

Abstract
Key points
The transient receptor potential ankyrin 1 (TRPA1) ion channel is expressed in nociceptive neurons and its activation causes ongoing pain and inflammation; TRPA1 is thought to play an important role in inflammation in the airways.
TRPA1 is sensitised by repeated stimulation with chemical agonists in a calcium‐free environment and this sensitisation is very long lasting following agonist removal.
We show that agonist‐induced sensitisation is independent of the agonist's binding site and is also independent of ion channel trafficking or of other typical signalling pathways.
We find that sensitisation is intrinsic to the TRPA1 protein and is accompanied by a slowly developing shift in the voltage dependence of TRPA1 towards more negative membrane potentials.
Agonist‐induced sensitisation may provide an explanation for sensitisation following long‐term exposure to harmful irritants and pollutants, particularly in the airways.
Abstract
The TRPA1 ion channel is expressed in nociceptive (pain‐sensitive) neurons and responds to a wide variety of chemical irritants, such as acrolein in smoke or isothiocyanates in mustard. Here we show that in the absence of extracellular calcium the current passing through TRPA1 gradually increases (sensitises) during prolonged application of agonists. Activation by an agonist is essential, because activation of TRPA1 by membrane depolarisation did not cause sensitisation. Sensitisation is independent of the site of action of the agonist, because covalent and non‐covalent agonists were equally effective, and is long lasting following agonist removal. Mutating N‐terminal cysteines, the target of covalent agonists, did not affect sensitisation by the non‐covalent agonist carvacrol, which activates by binding to a different site. Sensitisation is unaffected by agents blocking ion channel trafficking or by block of signalling pathways involving ATP, protein kinase A or the formation of lipid rafts, and does not require ion flux through the channel. Examination of the voltage dependence of TRPA1 activation shows that sensitisation is accompanied by a slowly developing shift in the voltage dependence of TRPA1 towards more negative membrane potentials, and is therefore intrinsic to the TRPA1 channel. Sensitisation may play a role in exacerbating the pain caused by prolonged activation of TRPA1.
Keywords: agonist, cation channel, hyperalgesia, inflammation, pain, trp channels
Key points
The transient receptor potential ankyrin 1 (TRPA1) ion channel is expressed in nociceptive neurons and its activation causes ongoing pain and inflammation; TRPA1 is thought to play an important role in inflammation in the airways.
TRPA1 is sensitised by repeated stimulation with chemical agonists in a calcium‐free environment and this sensitisation is very long lasting following agonist removal.
We show that agonist‐induced sensitisation is independent of the agonist's binding site and is also independent of ion channel trafficking or of other typical signalling pathways.
We find that sensitisation is intrinsic to the TRPA1 protein and is accompanied by a slowly developing shift in the voltage dependence of TRPA1 towards more negative membrane potentials.
Agonist‐induced sensitisation may provide an explanation for sensitisation following long‐term exposure to harmful irritants and pollutants, particularly in the airways.
Abbreviations
- AITC
allyl isothiocyanate
- DRG
dorsal root ganglion
- HEK293t
human embryonic kidney 293t cell line
- MβCD
methyl‐β‐cyclodextrin
- PKA
protein kinase A
- TRPA1
transient receptor potential ankyrin 1
- WTA
wheat germ agglutinin
- WT
wild type
Introduction
The transient receptor potential ankyrin 1 (TRPA1) channel is unique in its ability to respond to a wide range of irritant agonists, many found in the everyday environment, hence the term ‘irritant receptor’. The channel is expressed in neurons of somatosensory ganglia (Story et al. 2003; Jordt et al. 2004; Nagata et al. 2005; Anand et al. 2008) and its agonists trigger pain and inflammation. Electrophilic agonists, such as allyl‐isothiocyanate (AITC, mustard oil), cinnamaldehyde, formaldehyde and acrolein activate TRPA1 by forming covalent bonds with three intracellular N‐terminal cysteine residues (Story et al. 2003; Bandell et al. 2004; Bautista et al. 2006; Hinman et al. 2006; Macpherson et al. 2007; McNamara et al. 2007). Non‐electrophilic agonists, such as menthol, carvacrol (contained in oregano) and nicotine activate TRPA1 without causing modification of cysteines (Karashima et al. 2007; Lee et al. 2008; Talavera et al. 2009). TRPA1 is also activated by a range of endogenous ligands (Andersson et al. 2008; Eberhardt et al. 2012), protons (de la Roche et al. 2013), and possibly by noxious cold temperatures, although this remains controversial (Story et al. 2003; Jordt et al. 2004; Bautista et al. 2006; Zurborg et al. 2007; Karashima et al. 2009; Gentry et al. 2010). Intracellular and extracellular calcium activate TRPA1 and potentiate its response to several agonists; the activation is followed by long lasting inactivation of the channel (Jordt et al. 2004; Zurborg et al. 2007; Doerner et al. 2007; Wang et al. 2008 b).
Behavioural studies in rodents as well as genome wide association studies in humans demonstrate a role for TRPA1 in nociception, making the channel a promising target for the development of novel analgesics (Obata et al. 2005; Bautista et al. 2006; Kwan et al. 2006; Caceres et al. 2009; Kremeyer et al. 2010; Bell et al. 2014). As many of its agonists can potentially be inhaled, TRPA1 has become an important target for the treatment of inflammatory airway diseases.
Inflammation releases a range of inflammatory mediators which cause a lowering of the activation threshold of peripheral nociceptive nerve fibres. TRPA1 is sensitised by bradykinin or by activators of proteinase‐activated receptor 2 (PAR2) via activation of intracellular pathways involving protein kinase A (PKA) and phospholipase C (Dai et al. 2007; Wang et al. 2008 a; Schmidt et al. 2009). TRPA1 can also be sensitised by prolonged application of agonists in calcium‐free conditions (Raisinghani et al. 2011). Such agonist‐induced sensitisation could be important for the body's response during prolonged exposure to irritants and allergens. Elucidating the mechanism behind agonist‐induced sensitisation is important in understanding both the function of TRPA1 and its role in pathologies such as lung inflammation.
Here, we investigate agonist‐induced sensitisation of human TRPA1 and we show that this process is independent of the principal cell signalling pathways and also does not involve enhanced channel trafficking to the cell membrane. We find that agonist‐induced sensitisation is accompanied by a slow shift of the voltage dependence of channel activation towards more negative membrane potentials, leading to a slowly developing and long‐lasting gain in channel activation. The shift is rapidly reversed on removal of agonist but rapidly reappears on re‐presentation of the agonist. We suggest that activation by an agonist causes sensitisation through a long‐lasting and slowly‐reversible change in the structure of TRPA1.
Methods
Ethical approval
Studies involving animals were approved by the Home Office (UK) and by the Animal Welfare and Ethical Review Body, University of Cambridge.
Cell culture and transfection
Cell culture was performed as previously described (Fischer et al. 2013) with the following changes. Dorsal root ganglia (DRGs) were collected from C57BL/6 mice of either sex aged 6–16 weeks, killed by cervical dislocation. Harvested ganglia were transferred into calcium‐ and magnesium‐free Hank's Balanced Salt Solution and treated with 2 mg ml−1 papain (Sigma‐Aldrich, UK), followed by 2.5 mg ml−1 collagenase type IV (Invitrogen) for 30 min at 30°C. HEK293t cells were transfected using Metafectene (Biontex Laboratories GmbH, Martinsried/Planegg, Germany) according to the manufacturer's instructions. Electrophysiological recordings were performed on the day following the transfection. All experiments were performed at room temperature.
Plasmids, cloning and mutagenesis
Human TRPA1‐V5 was obtained from Dr Xuming Zhang (University of Aberdeen, UK). Human TRPA1 cDNA (hTRPA1) was subcloned into a pcDNA5/FRT‐mTRPA1‐IRES‐YFP vector, using HindIII and XbaI. The vector was a kind gift from Dr Ardem Patapoutian (Scripps Research Institute, La Jolla, CA, USA). The hTRPA1 cDNA containing three cysteine‐to‐serine mutations (C621S, C641S, C665S) was obtained from Dr Carla Nau (University Medical Center Schleswig‐Holstein, Lübeck, Germany) and was cloned into the same pcDNA5/FRT‐IRES‐YFP vector. All constructs were confirmed by sequencing.
Calcium imaging
Ratiometric calcium imaging was performed using the calcium‐sensitive dye fura‐2. Cells were loaded with fura‐2 AM (5 μm) in the presence of 0.02 % pluronic F‐127 (both Invitrogen) for 30 min at 37°C in serum‐ and antibiotic‐free DMEM. Medium was changed to Hepes‐buffered extracellular solution (in mm: 140 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 Hepes, and 5 glucose, adjusted to pH 7.4) and neurons were left to equilibrate at room temperature in the dark for 5 min and then mounted on a Nikon Eclipse Ti‐E inverted microscope with a ×10 Plan Fluor objective. Cells were continuously superfused with extracellular solution or test solutions through a common outlet. Cells were illuminated with a monochromator alternating between 340 ± 10 and 380 ± 10 nm (OptoScan; Cairn Research Ltd, Faversham, UK), controlled by WinFluor 3.2 software (Dr John Dempster, University of Strathclyde, Glasgow, UK). Ratiometric calcium imaging as described above was also used to identify DRG neurons expressing TRPA1 for patch clamp recordings from their responsiveness to AITC. Neurons were exposed to AITC (50 μm) for 40 s and responders identified. A selected neuron was then patched for whole‐cell voltage clamp recordings. Immediately after establishing the whole‐cell configuration perfusion was switched to calcium‐free extracellular solution for 2 min before beginning the voltage clamp recording. Note that at least 5 min passed between the end of the AITC application and the start of the voltage clamp recording.
Patch clamp electrophysiology
Whole‐cell voltage clamp was performed on transfected HEK293t cells or on DRG neurons selected by calcium imaging (see above). Membrane currents were acquired with an Axopatch 200B amplifier (Molecular Devices, Wokingham, UK), low‐pass filtered at 2 kHz and sampled at 10 kHz. The pClamp 9 or 10 software (Molecular Devices) was used for acquisition and offline analysis. Series resistance was compensated up to 75 %. Glass electrodes (GB150F‐8P; Science Products GmbH, Hofheim, Germany) were fabricated (P‐97; Sutter Instruments, Novato, USA) and heat polished on a microforge (MF‐830; Narishige International Ltd, Tokyo, Japan) to a final resistance of 2–5 MΩ. Cells were continuously superfused with extracellular solution or test solutions through a common outlet. Solution changes were controlled by an automated system using pinch‐valves designed by Dr V. Vellani (CV Scientific, Inc). Solution changes at the cell position were complete (10–90 % concentration change) within 100 ms, which was significantly more rapid than the time course of TRPA1 current activation in response to agonist (see below). All recordings were performed in calcium‐free Hepes‐buffered extracellular solution (see above). In experiments using ATP‐free intracellular solution the extracellular solution contained no glucose. In experiments designed to evoke maximal TRPA1 currents using 5 mm carvacrol, external sodium was reduced to 10 mm to reduce voltage‐clamp artefacts caused by large current flows. NaCl was substituted by choline‐chloride (130 mm), and titrated with CsOH. The internal solution contained the following (in mm): 140 KCl, 1.6 MgCl2, 2.5 MgATP, 0.5 NaGTP, 2 EGTA, and 10 Hepes, adjusted to pH 7.3. In ATP‐free experiments, MgATP and NaGTP were substituted with MgCl2 and NaCl2, respectively. All experiments were performed at room temperature and at a holding potential of −60 mV.
To investigate voltage dependence, a 500 ms voltage ramp protocol from −100 mV to +100 mV was applied every 5 s. Voltage dependence was also tested at given time points using 80 ms voltage pulses ranging from −140 mV to +220 mV in 30 mV increments from a holding potential of −60 mV, followed by a step to +60 mV for 80 ms for tail current analysis, and a final step back to −60 mV for 160 ms (see Fig. 6 B). TRPA1 currents were corrected for leak conductance obtained from a hyperpolarising pulse to −140 mV in the absence of agonist. Half‐maximal voltage gating (V ½) was obtained from tail currents, normalised to the maximum tail currents measured on each individual cell during the second application of carvacrol (I t/I t,max). Normalised tail current–voltage relationships were then fitted to a Boltzmann equation:
where I t,min and I t,max are the minimum and maximum tail currents, respectively, V is the membrane voltage and slope is an inverse measure of the steepness at inflection. Tail current analysis was restricted to the voltage domain below +100 mV in order to avoid channel inactivation at strong positive potentials (see Results section, Fig. 6).
Figure 6. Effect of repeated carvacrol application on the voltage dependence of TRPA1 .
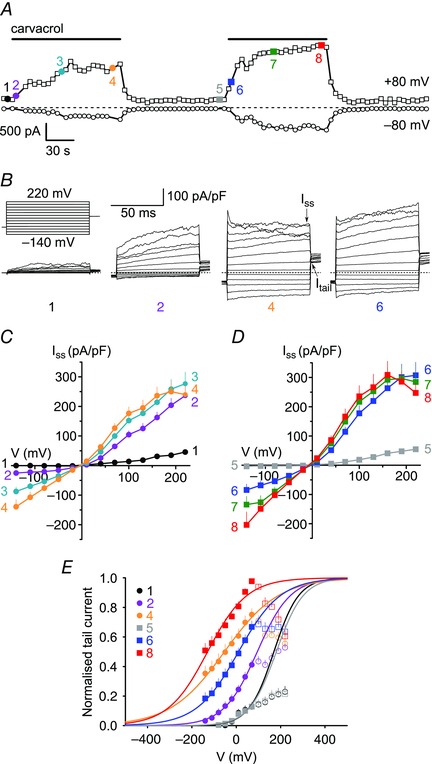
A, representative time course of whole‐cell currents elicited by two consecutive 120 s applications of 100 μm carvacrol, measured in hTRPA1‐expressing HEK293t cells at −80 mV (bottom trace) and +80 mV (top trace). Dashed line represents zero current level. At the coloured time points labelled 1–8, voltage dependence was tested using the step‐pulse protocol illustrated in B. B, currents recorded at the designated time points in A using the indicated step‐pulse protocol of 80 ms voltage steps from a holding potential of −60 mV ranging from −140 mV to +220 mV in 30 mV increments, followed by a final step to +60 mV. Time points for the measurement of steady‐state (I ss) and tail (I t) currents are indicated by arrows. C, I–V relationships obtained at the end (I ss) of the voltage steps illustrated in B. Shown are average I–V curves for time points 1–4 during the first carvacrol application. D, I–V relationships for time points 5–8 during the second carvacrol application. E, normalised tail currents measured at +60 mV at the indicated time points. Curve fitting was restricted to the voltage domain below +100 mV in order to avoid channel inactivation at strongly depolarised membrane potentials (see main text). Data points that were not included in the fit are shown as open symbols. Continuous lines are best fits to Boltzmann functions (n = 11 cells). The following V ½ values and slope factors (in brackets) were obtained for the given time points: time point 1, +170 mV (54 mV); time point 2, +94 mV (75 mV); time point 4, −36 mV (114 mV); time point 5, +179 mV (57 mV), time point 6, +22 mV (95 mV); time point 8, −132 mV (84 mV). [Colour figure can be viewed at wileyonlinelibrary.com]
Modelling
As an in silico model, four states and the transitions between these were modelled. To calculate the time course of channel opening or closing, the activating or deactivating component of the current traces were fitted by a mono‐exponential function:
where y(x) and C represent the current amplitude at time x and at steady state, respectively, A is the current amplitude, and τ is the time constant. To determine the time constant of the slow agonist‐induced effect, the V ½ shifts at 0 s, 10 s, 60 s, 120 s as well as 180 s and 240 s (cumulative) were considered and fitted by a double‐exponential function (Fig. 7 C):
where in addition to the above‐mentioned parameters, y 0 represents the current amplitude at time 0. The first exponential time constant τ1 was pre‐set to 3.1 s (see Results section). To reject outliers, log‐transformed time constants outside of mean ± 2SD were ignored.
Figure 7. Model of agonist‐induced sensitisation of TRPA1 .
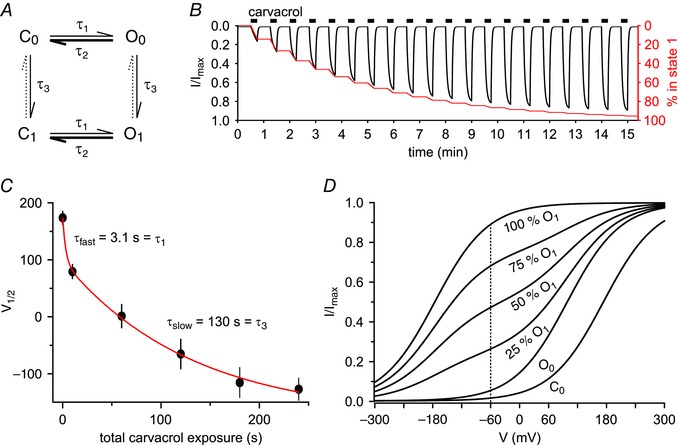
A, hypothetical model containing two open (O0, O1) and two closed (C0, C1) channel states. The gating of TRPA1 by agonist and voltage is displayed in the horizontal direction and is controlled by the opening and closing time constants τ1 and τ2 which depend on both membrane voltage and agonist concentration. The slow agonist‐induced sensitisation (vertical direction, τ3) is independent of channel gating and drives the transition from C0/O0 to the sensitised state C1/O1, leading to a slow and progressive accumulation of channels in the sensitised state (C1/O1). This accumulation means that the fraction of open channels at resting membrane potential is very different in the unsensitised and the sensitised states and this underlies the shift in the voltage dependence of channel activation shown in Fig. 6 E. Experimentally, the agonist‐dependent channel activation and deactivation rate was similar in both the unsensitised and the sensitised states (see main text), so the same values of τ1 and τ2 were used to describe agonist‐dependent activation and deactivation in the unsensitised and the sensitised states and at the resting membrane voltage. Agonist‐induced sensitisation is long lasting and no reversal was observed (dotted arrows). Upon washout of agonist channels rapidly close by reverting from O0 to C0 and from O1 to C1. The thickness of arrows reflects the transition rates of the respective process at physiological membrane potentials in the absence of agonist. B, based on the model shown in A, the expected inward currents upon repetitive carvacrol stimulation were simulated. In the model an increasing current (black trace, left axis) is generated by the agonist‐dependent transition from state 0 into the sensitised state 1. The fraction of channels in the sensitised state 1 is shown by the red trace (right axis). The overall increase in current in the model matches the experimental observation (see for example Fig. 1 E). Note that in voltage‐clamp recordings, the current reaches a plateau or briefly decreases within 15 s (see Fig. 5 C inset), reflecting a desensitising component which is omitted in the model for simplicity. C, average V ½ values of channel opening measured as shown in Fig. 6 E were fitted by a double‐exponential function (red line) to derive a time constant τ3 for the slow agonist‐induced sensitisation. The value of τ1 = 3.1 s, the measured time constant of current activation following agonist application (see Results section), provided a reasonable fit to the rapid phase of negative shift of V ½. The slow V ½ shift had a time constant of τ3 = 130 ± 27 s (n = 11 cells). D, we simulated the voltage‐dependent open probability of TRPA1 according to the four‐state model in A. Following carvacrol application, the voltage‐dependent open probability shows a transition from C0 to O0 with a time constant of 3.1 s (see above and Results section). As a result the current at −60 mV increases from 0.014 to 0.053 of the maximal current. Additional lines reflect the slow transition from O0 to O1. This transition, with a time constant of 130 s (see C), increases the current at −60 mV from 0.053 to 0.892 of the maximum. Note that the mixture of two states O0 and O1 means that the slope of the curve is decreased during the transition period, as was seen in Fig. 6 E. [Colour figure can be viewed at wileyonlinelibrary.com]
In our model, τ1 is the time constant of the net transition rate from the closed (C) to the open (O) channel state on application of agonist, τ2 is the time constant of the reverse process, occurring on removal of the agonist. τ3 is the time constant of the transition from state 0 (unsensitised) to state 1 (sensitised) in the presence of the agonist, applicable to both closed and open channels. Based on these transition time constants, the stochastically expected net transition was calculated for finite time periods of 0.1 s. The fraction of open channels was calculated for every state separately based on the Boltzmann equation above (I t/I t,max) and cumulated.
Immunocytochemistry
To examine trafficking of TRPA1 to the cell membrane, HEK 293t cells, transiently transfected with hTRPA1‐V5, were treated with 100 μm carvacrol or with vehicle (control) in calcium‐free extracellular solution for 5 min at room temperature prior to fixation. Immuncytochemistry was then performed as previously described (Btesh et al. 2013). The following antibodies were used: mouse anti‐V5 (1:1000; Invitrogen, Life Technologies Ltd, Paisley, UK) primary for 3 h, followed by Alexa Fluor 488 anti‐mouse secondary (1:1000; Life Technologies, Paisley, UK) for 1 h.
Chemicals
Carvacrol, AITC, MβCD and brefeldin A were all purchased from Sigma‐Aldrich (Gillingham, UK). H89 was purchased from Cayman Chemical (Ann Arbor, USA). HC030031 was a kind gift from Dr Sarah Skerratt, Neusentis, Pfizer (Granta Park, Cambridge, UK). AITC, H89 and HC030031 were dissolved in DMSO. The final concentration of DMSO did not exceed 0.1 %. Carvacrol was diluted directly in calcium‐free Hepes‐buffered extracellular solution and brefeldin A was dissolved in ethanol. MβCD was dissolved in cell culture medium and cells incubated prior to the start of the experiment.
Data analysis
Repetitive application of TRPA1 agonists such as carvacrol caused a steady increase in response amplitude (see for example Figs 1 and 2). In order to limit the time taken for recordings, and to reduce variability which may be caused by cell deterioration in very long experiments, we quantified fractional agonist‐induced sensitisation under different conditions as the peak current induced by the sixth agonist application divided by the peak current induced by the first application.
Figure 1. Repeated activation of TRPA1 by agonists, but not by voltage, sensitises the channel in the absence of extracellular calcium .
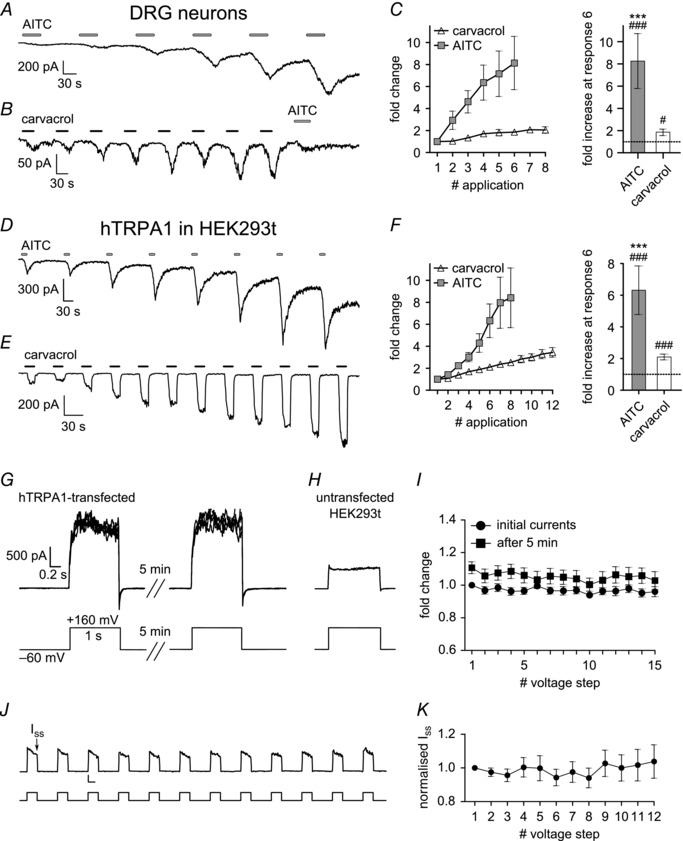
A, representative whole‐cell voltage‐clamp recording of a DRG neuron (holding potential −60 mV) showing that repeated application of AITC (50 μm; 40 s; 90 s gap) induces increasing inward currents in 0 [Ca2+]o. B, repeated application of carvacrol (100 μm; 30 s; 60 s gap) to a DRG neuron also has a sensitising effect. The TRPA1‐specific agonist AITC (50 μm; 40 s) was applied once at the end of the protocol. C, quantification of all experiments as in A and B. Graph (left) shows peak current of each response, measured as current increase on application of agonist, and normalised to first response. Bar chart (right) shows fractional sensitisation measured at sixth response. AITC induced an 8.3 ± 2.5‐fold sensitisation (P < 0.001 compared to first response; n = 9); carvacrol induced a 1.9 ± 0.3‐fold sensitisation (P = 0.01 compared to first response; n = 9). #Compared to first response. *Compared to carvacrol. D–F, similar recordings with HEK293t cells expressing hTRPA1 (holding potential −60 mV). Repeated application of AITC (50 μm; 15 s; 120 s gap) induced a 6.3 ± 1.5‐fold sensitisation (P < 0.001 compared to first response; n = 8) at the sixth response; carvacrol (100 μm; 15 s; 30 s gap) induced a 2.1 ± 0.2‐fold sensitisation (P < 0.001 compared to first response; n = 22). G, activation of TRPA1 by strong depolarisation has no sensitising effect. 15 voltage steps from −60 mV to +160 mV, duration 1 s, applied every 7 s. Same protocol repeated after a period of 5 min. Mean current 1624 ± 98 pA (67 ± 4 pA pF−1, n = 10). H, same protocol applied to untransfected HEK293t cells elicited much smaller background current. Scale bars as in G. Mean current 616 ± 41 pA (32 ± 3 pA pF−1; P < 0.001; n = 10). I, peak currents, normalised to the first response of the first protocol, do not increase with repeated activation of TRPA1 (circles), and there is no significant increase during the second protocol (squares) (all P ≥ 0.34; n = 8–10). J, voltage protocol similar to that used for agonist application. Voltage steps from −60 mV to +100 mV, duration 15 s (bottom trace) applied with 30 s intervals between steps. Time point for measurement of steady‐state (I ss) currents is indicated by arrow. Scale bars 50 pA and 10 s. K, steady‐state currents (I ss), normalised to the first response. No significant increase in current amplitude (all P ≥ 0.68; n = 6–8).
Figure 2. Agonist‐induced sensitisation does not rely on cysteine modification, ATP‐signalling, phosphorylation or the formation of lipid rafts .
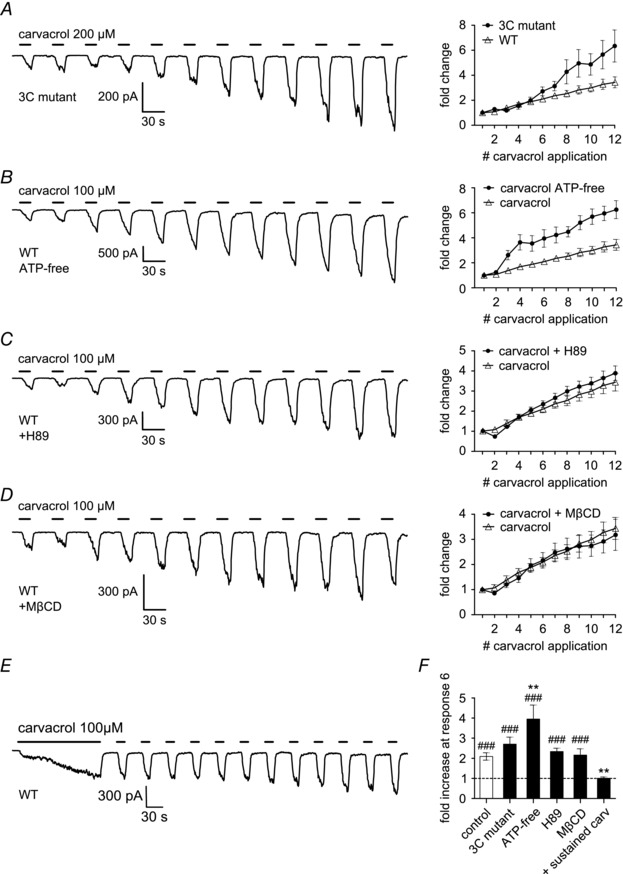
Whole‐cell voltage‐clamp experiments in HEK293t cells transiently transfected with hTRPA1. A, mutating the three intracellular cysteine residues modified by electrophilic agonists (C621, C641 and C665) to serine (3C mutant) does not reduce the sensitisation induced by repeated application of carvacrol (200 μm; 15 s). B, complete removal of ATP potentiated sensitisation induced by carvacrol (100 μm; 15 s). Recordings were performed using ATP‐ and GTP‐free intracellular solution and glucose‐free extracellular solution. C, inhibiting intracellular protein kinase A (PKA) does not reduce the effect of carvacrol (100 μm; 15 s). The PKA inhibitor H89 (10 μm) was applied extracellularly throughout the recording. D, disruption of lipid rafts by methyl‐β‐cyclodextrin (MβCD, 5 mm for 15 min before recording) does not inhibit sensitisation induced by carvacrol. E, an initial sustained carvacrol application (100 μm; 150 s) before switching to repetitive application stabilises the following repetitive responses. F, summary graph of fractional sensitisation measured at sixth response in all experiments as in A–F. The control group is the same as in Fig. 1 (E and F). In the 3C mutant, currents had increased 2.7 ± 0.4‐fold (P < 0.001 compared to first response; n = 14). In ATP‐free recordings, the increase was 4.0 ± 0.7‐fold (P < 0.001 compared to first response; n = 4). In the presence of H89, current increase was 2.4 ± 0.2‐fold (P < 0.001 compared to first response; n = 8). In the presence of MβCD, currents had increased 2.2 ± 0.3‐fold (P < 0.001 compared to first response; n = 8). With the initial sustained carvacrol application, currents had not increased at sixth response (1.0 ± 0.1‐fold; n = 14). #Compared to first response. *Compared to control.
Two groups of data containing < 10 samples were compared using the nonparametric U test. Two groups of data containing ≥ 10 samples were compared using the paired or unpaired t‐test as appropriate. Multiple groups were compared by one‐way or repeated measures ANOVA followed by either Dunnett's multiple comparison or Bonferroni post hoc test as appropriate. Recovery ratios in Fig. 5 D were statistically compared to the value of 1 using a one‐sample t‐test. Statistical analysis was performed using Statistica 8 (StatSoft, Bedford, UK) or Prism 6 (GraphPad Software, Inc., La Jolla, USA). Data are presented as mean ± SEM; P < 0.05 was considered significant. Significance levels are as follows: n.s., not significant; * P < 0.05; ** P < 0.01; *** P < 0.001.
Figure 5. Agonist‐induced sensitisation of TRPA1 is independent of ion flux through the channel and is long lasting .
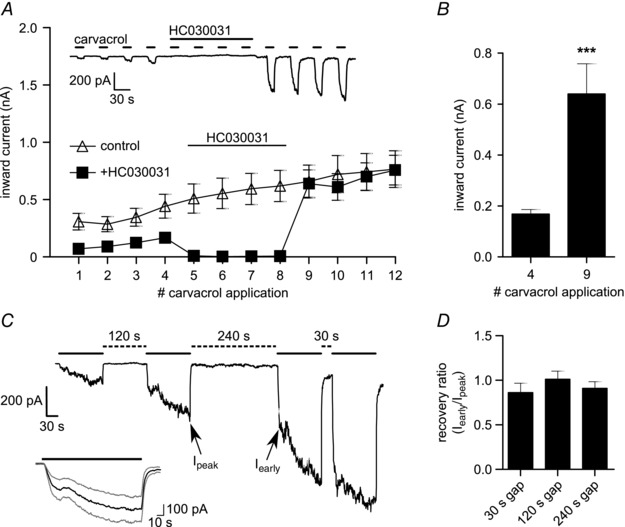
A, sensitisation of TRPA1 in HEK293t cells increases with agonist exposure even when the channel is blocked. Responses 5–8 to carvacrol (100 μm; 15 s) were blocked by the specific TRPA1 antagonist HC030031 (50 μm; 155 s) (see inset) and the measured inward currents (filled squares) are compared to control recordings without antagonist (open triangles). Responses 9–12 were comparable in amplitude to recordings where no antagonist had been applied (P ≥ 0.458; n = 7 for HC030031‐treated, n = 21 for control). B, TRPA1 is significantly sensitised by agonist exposure during channel block (P < 0.001; n = 7). C, TRPA1 sensitisation in HEK293t cells is long lasting. Gap periods of 30 s, 120 s or 240 s (dashed lines) were left between applications of carvacrol (100 μm; 120 s; continuous line). Sensitisation does not recover even with 240 s rest period. Sequence of gaps was changed at random between each cell. Arrows indicate time points for calculating the recovery ratio displayed in D. Inset: prolonged stimulation with carvacrol (100 μm; 150 s) induces two phases of current increase (average ± SEM trace; n = 16). D, the recovery ratio was calculated by measuring the early current at the end of the first steep increase (I early) of each response and comparing it to the peak current (I peak) of the previous response. None of the calculated recovery ratios were significantly different from 1 (P ≥ 0.24; n = 7–8).
Results
Repeated agonist‐induced activation sensitises TRPA1
Responses of TRPA1 in DRG neurons to a submaximal dose of AITC have been shown to increase with each application under calcium‐free conditions (Raisinghani et al. 2011). Here, we confirmed that repeated application of the electrophilic agonist AITC (50 μm) induces increasing inward currents in adult mouse DRG neurons expressing TRPA1 (Fig. 1 A and C). The non‐electrophilic TRPA1 agonist carvacrol (100 μm), which activates TRPA1 at a different agonist binding site (Lee et al. 2008) also induced increasing inward currents (Fig. 1 B and C). Both agonists also induced a similar sensitisation in HEK293t cells expressing hTRPA1 (Fig. 1 D–F).
TRPA1 can also be activated by strong depolarising voltage steps in the absence of agonist (Fig. 1 G; see also Fig. 6). The time‐dependent activation during the pulse, and the current deactivated on return to the holding potential, are both TRPA1 dependent as much smaller and time‐independent currents are observed in untransfected cells (Fig. 1 H). However, in contrast to the increasing current activated by repeated agonist application, we found that TRPA1 currents elicited by depolarisation were stable and did not show any sign of sensitisation, either during a train of activating pulses or when the train was repeated after 5 min (Fig. 1 I). Sensitisation was also not observed with longer voltage pulses, of the same duration as the exposures to carvacrol in Fig. 1 E (Fig. 1 J–K).
Sensitisation of TRPA1 is independent of N‐terminal cysteine modification or cell signalling pathways
Activation of TRPA1 by electrophilic agonists such as AITC depends on the covalent modification of three N‐terminal cysteine residues, C621, C641 and C665 (Hinman et al. 2006; Macpherson et al. 2007). We mutated all three cysteine residues to serine (hTRPA1‐C621S‐C641S‐C665S, 3C mutant), and used the non‐electrophilic agonist carvacrol to activate TRPA1. Repeated application of carvacrol still potently sensitised TRPA1 (Fig. 2 A and F), showing that the three cysteine residues are not required for agonist‐induced sensitisation.
Next, we tested if cell signalling pathways utilising intracellular ATP were responsible for sensitising TRPA1, a mechanism that is well known for the TRPV1 ion channel (Cesare & McNaughton, 1996; Tominaga et al. 2001; Vellani et al. 2001). However, removing all ATP and GTP from the intracellular solution and all glucose from the extracellular solution to prevent metabolic production of ATP did not reduce carvacrol‐induced sensitisation, and surprisingly sensitisation was significantly more potent compared to control experiments (Fig. 2 B and F).
TRPA1 has been reported to be sensitised by inflammatory mediators via activation of intracellular protein kinase A (PKA), although this has been disputed in a later study (Wang et al. 2008 b; Raisinghani et al. 2011). However, applying the PKA inhibitor H89 (10 μm) did not reduce agonist‐induced sensitisation of TRPA1 (Fig. 2 C and F). This concentration of H89 was chosen as it has previously shown to be effective in blocking bradykinin or forskolin‐induced activation of PKA (Wang et al. 2008 b; J. E. Meents, unpublished data).
Migration of ion channels into lipid rafts has been reported to potentiate their activation (Liu et al. 2006; Szőke et al. 2010). Lipid rafts can be disrupted by depletion of cholesterol from the cell membrane by methyl‐β‐cyclodextrin (MβCD). However, incubation of hTRPA1‐expressing HEK293t cells in MβCD before the recording did not influence the sensitisation induced by repeated application of carvacrol (Fig. 2 D and F). We used MβCD at a similar time of application and concentration that has been shown to diminish capsaicin‐induced responses in TRPV1‐expressing cells (Szőke et al. 2010).
Finally, we observed that an initial sustained carvacrol application causes slow sensitisation, as seen in a slow increase of inward current (Fig. 2 E). Once the channel has fully entered the sensitised state, little further sensitisation is seen during repetitive application of agonist (Fig. 2 E and F).
Trafficking of TRPA1 to the membrane is not involved in carvacrol‐induced sensitisation
It has previously been reported that TRPA1 ion channels can be trafficked to and inserted into the cell membrane in response to activation of the channel by AITC and it was argued that this mechanism contributes to increased sensitivity of neurons to TRPA1 agonists (Schmidt et al. 2009). We therefore investigated whether enhanced trafficking might be responsible for agonist‐induced sensitisation of TRPA1. HEK293t cells transfected with hTRPA1‐V5 were exposed to carvacrol (100 μm) for 5 min in a calcium‐free solution, similar to the carvacrol exposure in Fig. 2 E. TRPA1 was detected using a V5 antibody, and wheat germ agglutinin (WGA) was used to define the location of the plasma membrane (Btesh et al. 2013). However, no difference was observed in the presence of TRPA1 at the cell membrane after carvacrol treatment when compared to control cells treated with vehicle (Fig. 3 A–C).
Figure 3. Sensitisation of TRPA1 by carvacrol is not caused by increased trafficking to the membrane .
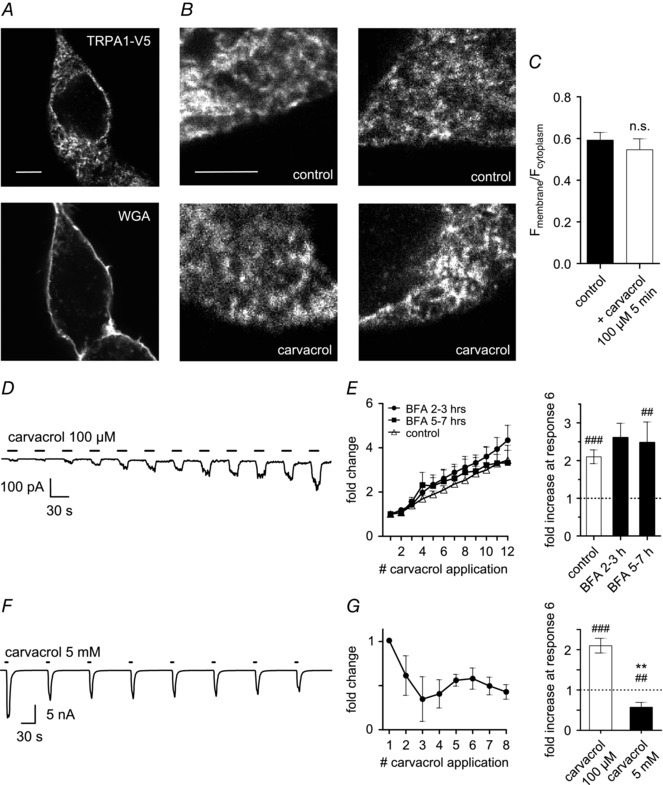
A, images show HEK293t cells transiently transfected with WT hTRPA1‐V5. Scale bar 5 μm is the same for both images. Membrane staining by WGA (bottom) was used to create a juxtamembrane ROI that was superimposed on the image of TRPA1 fluorescence (top) to measure TRPA1 levels at the membrane. A separate ROI obtained from the fluorescence within the cell defined the cytoplasm excluding the nucleus. B, cells were either untreated (control; top row) or stimulated with carvacrol (100 μm; 5 min; bottom row) before fixation. Scale bar (5 μm) is the same for all images. C, ratio of TRPA1 expression at the membrane compared to the cytoplasm was not different between control cells and those stimulated with carvacrol (P = 0.336; n = 7–8). D and E, TRPA1‐expressing HEK293t cells were incubated with brefeldin A (BFA, 5 μg ml−1) for 2–3 h (example trace) or 5–7 h to abolish protein trafficking to the membrane prior to start of voltage‐clamp experiments. Repetitive stimulation with carvacrol (100 μm; 15 s; 30 s gap) still induces pronounced sensitisation of inward currents. After 2–3 h BFA, sensitisation at sixth response was 2.6 ± 0.4‐fold (P = 0.08) and seventh response was increased significantly (2.9 ± 0.4; P = 0.015; both n = 7). After 5–7 h, sensitisation at sixth response was already significant (2.5 ± 0.6‐fold; P = 0.005; n = 6). F and G , repeated application of a saturating dose of carvacrol (5 mm; 6 s; 84 s gap) induces maximal currents that display tachyphylaxis. Third response is significantly smaller than the first (P = 0.034; n = 8). n.s. not significant. #Compared to first response. *Compared to 6th response in 100 μm carvacrol.
To extend these findings we repeated the electrophysiological recordings described above on hTRPA1‐expressing HEK293t cells that had been incubated with the protein transport inhibitor brefeldin A (5 μg ml−1). Brefeldin A has been successfully used to block trafficking of other ion channels, such as P2X (Fabbretti et al. 2006; Lalo et al. 2010) and TRPV1 (Johansen et al. 2006). However, blocking trafficking with brefeldin A had no effect on the sensitisation induced by repeated application of carvacrol (Fig. 3 D and E).
We next measured surface membrane TRPA1 expression directly by maximally activating TRPA1 with a saturating dose of 5 mm carvacrol. The large membrane currents observed with maximal activation were reduced by removing all but 10 mm extracellular Na+ in order to avoid patch clamp artefacts. Instead of the increasing inward currents that would be expected if exposure to agonist had induced trafficking of TRPA1 to the membrane, we instead observed tachyphylaxis of the induced responses (Fig. 3 F and G). These results are in accordance with those obtained by another group (Raisinghani et al. 2011) and argue against increased channel insertion into the cell membrane.
Sensitisation is independent of agonist site‐of‐action and of ion flux
Electrophilic agonists, such as AITC, activate the channel via intracellular N‐terminal cysteine modification (Hinman et al. 2006; Macpherson et al. 2007), while non‐electrophilic agonists such as menthol activate by binding to a site within transmembrane domain 5 (Xiao et al. 2008 a). Carvacrol also acts at the same site as menthol, because mutating two amino acids, S873 and T874, within transmembrane domain 5 of hTRPA1 to valine and leucine, respectively, completely abolished sensitivity to carvacrol but not to AITC (Fig. 4 C–E). We studied cross‐sensitisation between AITC and carvacrol to investigate whether the sensitisation induced by a specific agonist is limited to activation at its own binding site. Sensitisation was induced with either AITC or carvacrol, followed by a test of sensitisation by applying a pulse of carvacrol. Channel activation by either agonist caused equivalent sensitisation (Fig. 4 A), and in cells exposed to AITC, the following carvacrol response was significantly sensitised compared to control cells not exposed to AITC (Fig. 4 B). Sensitisation is therefore independent of the agonist's site of action.
Figure 4. Different agonists cross‐sensitise .
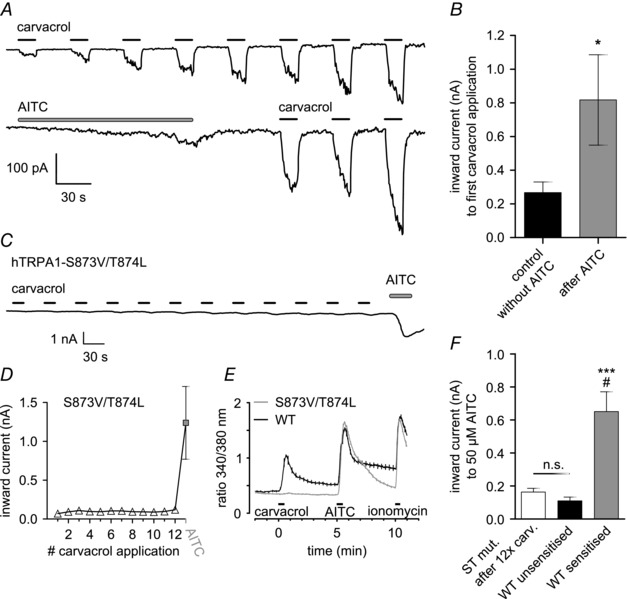
A, representative voltage‐clamp recordings in hTRPA1‐expressing HEK293t cells, showing cross‐sensitisation between AITC and carvacrol. Top: sensitisation of inward currents induced by carvacrol (100 μm; 15 s; 30 s gap). Bottom: recording in identical conditions but TRPA1 activated with AITC (50 μm; 150 s) before application of carvacrol. B, cross‐sensitisation of response to carvacrol by AITC. First carvacrol‐induced response is significantly larger following exposure to AITC (P = 0.016; n = 8 for AITC group, n = 24 for control). C, mutation S873V/T874L abolishes responses to carvacrol but not AITC. Representative recording (see Fig. 1 for protocol) in HEK293t cells expressing the hTRPA1‐S873V/T874L mutant. No response to carvacrol was observed. AITC 100 μm was applied at the end of the protocol and response was of normal amplitude. D, quantification of C, plotting the average inward current for each agonist application (n = 9). E, ratiometric calcium imaging confirms deletion of carvacrol response in hTRPA1‐S873V/T874L mutant. Experiments performed on HEK293t cells expressing either WT hTRPA1 (black) or hTRPA1‐S873V/T874L mutant (grey). Ratio of fura‐2 fluorescence excited at 340 nm and 380 nm plotted on vertical axis. Cells were exposed to carvacrol (12.5 μm) and AITC (50 μm) for 30 s each, followed by a 20 s application of ionomycin (5 μm) to induce a maximum response for comparison. Traces are averages of 30 (WT) and 167 (mutant) cells, SEM is represented by vertical lines. F, carvacrol does not cause sensitisation of responses to AITC in the S873V/T874L mutant. Responses of hTRPA1‐S873V/T874L mutant (white bar; n = 3) to 50 μm AITC following 12 applications of 100 μm carvacrol were of similar magnitude to naive responses to 50 μm AITC in WT channels (black bar; n = 9; P = 0,99). In contrast, WT channels that had been sensitised by repeated application of AITC (grey bar; n = 8) displayed significantly larger responses (P = 0,015 compared to white, P < 0,001 compared to black; one‐way ANOVA with Bonferroni post hoc analysis). Values for the unsensitised and sensitised WT channels are the same as in Fig. 1 D and F, 1st and 8th response, respectively. #Compared to S873V/T874L. *Compared to WT unsensitised.
The S873V/T874L mutant allowed us to examine the possibility that carvacrol might cause sensitisation by binding to a site other than that at which it activates the ion channel. Application of carvacrol to the S873V/T874L mutant failed to activate the channel (Fig. 4 C–E) and also failed to cause any sensitisation, as probed by a subsequent pulse of AITC (Fig. 4 F). Thus both activation and sensitisation are mediated by binding of carvacrol to an activation domain containing the S873V/T874L site.
We used the TRPA1 antagonist HC030031 to test if channel block could abolish carvacrol‐induced sensitisation. Channel block during a series of applications of carvacrol did not affect sensitisation because responses after channel block were comparable in amplitude to recordings where no antagonist had been applied (Fig. 5 A) and were significantly increased compared to responses before channel block (Fig. 5 B). To investigate the time course of recovery from carvacrol‐induced sensitisation, we applied carvacrol with variable gaps between each application (Fig. 5 C) and found no evidence for recovery over 240 s (Fig. 5 D).
Prolonged activation of TRPA1 by an agonist as shown in Fig. 5 C (see inset) showed the existence of two distinct processes during sustained channel activation. The initial current increase reached a plateau within a few seconds, which was followed by a delayed secondary current increase with a similar time course as the onset of sensitisation.
Taken together, the results described in this section show that sensitisation is driven when TRPA1 is activated by agonists but not by voltage. Sensitisation has a slow onset, reverses very slowly in the absence of agonist, and is independent of cell signalling pathways.
Sensitisation is accompanied by a change in dependence of TRPA1 on membrane voltage
To investigate whether sensitisation induced by carvacrol may be accompanied by changes in the intrinsic gating properties of TRPA1 we compared the voltage‐dependent activation of TRPA1 in unsensitised and sensitised states. Under calcium‐free conditions we applied carvacrol (100 μm) while testing for current activation using a rapid ramp protocol (Fig. 6 A). At the indicated time points before and during carvacrol, we used a protocol of repeated voltage pulses to examine the voltage dependence of activation (Fig. 6 B). Steady‐state current at the end of each test pulse was used to obtain the I–V relationship (Fig. 6 C and D) and the tail current at +60 mV following each pulse was used to calculate the voltage dependence of channel activation (Fig. 6 E).
Before application of carvacrol, voltage‐dependent activation was observed only at strongly positive potentials (Fig. 6 B and C; time point 1). Application of carvacrol (100 μm) induced an increase in steady‐state currents at all voltages (time point 2) which gradually increased (time points 3 and 4). Moreover, we observed channel inactivation at strongly depolarised voltages following prolonged application of carvacrol (time point 4). After washout of carvacrol, activation only by strongly positive membrane voltages was restored (Fig. 6 D; time point 5). Surprisingly, when we measured voltage dependence again during a second carvacrol application, there was a marked negative shift in the I–V relationship between each time point and its corresponding point during the first application (compare Fig. 6 C and D). Steady‐state current at +70 mV at the end of the second carvacrol application (time point 8) had increased by 33 % compared to the end of the first application (time point 4). Note that there is not an exact correspondence between current measured with the rapid ramp protocol in Fig. 6 A, which does not allow current to reach a steady‐state, and the steady‐state current measured with the step protocol in Fig. 6 B.
We next used tail currents at +60 mV as a measure of the voltage dependence of channel activation (Fig. 6 E). Corresponding to our observations above, TRPA1 was activated only at strongly depolarised membrane potentials in the absence of agonist (time point 1), which is in agreement with previous reports (Fajardo et al. 2008; Meseguer et al. 2008) although others have reported stronger activation at potentials above +100 mV (Karashima et al. 2007; Zurborg et al. 2007). With prolonged agonist application (see time points 4 and 8) tail currents were reduced following strong depolarisations, with currents reaching a maximum at +70 mV and then decreasing steadily (Fig. 6 E). Voltage‐dependent TRPA1 channel inactivation at strongly positive membrane potentials has been noted in previous publications (Karashima et al. 2007, 2008; Macpherson et al. 2007; Andersson et al. 2008; Fajardo et al. 2008; Talavera et al. 2009) and the basis for this inactivation is unknown. In analysing tail currents we therefore restricted analysis to the voltage domain below +100 mV. Voltage‐dependent activation of TRPA1 was quantified from tail currents normalised to the maximum tail current measured for each individual cell during either of the two responses. These tail current–voltage relationships were fitted by a Boltzmann function to calculate the voltage at which current was half‐activated, V ½ (Fig. 6 E). In the absence of agonist, we measured a V ½ of +170 ± 13 mV, which is similar albeit slightly higher than what has been previously reported for TRPA1 (Karashima et al. 2007; Zurborg et al. 2007; Kremeyer et al. 2010). During application of carvacrol, we found an initial rapid negative shift of the activation V ½; after 10 s exposure V ½ was +94 ± 16 mV (time point 2; P = 0.05 when compared to time point 1; n = 11). A slow negative shift followed; after 60 s exposure, V ½ had shifted to +34 ± 20 mV (not shown; P < 0.001 compared to time point 1; n = 11) and after 120 s exposure, V ½ had shifted even further to −36 ± 24 mV (time point 4; P < 0.001 compared to time point 1; n = 11; all repeated measures ANOVA with Bonferroni post hoc analysis) (Fig. 6 E). Corresponding to observations from steady‐state I–V relationships, tail current–voltage relationships returned to baseline values between carvacrol applications (time point 5; +179 ± 14 mV; P = 0.99 compared to time point 1), indicating that the shift of the activation curve was not due to factors such as progressive dialysis of the intracellular solution. The activation curves during the second carvacrol application, which had been shown in other experiments to cause significant sensitisation, were all shifted towards more negative potentials when compared to the corresponding time points in the first carvacrol application, indicating that part of the negative shift had been conserved between applications. We measured an activation V ½ of +22 ± 12 mV (P = 0.08), −93 ± 25 mV (P < 0.001) and −132 ± 18 mV (P = 0.003) after 10 s, 60 s and 120 s of the second application, respectively (all compared to corresponding time points in the first carvacrol application; n = 11; repeated measures ANOVA with Bonferroni post hoc analysis).
We next developed a model to explain the time course of sensitisation of TRPA1 activation during repetitive applications of carvacrol at a single agonist concentration and at a fixed membrane potential in experiments such as that shown in Fig. 1 E. First, the transition from the closed to the open channel state was compared between the first and the 20th carvacrol application (100 μm, 15 s, holding potential −60 mV), i.e. before and after agonist‐induced sensitisation. The time constant of channel opening determined by a mono‐exponential fit to the current traces was not significantly different between these applications (P = 0.43; paired t test). Therefore the transition to the open state was modelled with a single constant τ1 = 3.1 ± 1.1 s in both the basal and the sensitised state. Similarly, the time constant of channel closing after agonist removal was fitted after the first and the 20th application (P = 0.16; paired t test) and the closing time constant was determined as τ2 = 1.5 ± 0.5 s.
Based on these findings, we propose a model with two open and two closed conformations, either in the unsensitised (C0–O0) or the sensitised channel state (C1–O1) (Fig. 7 A). Because time constants for agonist‐dependent channel opening or closing are not different in the unsensitised or sensitised state (see above), we suggest that the rapid gating of TRPA1 by agonist and voltage (applicable for both C0–O0 and C1–O1) is independent of the additional slow agonist‐induced effect, which can be seen in the slow shift of voltage dependence (Fig. 6 E) and which is described by a transition to an additional sensitised channel closed–open state (C1–O1). We therefore suggest that sensitisation consists of a slow and progressive accumulation of channels in the sensitised state (C1–O1). Since this slow effect is agonist‐dependent, we modelled this with one time constant (τ3) for both transitions C0–C1 and O0–O1. To determine the time constants of this process, the change in V ½ at 0 s, 10 s, 60 s, 120 s as well as 180 s and 240 s (cumulative) after exposure to carvacrol was fitted by a double exponential with the fast time constant being equal to the time constant for transition τ1 = 3.1 s (see above) from the closed to the open state (Fig. 7 C). The slow time constant τ3 was taken to represent the transition from state 0 to state 1 and was determined as τ3 = 130 ± 27 s. In addition, this double‐exponential fit resulted in a V ½ shift from a baseline of +174 ± 11 mV to +99 ± 13 mV (75 mV shift) with time constant τ1 and an additional V ½ shift to ‐175 ± 24 mV with time constant τ3 (274 mV shift) (see Fig. 7 C).
With these parameters, a repetitive carvacrol application was modelled, considering the four possible channel states, the described transitions, and the V ½‐dependent open probability of the four channel states at −60 mV using the described Boltzmann equation (Fig. 7 B). The time to reach a rapid equilibrium of the open probability is an order of magnitude shorter than the slow V ½ shift and was therefore ignored in the model. The modelled result shows a good agreement with the experimental observations (see e.g. Fig. 1 E), and can therefore explain the slow current increase. At the end of the first application in the model 14 % of all channels are open, while a 4.7‐fold increase is predicted at the end of the 12th application (black trace). Figure 7 B overlays the percentage of channels in state 1 (red trace) to demonstrate how the observed current increase can be predicted by a slow transition to this state.
Because of the different values of V ½ in state O0 and O1, the model predicts that when both states are occupied there will be a mixture of values of V ½ and therefore a detectable decrease in the slope of conductance–voltage relationships. A minimum slope is to be expected when 50 % of channels are in each state (Fig. 7 D). Indeed, this decrease was observed in our experiments (see Fig. 6 E), the slope of the current–voltage relationship at 120 s (time point 4) was significantly lower compared to time points 2 and 8 (P < 0.001 and P = 0.002, respectively; repeated measures ANOVA with Bonferroni post hoc analysis; n = 11).
Discussion
We have shown that repeated activation of the irritant receptor TRPA1 by agonists, but not by membrane voltage, leads to a pronounced sensitisation of TRPA1 under calcium‐free conditions. This agonist‐induced sensitisation could be observed both in HEK293t cells expressing hTRPA1 and in adult mouse DRG neurons. Sensitisation is not limited to a specific class of agonist, as it is seen both with AITC and with carvacrol, which activate TRPA1 at different binding sites and by different mechanisms. In addition, the presence of intracellular ATP and the formation of lipid rafts, which are known to modulate TRPV1 (Tominaga et al. 2001; Liu et al. 2006; Szőke et al. 2010), as well as activation of PKA, which has been shown to enhance activation of TRPA1 (Wang et al. 2008 a), are not involved in agonist‐induced sensitisation of TRPA1. A further possibility is that exposure to agonists may increase trafficking and insertion of TRPA1 channels into the cell membrane, as has been proposed for sensitisation by AITC (Schmidt et al. 2009). In the present study, however, we were not able to find any support for enhanced trafficking as an explanation for the sensitisation induced by agonists of TRPA1. It should be noted that AITC‐induced trafficking of TRPA1, reported by Schmidt et al. (2009), was calcium‐dependent and did not occur in a calcium‐free environment, while the sensitisation reported in the present study was observed in the absence of extracellular calcium and is therefore likely to be a different phenomenon. Blocking intracellular protein transport with brefeldin A, which blocks trafficking of P2X and TRP channels (Fabbretti et al. 2006; Johansen et al. 2006; Lalo et al. 2010; Ma et al. 2010), did not reduce sensitisation caused by carvacrol. Furthermore, we did not see an enhancement in maximal current activated by a saturating dose of agonist, as would be expected if sensitisation was due to an increase in surface expression of TRPA1, and in fact observed tachyphylaxis of maximal responses.
Sensitisation is independent of the agonist's site of action, because channel activation by AITC sensitised responses to carvacrol, even though the two agonists activate TRPA1 at distinct sites. Furthermore, sensitisation is independent of ion flux as it occurs during channel block by HC030031. Current traces during prolonged activation showed a pattern of two distinct phases, with a secondary increase in inward current that has a surprisingly slow time constant that exceeds by far the time constant of channel activation and even of pore dilation (Chen et al. 2009; Meseguer et al. 2014). In the absence of evidence for other possible mechanisms, we hypothesised that the agonist‐induced sensitisation of TRPA1 may depend on changes in the intrinsic gating properties of the channel. A similar pattern was found in the TRPV3 ion channel, which is also sensitised by repeated activation by heat and by agonists (Chung et al. 2005; Xu et al. 2006; Liu et al. 2011). Voltage‐dependent gating of TRPA1 depends on a number of factors including the presence or absence of external calcium, the cell type and whether human or rodent TRPA1 is studied. Most studies used mouse TRPA1, expressed in Chinese hamster ovary cells (Karashima et al. 2007, 2009; Macpherson et al. 2007; Andersson et al. 2008; Fajardo et al. 2008; Meseguer et al. 2008; Talavera et al. 2009), before major species differences were discovered (Xiao et al. 2008 a). In the present study we used the human isoform, transiently transfected into HEK293t cells.
Synergy between activation of TRPA1 by agonists and by membrane voltage
TRPA1 was found to be activated by strong depolarisation even in the absence of agonist, and carvacrol caused a shift of the voltage dependence of channel gating to more negative membrane potentials, as has been reported for other TRPA1 agonists and for cold temperatures (Karashima et al. 2007, 2009; Zurborg et al. 2007; Fajardo et al. 2008; Meseguer et al. 2008; Talavera et al. 2009). Here, we measured a V ½ of +170 mV under agonist‐free conditions. The voltage sensitivity of TRPA1 activation was very weak, with a mean value for the slope factor of 54.9 mV (Fig. 6 E). This corresponds to a gating charge movement of z = 0.46 for all four subunits across the full electric field, or z = 0.12 per subunit. The value of z obtained here is similar to that previously reported (0.375, Karashima et al. 2009) and corresponds to movement of a single charge per subunit across only a small fraction (12 %) of the membrane field.
The V ½ was rapidly shifted in the negative direction by agonist, to +94 mV following 10 s application of carvacrol, and then became more negative with a slow time course, consistent with the slow development of sensitisation, reaching a value of −36 mV after 120 s exposure to carvacrol. The shift is rapidly and completely reversed on removal of agonist, but sensitisation can be seen to be conserved even if agonist is removed for times exceeding 240 s (Fig. 5 C and D), a property that is shared by the negative shift of V ½, because both current and the shift in V ½ are seen to be enhanced following a second application of agonist (Fig. 6 E).
In contrast to other voltage‐dependent TRP channels, TRPA1 is inactivated by strong depolarisation (see Fig. 6). A similar inactivation has been reported by some studies (Karashima et al. 2007, 2008; Macpherson et al. 2007; Andersson et al. 2008; Fajardo et al. 2008; Talavera et al. 2009) but not by others (Doerner et al. 2007; Meseguer et al. 2008; Banke et al. 2010; Wan et al. 2013). It has been proposed that this discrepancy is due to differing factors in recording conditions that negatively shift the V ½ for inactivation to varying degrees (Wan et al. 2013). Our results clearly identify inactivation of TRPA1, both in steady‐state I–V relations (Fig. 6 C and D) and in tail currents (Fig. 6 E), and show that channel inactivation is enhanced by prolonged agonist exposure (Fig. 6 C and D). In the absence of a clear explanation for TRPA1 inactivation at strongly positive voltages we restricted analysis of activation curves to membrane voltages below +100 mV, where inactivation is minimal.
A model for sensitisation of TRPA1
How is agonist‐induced sensitisation caused? One possible explanation could be relief of voltage‐dependent pore block by calcium, as has been reported for TRPV3 (Chung et al. 2005; Xiao et al. 2008 b), although a more recent study has shown that a component of sensitisation of TRPV3 is calcium‐independent and therefore resembles the sensitisation studied in the present paper (Liu et al. 2011). Another possibility is a time‐dependent increase in pore diameter, which is enhanced under calcium‐free conditions (Chen et al. 2009; Banke et al. 2010; Karashima et al. 2010; Bobkov et al. 2011). However, pore dilation occurs within less than 15 s of AITC application and well before maximal channel activation (Chen et al. 2009; Banke et al. 2010), which challenges the contribution of pore dilation for the long‐lasting agonist‐induced sensitisation that was shown here. A further possibility is that agonists may induce a transformation of the channel conformation leading to enhanced sensitivity to membrane voltage and therefore to sensitisation. We propose a model for TRPA1 sensitisation (Fig. 7 A) in which the channel is not only reversibly gated by agonists but also, with a time constant of more than one minute, enters a sensitised conformational state ‘1’ that allows enhanced gating. This conformational change is likely to involve a slow shift in charge distribution within the TRPA1 molecule such that the gating charge responsible for voltage‐dependent channel activation is activated at progressively more negative membrane potentials (Fig. 6 E). Finally, with prolonged agonist application, more and more channels accumulate in this sensitised state ‘1’, explaining the observed slow shift in voltage dependence and the accompanying change in slope factor as well as the increased inward current (Fig. 7 D).
Modelling of repetitive agonist application using experimentally determined parameters resembles our experimental observations. Even if the underlying biology is more complex, a simple model with four states and three transition time constants is sufficient to produce the major features of the slow current increase in response to agonist (Fig. 7 B–D) and the shift in V ½ (Fig. 7 D).
Alternative models which could generate a slow current increase have been considered, e.g. as described for TRPV3 where this was termed ‘hysteresis’ (Liu et al. 2011). Adopting that model, two sequential closed states before transition to one open state could generate hysteresis in the case of a longer time constant between the two closed states compared to the transition to the open state. In such a scenario only the slowly rising amount of channels which can open would explain the hysteresis, and the observed V ½ shift would not be reflected. Alternatively, one closed and two sequential open states could also generate hysteresis, but a different kinetic upon agonist removal should be expected in this case. Finally, if in our model the transition C1–O1 was at least an order of magnitude faster than C0–O0, hysteresis could also occur, but this was excluded by our observations that activation and deactivation time constants are similar in the basal and sensitised states (see Results section).
Finally, we note that while activation of TRP channels by agonists or by voltage were initially thought to be equivalent (Voets et al. 2004), a more recent study has demonstrated that these two modes of activation are in fact not equivalent (Matta & Ahern, 2007). In the case of TRPV1 and TRPM8, membrane voltage acts only as a partial agonist, in that the increase in channel open probability caused by voltage is much smaller than the increase in open probability caused by thermal activation, and at high agonist concentrations gating is voltage independent.
Physiological relevance of sensitisation of TRPA1
Considering the physiological relevance of agonist‐induced sensitisation of TRPA1, it is likely that it would initially be masked by calcium‐mediated inhibition of the channel. However, the sensitising effect of agonists increases over time and sensitisation may eventually overcome the inhibition of the channel by calcium, leading in total to a sensitised channel state. Agonist‐induced sensitisation could provide a mechanism for the detection of repeated or long‐term exposure to harmful irritants. Such a mechanism would be especially appealing in the airways, as many TRPA1 agonists can be ingested or inhaled and the channel is implicated in the development of inflammatory airway diseases (Andrè et al. 2008; Birrell et al. 2009). This hypothesis is supported by recent findings of Kunkler et al. (2015) showing that long‐term inhalation exposure of rats to sub‐activating doses of the TRPA1 agonist acrolein led to increased meningeal blood flow in response to acute nasal challenge with mustard oil. Animals had been pre‐exposed to acrolein 4 h per day for 4 days, which agrees with our findings that agonist‐induced sensitisation of TRPA1 is very long lasting and potentially irreversible. This mechanism of TRPA1 sensitisation may therefore represent an important physiological function.
In summary, we have shown that prolonged agonist application sensitises the TRPA1 channel both in nociceptive neurons and when heterologously expressed and that this slow process is agonist‐driven and is intrinsic to the channel itself. Sensitisation is accompanied by a slowly developing shift of voltage‐dependent activation towards more negative membrane potentials and thus represents a slow gain of function. Agonist‐induced sensitisation of TRPA1 may provide a mechanism for the detection of long‐term exposure to harmful stimuli, which would be especially valuable in the airways, where many irritants activate TRPA1.
Additional information
Competing interests
The authors declare no competing financial interests.
Author contributions
J.E.M., M.J.M.F. and P.A.M. conceptualised and designed the experiments; J.E.M. and M.J.M.F. collected and assembled the data; J.E.M., M.J.M.F. and P.A.M. analysed and interpreted the data; J.E.M. drafted the manuscript; M.J.M.F. and P.A.M. revised the manuscript. All authors approved the final version of the manuscript. All persons designated as authors qualify for authorship and all those who qualify for authorship are listed.
Funding
Funded by grants from the Biotechnology and Biological Sciences Research Council, UK (BBSRC) to P.A.M., PhD studentship support from the Department of Pharmacology Cambridge, the Ernst Schering Foundation, the Deutsche Schmerzgesellschaft e.V. and the German Academic Exchange Service to J.E.M. and a fellowship from the Alexander von Humboldt Foundation to M.J.M.F.
This is an Editor's Choice article from the 15 November 2016 issue.
References
- Anand U, Otto WR, Facer P, Zebda N, Selmer I, Gunthorpe MJ, Chessell IP, Sinisi M, Birch R & Anand P (2008). TRPA1 receptor localisation in the human peripheral nervous system and functional studies in cultured human and rat sensory neurons. Neurosci Lett 438, 221–227. [DOI] [PubMed] [Google Scholar]
- Andersson DA, Gentry C, Moss S & Bevan S (2008). Transient receptor potential A1 is a sensory receptor for multiple products of oxidative stress. J Neurosci 28, 2485–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrè E, Campi B, Materazzi S, Trevisani M, Amadesi S, Massi D, Creminon C, Vaksman N, Nassini R, Civelli M, Baraldi PG, Poole DP, Bunnett NW, Geppetti P & Patacchini R (2008). Cigarette smoke‐induced neurogenic inflammation is mediated by alpha,beta‐unsaturated aldehydes and the TRPA1 receptor in rodents. J Clin Invest 118, 2574–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandell M, Story GM, Hwang SW, Viswanath V, Eid SR, Petrus MJ, Earley TJ & Patapoutian A (2004). Noxious cold ion channel TRPA1 is activated by pungent compounds and bradykinin. Neuron 41, 849–857. [DOI] [PubMed] [Google Scholar]
- Banke TG, Chaplan SR & Wickenden AD (2010). Dynamic changes in the TRPA1 selectivity filter lead to progressive but reversible pore dilation. Am J Physiol Cell Physiol 298, C1457–C1468. [DOI] [PubMed] [Google Scholar]
- Bautista DM, Jordt S‐E, Nikai T, Tsuruda PR, Read AJ, Poblete J, Yamoah EN, Basbaum AI & Julius D (2006). TRPA1 mediates the inflammatory actions of environmental irritants and proalgesic agents. Cell 124, 1269–1282. [DOI] [PubMed] [Google Scholar]
- Bell JT, Loomis AK, Butcher LM, Gao F, Zhang B, Hyde CL, Sun J, Wu H, Ward K, Harris J, Scollen S, Davies MN, Schalkwyk LC, Mill J; MuTHER Consortium, Williams FM, Li N, Deloukas P, Beck S, McMahon SB, Wang J, John SL & Spector TD (2014). Differential methylation of the TRPA1 promoter in pain sensitivity. Nat Commun 5, 2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell MA, Belvisi MG, Grace M, Sadofsky L, Faruqi S, Hele DJ, Maher SA, Freund‐Michel V & Morice AH (2009). TRPA1 agonists evoke coughing in guinea pig and human volunteers. Am J Respir Crit Care Med 180, 1042–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bobkov YV, Corey EA & Ache BW (2011). The pore properties of human nociceptor channel TRPA1 evaluated in single channel recordings. Biochim Biophys Acta 1808, 1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Btesh J, Fischer MJM, Stott K & McNaughton PA (2013). Mapping the binding site of TRPV1 on AKAP79: Implications for inflammatory hyperalgesia. J Neurosci 33, 9184–9193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres AI, Brackmann M, Elia MD, Bessac BF, Del Camino D, D'Amours M, Witek JS, Fanger CM, Chong JA, Hayward NJ, Homer RJ, Cohn L, Huang XZ, Moran MM & Jordt SE (2009). A sensory neuronal ion channel essential for airway inflammation and hyperreactivity in asthma. Proc Natl Acad Sci USA 106, 9099–9104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P & McNaughton P (1996). A novel heat‐activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci USA 93, 15435–15439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Kim D, Bianchi BR, Cavanaugh EJ, Faltynek CR, Kym PR & Reilly RM (2009). Pore dilation occurs in TRPA1 but not in TRPM8 channels. Mol Pain 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung M‐K, Güler AD & Caterina MJ (2005). Biphasic currents evoked by chemical or thermal activation of the heat‐gated ion channel, TRPV3. J Biol Chem 280, 15928–15941. [DOI] [PubMed] [Google Scholar]
- Dai Y, Wang S, Tominaga M, Yamamoto S, Fukuoka T, Higashi T, Kobayashi K, Obata K, Yamanaka H & Noguchi K (2007). Sensitization of TRPA1 by PAR2 contributes to the sensation of inflammatory pain. J Clin Invest 117, 1979–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Roche J, Eberhardt MJ, Klinger AB, Stanslowsky N, Wegner F, Koppert W, Reeh PW, Lampert A, Fischer MJM & Leffler A (2013). The molecular basis for species‐specific activation of human TRPA1 protein by protons involves poorly conserved residues within transmembrane domains 5 and 6. J Biol Chem 288, 20280–20292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerner JF, Gisselmann G, Hatt H & Wetzel CH (2007). Transient receptor potential channel A1 is directly gated by calcium ions. J Biol Chem 282, 13180–13189. [DOI] [PubMed] [Google Scholar]
- Eberhardt MJ, Filipovic MR, Leffler A, de la Roche J, Kistner K, Fischer MJ, Fleming T, Zimmermann K, Ivanovic‐Burmazovic I, Nawroth PP, Bierhaus A, Reeh PW & Sauer SK (2012). Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1) a possible mechanism of metabolic neuropathies. J Biol Chem 287, 28291–28306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbretti E, D'Arco M, Fabbro A, Simonetti M, Nistri A & Giniatullin R (2006). Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene‐related peptide. J Neurosci 26, 6163–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo O, Meseguer V, Belmonte C & Viana F (2008). TRPA1 channels: Novel targets of 1,4‐dihydropyridines. Channels 2, 429–438. [DOI] [PubMed] [Google Scholar]
- Fischer MJM, Btesh J & McNaughton PA (2013). Disrupting sensitization of transient receptor potential vanilloid subtype 1 inhibits inflammatory hyperalgesia. J Neurosci 33, 7407–7414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry C, Stoakley N, Andersson DA & Bevan S (2010). The roles of iPLA2, TRPM8 and TRPA1 in chemically induced cold hypersensitivity. Mol Pain 6, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman A, Chuang H, Bautista DM & Julius D (2006). TRP channel activation by reversible covalent modification. Proc Natl Acad Sci USA 103, 19564–19568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen ME, Reilly CA & Yost GS (2006). TRPV1 Antagonists elevate cell surface populations of receptor protein and exacerbate TRPV1‐mediated toxicities in human lung epithelial cells. Toxicol Sci 89, 278–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordt SE, Bautista DM, Chuang HH, Mckemy DD, Zygmunt PM, Hogestatt ED, Meng ID & Julius D (2004). Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature 427, 260–265. [DOI] [PubMed] [Google Scholar]
- Karashima Y, Damann N, Prenen J, Talavera K, Segal A, Voets T & Nilius B (2007). Bimodal action of menthol on the transient receptor potential channel TRPA1. J Neurosci 27, 9874–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karashima Y, Prenen J, Meseguer V, Owsianik G, Voets T & Nilius B (2008). Modulation of the transient receptor potential channel TRPA1 by phosphatidylinositol 4,5‐biphosphate manipulators. Pflügers Arch 457, 77–89. [DOI] [PubMed] [Google Scholar]
- Karashima Y, Prenen J, Talavera K, Janssens A, Voets T & Nilius B (2010). Agonist‐induced changes in Ca2+ permeation through the nociceptor cation channel TRPA1. Biophys J 98, 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karashima Y, Talavera K, Everaerts W, Janssens A, Kwan KY, Vennekens R, Nilius B & Voets T (2009). TRPA1 acts as a cold sensor in vitro and in vivo . Proc Natl Acad Sci USA 106, 1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, Woods CG, Jones NG, Paterson KJ, Fricker FR, Villegas A, Acosta N, Pineda‐Trujillo NG, Ramírez JD, Zea J, Burley MW, Bedoya G, Bennett DL, Wood JN & Ruiz‐Linares A (2010). A gain‐of‐function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 66, 671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkler PE, Zhang L, Pellman JJ, Oxford GS & Hurley JH (2015). Sensitization of the trigeminovascular system following environmental irritant exposure. Cephalalgia 35, 1192–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan KY, Allchorne AJ, Vollrath MA, Christensen AP, Zhang DS, Woolf CJ & Corey DP (2006). TRPA1 contributes to cold, mechanical, and chemical nociception but is not essential for hair‐cell transduction. Neuron 50, 277–289. [DOI] [PubMed] [Google Scholar]
- Lalo U, Allsopp RC, Mahaut‐Smith MP & Evans RJ (2010). P2X1 receptor mobility and trafficking; regulation by receptor insertion and activation. J Neurochem 113, 1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SP, Buber MT, Yang Q, Cerne R, Cortes RY, Sprous DG & Bryant RW (2008). Thymol and related alkyl phenols activate the hTRPA1 channel. Br J Pharmacol 153, 1739–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yao J, Zhu MX & Qin F (2011). Hysteresis of gating underlines sensitization of TRPV3 channels. J Gen Physiol 138, 509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Huang W, Wu D & Priestley JV (2006). TRPV1, but not P2X3, requires cholesterol for its function and membrane expression in rat nociceptors. Eur J Neurosci 24, 1–6. [DOI] [PubMed] [Google Scholar]
- Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS & Yao X (2010). Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4‐c1 heteromeric channels to the plasma membrane. Arterioscler Thromb Vasc Biol 30, 2249–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara CR, Mandel‐Brehm J, Bautista DM, Siemenst J, Deranian KL, Zhao M, Hayward NJ, Chong JA, Julius D, Moran MM & Fanger CM (2007). TRPA1 mediates formalin‐induced pain. Proc Natl Acad Sci USA 104, 13525–13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macpherson LJ, Dubin AE, Evans MJ, Marr F, Schultz PG, Cravatt BF & Patapoutian A (2007). Noxious compounds activate TRPA1 ion channels through covalent modification of cysteines. Nature 445, 541–545. [DOI] [PubMed] [Google Scholar]
- Matta JA & Ahern GP (2007). Voltage is a partial activator of rat thermosensitive TRP channels. J Physiol 585, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseguer V et al (2014). TRPA1 channels mediate acute neurogenic inflammation and pain produced by bacterial endotoxins. Nat Commun 5, 3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meseguer V, Karashima Y, Talavera K, D'Hoedt D, Donovan‐Rodríguez T, Viana F, Nilius B & Voets T (2008). Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J Neurosci 28, 576–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Duggan A, Kumar G & Garcia‐Anoveros J (2005). Nociceptor and hair cell transducer properties of TRPA1, a channel for pain and hearing. J Neurosci 25, 4052–4061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Katsura H, Mizushima T, Yamanaka H, Kobayashi K, Dai Y, Fukuoka T, Tokunaga A, Tominaga M & Noguchi K (2005). TRPA1 induced in sensory neurons contributes to cold hyperalgesia after inflammation and nerve injury. J Clin Invest 115, 2393–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisinghani M, Zhong L, Jeffry JA, Bishnoi M, Pabbidi RM, Pimentel F, Cao DS, Evans MS & Premkumar LS (2011). Activation characteristics of transient receptor potential ankyrin 1 and its role in nociception. Am J Physiol Cell Physiol 301, C587–C600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Dubin AE, Petrus MJ, Earley TJ & Patapoutian A (2009). Nociceptive signals induce trafficking of TRPA1 to the plasma membrane. Neuron 64, 498–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, McIntyre P, Jegla T, Bevan S & Patapoutian A (2003). ANKTM1, a TRP‐like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829. [DOI] [PubMed] [Google Scholar]
- Szőke É, Börzsei R, Tóth DM, Lengl O, Helyes Z, Sándor Z & Szolcsányi J (2010). Effect of lipid raft disruption on TRPV1 receptor activation of trigeminal sensory neurons and transfected cell line. Eur J Pharmacol 628, 67–74. [DOI] [PubMed] [Google Scholar]
- Talavera K, Gees M, Karashima Y, Meseguer VM, Vanoirbeek JA, Damann N, Everaerts W, Benoit M, Janssens A, Vennekens R, Viana F, Nemery B, Nilius B & Voets T (2009). Nicotine activates the chemosensory cation channel TRPA1. Nat Neurosci 12, 1293–1299. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M & Masu M (2001). Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP‐evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98, 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB & McNaughton PA (2001). Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol 534, 813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Droogmans G, Wissenbach U, Janssens A, Flockerzi V & Nilius B (2004). The principle of temperature‐dependent gating in cold‐ and heat‐sensitive TRP channels. Nature 430, 748–754. [DOI] [PubMed] [Google Scholar]
- Wang S, Dai Y, Fukuoka T, Yamanaka H, Kobayashi K, Obata K, Cui X, Tominaga M & Noguchi K (2008. a). Phospholipase C and protein kinase A mediate bradykinin sensitization of TRPA1: a molecular mechanism of inflammatory pain. Brain 131, 1241–1251. [DOI] [PubMed] [Google Scholar]
- Wang YY, Chang RB, Waters HN, Mckemy DD & Liman ER (2008. b). The nociceptor ion channel TRPA1 is potentiated and inactivated by permeating calcium ions. J Biol Chem 283, 32691–32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Lu Y, Chen X, Xiong J, Zhou Y, Li P, Xia B, Li M, Zhu MX & Gao Z (2013). Bimodal voltage dependence of TRPA1: mutations of a key pore helix residue reveal strong intrinsic voltage‐dependent inactivation. Pflügers Arch 466, 1273–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Dubin AE, Bursulaya B, Viswanath V, Jegla TJ & Patapoutian A (2008. a). Identification of transmembrane domain 5 as a critical molecular determinant of menthol sensitivity in mammalian TRPA1 channels. J Neurosci 28, 9640–9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R, Tang J, Wang C, Colton CK, Tian J & Zhu MX (2008. b). Calcium plays a central role in the sensitization of TRPV3 channel to repetitive stimulations. J Biol Chem 283, 6162–6174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Delling M, Jun JC & Clapham DE (2006). Oregano, thyme and clove‐derived flavors and skin sensitizers activate specific TRP channels. Nat Neurosci 9, 628–635. [DOI] [PubMed] [Google Scholar]
- Zurborg S, Yurgionas B, Jira JA, Caspani O & Heppenstall PA (2007). Direct activation of the ion channel TRPA1 by Ca2+ . Nat Neurosci 10, 277–279. [DOI] [PubMed] [Google Scholar]