude conscious perception.
Here, using a well‐recognized genetic model of absence epilepsy, we investigated in vivo how information processing was impaired in the ictogenic neurons, i.e. the population of cortical neurons responsible for seizure initiation.
In between seizures, ictogenic neurons were more prone to generate bursting activity and their firing response to weak depolarizing events was considerably facilitated compared to control neurons.
In the course of seizures, information processing became unstable in ictogenic cells, alternating between an increased and a decreased responsiveness to excitatory inputs, depending on the spike and wave patterns.
The state‐dependent modulation in the excitability of ictogenic neurons affects their inter‐seizure transfer function and their time‐to‐time responsiveness to incoming inputs during absences.
Abstract
Epileptic seizures result from aberrant cellular and/or synaptic properties that can alter the capacity of neurons to integrate and relay information. During absence seizures, spike‐and‐wave discharges (SWDs) interfere with incoming sensory inputs and preclude conscious experience. The Genetic Absence Epilepsy Rats from Strasbourg (GAERS), a well‐established animal model of absence epilepsy, allows exploration of the cellular basis of this impaired information processing. Here, by combining in vivo electrocorticographic and intracellular recordings from GAERS and control animals, we investigated how the pro‐ictogenic properties of seizure‐initiating cortical neurons modify their integrative properties and input–output operation during inter‐ictal periods and during the spike (S‐) and wave (W‐) cortical patterns alternating during seizures. In addition to a sustained depolarization and an excessive firing rate in between seizures, ictogenic neurons exhibited a pronounced hyperpolarization‐activated depolarization compared to homotypic control neurons. Firing frequency versus injected current relations indicated an increased sensitivity of GAERS cells to weak excitatory inputs, without modifications in the trial‐to‐trial variability of current‐induced firing. During SWDs, the W‐component resulted in paradoxical effects in ictogenic neurons, associating an increased membrane input resistance with a reduction in the current‐evoked firing responses. Conversely, the collapse of cell membrane resistance during the S‐component was accompanied by an elevated current‐evoked firing relative to W‐sequences, which remained, however, lower compared to inter‐ictal periods. These findings show a dynamic modulation of ictogenic neurons’ intrinsic properties that may alter inter‐seizure cortical function and participate in compromising information processing in cortical networks during absences.
Key points
Absence seizures are accompanied by spike‐and‐wave discharges in cortical electroencephalograms. These complex paroxysmal activities, affecting the thalamocortical networks, profoundly alter cognitive performances and preclude conscious perception.
Here, using a well‐recognized genetic model of absence epilepsy, we investigated in vivo how information processing was impaired in the ictogenic neurons, i.e. the population of cortical neurons responsible for seizure initiation.
In between seizures, ictogenic neurons were more prone to generate bursting activity and their firing response to weak depolarizing events was considerably facilitated compared to control neurons.
In the course of seizures, information processing became unstable in ictogenic cells, alternating between an increased and a decreased responsiveness to excitatory inputs, depending on the spike and wave patterns.
The state‐dependent modulation in the excitability of ictogenic neurons affects their inter‐seizure transfer function and their time‐to‐time responsiveness to incoming inputs during absences.
Abbreviations
- AP
action potential
- CV
coefficient of variation
- ECoG
electrocorticogram
- EEG
electroencephalogram
- <F>
mean firing frequency
- FF
Fano factor
- F–I
firing frequency versus injected current
- GAERS
Genetic Absence Epileptic Rats from Strasbourg
- γ
gain
- Ih
hyperpolarization‐activated inward cationic current
- IB
intrinsic bursting
- IDC
injected direct current
- I‐I
interictal
- Ith
current threshold
- ISI
interspike interval
- Rm
membrane input resistance
- S1
primary somatosensory
- S‐
spike
- W‐
wave
- SWD
spike‐and‐wave discharge
- V–I
voltage versus injected current
- Vm
membrane potential
Introduction
Typical absence seizures occur in various syndromes of idiopathic generalized epilepsies with onset in childhood and adolescence (Panayiotopoulos, 2008; Matricardi et al. 2014). During the absence attack, lasting usually a few seconds, patients experience a sudden clouding of consciousness combining fading of awareness, unresponsiveness to mild external stimuli and interruption of ongoing behaviours (Blumenfeld, 2005; Panayiotopoulos, 2008; Cavanna & Monaco, 2009; Chipaux et al. 2013). Human absences are reflected in the electroencephalogram (EEG) by a typical spatiotemporal pattern of activity consisting of bilateral and symmetrical 3–4 Hz spike‐and‐wave discharges (SWDs), driven by synchronized oscillatory activity in reciprocally connected cortical and thalamic networks (Williams, 1953; Panayiotopoulos, 2008; Chipaux et al. 2013). EEG, magnetoencephalographic and brain metabolism investigations in human patients (Holmes et al. 2004; Westmijse et al. 2009; Bai et al. 2010) and rodent genetic models (Meeren et al. 2002; Polack et al. 2007; David et al. 2008; Depaulis et al. 2015; Lüttjohann & Luijtelaar, 2015) showed that SWDs accompanying absence seizures have a cortical focal onset, suggesting that they could originate from a population of cortical neurons with pro‐ictogenic properties, i.e. which are prone to initiate and spread paroxysmal oscillations through synaptic networks (Meeren et al. 2005; Polack et al. 2009; Lüttjohann & Luijtelaar, 2015).
Recently, neurons with pro‐ictogenic properties have been identified in the cortical focus of the Genetic Absence Epilepsy Rats from Strasbourg (GAERS), a well‐documented rodent model that closely phenocopies the human absence seizures (Depaulis & van Luijtelaar, 2006). These seizure‐initiating neurons are a subset of pyramidal cells located in layers 5 and 6 of the facial somatosensory cortex (Polack et al. 2007, 2009). As previously described (Polack et al. 2007, 2009; Polack & Charpier, 2009; Chipaux et al. 2011, 2013), ictogenic neurons display an excessively depolarized membrane potential (V m) associated with an intense and regular spontaneous firing during inter‐ictal periods. During SWDs, their rhythmic brisk firing precedes that of distant cortical and thalamic neurons, which are secondarily recruited to sustain synchronized paroxysmal activity in corticothalamic loops (Polack et al. 2007, 2009).
It is proposed that the ability of the cortex to process ongoing physiological inputs is profoundly impaired during seizures by the excessive level of synchrony in synaptic networks, notably in the core territory of paroxysms (Trevelyan et al. 2013). However, the cellular mechanisms underlying this alteration of cortical functions are not fully understood. In a cat pharmacological model of absence epilepsy, a seizure‐dependent increase in the membrane conductance of cortical neurons resulted in an attenuation of voltage responses to depolarizing current pulses (Steriade & Amzica, 1999), a reduced cell excitability that could disrupt the normal integration of synaptic inputs. In genetically determined absence epilepsy, the functional consequences of epileptic discharges on information processing by the cortical neurons generating seizures remain unclear. Indeed, although sensory inputs are propagated in the thalamocortical pathways of epileptic rodents and patients during seizures, their integration at the cortical level remains inefficient to produce conscious experience (Inoue et al. 1992; Chipaux et al. 2013). Here, by the means of in vivo simultaneous electrocorticographic (ECoG) and intracellular recordings in the GAERS, we first investigated how the pro‐ictogenic properties of seizure‐initiating cortical neurons, latent but operating during inter‐ictal periods, affect their integrative properties, input–output function and time‐to‐time firing dynamics compared to homologous cortical neurons recorded in normal rats. We next examined how the cellular events underlying the two ECoG components of the spike‐and‐wave complexes (i.e. the ‘spike’ and the ‘wave’ components) differentially impact the excitability and current‐evoked firing responses of ictogenic cortical neurons.
Methods
Ethics approval
The care and experimental manipulation of the animals were carried out in accordance with the European Union guidelines (directive 2010/63/EU) and approved by the Charles Darwin Ethical Committee on Animal Experimentation (C2EA‐05). Every precaution was taken to minimize the suffering and the number of animals used in each series of experiments.
Animal preparation for in vivo electrophysiology
Experiments were conducted on 16 GAERS (7 females and 9 males, 5–15 months of age; animal facility of the Brain and Spine Institute, Paris, France) presenting spontaneously recurrent SWDs, and 14 non‐epileptic Wistar rats (8 females and 6 males, 3–10 months of age; Charles River Laboratories, L'Arbresle, France). All the Wistar rats included in this study were devoid of SWDs. For surgical purpose, both rat strains were initially anaesthetized by intraperitoneal (i.p.) injections of sodium pentobarbital (40 mg kg–1; Centravet, Plancoët, France) and ketamine (50 mg kg–1; Centravet), or by inhalation of isoflurane (2.5–3%; Centravet). The animals were then cannulated after incision of the trachea and placed in a stereotaxic apparatus. Incisions and pressure points were repeatedly infiltrated with lidocaine (2%; Centravet) throughout the experiment. A small trepanation (∼1 mm of diameter) was made over the facial region of the primary somatosensory (S1) cortex (0.5–2.4 mm posterior to the bregma, 4.1–6.5 mm lateral to the midline) previously identified in the GAERS (Polack et al. 2007, 2009; David et al. 2008; Depaulis et al. 2015) and another genetic model of absence epilepsy (Meeren et al. 2002; Lüttjohann & Luijtelaar, 2015) as the cortical focus for SWD initiation. To obtain long‐lasting stable intracellular recordings, rats were immobilized by intramuscular injection of gallamine triethiodide (40 mg (2 h)–1; Sigma‐Aldrich, Saint‐Quentin‐Fallavier, France) and artificially ventilated. Sedation and analgesia were maintained throughout the recording sessions by repeated injections of the synthetic opioid fentanyl (3 μg kg–1, i.p.; Janssen‐Cilag, Issy‐les‐Moulinaux, France). This neuroleptanalgesia did not alter the spatiotemporal dynamics or the morphology of SWDs compared to freely moving GAERS (Polack et al. 2007) and induced inter‐ictal cortical patterns characterized by fast and apparently desynchronized waves of small amplitude comparable to the ‘activated’ waking pattern (Constantinople & Bruno, 2011; Altwegg‐Boussac et al. 2014). Moreover, the use of fentanyl did not, by itself, significantly impact the intrinsic electrophysiological properties of cortical pyramidal neurons (Mahon et al. 2001; Altwegg‐Boussac et al. 2014). The absence of concurrent effect of fentanyl on cortical network activity and cellular excitability was of a crucial importance in this study where we investigated the functional consequences of inter‐ictal and ictal activity at single cell level (Depaulis et al. 2015). Body temperature was maintained (36.5–37.5°C) with a homoeothermic blanket. At the end of the experiments, animals received an overdose of sodium pentobarbital (200 mg kg–1, i.p.; Centravet).
Coupled recordings of ECoG and intracellular activities
Spontaneous ECoG activity was recorded from GAERS and non‐epileptic rats by the means of a low impedance (∼60 kΩ) silver electrode delicately placed on the dura above the facial part of S1 cortex and a reference electrode inserted in a muscle at the opposite side of the head. Surface cortical signals were amplified using a differential AC amplifier (Model 1700; A‐M Systems, Carlsborg, WA, USA), filtered at 1 Hz–5 kHz, and digitized at 3 kHz (CED 1401plus; Cambridge Electronic Design, Cambridge, UK). Intracellular recordings were made close (< 200 μm) to the surface macroelectrode using glass micropipettes filled with 2 m potassium acetate (50–70 MΩ). Current‐clamp recordings were amplified using an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA, USA) operating in bridge mode, filtered at 1 Hz–3 kHz and digitized at 25 kHz. Intracellularly recorded neurons, identified as pyramidal cells by their morphological and electrophysiological characteristics, were located in the layers 5–6 of S1 cortex at depths ranging from 903 to 3328 μm below the cortical surface (Paxinos & Watson, 1986; Wilent & Contreras, 2004).
All electrophysiological recordings were acquired and analysed using Spike2 software (Spike2 version 7.06; Cambridge Electronic Design). Additional data analysis was performed using Origin 8.1 (OriginLab Corp., Northampton, MA, USA) and Matlab (The MathWorks Inc., Natick, MA, USA).
Neuron staining
Recorded neurons were labelled by intracellular injection of neurobiotin (1.5% added to the pipette solution; Vector Laboratories, Burlingame, CA, USA). Depolarizing current pulses (0.2–1 nA; 50–100 ms) were applied at 2.5 Hz during 10–15 min to obtain a reliable labelling of neuronal processes (Polack et al. 2007). At the end of the experiment, rats were killed with sodium pentobarbital and perfused with 0.3 % glutaraldehyde (VWR, Fontenay‐sous‐Bois, France)–4% paraformaldehyde (Carlo Erba, Val‐de‐Reuil, France) in 0.1 m phosphate‐buffered saline solution (PBS; pH 7.4). Brains were post‐fixed into 4% paraformaldehyde for at least 2 h. After cryoprotection with 30% sucrose, brains were frozen in isopentane (−50 ± 5°C; VWR). 50‐μm‐thick sections were cut on a freezing microtome (Microm HM450, Thermo Fisher Scientific, Waltham, MA, USA) and incubated overnight in PBS–0.4 % Triton X‐100 (Sigma‐Aldrich) + 0.4% of each reagent from Vectastain ABC Elite kit (Vector Laboratories, Burlingame, CA, USA) to create an avidin‐biotin complex on labelled neurons. After three washes in PBS, brain sections were pre‐incubated 10 min in 0.5 mg ml–1 diaminobenzidine (DAB dissolved in PBS; Sigma‐Aldrich) and 0.02% nickel sulfate (Sigma‐Aldrich). Hydrogen peroxide at 0.04% (30% aqueous solution; Merck Millipore, Molsheim, France) was subsequently added for 15 min to stain labelled neurons. Finally, sections were washed four times in PBS, mounted on slides and air dried. Counterstaining was accomplished with safranine (RAL Diagnostics, Martillac, France). Slides were dehydrated in ethanol baths and coverslipped in Eukitt (Sigma‐Aldrich). Microphotographs of labelled neurons were taken with a Zeiss Axioskop 2 Plus microscope coupled to a Leica DFC 310 FX camera (Leica Application Suite software) and neuronal reconstruction was performed using PaintNet software. The position and depth of labelled neurons within the S1 cortex were confirmed using the atlas of Paxinos & Watson (1986).
Analysis of ECoG and intracellular signals
The start (or end) of a SWD in the ECoG was taken as the first (or last) spike–wave complex which amplitude of the spike component was at least two times the peak‐to‐peak amplitude of the baseline ECoG activity. To extract the frequency content of ECoG signals, we performed time–frequency (TF) maps using wavelet transform analysis made with custom‐written functions in Matlab. A signal x(t) was convolved with a complex Morlet's wavelet function defined as:
Wavelets were normalized and thus:
The width of each wavelet function (m = f 0 /σ f) was chosen to be 7; where σf = 1/2πσt.
TF contents were represented as the energy of the convolved signal:
Average membrane potential (V m) values were calculated in each neuron, in between seizures in GAERS and during quiescent periods in control Wistar rats, as the mean of the distribution of spontaneous intracellular subthreshold activity recorded for at least 10 s. When necessary, they were corrected by subtracting the extracellular tip potential measured immediately after the end of the recording. Voltage threshold of action potentials (APs) was measured as the V m at which the dV/dt first exceeded 10 V s–1 (Fricker et al. 1999; Mahon et al. 2003). The amplitude of APs was calculated as the potential difference between the voltage threshold and the peak after averaging at least 10 waveforms and their duration was measured as the width at half‐maximal AP amplitude (half‐width duration). The mean spontaneous firing rate (<F>) of cortical neurons was calculated in each cell from continuous records of at least 10 s. To estimate the variability of inter‐spike intervals (ISIs) during spontaneous and current‐evoked firing, we used the CV2 method, which compares adjacent intervals and is independent of variations in average firing rates (Holt et al. 1996). Assuming that APs in a train occur at times ti (0 ≤ i ≤ N), the ISIi can be defined as:
The CV2 of ISIs was calculated using the following equation:
CV2 measurements were restricted to neurons having a mean firing rate exceeding 1.5 Hz (Mahon et al. 2006).
Measurements of apparent membrane input resistance (R m) were based on the linear electrical cable theory applied to an idealized isopotential neuron (Rall, 1969). During baseline periods in GAERS and control neurons, voltage–current (V–I) relationships were constructed from the mean V m drops (ΔV m) evoked by hyperpolarizing current pulses of varying intensity (−0.2 to −1 nA; 100 ms duration; every 0.5–1 s; n ≥ 20 trials for each intensity). ΔV m measurements were made at the peak of the hyperpolarizing voltage deflection, immediately after the end of the membrane capacitance charge, to avoid interference with a hyperpolarization‐activated depolarizing sag potential (see Fig. 2 Aa). Once the linearity of the V–I relations was attested, R m was calculated as the slope of the corresponding linear fit (see Fig. 2 Ab and Bb, dashed lines). Sag ratio was calculated by dividing the steady‐state voltage drop by the peak voltage response to hyperpolarizing current pulses of high intensity (−0.8 nA).
Figure 2. GAERS S1 cortex neurons display time‐dependent inward rectification .
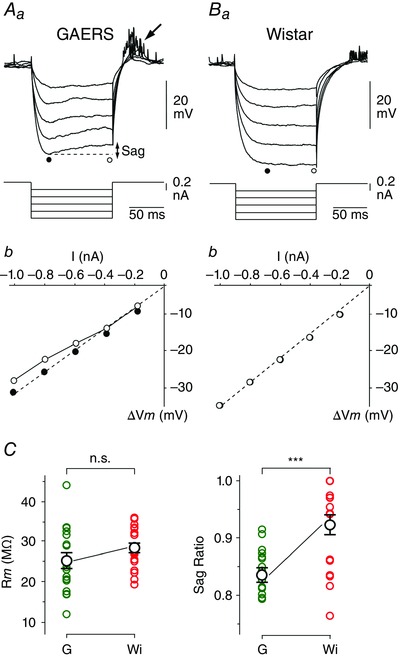
A and B, average (n ≥ 20 successive trials) voltage responses (top records) in a GAERS (Aa) and a non‐epileptic rat (Ba) neuron to hyperpolarizing current pulses of increasing intensity (bottom traces). Measurements of voltage deflection (ΔV m) were made after the charge of the membrane capacitance (filled circles) and at steady‐state of the responses (open circles). Note the depolarizing sag potential during the course of the largest voltage drops in the GAERS cell and the subsequent post‐anodal break excitation (arrow). The corresponding plots of ΔV m as a function of the injected current (I) are shown for the GAERS (Ab) and control (Bb) neuron. Dashed lines represent linear regressions of V–I relations computed from the peak of the responses (r ≥ 0.99). C, summary data of membrane resistance (R m), calculated as the slope of V–I relations, and sag ratio values measured in GAERS (G) and control rat (Wi) neurons. *** P < 0.001; n.s., non‐significant.
To assess the differential impact of wave and spike components on the integrative properties and excitability of ictogenic neurons, we applied brief repeated hyperpolarizing (−0.8 nA, 10 ms duration, every 50 ms) or depolarizing (+0.4 nA, 50 ms duration, every 150 ms) current pulses during seizures and measured the corresponding mean ΔV m or averaged evoked firing rate.
The neuronal transfer function, i.e. the rate‐coded output firing as a function of excitatory inputs of increasing magnitude (Silver, 2010), was quantified in control and GAERS neurons during baseline period as the relation linking the intensity of intracellularly injected currents to the evoked firing frequency (F–I relationships) (Mahon & Charpier, 2012; Altwegg‐Boussac et al. 2014). The mean firing rate (<F>) was measured in response to depolarizing current pulses of increasing intensity (200 ms, 0.1–1.5 nA) delivered with an inter‐stimulus interval of at least 1 s. Each current intensity was applied 20–25 times and the corresponding firing responses were averaged. Linear regressions were applied to F–I curves to determine the threshold current for AP generation (I th), extrapolated as the x‐intercept of the linear fit (see Fig. 3 B), and the neuronal gain (γ), defined as the slope of the F–I curve (Mahon & Charpier, 2012; Altwegg‐Boussac et al. 2014). Current‐evoked firing was limited to frequency less than 50 Hz in order to avoid possible activity‐dependent intrinsic plasticity (Mahon & Charpier, 2012) that may alter cortical neurons excitability during the course of the recordings. The temporal dynamics and trial‐to‐trial variability of current‐evoked firing responses were compared in GAERS and non‐epileptic neurons. First, to estimate the temporal variability of firing pattern during a train of evoked activity, we used the CV2 method (see above). The mean CV2 was calculated for a series of stimuli (n = 20–25 current injections) that generated approximately the same mean firing rate (∼30 Hz) in the two cell populations. Second, to assess the trial‐to‐trial variability in the number of current‐evoked APs, we applied the Fano factor (FF) method (Teich, 1992; Altwegg‐Boussac et al. 2014). FF was calculated as the variance of the number of evoked APs, for a given intensity, divided by the corresponding mean number of spikes generated during the series of stimulations. FF and CV2 measurements were made on the same current‐evoked responses.
Figure 3. GAERS S1 cortex neurons exhibit in between seizures an increased sensibility to weak excitatory inputs .
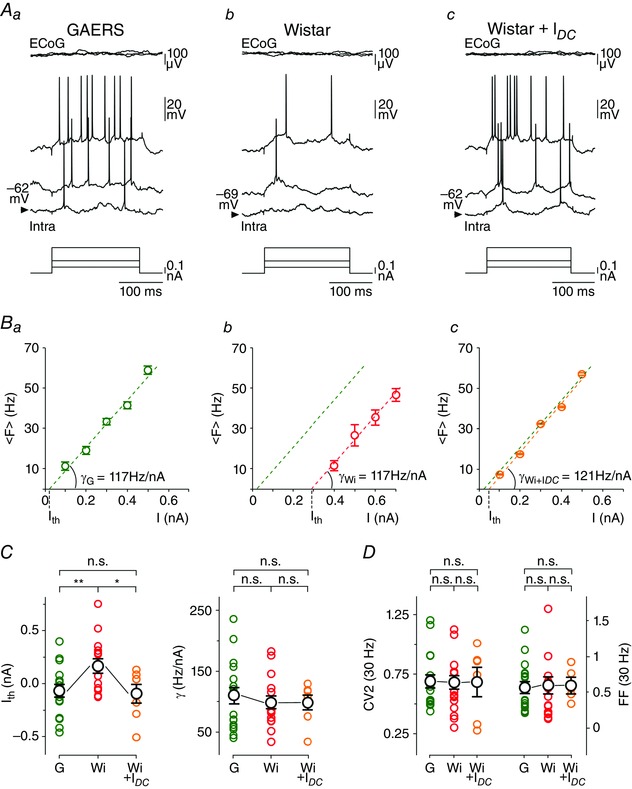
Aa–c, current‐evoked (bottom traces) voltage responses (Intra) recorded from S1 cortex neurons in GAERS during inter‐ictal period (Aa) and in a control Wistar rat at V m (Ab) and during DC depolarization (Ac). Superimposed top traces are the simultaneous recorded ECoG activity. Ba–c, corresponding F–I curves from the neurons illustrated in A. Each symbol corresponds to the mean (± SEM) firing rate from 20–25 successive trials. Values of the neuronal gain (γ) and threshold current (I th) calculated in GAERS (G) and Wistar rat neurons in control (Wi) and during DC depolarization (Wi + I DC) are indicated. The green dashed line in Ba–c is the linear fit of the F–I curve computed in the GAERS neuron. C, population data of neuronal sensitivity (I th) and gain (γ) for neurons recorded in GAERS (G, green circles), control Wistar rats in control condition (Wi, red circles) and during DC depolarization (Wi + I DC, orange circles). D, pooled data comparing Fano factor (FF) values of spike counts and mean CV2 of ISIs between GAERS (G) and Wistar rat neurons at V m (Wi) and during artificial depolarization (Wi + I DC). Measurements were made on current pulses evoking on average a firing rate of 30 Hz. * P < 0.05; ** P < 0.01; n.s., non‐significant.
Statistics
Differences between groups were assessed with Student's t tests (paired or unpaired), the non‐parametric Wilcoxon signed rank test and Mann–Whitney rank sum test or Fisher's exact test when appropriate (SigmaStat version 3.5, Systat Software Inc., Erkrath, Germany). Differences were considered statistically significant if P < 0.05. Numerical values are presented as the mean ± SEM.
Results
GAERS cortical neurons are hyperactive in between seizures and display anomalous membrane rectification
In the first part of this study, we examined how the pro‐ictogenic properties of GAERS cortical neurons shape their integrative properties and transfer function during baseline periods. We thus compared the electrical membrane properties of cortical neurons initiating absence seizures in the GAERS to those measured from ‘homologous’ neurons (same neuronal type and cortical location) in non‐epileptic rats (Fig. 1 A and B).
Figure 1. Comparison of basic morpho‐functional properties of GAERS S1 cortex neurons with their counterparts in Wistar control rats .
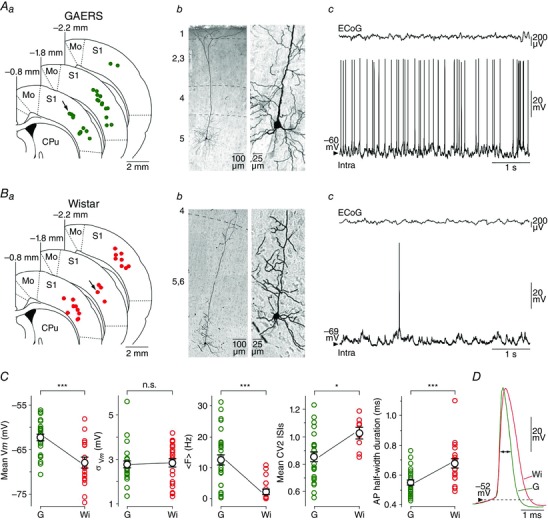
Aa and Ba, superimposed slice drawings, derived from the stereotaxic rat brain atlas (Paxinos & Watson, 1986), at the indicated distances from the bregma. Green and red circles indicate the location of intracellularly recorded neurons from the S1 cortex of GAERS (Aa) and control Wistar rats (Ba), respectively. CPu, caudate‐putamen; Mo, motor cortex; S1, primary somatosensory cortex. Ab and Bb, photomicrographs of neurobiotin‐filled neurons in the S1 cortex of GAERS (Ab) and control Wistar rats (Bb). The location of these neurons is indicated by the arrows in Aa and Ba. The numbers at the left correspond to the different cortical layers. As shown by the expansion of the somatodendritic regions (right part), neurons had the typical morphology of pyramidal cells. Ac and Bc, simultaneous monitoring of S1 cortex spontaneous ECoG waves (top records) and corresponding intracellular activities (bottom records) recorded from the neurons shown in Ab and Bb. C, population data of V m, magnitude fluctuation of V m (σV m), mean spontaneous firing rate (<F>), variability of ISIs (CV2 ISI) and half‐duration of APs for GAERS (G) and control Wistar rat (Wi) neurons. Here and in similar figure panels, each circle represents an individual neuron and the larger black circles represent mean values ± SEM. D, DC superimposition of average (n > 9) APs recorded from the GAERS (green trace) and control (red trace) neurons illustrated in Ab and Ac and Bb and Bc. * P < 0.05; *** P < 0.001; n.s., non‐significant.
We made in vivo intracellular recordings of S1 cortex neurons from GAERS (n = 27 neurons from 16 GAERS) and non‐epileptic control Wistar rats (n = 21 neurons from 14 Wistar rats), simultaneously with the corresponding surface ECoG (Fig. 1 Ac and Bc). Stereotaxic coordinates indicated that recorded cortical neurons, in both GAERS and control rats, were localized within the same region of S1 cortex and similarly distributed in the deep layers, from the superficial part of layer 5 to layer 6 (Fig. 1 Aa and Ba). This was confirmed by subsequent histological analysis of intracellularly labelled neurons in GAERS (n = 9 neurons from 7 GAERS) and control rats (n = 4 neurons from 4 Wistar rats) (Fig. 1 Ab and Bb, left panels), which exhibited the typical morphology of pyramidal neurons (Feldman, 1984), including a triangular cell body, a prominent apical dendrite extending vertically toward the pial surface and basal dendrites radiating out from the base of the soma (Fig. 1 Ab and Bb, right panels). Basic morphometric analysis of the labelled neurons did not reveal evident differences in the somatodendritic arborizations of GAERS and control neurons. Consistent with their morphological identification as deep‐layer pyramidal cells, recorded neurons displayed current‐evoked firing patterns characteristic of regular spiking or intrinsic bursting (IB) cells (Steriade, 2004). The relative proportion of IB neurons in the S1 cortex of GAERS (44.4 %) was found to be larger compared to control animals (23.8 %), but not significantly (P > 0.2) (see Figs 6 Aa and 8 Aa). Because we did not find any specific relation between the neuronal type and the membrane excitability parameters, numerical values obtained from these two cell populations were pooled for both GAERS and control rats.
Figure 6. The excitability of cortical neurons is reduced during the W‐component .
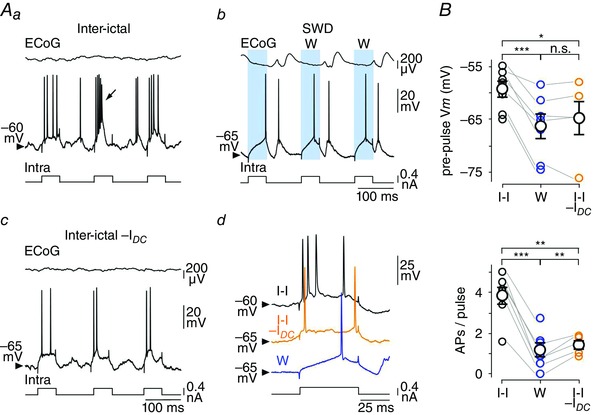
A, voltage responses of a S1 cortical neuron to repetitive injections of depolarizing current pulses (+0.4 nA), in between seizures (Aa), during SWDs (Ab) and inter‐ictal epochs associated with a DC hyperpolarization (–0.2 nA) (Ac). Suprathreshold responses obtained during the W‐component are shaded blue in Ab. The arrow indicates the occurrence of an intrinsic burst in the stimulated neuron. In each panel, the top trace is the corresponding ECoG activity. The individual responses shown in Ad are representative examples obtained during inter‐ictal (black trace) and wave (blue trace) periods, and during the course of inter‐ictal epochs together with DC injection (orange trace). Calibrations in Ab apply also to Aa. B, population data indicating the V m values just before the current pulse (top) during inter‐ictal epochs (I‐I), the W‐component (W) and inter‐ictal period coupled with DC hyperpolarization (I‐I −I DC) (top). The bottom graph provides the corresponding number of current‐evoked APs in the three conditions. * P < 0.05; ** P < 0.01; *** P < 0.001; n.s., non‐significant.
Figure 8. The membrane excitability of cortical neurons is markedly reduced during the S‐component of the SWD .
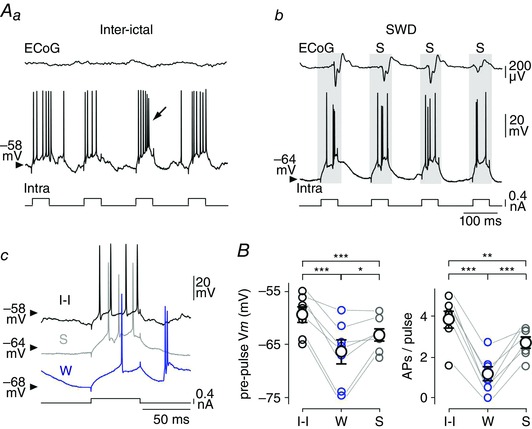
A, firing responses of a S1 cortex neuron to iterative injections of depolarizing current pulses (+0.4 nA) in between seizure activity (Aa, Inter‐ictal) and in conjunction with the ECoG spikes during a SWD (Ab, grey shading). Typical cell responses obtained during the inter‐ictal period (I‐I), the S‐component (S) and the W‐component (W) are shown in Ac. The intracellular stimulation could generate an intrinsic burst of APs (arrow in Aa). Calibrations in Ab apply also to Aa. B, pooled data showing the pre‐pulse V m values (left) and the corresponding number of APs generated by the current pulses (right). * P < 0.05; ** P < 0.01; *** P < 0.001.
As previously described (Polack et al. 2007; Constantinople & Bruno, 2011), the background ECoG activity recorded from S1 cortex in GAERS between seizures (Fig. 1 Ac, top record) and in control rats (Fig. 1 Bc, top record) under fentanyl was composed of low amplitude, apparently desynchronized, waves resembling those observed during wakefulness. The intracellular counterpart, recorded from GAERS and non‐epileptic rats, was characterized by irregular and small‐amplitude voltage fluctuations representing a mixture of excitatory and inhibitory synaptic events (Fig. 1 Ac and Bc, bottom records). The corresponding V m values were unimodally distributed in both cell populations (Fig. 4 Ba), with a mean V m significantly more depolarized in GAERS neurons (GAERS, V m = −62.2 ± 0.7 mV, n = 27 neurons from 16 rats; control Wistar rats, V m = −68.1 ± 1.1 mV, n = 21 neurons from 12 rats; P < 0.001) (Fig. 1 C). Despite the relative hyperpolarization of control neurons, expected to increase the driving force of excitatory synaptic currents, the magnitude of V m fluctuations in the two neuronal groups was similar (GAERS, σVm = 2.8 ± 0.2 mV, n = 27 neurons from 16 rats; control Wistar rats, σVm = 2.9 ± 0.2 mV, n = 21 neurons from 12 rats; P > 0.7).
Figure 4. Spontaneous activity of GAERS S1 cortex neurons in between and during seizures .
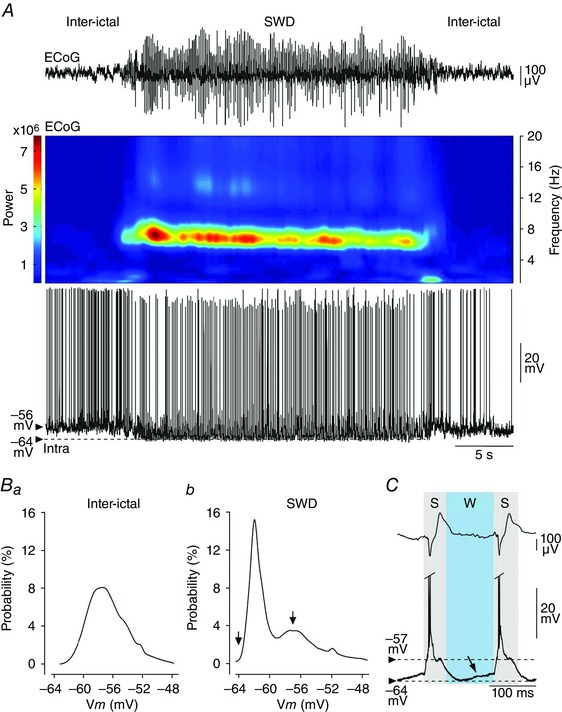
A, simultaneous recordings (40 s duration) of S1 cortex ECoG waves (top) and intracellular activity of a layer 5 pyramidal neuron (bottom), during inter‐ictal and seizure (SWD) periods. The corresponding time–frequency analysis of the ECoG signal (energy density for the 0–20 Hz band) is depicted. Note the sustained hyperpolarization of the neuron (dashed line) throughout the SWD. B, probability density of V m (bin: 0.5 mV) computed from the neuron illustrated in A, inter‐seizure epochs (Ba, 30 s of spontaneous activity) and during the SWD (Bb). The left arrow in Bb indicates the maximal level of polarization reached during the seizure‐associated envelope, and the right one corresponds to the mean subthreshold potential during the rhythmic depolarizations concomitant with the ECoG spikes. C, segment of the paired recordings shown in A during the ictal period. The spike (S) and wave (W) components of the SWD and their intracellular correlates are delimitated by the grey and blue boxes, respectively. The arrow shows the ramp‐like depolarization preceding the intracellular paroxysmal excitation. APs are truncated.
Consistent with their relative membrane depolarization, GAERS ictogenic neurons had a higher mean spontaneous firing rate (GAERS, <F> = 12.5 ± 1.8 Hz, n = 27 neurons from 16 rats) compared to control homologous cells (<F> = 2.3 ± 0.8 Hz, n = 21 neurons from 12 rats; P < 0.001) (Fig. 1 Ac, Bc and C). The spontaneous firing pattern of GAERS cortical neurons was also more regular, as attested by the lower values of ISI CV2 (GAERS, ISI CV2 = 0.85 ± 0.04, n = 24 neurons from 14 rats; control Wistar rats, ISI CV2 = 1.03 ± 0.05, n = 8 neurons from 8 rats; P < 0.05) (Fig. 1 C). These altered firing properties in ictogenic neurons were associated with a reduced duration of individual APs (Fig. 1 D) measured at half‐amplitude (GAERS, 0.55 ± 0.02 ms, n = 27 neurons from 16 rats; control Wistar rats, 0.68 ± 0.03 ms, n = 19 neurons from 11 rats; P < 0.001) (Fig. 1 C). The other AP properties, including voltage threshold (GAERS, −53.5 ± 0.7 mV, n = 27 neurons from 16 rats; control Wistar rats, −52.9 ± 1.2 mV, n = 19 neurons from 11 rats) and full amplitude (GAERS, 59.0 ± 1.2 mV, n = 27 neurons from 16 rats; control Wistar rats, 58.1 ± 1.7 mV, n = 19 neurons from 11 rats) did not significantly differ between the two cell groups (P > 0.6 for each parameter).
The increased level of membrane polarization and firing rate of ictogenic neurons likely result from abnormal intrinsic and/or synaptic properties that could alter their integrative properties. We thus characterized the input–output relationships of GAERS and control neurons and compared their R m and membrane rectification values. V–I relationships were quantified by measuring V m changes (ΔV m) in response to intracellular hyperpolarizing current pulses of increasing intensity (Fig. 2 Aa and Ba). When measurements were made at the end of the membrane capacitance charge (Fig. 2 Aa and Ba, filled circles), data points were best fitted by linear regression (Fig. 2 Ab and Bb). R m values, calculated as the slopes of V–I relations, were similar in the two cell populations (GAERS, R m = 25.2 ± 1.9 MΩ, n = 18 neurons from 10 rats; control Wistar rats, R m = 28.3 ± 1.3 MΩ, n = 18 neurons from 8 rats; P > 0.1) (Fig. 2 C). In all GAERS neurons, injection of negative current pulses of high magnitude (−0.8 nA) resulted in a depolarizing ‘sag’ of V m (Fig. 2 Aa), systematically followed by a post‐anodal break depolarization that could generate one or multiple APs (Fig. 2 Aa, arrow). This sequence of cell response was indicative of the presence of the hyperpolarization‐activated inward cationic current I h (Pape, 1996). The comparison of sag ratio (SR) values in the two cell groups (GAERS, SR = 0.84 ± 0.01, n = 18 neurons from 10 rats; control Wistar rats, SR = 0.92 ± 0.02, n = 18 neurons from 8 rats; P < 0.001) indicated that the hyperpolarization‐activated anomalous rectification was more prominent on average in seizure‐initiating neurons (Fig. 2 C).
These findings confirm and extend the specific electrophysiological alterations found in GAERS S1 cortex deep‐layer pyramidal neurons as compared to their control counterparts. In addition to the previously reported sustained depolarization and hyperactivity, we now uncovered that seizure‐initiating neurons display shorter APs, a more pronounced inward membrane rectification and a higher propensity to generate intrinsic bursts of APs.
Transfer function of GAERS cortical neurons is altered during inter‐ictal periods
To investigate whether the suprathreshold input–output operations performed by seizure‐initiating neurons were modified during inter‐ictal periods, we first compared F–I relationships in GAERS and control neurons. In both cell populations, the mean firing rate increased linearly with stimulus magnitude over the range of tested intensities (Fig. 3 A and B). The threshold current values (I th), extrapolated from the x‐intercept of the linear fits (Fig. 3 B), showed a high cell‐to‐cell variability within both groups (GAERS, from −0.5 to 0.4 nA, n = 18 neurons; control Wistar rats, from −0.3 to 0.8 nA, n = 18 neurons) and were significantly decreased on average in the epileptic animals (GAERS, I th = −0.07 ± 0.05 nA, n = 18 neurons from 10 rats; control Wistar rats, I th = 0.17 ± 0.07 nA, n = 18 neurons from 11 rats; P < 0.01) (Fig. 3 C). The neuronal gain, evaluated by the slope of the F–I relation, was not different in GAERS and control neurons (GAERS, γ = 110.0 ± 13.4 Hz nA–1, n = 18 neurons from 10 rats; control Wistar rats, γ = 100.0 ± 10.7 Hz nA–1, n = 18 neurons from 11 rats, P > 0.5) (Fig. 3 Ba and b, and C).
We next examined if the potentiation of current‐induced firing responses in GAERS deep‐layer neurons was accompanied by modifications in the moment‐to‐moment variability in the rate and temporal pattern of APs when neurons are submitted to repeated excitatory stimuli (Ermentrout et al. 2008; Civillico & Contreras, 2012; Altwegg‐Boussac et al. 2014). We thus compared the variability of current‐induced AP responses in S1 cortical neurons of epileptic and normal rats during baseline periods. Measurements were made for stimulus intensities that generated a similar mean firing rate (∼30 Hz) in the two cell groups to allow a reliable comparison between GAERS and control neurons. The reliability of the firing rate during repeated trials, assessed by measuring the FF, was quite variable from cell to cell (GAERS, from 0.2 to 1.4, n = 17 neurons; control Wistar rats, from 0.1 to 1.8, n = 17 neurons), but did not differ on average between GAERS and control neurons (GAERS, FF = 0.56 ± 0.09, n = 17 neurons from 10 rats; control Wistar rats, FF = 0.61 ± 0.12, n = 17 neurons from 11 rats; P > 0.8) (Fig. 3 D). The temporal regularity of firing patterns in the two groups of cells was assessed by measuring ISI variability across trials. Again, average values of ISI CV2 were similar in neurons recorded in epileptic and non‐epileptic animals (GAERS, CV2 = 0.69 ± 0.06, n = 17 neurons from 10 rats; control Wistar rats, CV2 = 0.68 ± 0.06, n = 17 neurons from 11 rats; P > 0.9), despite a large inter‐cell variability (GAERS, from 0.4 to 1.2, n = 17 neurons from 10 rats; control Wistar rats, from 0.31 to 1.1, n = 17 neurons from 11 rats) (Fig. 3 D). Values of FF and ISI CV2 calculated in this study closely match those previously measured in S1 cortex layer 5 pyramidal neurons from normal rats recorded under fentanyl (Altwegg‐Boussac et al. 2014).
As for the whole population of cells, GAERS neurons included in this series of experiments had a V m significantly more depolarized compared to the corresponding control group (GAERS, V m = –62.5 ± 0.9 mV, n = 18 neurons from 10 rats; control Wistar rats, V m = –68.0 ± 1.2 mV, n = 18 neurons from 11 rats, P < 0.001). We thus examined the impact of the level of membrane polarization on the lateral translation of the transfer function by manipulating the V m of control neurons. Before applying F–I protocols, DC injection (+0.2–0.5 nA) in S1 cortical neurons of non‐epileptic rats was adjusted to bring the cells at a V m (–60.1 ± 1.8 mV, n = 7 neurons from 7 rats) matching that of GAERS cells (P > 0.1) (Fig. 3 Ac). This V m manipulation of control neurons led to a significant reduction of I th values (control Wistar rats, I th = 0.17 ± 0.07 nA, n = 18 neurons from 11 rats; control Wistar rats + I DC, I th = –0.09 ± 0.10 nA, n = 7 neurons from 7 rats; P < 0.05), which became similar to those measured in GAERS neurons (P > 0.8) (Fig. 3 Bc and C). In contrast, the neuronal gain was not affected by the V m depolarization (γ = 98.9 ± 13.5 Hz nA–1, n = 7 neurons from 7 rats) and remained comparable to that measured in GAERS neurons (P > 0.6 for both comparisons) (Fig. 3 Bc and C). Consistent with the lack of differences in the regularity of AP number and temporal precision between cortical neurons from GAERS and non‐epileptic rats at V m, DC depolarization of control neurons did not lead to modifications in FF values (control Wistar rats, FF = 0.61 ± 0.12, n = 17 neurons from 11 rats; control Wistar rats + I DC, FF = 0.62 ± 0.09, n = 6 neurons from 6 rats, P > 0.9) or CV2 of ISIs (control Wistar rats, CV2 = 0.68 ± 0.06, n = 17 neurons from 11 rats; control Wistar rats + I DC, CV2 = 0.68 ± 0.14, n = 6 neurons from 6 rats, P > 0.9) (Fig. 3 D).
Differential modulation of intrinsic excitability in GAERS cortical neurons during the ‘spike’ and ‘wave’ components of the seizure
Our findings showed that S1 cortex deep‐layer pyramidal neurons from GAERS displayed in between seizures an increased sensitivity to weak excitatory inputs, which probably resulted from their abnormal membrane depolarization. We also found that these cells exhibited a prominent depolarizing sag potential activated during hyperpolarization that could participate in their oscillatory behaviour during SWDs. We next examined how seizure activity impacts their integrative properties and intrinsic excitability, differentiating the effect of the two main waveforms composing the paroxysms, i.e. the ‘spike’ (S) and the ‘wave’ (W) components, which are associated with two opposed profiles of intracellular activity.
At the occurrence of seizures, the inter‐ictal desynchronized ECoG was abruptly replaced by SWDs composed of successive spike‐and‐wave complexes. The frequency of spike‐and‐wave complexes, which peaked at 8–10 Hz during the first 1–2 s of the seizures, slowed down and stabilized at 7–8 Hz (mean = 7.5 ± 0.1 Hz, n = 288 SWDs from 14 GAERS) for the rest of the epileptic episode (Fig. 4 A, top panels). The mean duration of SWDs was 20.6 ± 4.1 s, ranging from 5 to 47 s (n = 288 SWDs from 14 GAERS). Seizure activity was associated in cortical neurons with rhythmic synaptic depolarizations superimposed on a sustained hyperpolarizing envelope (–8.2 ± 0.8 mV, n = 17 neurons from 13 GAERS), which brought the cells to a membrane potential (–69.3 ± 1.0 mV, n = 17 neurons from 13 GAERS) significantly more polarized compared to inter‐ictal periods (P < 0.001) (Fig. 4 A, bottom record). The unimodal distribution of V m values outside seizures was consequently replaced, during ictal activity, by a bimodal pattern (Fig. 4 Ba and b) reflecting the alternation of the two distinct intracellular profiles respectively correlated with the S‐ and W‐components of the ECoG (Fig. 4 C). During the S‐components, cortical neurons displayed sharp and large amplitude synaptic membrane depolarizations usually suprathreshold for AP firing (Fig. 4 A and C, grey boxes). In contrast, W‐elements correlated with periods of hyperpolarization, a lack of AP firing and network quiescence in cortical neurons (Fig. 4 C, blue box). Transitions from W‐ to S‐components were accompanied by a slow depolarizing ramp in most (15 out of 17) recorded neurons (Fig. 4 C, arrow; see also Fig. 3C in Polack et al. 2007).
Paradoxical effect of the Wave component
To investigate the consequence of the W‐component on R m of S1 cortex neurons, we applied repeated hyperpolarizing current pulses of short duration (10 ms in duration, −0.8 nA, every 50 ms, n = 7 neurons from 5 GAERS) in between and during seizures (Fig. 5 Aa and Ab). Although the voltage responses did not reach steady‐state during the course of the current injection due to the membrane time constant of cortical neurons, measurements of voltage deflections (ΔV m) provided an indirect estimation of R m changes. Cell responses were considered as occurring during the W‐component (‘W‐responses’) when stimuli occurred between 50 and 70 ms after the peak of the ECoG spikes (Fig. 5 Ab, bottom panel). This temporal window corresponded to a period of membrane hyperpolarization devoid of synaptic events, with no overlap with the preceding or following S‐dependent depolarizing shift (Fig. 5 Ab). The mean V m, measured just before (3 ms) the beginning of current pulses (W: pre‐pulse V m = –65.8 ± 1.6 mV, n = 3037 W‐components from 51 SWDs, n = 7 neurons from 5 GAERS), was close to that reached during the seizure‐related hyperpolarizing envelope (V m envelope = –67 ± 1.9 mV, n = 51 SWDs from 7 neurons from 5 GAERS; P > 0.1) and was significantly more negative than the mean pre‐pulse V m during inter‐ictal (I‐I) periods (I‐I: pre‐pulse V m = –60.5 ± 1.4 mV, n = 7 neurons from 5 GAERS; P < 0.001) (Fig. 5 Aa, b and d, and B). The mean voltage drop induced by the negative current pulses during the W‐components (W: ΔV m = –11.9 ± 0.7 mV, n = 3037 W‐components from 51 SWDs, n = 7 neurons from 5 GAERS) was significantly larger compared to the corresponding inter‐ictal periods (I‐I: ΔV m = –11.0 ± 0.9 mV, n = 7 neurons from 5 GAERS; P < 0.05) (Fig. 5 Ad and B). This increase in R m could result from a decrease in synaptic conductance and/or a modulation of voltage‐dependent intrinsic ionic channels during the sustained hyperpolarization associated with the seizure. To test the latter possibility, we hyperpolarized neurons during inter‐ictal periods (I‐I −I DC: pre‐pulse V m = –65.4 ± 1.9 mV, n = 6 neurons from 4 GAERS; I DC = –0.1 to –0.4 nA), to match the V m reached during the W‐component (P > 0.5), and measured the voltage deflections induced by the same current steps (Fig. 5 Ac). At hyperpolarized V m, the mean current‐induced voltage drop during inter‐ictal periods was comparable to that measured in absence of DC injection (I‐I −I DC: ΔV m = –10.9 ± 0.9 mV, n = 6 neurons from 4 GAERS; P > 0.2) and remained smaller than that measured during the ECoG wave (P < 0.05) (Fig. 5 Ad,B).
Figure 5. The W‐component is associated with an increase in the membrane input resistance of cortical neurons .
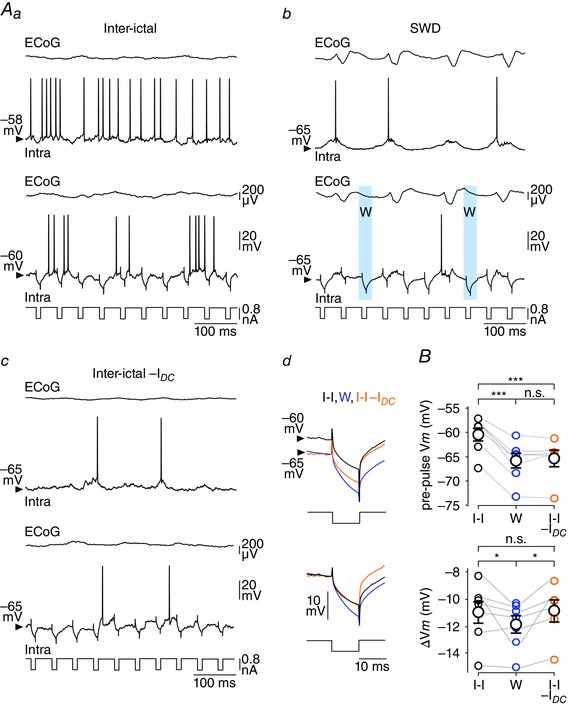
A, coupled recordings of S1 cortex ECoG and intracellular activities in between SWDs (Aa), during seizure activity (Ab) and during an inter‐ictal period associated with DC hyperpolarization (I DC = −0.3 nA) to approach the minimal V m reached during the seizure. Differences in membrane conductance between these three conditions were assessed from voltage responses (bottom records) to negative (−0.8 nA) current pulse injections (lowest traces). Examples of cell responses obtained during the W‐component are shaded blue in Ab. Ad, DC superimposition of the average current‐evoked voltage changes obtained during the inter‐ictal period (I‐I, black trace, n = 1250 responses), the W‐component (W, blue trace, n = 152 responses from 12 SWDs) and DC hyperpolarization in between seizures (I‐I –I DC, orange traces, n = 224 responses). The bottom panel illustrates the same three records superimposed by matching the V m before the start of the pulse. B, population data showing the mean V m values calculated just before the onset of current injection, during inter‐ictal period (I‐I), the W‐component (W) and DC hyperpolarization during inter‐ictal period (I‐I–I DC) (top). The bottom graph shows the corresponding values of current‐induced voltage deflections (ΔV m). Here and in the following population graphs, the grey lines connect measurements made from the same neurons. * P < 0.05; *** P < 0.001; n.s., non‐significant.
We next investigated the impact of the W‐element on cell excitability, i.e. the intrinsic ability of cortical neurons to generate APs in response to a given excitatory input. Repeated depolarizing current pulses were applied (50 ms in duration, +0.4 nA, every 150 ms) and the firing responses obtained during the W‐components and the corresponding inter‐ictal periods were compared (Fig. 6 Aa and b). Again, the W‐associated pre‐pulse V m (W: pre‐pulse V m = –66.3 ± 2.5 mV, n = 763 W‐components from 26 SWDs, n = 7 neurons from 4 GAERS) was significantly hyperpolarized compared to inter‐ictal epochs (I‐I: pre‐pulse V m = –59.3 ± 1.6 mV, n = 7 neurons from 4 GAERS; P < 0.001) (Fig. 6 Aa and b, and B) and similar to the V m reached during the hyperpolarizing envelope (V m envelope = –68.4 ± 2.1 mV, n = 7 neurons from 4 GAERS; P > 0.7). The number of current‐evoked APs was markedly diminished during the W‐component compared to the corresponding inter‐ictal periods (W: 1.2 ± 0.4 APs/pulse, n = 763 W‐components from 26 SWDs, n = 7 neurons versus I‐I: 3.8 ± 0.5 APs/pulse, n = 7 neurons from 4 GAERS; P < 0.001) (Fig. 6 Aa and b, and B). The difference in firing responses could result from the relative membrane depolarization of S1 cortical neurons during inter‐ictal periods and their elevated spontaneous firing rate (<F> = 15.2 ± 3.9 Hz, n = 7 neurons from 4 GAERS) as compared to W‐related periods (see Fig. 5 Aa versus Ab). To assess the impact of the level of membrane polarization, we made DC injection during the inter‐seizure periods to bring neurons at a V m (I‐I −I DC: pre‐pulse V m = –64.8 ± 3.5 mV, n = 5 neurons from 3 GAERS; I DC = −0.1 to −0.3 nA) similar to that associated with the W‐event (P > 0.7), and applied the same train of depolarizing current pulses (Fig. 6 Aa–c and B). This resulted in a considerable reduction in the number of current‐induced APs (1.4 ± 0.2 APs/pulse, n = 5 neurons from 3 GAERS) compared to inter‐ictal epochs in absence of DC hyperpolarization (P < 0.01), which, however, remained above that calculated during the W‐periods (P < 0.01) (Fig. 6 A and B).
These data demonstrate a paradoxical effect of the W‐component on information processing by cortical neurons, combining an increase in R m, expected to amplify cell responsiveness, with a decrease in firing responses to depolarizing inputs.
Inhibitory effect of the spike component
The S‐component in the ECoG during seizure was reflected in S1 cortex neurons by an oscillatory‐like depolarization, lasting 50–60 ms and giving rise to single or multiple discharge of APs (Fig. 4 C). We first examined the effect of this paroxysmal activity on R m of GAERS neurons by applying iterative intracellular injections of brief hyperpolarizing current pulses (10 ms in duration, −0.8 nA, every 50 ms, n = 7 neurons from 5 GAERS) (Fig. 7 Aa and b). The current‐induced ΔV m concomitant with the ECoG spikes were compared to those obtained during the corresponding W‐components and inter‐ictal periods (Fig. 7 Ac). The mean V m just preceding the current pulses applied during the S‐component (S: pre‐pulse V m = –60.5 ± 1.2 mV, n = 1310 ECoG spikes from 51 SWDs, n = 7 neurons from 5 GAERS) was more depolarized than the pre‐pulse V m during W‐periods (W: pre‐pulse V m = −65.8 ± 1.6 mV, 3037 W‐components, 51 SWDs, n = 7 neurons from 5 GAERS; P < 0.001) and comparable to that of inter‐ictal periods (I‐I: pre‐pulse V m = −60.5 ± 1.4 mV, n = 7 neurons from 5 GAERS; P > 0.8) (Fig. 7 Ac and B). The current‐evoked voltage changes during the S‐components (S: ΔV m = –4.4 ± 0.5 mV, n = 1310 ECoG spikes from 51 SWDs, n = 7 neurons from 5 GAERS) were considerably reduced compared to the corresponding W‐components and inter‐ictal epochs (P < 0.001 for each paired comparison) (Fig. 7 Ac and B).
Figure 7. The S‐component of the seizure results in a powerful shunting inhibition of cortical neurons .
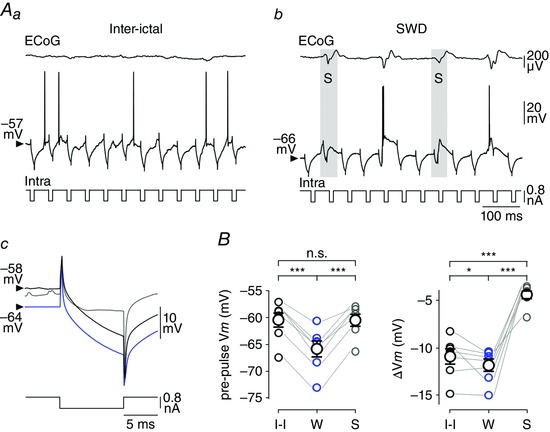
A, voltage responses of a cortical neuron to hyperpolarizing current pulses (–0.8 nA) during inter‐ictal period (Aa) and seizure activity (Ab). The cell responses occurring in phase with ECoG spikes are shaded grey. The corresponding mean current‐induced voltage drops obtained during the inter‐ictal period (I‐I, black trace, n = 373 responses), the W‐component (W, blue trace, n = 383 responses from 4 SWDs) and the ECoG spikes (S, grey trace, n = 111 responses from 4 SWDs) are shown in Ac. Calibrations in Ab apply also to Aa. B, population data providing the mean pre‐pulse V m values (left) and the current‐induced voltage changes (right) during inter‐ictal epochs (I‐I), the W‐ and S‐components. * P < 0.05; *** P < 0.001; n.s., non‐significant.
Despite the membrane depolarization concomitant with the S‐component, the large decrease in R m could negatively impact cortical excitability. To assess the overall change in neuronal excitability during the S‐component, we injected a series of depolarizing current pulses (50 ms in duration, +0.4 nA, every 150 ms, n = 7 neurons from 4 GAERS) and compared, in the same cells, the firing responses elicited during the S‐component, the W‐component and the corresponding inter‐ictal periods (Fig. 8 A). Only cell responses overlapping with the synaptic depolarizations associated with the S‐component were considered as ‘S‐responses’. In this set of experiments, the pre‐pulse V m during the S‐component (S: pre‐pulse V m = –63.2 ± 1.3 mV, n = 1135 ECoG spikes from 26 SWDs, n = 7 neurons from 4 GAERS) was more depolarized than during the ECoG wave (W: pre‐pulse V m = –66.3 ± 2.5 mV, n = 763 ECoG waves from 26 SWDs, n = 7 neurons from 4 GAERS; P < 0.05) and slightly hyperpolarized relative to the corresponding inter‐ictal periods (I‐I: pre‐pulse V m = –59.3 ± 1.6 mV, n = 7 neurons from 4 GAERS; P < 0.001) (Fig. 8 Ac and B). In each tested neuron, the current‐evoked firing was noticeably augmented during the S‐component (S: 2.7 ± 0.3 APs/pulse, n = 1135 ECoG spikes from 26 SWDs, n = 7 neurons from 4 GAERS) compared to the W‐component (W: 1.2 ± 0.4 APs/pulse, n = 763 ECoG waves from 26 SWDs, n = 7 neurons from 4 GAERS; P < 0.001). However, it remained significantly lower than that calculated during the corresponding inter‐ictal epochs (I‐I: 3.8 ± 0.5 APs/pulse, n = 7 neurons from 4 GAERS; P < 0.01). It is worth noting that the current‐evoked firing rates during the ECoG spikes and inter‐ictal periods are likely to be slightly overestimated due to the concurrent spontaneous firing during these two phases of activity.
Altogether, these findings indicate a moment‐to‐moment change in the responsiveness of GAERS cortical neurons during SWDs, with a relative increased excitability during the S‐components, despite a dramatic increase in membrane conductance, compared to the wave‐sequences. The excitability of cortical cells during the depolarizing element of the seizure remained however lower than during inter‐ictal periods.
Discussion
The aim of the present study was to determine how the specific electrophysiological features of GAERS ictogenic neurons impact their integrative properties and input–output operations during inter‐ictal periods and during the different components of SWDs. In addition, to confirm their sustained depolarization and elevated spontaneous firing in between seizures, we demonstrated that ictogenic neurons display a prominent inward rectification and post‐inhibitory rebound of excitation, probably caused by the activation of I h. Analysis of F‐I relations indicated that their sensitivity to weak excitatory inputs is increased compared to homologous neurons from non‐epileptic rats, without changes in the neuronal gain and trial‐to‐trial variability of firing responses. We also showed that the current‐evoked firing of ictogenic neurons is reduced during the W‐component of the seizure compared to inter‐ictal epochs despite an augmentation of their R m. Conversely, while R m of cortical cells is considerably attenuated during the S‐component, the level of intrinsic excitability is enhanced relative to W‐phases and stands just below that reached during inter‐ictal periods.
Increased activity and sensitivity of ictogenic neurons in between seizures
A consistent and specific feature of GAERS ictogenic neurons during inter‐ictal periods is their depolarized V m coupled with a sustained and regular spontaneous firing (Polack et al. 2007, 2009; Polack & Charpier, 2009; Chipaux et al. 2011, 2013; present study). This could result from an increase in the background depolarizing synaptic drive, as suggested by the synaptic alterations found in the S1 cortex of WAG/Rij rats, another genetic model of absence epilepsy (Depaulis & van Luijtelaar, 2006), combining an increase in NMDA receptor‐mediated activity (Luhmann et al. 1995; D'antuono et al. 2006), an augmented expression of mGlu2/3 receptors (Ngomba et al. 2005) and a decrease in the efficacy of GABAergic synaptic transmission (D'antuono et al. 2006; Inaba et al. 2009). Alternatively, the persistent membrane depolarization could be caused by an upregulation of voltage‐gated sodium channels as previously reported (Klein et al. 2004). This hypothesis is supported by the effect of ethosuximide, an anti‐absence medicine that attenuates the non‐inactivating sodium currents (Crunelli & Leresche, 2002) and leads to a recovery of physiological values of V m and firing rate in ictogenic neurons (Polack & Charpier, 2009).
Our findings indicate that the depolarized V m of ictogenic neurons is responsible for a modification in their input–output function during inter‐ictal periods. Indeed, F–I relations computed from GAERS neurons displayed a significant leftward shift without changes in neuronal gain, which could be reproduced by artificial depolarization in control neurons. This dependence of neuronal sensitivity on the level of membrane polarization is consistent with a recent study showing a transition to the left of F–I relations in S1 cortex neurons from normal rats following DC depolarization, in the presence or absence of background synaptic activity (Altwegg‐Boussac et al. 2014). Similar sustained membrane depolarization has not been observed in other cortical and thalamic neurons (Slaght et al. 2004; Paz et al. 2007; Polack et al. 2009), suggesting a distinctive, probably pro‐ictogenic, feature of deep‐layer pyramidal neurons of GAERS S1 cortex. The narrow difference between V m and AP voltage threshold in ictogenic neurons during inter‐ictal periods implies that weak and uncorrelated excitatory inputs are likely to substantially influence their output firing and augment their tonic and bursting activities (Parri & Crunelli, 1998), which are critically involved in the generation of corticothalamic SWDs (Blumenfeld & McCormick, 2000).
We also provided evidence that ictogenic neurons present a prominent hyperpolarization‐induced depolarizing sag in response to large‐amplitude negative current pulses, which was followed by a robust post‐inhibitory rebound. This finding, which is indicative of an overexpression of I h, contrasts with recent in vitro investigations showing a reduction of h‐channels in S1 cortical neurons of WAG/Rij rats (Strauss et al. 2004; Kole et al. 2007). In addition to promoting seizure activity by taking part to the slow ramp depolarization preceding paroxysmal depolarizations during SWDs (Timofeev et al. 2002; see Fig. 4 C), the increase in I h currents could participate in the regularity of firing patterns during inter‐ictal periods (Kole et al. 2006).
Dynamic modulation of membrane excitability and integrative properties of ictogenic neurons during seizures
To our knowledge, the present study provides the first demonstration that the intrinsic properties of neurons initiating genetically determined absence seizures are differentially modulated during the S‐ and W‐components of seizures. During the W‐component, ictogenic neurons are hyperpolarized, silent and deprived of synaptic events, resulting in an increased R m compared to inter‐ictal epochs. This is consistent with previous reports showing higher values of R m during seizures in the GAERS motor cortex (Charpier et al. 1999; Slaght et al. 2002) and cat suprasylvian areas (Neckelmann et al. 2000). This result was attributed to a process of disfacilitation during the W‐event, i.e. a transient interruption of the sustained synaptic drive, leading to a global decrease in membrane conductance and a passive return to the resting V m. The initial and last part of the W‐phase in ictogenic neurons involve the activation of synaptic and intrinsic conductances that could have led, in theory, to a reduction in R m. First, Cl−‐dependent GABAergic synaptic inputs are responsible for a shunting inhibition that participates in the termination of the S‐associated synaptic depolarizations and lasts for a few tens of milliseconds during the subsequent W‐component (Chipaux et al. 2011). Second, paroxysmal synaptic depolarizations are preceded in most neurons by a slow ramp depolarization, reminiscent of the activation of h‐channels (see Fig. 4 C). However, the net increase in R m indicates that the transient interruption of synaptic conductance has an overwhelming impact compared to the other possible sources of R m modulation. Despite this reduction in membrane conductance, the number of current‐evoked APs was considerably reduced during the W‐element compared to inter‐ictal periods. This suggests that the increase in R m was compensated by the W‐related membrane hyperpolarization that moved the V m away from the voltage threshold for AP generation and, consequently, diminished the firing responses of neurons to a given excitatory input.
The S‐component is associated in ictogenic neurons with large‐amplitude synaptic depolarizations, likely to be glutamatergic, leading to a transient AP discharge interrupted by a Cl−‐dependent synaptic conductance due to the recruitment of local GABAergic interneurons (Chipaux et al. 2011). This activation of combined excitatory and inhibitory synaptic conductances is likely to be responsible for the dramatic reduction in R m during the ECoG spike. An increase in cortical neurons’ membrane conductance during the cell depolarizations associated with the S‐components has also been reported in the cat (Neckelmann et al. 2000). The current‐evoked firing during the S‐component, despite the considerable attenuation of R m, was higher than during the ECoG wave, but remained significantly lower compared to inter‐seizures epochs.
Altogether, these data indicate that the synaptic depolarization during the S‐component is sufficient to partially restore cortical excitability relative to the W‐element, but remained ineffective to fully recover the level of excitability reached outside seizures.
Potent mechanisms for aberrant cortical function in between and during absence seizures
The pro‐ictogenic properties of GAERS S1 cortex neurons, already operating during inter‐ictal periods, are primarily responsible for the propensity of the cortex to generate seizures. They may also partly underlie the behavioural comorbidities accompanying absence seizures in GAERS, such as anxiety, depression and psychosis (Jones et al. 2008, 2010). Consistently, it has been shown that treating GAERS with ethosuximide produces anti‐epileptogenic effects, reduces the behavioural comorbidity (Dezsi et al. 2013) and restores normal electrophysiological features in individual cortical neurons (Polack & Charpier, 2009). Moreover, the increase in the power of gamma (30–80 Hz) oscillations in the GAERS fronto‐parietal ECoG activity (Jones et al. 2010), proposed as a neurophysiological correlate of psychosis (Lee et al. 2003), is consistent with the high‐frequency depolarizing synaptic barrage we observed in ictogenic cortical neurons.
The abnormal activity of cortical neurons in the course of the seizure, which results in time‐to‐time changes in cellular excitability and integrative properties, might contribute to the impaired integration of sensory information during absences. Convergent findings from human patients and rodent genetic models have shown the persistence of cortical responses to sensory stimulations during SWDs (Inoue et al. 1992; Chipaux et al. 2013), indicating that the thalamo‐cortical system still integrates external stimuli during absences. However, cortical processing of neutral environmental inputs remains ineffective in producing conscious perception during SWDs (Blumenfeld, 2005; Chipaux et al. 2013), while positively reinforced stimuli seem more able to perturb cortical paroxysms until recovery of conscious processes (Drinkenburg et al. 2003). The lack of conscious experiences during absences may thus mainly result from a transient functional disorder in large‐scale networks, including bilateral associative cortices and related subcortical structures (Blumenfeld, 2005; Cavanna & Monaco, 2009; Chipaux et al. 2013). The dynamic alterations in the intrinsic properties of S1 cortex neurons suggest an additional cellular mechanism that could dramatically perturb the flow of sensory inputs in primary sensory cortices. Indeed, the moment‐to‐moment variability of cortical intrinsic excitability, dynamically influenced by the elementary epileptic pattern (wave or spike) at the time of the stimulus, is likely to introduce a severe uncertainty in sensory responsiveness during the course of the seizure, disrupting the stability of cortical network activity required for conscious experience (Schurger et al. 2015).
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
S.C., S.M. and M.S.W. designed the research. M.S.W. and T.A.B. performed the experiments. M.S.W., T.A.B. and M.C. performed data analysis and S.C., S.M. and M.S.W. participated in the interpretation of the data. S.L. performed the histological processing. S.C. and S.M. wrote the manuscript and M.S.W., T.A.B., M.C. and S.L. revised and improved it. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the Investissements d'avenir ANR‐10‐IAIHU‐06, the Institut National de la Santé et de la Recherche Médicale (INSERM), and the Université Pierre et Marie Curie (UPMC).
Acknowledgements
Part of this work was carried out on the Histomics platform of the ICM and we thank all technical staff involved.
S. Mahon and S. Charpier co‐supervised the study.
Linked articles This article is highlighted by a Perspective by Polack. To read this Perspective, visit http://dx.doi.org/10.1113/JP272921.
References
- Altwegg‐Boussac T, Chavez M, Mahon S & Charpier S (2014). Excitability and responsiveness of rat barrel cortex neurons in the presence and absence of spontaneous synaptic activity in vivo . J Physiol 592, 3577–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, Desalvo M, Novotny EJ, Constable RT & Blumenfeld H (2010). Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci 30, 5884–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H (2005). Consciousness and epilepsy: why are patients with absence seizures absent? Prog Brain Res 150, 271–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenfeld H & McCormick DA (2000). Corticothalamic inputs control the pattern of activity generated in thalamocortical networks. J Neurosci 20, 5153–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanna AE & Monaco F (2009). Brain mechanisms of altered conscious states during epileptic seizures. Nat Rev Neurol 5, 267–276. [DOI] [PubMed] [Google Scholar]
- Charpier S, Leresche N, Deniau JM, Mahon S, Hughes SW & Crunelli V (1999). On the putative contribution of GABA(B) receptors to the electrical events occurring during spontaneous spike and wave discharges. Neuropharmacology 38, 1699–1706. [DOI] [PubMed] [Google Scholar]
- Chipaux M, Charpier S & Polack PO (2011). Chloride‐mediated inhibition of the ictogenic neurones initiating genetically determined absence seizures. Neuroscience 192, 642–651. [DOI] [PubMed] [Google Scholar]
- Chipaux M, Vercueil L, Kaminska A, Mahon S & Charpier S (2013). Persistence of cortical sensory processing during absence seizures in human and an animal model: evidence from EEG and intracellular recordings. PLoS One 8, e58180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civillico EF & Contreras D (2012). Spatiotemporal properties of sensory responses in vivo are strongly dependent on network context. Front Syst Neurosci 6, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinople C & Bruno RM (2011). Effects and mechanisms of wakefulness on local cortical networks. Neuron 69, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V & Leresche N (2002). Block of thalamic T‐type Ca2+ channels by ethosuximide is not the whole story. Epilepsy Curr 2, 53–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antuono M, Inaba Y, Biagini G, D'Arcangelo G, Tancredi V & Avoli M (2006). Synaptic hyperexcitability of deep layer neocortical cells in a genetic model of absence seizures. Genes Brain Behav 5, 73–84. [DOI] [PubMed] [Google Scholar]
- David O, Guillemain I, Saillet S, Reyt S, Deransart C, Segebarth C & Depaulis A (2008). Identifying neural drivers with functional MRI: an electrophysiological validation. PLoS Biol 6, 2683–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis A, David O & Charpier S (2015). The genetic absence epilepsy rat from Strasbourg as a model to decipher the neuronal and network mechanisms of generalized idiopathic epilepsies. J Neurosci Methods 260, 159–174. [DOI] [PubMed] [Google Scholar]
- Depaulis A & van Luijtelaar G (2006). Genetic models of absence epilepsy in the rat In Models of Seizures and Epilepsy, ed. Pitkänen A, Schwartzkroin P. & Moshe S, pp. 233–248. Elsevier Academic Press. [Google Scholar]
- Dezsi G, Ozturk E, Stanic D, Powell KL, Blumenfeld H, O'Brien TJ & Jones NC (2013). Ethosuximide reduces epileptogenesis and behavioural comorbidity in the GAERS model of genetic generalized epilepsy. Epilepsia 54, 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinkenburg WH, Schuurmans ML, Coenen AM, Vossen JM & van Luijtelaar EL (2003). Ictal stimulus processing during spike‐wave discharges in genetic epileptic rats. Behav Brain Res 143, 141–146. [DOI] [PubMed] [Google Scholar]
- Ermentrout GB, Galan RF & Urban NN (2008). Reliability, synchrony and noise. Trends Neurosci 31, 428–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman MD (1984). Morphology of the neocortical pyramidal neuron In Cerebral Cortex, ed. Peters A. & Jones EG, pp. 123–200. Plenum Press, New York. [Google Scholar]
- Fricker D, Verheugen JA & Miles R (1999). Cell‐attached measurements of the firing threshold of rat hippocampal neurones. J Physiol 517, 791–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MD, Brown M & Tucker DM (2004). Are ‘‘generalized’’ seizures truly generalized? Evidence of localized mesial frontal and frontopolar discharges in absence. Epilepsia 45, 1568–1579. [DOI] [PubMed] [Google Scholar]
- Holt GR, Softky WR, Koch C & Douglas RJ (1996). Comparison of discharge variability in vitro and in vivo in cat visual cortex neurons. J Neurophysiol 75, 1806–1814. [DOI] [PubMed] [Google Scholar]
- Inaba Y, D'Antuono M, Bertazzoni G, Biagini G & Avoli M (2009). Diminished presynaptic GABA(B) receptor function in the neocortex of a genetic model of absence epilepsy. Neurosignals 17, 121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, van Luijtelaar EL, Vossen JM & Coenen AM (1992). Visual evoked potentials during spontaneously occurring spike‐wave discharges in rats. Electroencephalogr Clin Neurophysiol 84, 172–179. [DOI] [PubMed] [Google Scholar]
- Jones NC, Martin S, Megatia I, Hakami T, Salzberg MR, Pinault D, Morris MJ, O'Brien TJ & van den Buuse M (2010). A genetic epilepsy rat model displays endophenotypes of psychosis. Neurobiol Dis 39, 116–125. [DOI] [PubMed] [Google Scholar]
- Jones NC, Salzberg MR, Kumar G, Couper A, Morris MJ & O'Brien TJ (2008). Elevated anxiety and depressive‐like behaviour in a rat model of genetic generalized epilepsy suggesting common causation. Exp Neurol 209, 254–260. [DOI] [PubMed] [Google Scholar]
- Klein JP, Khera DS, Nersesyan H, Kimchi EY, Waxman SG & Blumenfeld H (2004). Dysregulation of sodium channel expression in cortical neurons in a rodent model of absence epilepsy. Brain Res 1000, 102–109. [DOI] [PubMed] [Google Scholar]
- Kole MHP, Brauer AU & Stuart GJ (2007). Inherited cortical HCN1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J Physiol 578, 507–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MHP, Hallermann S & Stuart GJ (2006). Single I h channels in pyramidal neuron dendrites: properties, distribution, and impact on action potential output. J Neurosci 26, 1677–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Williams LM, Breakspear M & Gordon E (2003). Synchronous Gamma activity: a review and contribution to an integrative neuroscience model of schizophrenia. Brain Res Rev 41, 57–78. [DOI] [PubMed] [Google Scholar]
- Luhmann HJ, Mittmann T, van Luijtelaar G & Heinemann U (1995). Impairment of intracortical GABAergic inhibition in a rat model of absence epilepsy. Epilepsy Res 22, 43–51. [DOI] [PubMed] [Google Scholar]
- Lüttjohann A & van Luijtelaar G (2015). Dynamics of networks during absence seizure's on‐ and offset in rodents and man. Front Physiol 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Casassus G, Mulle C & Charpier S (2003). Spike‐dependent intrinsic plasticity increases firing probability in rat striatal neurons in vivo . J Physiol 550, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S & Charpier S (2012). Bidirectional plasticity of intrinsic excitability controls sensory inputs efficiency in layer 5 barrel cortex neurons in vivo. J Neurosci 32, 11377–11389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahon S, Deniau JM & Charpier S (2001). Relationship between EEG potentials and intracellular activity of striatal and cortico‐striatal neurons: an in vivo study under different anaesthetics. Cereb Cortex 11, 360–373. [DOI] [PubMed] [Google Scholar]
- Mahon S, Vautrelle N, Pezard L, Slaght SJ, Deniau JM, Chouvet G & Charpier S (2006). Distinct patterns of striatal medium spiny neuron activity during the natural sleep–wake cycle. J Neurosci 26, 12587–12595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matricardi S, Verrotti A, Chiarelli F, Cerminara C & Curatolo P (2014). Current advances in childhood absence epilepsy. Pediatr Neurol 50, 205–212. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM & Lopes da Silva FH (2002). Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci 22, 1480–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeren H, van Luijtelaar G, Lopes da Silva F & Coenen A (2005). Evolving concepts on the pathophysiology of absence seizures: the cortical focus theory. Arch Neurol 62, 371–376. [DOI] [PubMed] [Google Scholar]
- Neckelmann D, Amzica F & Steriade M (2000). Changes in neuronal conductance during different components of cortically generated spike‐wave seizures. Neuroscience 96, 475–485. [DOI] [PubMed] [Google Scholar]
- Ngomba RT, Biagioni F, Casciato S, Willems‐Van Bree E, Battaglia G, Bruno V, Nicoletti F & van Luijtelaar EL (2005). The preferential mGlu2/3 receptor antagonist, LY341495, reduces the frequency of spike‐wave discharges in the WAG/Rij rat model of absence epilepsy. Neuropharmacology 49, 89–103. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP (2008). Typical absence seizures and related epileptic syndromes: assessment of current state and directions for future research. Epilepsia 49, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Pape HC (1996). Queer current and pacemaker: the hyperpolarization‐activated cation current in neurons. Annu Rev Physiol 58, 299–327. [DOI] [PubMed] [Google Scholar]
- Parri HR & Crunelli V (1998). Sodium current in rat and cat thalamocortical neurons: role of a non‐inactivating component in tonic and burst firing. J Neurosci 18, 854–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G & Watson C (1986). The Brain in Stereotaxic Coordinates Academic Press, Sydney. [Google Scholar]
- Paz JT, Chavez M, Saillet S, Deniau JM & Charpier S (2007). Activity of ventral medial thalamic neurons during absence seizures and modulation of cortical paroxysms by the nigrothalamic pathway. J Neurosci 27, 929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO, Guillemain I, Hu E, Deransart C, Depaulis A & Charpier S (2007). Deep layer somatosensory cortical neurons initiate spike‐and‐wave discharges in a genetic model of absence seizures. J Neurosci 27, 6590–6599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polack PO & Charpier S (2009). Ethosuximide converts ictogenic neurons initiating absence seizures into normal neurons in a genetic model. Epilepsia 50, 1816–1820. [DOI] [PubMed] [Google Scholar]
- Polack PO, Mahon S, Chavez M & Charpier S (2009). Inactivation of the somatosensory cortex prevents paroxysmal oscillations in cortical and related thalamic neurons in a genetic model of absence epilepsy. Cereb Cortex 19, 2078–2091. [DOI] [PubMed] [Google Scholar]
- Rall W (1969). Time constants and electrotonic length of membrane cylinders and neurons. Biophys J 9, 1483–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurger A, Sarigiannidis I, Naccache L, Sitt JD & Dehaene S (2015). Cortical activity is more stable when sensory stimuli are consciously perceived. Proc Natl Acad Sci USA 112, E2083–E2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA (2010). Neuronal arithmetic. Nat Rev Neurosci 11, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaght SJ, Paz JT, Chavez M, Deniau JM, Mahon S & Charpier S (2004). On the activity of the corticostriatal networks during spike‐and‐wave discharges in a genetic model of absence epilepsy. J Neurosci 24, 6816–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaght SJ, Paz JT, Mahon S, Maurice N, Charpier S & Deniau JM (2002). Functional organization of the circuits connecting the cerebral cortex and the basal ganglia: implications for the role of the basal ganglia in epilepsy. Epileptic Disord 4 (Suppl. 3), S9–S22. [PubMed] [Google Scholar]
- Steriade M (2004). Neocortical cell classes are flexible entities. Nat Rev Neurosci 5, 121–134. [DOI] [PubMed] [Google Scholar]
- Steriade M & Amzica F (1999). Intracellular study of excitability in the seizure‐prone neocortex in vivo. J Neurophysiol 82, 3108–3122. [DOI] [PubMed] [Google Scholar]
- Strauss U, Kole MH, Brauer AU, Pahnke J, Bajorat R, Rolfs A, Nitsch R & Deisz RA (2004). An impaired neocortical I h is associated with enhanced excitability and absence epilepsy. Eur J Neurosci 19, 3048–3058. [DOI] [PubMed] [Google Scholar]
- Teich MC (1992). Fractal neuronal firing patterns In Single Neuron Computation, ed. McKenna T, Davis J. & Zormetzer SF, pp. 589–625. Academic Press, Boston. [Google Scholar]
- Timofeev I, Bazhenov M, Sejnowski T & Steriade M (2002). Cortical hyperpolarization‐activated depolarizing current takes part in the generation of focal paroxysmal activities. Proc Natl Acad Sci USA 99, 9533–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Bruns W, Mann EO, Crepel V & Scanziani M (2013). The information content of physiological and epileptic brain activity. J Physiol 591, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmijse I, Ossenblok P, Gunning B & van Luijtelaar G (2009). Onset and propagation of spike and slow wave discharges in human absence epilepsy: A MEG study. Epilepsia 50, 2538–2548. [DOI] [PubMed] [Google Scholar]
- Wilent WB & Contreras D (2004). Synaptic responses to whisker deflections in rat barrel cortex as a function of cortical layer and stimulus intensity. J Neurosci 24, 3985–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams D (1953). A study of thalamic and cortical rhythms in petit mal. Brain 76, 50–69. [DOI] [PubMed] [Google Scholar]