

Abstract
Key points
Kv1.2 and related voltage‐gated potassium channels have a highly conserved N‐linked glycosylation site in the first extracellular loop, with complex glycosylation in COS‐7 cells similar to endogenous Kv1.2 glycosylation in hippocampal neurons.
COS‐7 cells expressing Kv1.2 show a crucial role of this N‐linked glycosylation in the forward trafficking of Kv1.2 to the cell membrane.
Although both wild‐type and non‐glycosylated mutant Kv1.2 channels that have reached the cell membrane are internalized at a comparable rate, mutant channels are degraded at a faster rate.
Treatment of wild‐type Kv1.2 channels on the cell surface with glycosidase to remove sialic acids also results in the faster degradation of internalized channels.
Glycosylation of Kv1.2 is important with respect to facilitating trafficking to the cell membrane and enhancing the stability of channels that have reached the cell membrane.
Abstract
Studies in cultured hippocampal neurons and the COS‐7 cell line demonstrate important roles for N‐linked glycosylation of Kv1.2 channels in forward trafficking and protein degradation. Kv1.2 channels can contain complex N‐linked glycans, which facilitate cell surface expression of the channels. Additionally, the protein stability of cell surface‐expressed Kv1.2 channels is affected by glycosylation via differences in the degradation of internalized channels. The present study reveals the importance of N‐linked complex glycosylation in boosting Kv1.2 channel density. Notably, sialic acids at the terminal sugar branches play an important role in dampening the degradation of Kv1.2 internalized from the cell membrane to promote its stability.
Keywords: channel stability, channel trafficking, glycosylation, potassium channel, voltage‐gated channels
Key points
Kv1.2 and related voltage‐gated potassium channels have a highly conserved N‐linked glycosylation site in the first extracellular loop, with complex glycosylation in COS‐7 cells similar to endogenous Kv1.2 glycosylation in hippocampal neurons.
COS‐7 cells expressing Kv1.2 show a crucial role of this N‐linked glycosylation in the forward trafficking of Kv1.2 to the cell membrane.
Although both wild‐type and non‐glycosylated mutant Kv1.2 channels that have reached the cell membrane are internalized at a comparable rate, mutant channels are degraded at a faster rate.
Treatment of wild‐type Kv1.2 channels on the cell surface with glycosidase to remove sialic acids also results in the faster degradation of internalized channels.
Glycosylation of Kv1.2 is important with respect to facilitating trafficking to the cell membrane and enhancing the stability of channels that have reached the cell membrane.
Abbreviations
- DIV
days in vitro
- DMEM
Dulbecco's modified Eagle's medium
- EGFP
enhanced green fluorescent protein
- ER
endoplasmic reticulum
- HA
haemagglutinin
- MTX
maurotoxin
- PVDF
polyvinylidene difluoride
Introduction
Potassium channels serve many important physiological functions (Hille, 2001). The large diversity of voltage‐gated potassium channels (Jan et al. 1997; Coetzze et al. 1999) is crucial for controlling neuronal signalling and the action potential waveform. There are ∼40 mammalian genes that encode Kv channel α subunits, which are grouped into 12 families with the nomenclature Kv1 to Kv12 (Gutman et al. 2005). Four α subunits associate with one another as homo‐ or heteromeric tetramers to form the voltage‐gated potassium channel. Each α subunit consists of six membrane‐spanning segments (Fig. 1). The fourth transmembrane segment (S4) contains multiple arginine residues that sense changes in the membrane potential. A pore loop exists between the fifth and sixth transmembrane segments (S5 and S6), which forms the narrowest portion of the pore. The tetramers of α subunits may also be associated with auxiliary subunits. For Kv1 channels, auxiliary β subunits are globular proteins that interact with the cytoplasmic N‐terminal domain and have been implicated in surface expression and axonal targeting (Campomanes et al. 2002; Shi et al. 1996; Gu et al. 2006).
Figure 1. Kv1.2 N‐linked glycosylation is important for plasma membrane expression in cultured hippocampal neurons .
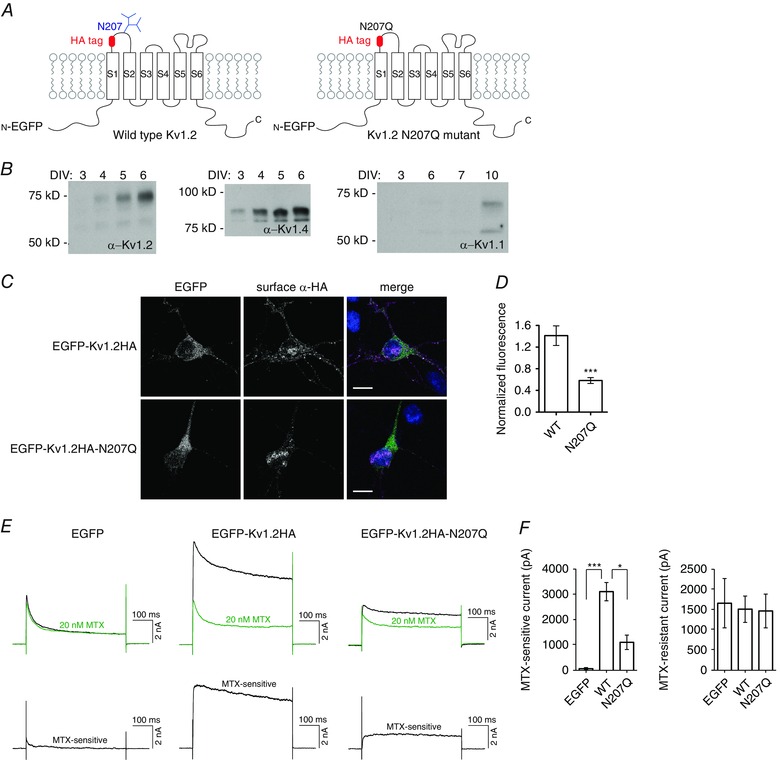
A, schematic representation of voltage‐gated potassium channel Kv1.2 constructs, showing transmembrane segments S1 to S6, N‐linked glycosylation site N207 and position of HA tag insertion. B, western blot of lysate from cultured hippocampal neurons from different DIV. C, cultured hippocampal neurons were transfected with EGFP‐Kv1.2HA constructs at 1 DIV, and non‐permeabilized surface immunocytochemistry with rat anti‐HA antibody (AlexaFluor 555‐conjugated anti‐rat secondary antibody labelling) was performed at 3 DIV. Confocal images were obtained and quantified to determine the fluorescence of surface anti‐HA antibody signal normalized to EGFP expression (as a measure of transfection efficiency). Merged images show EGFP‐Kv1.2HA (green), surface anti‐HA antibody (magenta) and DAPI (blue). Scale bar = 10 μm. D, quantification of fluorescence intensity for wild‐type (WT) (n = 9) and N207Q (n = 9) transfected neurons. *** P < 0.001, Student's t test. E, top traces are representative sample traces of whole cell patch clamp recording, stepping to +20 mV from a holding potential of −100 mV, of potassium current before (black) and after (green) application of 20 nm MTX. The MTX‐sensitive current was computed as the difference in current and plotted in the bottom traces. F, quantification of MTX‐sensitive and MTX‐resistant current for EGFP (n = 4), EGFP‐Kv1.2HA (n = 7) and EGFP‐Kv1.2HA‐N207Q (n = 8) transfected neurons. *** P < 0.001, * P < 0.05, one‐way ANOVA.
Besides diversity through association of different subunits, post‐translational modifications, such as phosphorylation, glycosylation or lipidation, may contribute to Kv channel diversity (Martens et al. 1999; Sutachan et al. 2005). The role of glycosylation in the function of proteins has not been fully clarified, although reported roles for membrane proteins include an involvement in protein folding in the endoplasmic reticulum (ER), trafficking to the cell surface, the lifetime of cell surface expression and activity modulation (Lennarz, 1983). Glycosylation is also important for animal physiology because deficiencies in glycosylation in the ER and Golgi cause carbohydrate‐deficient glycoprotein syndromes I–IV in humans (Jaeken et al. 1991; Carchon et al. 1999). Additionally, glycosylation of cardiac sodium and potassium channels may be important for preventing cardiac arrhythmias (Fozzard and Kyle, 2002).
In the present study, we describe our studies of N‐linked glycosylation of Kv1.2 channels for trafficking to, as well as stability on, the cell surface. Although by no means an exhaustive review of the literature, a few examples are provided from the many reports concerning these types of roles of N‐linked glycosylation for ion channels. Reduced cell surface expression has been observed for some non‐glycosylated mutant channels, including HERG (Gong et al. 2002 ), HCN2 (Li et al. 2015), Kv1.2 (Watanabe et al. 2007), Kv1.3 (Zhu et al. 2012), Kv1.4 (Watanabe et al. 2004, Watanabe et al. 2015), Trm4b (Woo et al. 2013) and CFTR (Glozman et al. 2009). A lack of N‐linked glycosylation is also linked to faster degradation in HERG, HCN, Kv1.4 and CFTR channels. Additionally, N‐linked glycosylation of Kv3.1 splice variants affects the plasma membrane arrangement in neuronal‐derived cells (Hall et al. 2015). Given that neuronal ion channels expressed in heterologous systems sometimes exhibit N‐linked oligomannose forms of the glycosylation characteristic of glycans introduced in the ER, it is important to examine the contributions of glycosylation in neurons and other cell types that display N‐linked complex or hybrid glycans indicative of Golgi apparatus processing.
In the present study, we report our investigations of the role of glycosylation in Kv1.2 channels. Kv1.2 α subunits have a single N‐linked glycosylation site at residue N207 in the first extracellular loop between transmembrane segments S1 and S2. Studies of channel glycosylation using heterologous expression systems are often confronted with the problem of the expressed neuronal channels displaying glycan structures that are not only different from those of channels expressed in neurons, but also less complex, as well as characteristic of glycans attached in the ER rather than the Golgi apparatus (Shi and Trimmer, 1999). To circumvent this problem, we used primary cultured hippocampal neurons to study the endogenous glycosylation of these channels and also used COS‐7 cells for heterologous expression. Although we found that the glycan structures of Kv1.2 channels were different in lysates of neuronal cultures compared to the COS‐7 cell culture, we found a significant expression of Golgi‐processed complex glycans on the plasma membrane of COS‐7 cells. Furthermore, we found that N‐linked glycosylation was important for cell surface expression of Kv1.2 in cultured neurons and COS‐7 cells.
We also found that glycosylation affects the protein stability of channels expressed on the cell surface. Although wild‐type and glycosylation‐deficient channels were internalized at similar rates from the plasma membrane, the glycosylation‐deficient channels were degraded faster than wild‐type channels. To clarify the role of the glycosylation of channels on their stability, we tested for the effect of enzymes that remove sugars from channels on the cell surface. Further analysis using sialidase V revealed the faster degradation of surface expressed channels lacking sialic acid. Although sialylation of ion channels is known to be important for channel function (Ednie et al. 2012, Baycin‐Hizal et al. 2014), our observation of a relationship between sialylation of a channel and its cell surface stability represents a novel glycan structure‐specific role for glycosylation.
Methods
Ethical approval
All experiments described in the present study involving animals were approved by the University of California San Francisco Institutional Animal Care and Use Committee (IACUC) under protocol AN100730 and were conducted in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. In preparation for primary cultures of embryonic hippocampal neurons (see below), a pregnant female Sprague–Dawley rat with 18‐day‐old embryos was killed by carbon dioxide inhalation (as monitored by observing respiration and confirmed by performing a firm toe pinch) and bilateral thoracotomy just prior to removal of embryos by caesarian section. Embryos were physically killed by decapitation, followed by immediate removal of brain tissue. The investigators of the present study understand the ethical principles that The Journal of Physiology operates under, and their work complied with the animal ethics checklist reported by Grundy (2015).
Animal care
Donor pregnant female Sprague–Dawley rats were obtained from Charles River (Wilmington, MA, USA). All rats were singly housed at the University of California San Francisco barrier vivarium in accordance with the institutional requirements for animal care. All rats used in the present study were maintained under a standard 12:12 h light/dark cycle and were allowed ad libitum access to water and chow.
Materials
Chemical reagents used include sulfo‐NHS‐SS‐biotin (Life Technologies, Grand Island, NY, USA) and maurotoxin (Alomone Labs, Jerusalem, Israel). The glycosidases PNGase F, Endo H, sialidase T and sialidase V (Prozyme, Hayward, CA, USA) and fetuin (Sigma‐Aldrich, St Louis, MO, USA) were also used. Immunological reagents used include rat anti‐haemagglutinin (HA) (3F10; Roche, Basel, Switzerland), mouse anti‐HA (12CA5; Roche), mouse anti‐Kv1.2 (K14/16; NeuroMab, Davis, CA, USA), mouse anti‐Kv1.4 (K13/31; NeuroMab), mouse anti‐Kv1.1 (K20/78; NeuroMab), mouse anti‐GAPDH (GAPDH‐71.1 IgM; Sigma‐Aldrich) and mouse anti‐EEA1 (14/EEA1; BD Biosciences, San Jose, CA, USA) monoclonal antibodies and rat anti‐HA affinity matrix (Roche). Secondary Alexa Fluor‐conjugated antibodies (Life Technologies) and unlabelled AffiniPure goat anti‐mouse and AffiniPure goat anti‐rat (Jackson ImmunoResearch, West Grove, PA, USA) were also used.
DNA constructs and mutagenesis
EGFP‐Kv1.2HA was reported by Gu et al. (2003). EGFP‐Kv1.2HA‐N207Q was made by site‐directed mutagenesis of EGFP‐Kv1.2HA using high fidelity Pfu turbo DNA polymerase (Stratagene, La Jolla, CA, USA) and verified by sequencing.
Kv1.2N207Q was constructed by site‐directed mutagenesis of Kv1.2‐pcDNA3 with the QuikChange strategy using high fidelity Pfu turbo DNA polymerase (Stratagene) and verified by sequencing. The HA tag was inserted into the coding sequence between residues 201 and 202 with the QuikChange strategy.
Primary cultures of hippocampal neurons
Primary cultures of hippocampal neurons from 18‐day embryonic rats were prepared as previously described (Shi et al. 2003). Coverslips (Warner Instruments, Hamden, CT, USA) were pretreated with nitric acid and precoated with poly‐l‐lysine (0.1 ml ml−1; Sigma‐Aldrich). Each 12 mm coverslip was plated with 5 × 104 neurons. Cultures were maintained in Neurobasal medium (Life Technologies) containing B27 extract (Life Technologies), 0.5 mm glutamine, 100 units of penicillin and 100 μg mL–1 streptomycin. For transient transfection, neurons in culture were treated with Neurobasal medium containing the cDNA plasmid and Lipofectamine 2000 (Life Technologies).
COS‐7 cell cultures
COS‐7 cells were grown in Dulbecco's modified Eagle's medium (DMEM) high glucose/F12 (50%/50%), 10% fetal bovine serum, 2 mm glutamine, 100 units of penicillin and 100 mg ml−1 streptomycin. Coverslips (Warner Instruments) or plastic six‐ or 12‐well cell culture plates were precoated with poly‐l‐lysine. For transient transfection, cells were treated with DMEM I medium containing the cDNA plasmid and Lipofectamine 2000 (Life Technologies).
Immunoblot analysis
Cultured cells were lysed with lysis buffer (50 mm Tris, 300 mm NaCl, 5 mm EDTA, 1% Triton X‐100, pH 7.4, plus complete EDTA‐free protease inhibitor cocktail tablet) for 15 min on ice. Lysate was cleared by centrifugation at 20,000 g for 15 min. Proteins were denatured in SDS protein gel sample loading buffer with the addition of 100 mm dithiothreitol and 10 mm tris(2‐carboxyethyl)phosphine for 30 min at 50°C. Proteins were separated by SDS‐PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon‐P; Millipore, Billerica, MA, USA). The membrane was blocked with blocking buffer (5% milk in Tris‐buffered saline: 50 mm Tris, 150 mm NaCl, pH 7.4) for at least 1 h then incubated with primary antibodies in blocking buffer with 0.1% Tween‐20 (at room temperature for at least 1 h or at 4°C overnight) with mouse anti‐Kv1.2 (dilution 1:2000), mouse anti‐Kv1.4 (dilution 1:1000), mouse anti‐Kv1.1 (dilution 1:1000) and mouse anti‐GAPDH (dilution 1:1000). After washing three times with washing buffer (0.1% Tween‐20 in Tris‐buffered saline), the membrane was probed with HRP‐conjugated secondary antibodies (1:1000) for 1 h. The membrane was washed three times with washing buffer and visualized by ECL development (Amersham Biosciences, Little Chalfont, UK). Exposed film was scanned and quantification was performed using Fiji software (http://fiji.sc).
Immunocytochemistry
For non‐permeabilized immunostaining of surface proteins, transfected primary hippocampal neurons or COS‐7 cells were fixed with 4% formaldehyde/4% sucrose in PBS for 5 min then immediately washed once with PBS for 10 s and three times with PBS for 5 min each. Samples were blocked with blocking solution (5% normal goat serum in PBS) for at least 1 h and then incubated with rat anti‐HA antibody (dilution 1:200) in blocking solution (at room temperature for at least 2 h or at 4°C overnight). After washing six times with PBS for 5 min each, cells were incubated for 1 h with goat anti‐rat Alexa Fluor 555 secondary antibody (dilution 1:1000). After washing three times with PBS for 5–10 min each, the coverslips were mounted on Superfrost microslides (VWR International Ltd, Lutterworth, UK) using DAPI Fluoromount‐G (SouthernBiotech, Birmingham, AL, USA). Images were acquired on a SP5 confocal microscope (Leica Microsystems, Wetzlar, Germany) and analysis was performed using Fiji software.
Electrophysiological recordings
Two days after the transfection of primary hippocampal neurons or COS‐7 cells, membrane currents and membrane capacitance were measured in the standard whole‐cell, patch clamp configuration. Datawere acquired at 5 kHz with Clampex, version 10 (Molecular Devices, Sunnyvale, CA, USA). Data with a series resistance lower than 25 MΩ were included in the present study and series resistance was compensated by 65–75%. The extracellular solution contained (in mm): 140 NaCl, 10 Hepes, 2 CaCl2 and 1 MgCl2 and the pH was adjusted to 7.2 with NaOH. The intracellular solution contained (in mm): 140 KCl, 10 Hepes, 5 Mg2ATP, 1 Na3GTP, 10 sodium phosphocreatine and 0.05 EGTA and the pH was adjusted to 7.2 with KOH. Pipettes were pulled from 1.5 mm borosilicate glass capillaries and had a resistance of 2–4 MΩ when filled with the intracellular solution. All of the experiments were performed at room temperature. KV currents were activated by a 500 ms voltage pulse stepping to +20 mV from a holding potential of −70 mV and currents that were sensitive to extracellular 20 nm maurotoxin were defined as Kv1.2 currents. The activation and deactivation time constants (τ) were determined by single exponential fittings using Clampfit, version 10 (Molecular Devices).
Glycosidase analysis
For Kv1.2HA expressed in COS‐7 cells, 2 days after transfection, cells were lysed and HA‐tagged Kv1.2 proteins were immunoprecipitated with rat anti‐HA affinity matrix. Glycosidase reactions were performed on‐resin in accordance with the manufacturer's (Prozyme) recommended protocol with the supplied buffers at 37°C for 4 h. Proteins were eluted from the HA‐affinity matrix in SDS protein gel sample loading buffer with the addition of 100 mm dithiothreitol and 10 mm tris(2‐carboxyethyl)phosphine for 30 min at 50°C. Proteins were separated by SDS‐PAGE, transferred to PVDF membrane, and immunoblot analysis was continued as described above.
Glycosidase reactions were also performed on fetuin as a control glycoprotein. Fetuin samples were separated by SDS‐PAGE and visualized by Coomasie staining.
For glycosidase analysis of surface Kv1.2 and Kv1.2HA channels, biotinylation reactions were performed as described below. Before elution from the NeutrAvidin resin (Thermo Fisher Scientific, Waltham, MA, USA), glycosidase reactions were performed on‐resin. Proteins were eluted from the resin in SDS protein gel sample loading buffer with the addition of 100 mm dithiothreitol and 10 mm tris(2‐carboxyethyl)phosphine for 30 min at 50°C. Proteins were separated by SDS‐PAGE, transferred to PVDF membrane, and immunoblot analysis was continued as described above.
Surface protein biotinylation
COS‐7 cells were plated in six‐ or 12‐well plates and transfected with Kv1.2, Kv1.2N207Q, Kv1.2HA or Kv1.2HA‐N207Q cDNA construct (in pcDNA3) for 2 days. For protein degradation studies, cells were incubated at 4°C in recording solution (140 mm NaCl, 10 mm Hepes, 2 mm CaCl2 and 1 mm MgCl2, pH 7.2) with or without sialidase V (Prozyme) for 2 h, washed twice with cold 2.5% fetal bovine serum in recording solution for 5 min each to remove sialidase V, washed four times with cold recording solution, then incubated at 4°C or 37°C for 2 h, and finally washed four times with cold PBS for 5 min each. Biotinylation reactions were performed on live cells with sulfo‐NHS‐SS‐biotin (Thermo Fisher Scientific, Waltham, MA, USA) in PBS (1 mg ml−1, 0.13 mg cm–2) at 0–4°C for 20 min, with gentle agitation every 5 min. Reactions were quenched twice with ice‐cold 100 mm glycine in PBS for 5 min each, and then cells were washed twice with ice‐cold PBS for 5 min each. For protein degradation studies, cells were returned to 37°C for 0–120 min, then washed twice with ice‐cold PBS before lysis. Cells were lysed with lysis buffer on ice for 15 min, and then the lysate was cleared by 15 min cenrifugation at 20,000 g. One tenth of the lysate was saved on ice. The remaining lysate was diluted with lysis buffer (0.5 ml for 12‐well and 1 ml for six‐well), and biotinylated proteins were captured by NeutrAvidin resin for 2–3 h at 4°C with gentle agitation. Resin was collected by centrifugation at 2,400 g for 3 min and aspiration of supernatant. Resin was washed four times with lysis buffer, collecting resin by centrifugation at 2,400 g for 3 min and aspiration of supernatant between washes. Proteins were eluted from the resin in SDS protein gel sample loading buffer with the addition of 100 mm dithiothreitol and 10 mm tris(2‐carboxyethyl)phosphine for 30 min at 50°C. Proteins were separated by SDS‐PAGE, transferred to PVDF membrane, and immunoblot analysis was continued as described above.
Internalization immunoassay
Two days after transfection with EGFP‐Kv1.2HA or EGFP‐Kv1.2HA‐N207Q, COS‐7 cells were washed twice with ice‐cold PBS and incubated with blocking solution (5% normal goat serum in PBS) for 20 min at 4°C. Surface exposed HA epitopes were blocked with mouse anti‐HA antibody (dilution 1:200) in blocking solution for 2 h at 4°C. Cells were extensively washed five times with cold PBS for 5 min each. Cells were then returned to 37°C for 0, 15, 30 or 60 min to allow internalization of surface proteins. Cells were returned to 4°C and washed twice with cold PBS for 5 min each. Remaining surface anti‐HA antibodies were blocked with goat anti‐mouse antibody (dilution 1:200) for 3 h at 4°C. Cells were washed five times at 4°C with cold PBS for 5 min each. Fixation was performed with 4% sucrose/4% formaldehyde in PBS for 10 min, then immediately washed once with PBS for 10 s and three times with PBS for 5 min each.
Fixed cells were blocked and permeabilized with 0.1% Triton X‐100 in blocking solution for at least 1 h and then incubated with goat anti‐mouse Alexa Fluor 555 antibody (dilution 1:1000) in blocking solution at room temperature for 1 h. After washing three times with PBS for 5–10 min each, the coverslips were mounted on Superfrost microslides (VWR) using DAPI Fluoromount‐G (SouthernBiotech). Images were acquired on a SP5 confocal microscope (Leica Microsystems) and analysis was performed using Fiji software.
For co‐localization studies with anti‐EEA1 antibody, the internalization immunoassay was performed as described but internalization at 37°C was limited to 15 min. Additionally, rat anti‐HA antibody was used instead of mouse anti‐HA, goat anti‐rat antibody was used instead of goat anti‐mouse antibody, and goat anti‐rat Alexa Fluor 555 antibody was used instead of goat anti‐mouse Alexa Fluor 555 antibody. After permeabilization, cells were incubated with mouse anti‐EEA1 (dilution 1:500) for 1 h, washed three times with PBS for 5 min each, and then incubated with goat anti‐mouse Alexa Fluor 647 (dilution 1:1000) and goat anti‐rat Alexa Fluor 555 (dilution 1:1000) antibodies for 1 h.
Statistical analysis
The results are reported as the mean ± SEM. Statistical analysis was performed using Prism, version 5 (GraphPad Software Inc., San Diego, CA, USA). P < 0.05 was considered statistically significant.
Results
N‐linked glycosylation on Kv1.2 channels is important for neuronal surface expression
To monitor surface expression of Kv1.2, we used a construct of Kv1.2 carrying an extracellular HA tag and fusion of its cytoplasmic domain to enhanced green fluorescent protein (EGFP) (EGFP‐Kv1.2HA) (Gu et al. 2003) (Fig. 1 A). The glycosylation‐deficient mutant N207Q construct (EGFP‐Kv1.2HA‐N207Q) was made by site‐directed mutagenesis of the wild‐type sequence.
To avoid complications as a result of the presence of heteromeric channels formed by transfected EGFP‐Kv1.2HA and endogenous Kv1.1, Kv1.2 and Kv1.4, we determined the time course of endogenous expression of these Kv1 channels in primary rat hippocampal neuron cultures (Fig. 1 B). Antibodies used for detection of Kv1.1, Kv1.2 and Kv1.4 have known specificity for western blots, as reported in the supplier's (NeuroMab) datasheets online. The protein bands detected match with the expected sizes from endogenous and heterologous expression as reported by NeuroMab. We decided to transfect neurons at 1 days in vitro (DIV) and examine the expression at 3 DIV, when Kv1.1 and Kv1.2 expression is undetectable and Kv1.4 expression is very low. Surface expression for individual neurons was quantified from confocal images by measuring the non‐permeabilized anti‐HA immunocytochemical staining normalized to the EGFP expression level. We observed a decrease in surface expression for the Kv1.2 glycosylation‐deficient mutant N207Q compared to wild‐type, with normalized fluorescence of 0.6 ± 0.05 for N207Q and 1.4 ± 0.2 for wild‐type (Fig. 1 C and D).
Having found a difference in surface expression for cultured neurons by immunocytochemistry, we used whole cell electrophysiological recordings to measure functional surface expression. We quantified the maurotoxin (MTX) sensitive current because this blocker is specific for Kv1.2 at low nanomolar concentration (Kharrat et al. 1997). MTX binds to two Kv1.2 α subunits within a tetrameric channel (Chen and Chung, 2012) and gives the most reliable quantification for homomeric channels. We performed electrophysiological recordings on 3 DIV transfected neurons and confirmed the reduction of surface expression of the N207Q mutant channels compared to wild‐type channels by observing less MTX‐sensitive current, with 1100 ± 280 pA for N207Q and 3100 ± 360 pA for wild‐type (Fig. 1 E and F). We next examined whether this N‐linked glycosylation on Kv1.2 channels would also alter the basic biophysical properties of the MTX‐sensitive currents in neurons. We found that both activation and deactivation kinetics were comparable to earlier studies (Rezazadeh et al. 2007) and we did not detect any apparent difference in channel activation and deactivation kinetics between wild‐type and N207Q channels (Table 1). These findings provide evidence for the importance of channel glycosylation in neurons with respect to facilitating channel expression on the neuronal cell membrane.
Table 1.
Biophysical properties of Kv1.2 (MTX‐sensitive) currents in COS7 and neurons
| COS‐7 cells | Neurons | |||||
|---|---|---|---|---|---|---|
| Activation (ms) | Deactivation (ms) | Activation (ms) | Deactivation (ms) | |||
| Wild‐type | 2.195 ± 0.2985 | 15.19 ± 2.087 | (n = 17) | 2.004 ± 0.4638 | 15.40 ± 2.553 | (n = 11) |
| N207Q | 2.016 ± 0.2798 | 17.63 ± 2.588 | (n = 20) | 1.810 ± 0.5706 | 15.41 ± 3.729 | (n = 11) |
The results are shown as the mean ± SEM. No difference in activation and deactivation kinetics is detected between wild‐type and N207Q mutant, in either COS7 cells or neurons (P > 0.05; two‐way ANOVA).
The N‐linked glycosylation of Kv1.2 varies in different cell types
Because we were interested in studying the endogenous N‐linked glycosylation of Kv1.2 channels, for the experiments described above, we used cultured hippocampal neurons. Rat brain is known to express Kv1.2 with N‐linked complex glycosylation (Shi and Trimmer, 1999), which we observed in our cultured neurons (Fig. 2 A). However, as a result of the low transfection efficiency of primary hippocampal neuron cultures, the type of glycan expressed on EGFP‐Kv1.2HA could not be evaluated. Additional attempts using electroporation and lentivirus and Sindbis virus were not feasible because of the observed cytotoxicity in cultured neurons after transfection or transduction. Because of these limitations, we looked for a cell line with complex glycosylation of Kv1.2 similar to endogenous Kv1.2 in neurons.
Figure 2. Kv1.2 undergoes cell‐type specific N‐linked glycosylation .
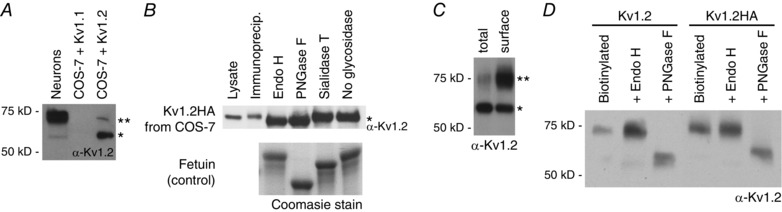
A, mature 21 DIV cultured hippocampal neurons (lane 1) express mostly complex glycosylation (** band), whereas transiently transfected Kv1.2 in COS‐7 cells (lane 3) express more of smaller glycoforms (* band). B, these glycoforms in transfected COS‐7 cells were determined to be oligomannose type as a result of sensitivity to PNGase F and Endo H (gel shift) and resistance to sialidase T. Kv1.2HA was immunoprecipitated with anti‐HA affinity matrix, and the glycosidase reactions were performed on‐resin, then analysed by immunoblot. Parallel glycosidase reactions were also performed on the control glycoprotein fetuin and analysed by Coomasie staining of SDS‐PAGE. C, surface biotinylation of COS‐7 cells transfected with Kv1.2 shows enrichment of the complex glycosylation (** band) on the cell surface compared to total lysate. D, biotinylated Kv1.2 channels expressed in COS‐7 cells were analysed by glycosidase reactions with Endo H and PNGase F. Resistance to Endo H and sensitivity to PNGase F (gel shift) confirm complex glycosylation.
We examined COS‐7 cells transfected with Kv1.2 and predominantly found expression of glycans of a smaller apparent molecular weight. Glycosidase analysis of Kv1.2HA immunoprecipitated from COS‐7 cells via anti‐HA antibody showed sensitivity to Endo H and PNGase F and insensitivity to sialidase T [releases α(2‐3,6,8)‐linked sialic acid] (Fig. 2 B). Therefore, COS‐7 cells express mostly N‐linked oligomannose type glycoforms of Kv1.2, although the total lysate from transfected COS‐7 cells shows some complex glycosylation (Fig. 2 A and D). To test whether Kv1.2 with this complex glycosylation is enriched on the cell surface, we performed selective biotinylation of surface proteins on COS‐7 cells transfected with Kv1.2 with or without the HA tag (Fig. 2 C). We confirmed that Kv1.2 expressed on the surface of COS‐7 cells exhibited N‐linked complex glycans by glycosidase analysis. The surface Kv1.2 or Kv1.2HA channels were sensitive to PNGase F and resistant to Endo H (Fig. 2 D), as well as sensitive to sialidase V (see below).
N‐linked glycosylation on Kv1.2 channels is important for surface expression in COS‐7 cells
Knowing that Kv1.2 is processed to express complex glycosylation on the cell surface of COS‐7 cells, we decided to perform subsequent experiments in COS‐7 cells, which permit additional biochemical experiments that are not feasible in cultured neurons as a result of a low efficiency of lipid‐based transfection and cytotoxicity from electroporation or viral infection attempted for greater channel expression. First, we repeated the non‐permeabilized immunocytochemical staining of extracellularly tagged HA constructs EGFP‐Kv1.2HA and EGFP‐Kv1.2HA‐N207Q in transfected COS‐7 cells (Fig. 3 A and B). We observed less surface expression of the glycosylation mutant N207Q compared to wild‐type Kv1.2, with normalized fluorescence of 0.4 ± 0.1 for N207Q and 2.2 ± 0.3 for wild‐type.
Figure 3. N‐linked glycosylation is important for cell membrane expression in heterologous COS‐7 cells .
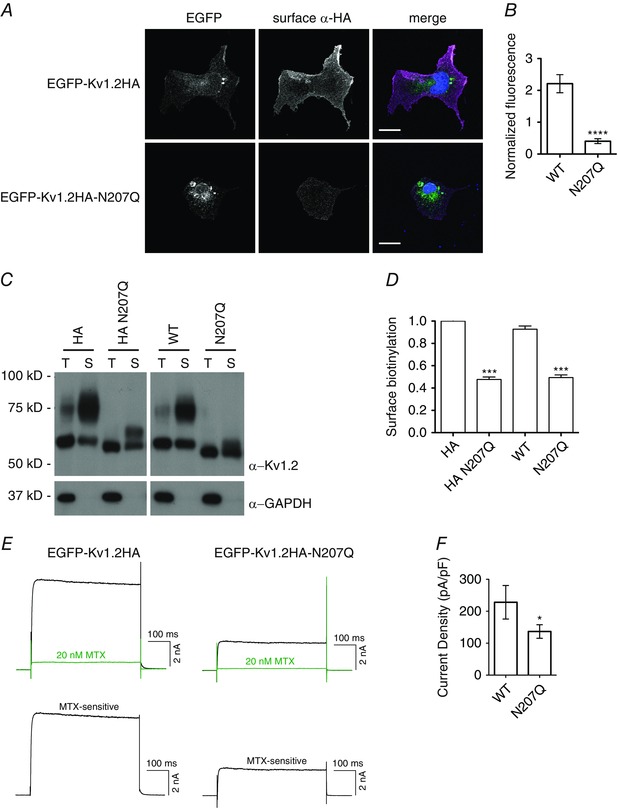
A, COS‐7 cells were transfected with EGFP‐Kv1.2HA constructs, and non‐permeabilized surface immunocytochemistry with rat anti‐HA antibody (AlexaFluor 555‐conjugated anti‐rat secondary antibody labelling) was performed 2 days later. Confocal images were obtained and quantified to determine the fluorescence of surface anti‐HA antibody signal normalized to EGFP expression (as a measure of transfection efficiency). Merged images show EGFP‐Kv1.2HA (green), surface anti‐HA antibody (magenta) and DAPI (blue). Scale bar = 10 μm. B, quantification of fluorescence intensity for wild‐type (WT) (n = 19) and N207Q (n = 13) transfected cells. **** P < 0.0001, Student's t test. C, COS‐7 cells were transfected with Kv1.2 or Kv1.2HA constructs, and surface biotinylation reactions were performed 2 days later on live cells at 0°C. Immunoblot of a portion of the total lysate (T) and biotinylated surface proteins (S) were probed with anti‐Kv1.2 antibody for quantification of Kv1.2 surface expression. The immunoblot was also probed with anti‐GAPDH antibody as a cytoplasmic protein control. All samples were run on the same gel and immunoblot, though a space is indicated between HA and non‐HA constructs to indicate unnecessary lanes were removed digitally. D, quantification of surface biotinylation from immunoblots, normalized to Kv1.2HA, as fraction of surface anti‐Kv1.2 signal relative to the total anti‐Kv1.2 signal in lysate (n = 2). *** P < 0.001, ANOVA with Bonferroni multiple comparison test. E, COS‐7 cells were transfected with EGFP‐Kv1.2HA constructs, and whole cell patch clamp recording, stepping to +20 mV from holding potential of −100 mV, was performed 2 days later. Sample traces for WT and N207Q mutant Kv1.2 current are shown before (black) and after application of 20 nm MTX (green). MTX‐sensitive current is also shown as determined by substraction of the green trace from the black trace. F, quantification of current density for WT (n = 19) and N207Q (n = 28) cells. * P < 0.05, Student's t test.
Additionally, we performed surface biotinylation of cell surface proteins expressed in COS‐7 cells transfected with several Kv1.2 constructs (Fig. 3 C). We examined wild‐type and glycosylation‐deficient N207Q constructs of Kv1.2 and their HA‐tagged counterparts. Quantification of biotinylated Kv1.2 by western blotting revealed no significant effect of the HA tag on surface expression. Regardless of the presence or absence of the HA tag, wild‐type Kv1.2 was expressed on the cell surface about twice as much as the glycosylation deficient N207Q mutant (Fig. 3 D). The remaining higher apparent molecular weight band present in the N207Q mutant is a result of phosphorylation (Zhu et al. 2003).
The decrease in surface expression was also observed by electrophysiological recordings using whole cell patch clamp. We measured the MTX‐sensitive current in COS‐7 cells transfected with wild‐type and N207Q constructs (Fig. 3 E). We found decreased current density for the non‐glycosylated mutant N207Q, 140 ± 20 pA pF–1 for N207Q compared to 230 ± 50 pA pF–1 for wild‐type (Fig. 3 F). We also analysed the biophysical properties of the MTX‐sensitive currents in COS7 cells, and we did not detect any difference in channel activation and deactivation kinetics between wild‐type and N207Q channels (Table 1). Taken together, these results reveal a strong dependence on glycosylation for the surface expression of Kv1.2 channels in COS‐7 cells, as well as hippocampal neurons.
Glycosylation has a role in the stability of internalized Kv1.2
To test whether the glycosylation of Kv1.2 modulates its surface stability, we performed channel internalization immunoassays to follow the internalization and degradation of surface Kv1.2 channels. Wild‐type and N207Q mutant Kv1.2HA channels were expressed in COS‐7 cells by transient transfection. Two days after transfection, surface channels were labelled with mouse anti‐HA antibody at 4°C, and then the cells were returned to 37°C to allow endocytosis. Remaining HA antibody‐tagged surface channels were blocked by secondary goat anti‐mouse antibody, and then cells were fixed and internalized channels were labelled with fluorescently tagged goat anti‐mouse antibody (Fig. 4 A). The intensity of fluorescence was quantified for internalized Kv1.2HA from confocal microscopy images. Figure 4(B) shows sample images from 60 min of internalization at 37°C. Although both wild‐type and N207Q mutant Kv1.2 showed similar rates of internalization over 30 min, internalized wild‐type Kv1.2 remained present in the cells from 30 to 60 min, whereas N207Q channels were eliminated during this 30 min time period (Fig. 4 C). Therefore, N‐linked glycosylation does not affect cell surface stability of Kv1.2 channels but does influence the rate of degradation of internalized Kv1.2 proteins.
Figure 4. Internalization immunoassay in COS‐7 cells reveals role for glycosylation in the stability of internalized Kv1.2 channels .
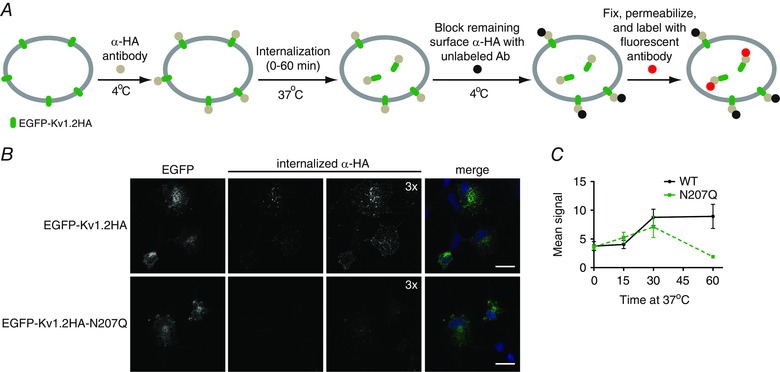
A, schematic of experimental design for quantitative immunoassay of channel internalization. COS‐7 cells were transfected with EGFP‐Kv1.2HA constructs and surface channels were labelled in live cells at 4°C with mouse anti‐HA antibody. Cells were placed at 37°C for 0 to 60 min to allow channel internalization. EGFP‐Kv1.2HA constructs remaining on the plasma membrane were blocked with excess unlabelled anti‐mouse IgG at 4°C. After fixation and permeabilization, internalized EGFP‐Kv1.2HA proteins were labelled with AlexaFluor 555‐conjugated anti‐mouse secondary antibodies. B, sample images of Kv1.2HA internalization for 60 min at 37°C. Merged images show EGFP‐Kv1.2HA (green), internalized anti‐HA (magenta) and DAPI (blue). Scale bar = 20 μm. C, quantification of fluorescence intensity for WT (n = 5, 4, 6, 7 for 0, 15, 30 and 60 min, respectively) and N207Q (n = 4, 4, 6, 6 for 0, 15, 30 and 60 min, respectively) cells. ** P < 0.01, two‐way ANOVA with Bonferroni multiple comparison test.
To further validate these observations, we first showed that both the wild‐type and mutant Kv1.2 constructs are internalized to endosomes. We observed co‐localization of internalized channels with the endosomal marker EEA‐1 after 15 min of endocytosis at 37°C (Fig. 5). We also used surface biotinylation to quantify the degradation of cell surface Kv1.2 channels. Surface proteins were biotinylated on ice and then returned to 37°C for 0 to 120 min. Cells were then lysed and remaining biotinylated Kv1.2 was determined by quantitative western blot analysis (Fig. 6 A). Similar to our observations with the channel internalization immunoassays, we found surface expressed N207Q mutants to have a shorter protein half‐life compared to wild‐ type (Fig. 6 B).
Figure 5. Surface Kv1.2 channels are internalized to endosomes .
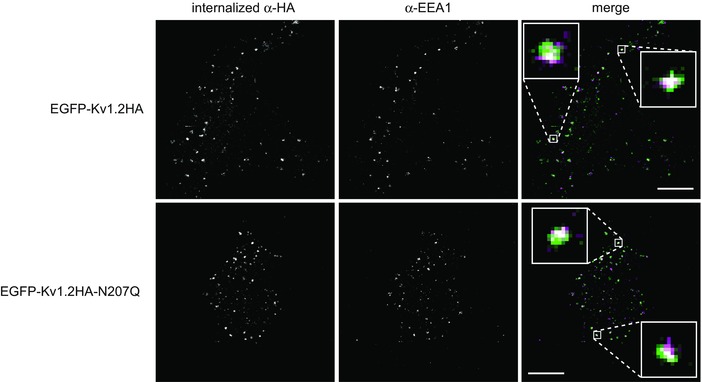
Internalization immunoassay was performed as shown in Fig. 4(A) for 15 min at 37°C. Surface channels were labelled in live cells at 4°C with rat anti‐HA antibody. After fixation and permeabilization, endosomes were labelled with mouse anti‐EEA1 antibody, and then internalized EGFP‐Kv1.2HA proteins were labelled with AlexaFluor 555‐conjugated anti‐rat secondary antibodies and endosomal protein EEA‐1 was labelled with AlexaFluor 647‐conjugated anti‐mouse secondary antibodies. Merged images show internalized anti‐HA (green) and anti‐EEA1 (magenta). Scale bar = 10 μm. Enlarged windows (2 μm square) show co‐localization (white circles).
Figure 6. Glycosylation affects degradation of cell surface Kv1.2 channels .
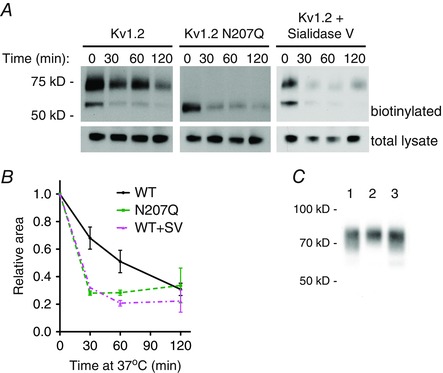
A, COS‐7 cells were transfected with Kv1.2 wild‐type and N207Q constructs. Two days later, live cells were incubated at 4°C for 2 h with sialidase V (WT+SV) or without enzyme (WT and N207Q) and then surface biotinylation reactions were performed, also at 4°C. Cells were placed at 37°C for 0 to 120 min to allow channel internalization and degradation. Immunoblot of a portion of the total lysate and remaining biotinylated proteins were probed with anti‐Kv1.2 antibody for quantification of surface Kv1.2 degradation. B, quantification of remaining biotinylated Kv1.2 from immunoblots (n = 3). Relative area is fraction of surface anti‐Kv1.2 signal relative to total anti‐Kv1.2 signal in lysate, normalized to initial amounts. Signals were calculated from gel analysis using Fiji software. * P < 0.05, ** P < 0.01, *** P < 0.001, two‐way ANOVA with Bonferroni multiple comparison test. C, COS‐7 cells were transfected with Kv1.2 wild‐type construct. Two days later, live cells were incubated with or without sialidase V at 4°C for 2 h. After extensive washing to ensure removal of sialidase V, surface biotinylation was performed to analyse cell surface Kv1.2 by immunoblot. Lane 1, sialidase V incubation followed by washes and additional 2 h incubation at 4°C in recording solution only. Lane 2, incubation in recording solution only followed by washes and an additional 2 h of incubation at 4°C in recording solution only. Lane 3, sialidase V incubation followed by washes and no additional incubation. Gel shift of lane 1 compared to lane 2 shows enzymatic removal of sialic acid residues by sialidase V. Lane 1 compared to lane 3 shows no additional sialic acid removal upon further incubation, demonstrating efficient removal of sialidase V by washes. [Colour figure can be viewed at wileyonlinelibrary.com]
Although the genetically encoded N207Q mutant is useful for a variety of studies, this mutation results in a lack of the entire glycan. We decided to explore methods aiming to modify the existing glycan expressed on cultured cells. We chose glycosidases because, compared to chemical methods, enzymatic conditions are often more compatible with live cultured cells. We found sialidase V, which releases α(2‐3,6,8)‐linked sialic acid, was amenable to the removal of terminal sialic acid residues from Kv1.2 expressed in COS‐7 cells. Other glycosidases tested were not amenable for use in non‐denaturing conditions on live cells. Although use of sialidase V at 37°C results in the most efficient enzymatic removal of sialic acid, sialidase V is internalized into cells at this temperature. However, we found that sialidase V remains active at 4°C, whereas its endocytosis is blocked (Fig. 6 C). Although sialidase V could remove terminal sialic acid residues from the extracellular domains of all membrane proteins, we were able to observe a specific effect on Kv1.2. Removal of sialic acid residues from the N‐linked glycan expressed on Kv1.2 channels on the surface of COS‐7 cells resulted in a shorter protein half‐life, which was comparable to the glycosylation‐deficient mutant N207Q (Fig. 6 A and B). Thus, features of complex glycosylation such as sialic acids are a factor in the Kv1.2 channel stability.
Discussion
In the present study, we report roles of N‐linked glycosylation of Kv1.2 channels, including forward trafficking to the cell surface and degradation of internalized channels.
In light of reports of differential glycosylation of Kv1.2 depending on cell types (Shi and Trimmer, 1999), we were interested in studying endogenous glycosylation of the channel. We began our study using primary cultured hippocampal neurons because Kv1.2 is expressed in the hippocampus in the brain. We also examined the glycosylation of Kv1.2 in transfected COS‐7 cells to establish this expression system for studies of N‐linked complex glycosylation on channel surface expression and stability. Transient transfection of COS‐7 cells has the limitation of overexpression of Kv1.2, which may be mitigated partially in future experiments by generating stable cell lines. Although most Kv1.2 proteins expressed in COS‐7 cells contain oligomannose type glycosylation, we found significant cell surface expression of complex glycans. As such, we used both primary cultured neurons and the COS‐7 cell line to study the trafficking of Kv1.2 channels to the cell surface. We observed decreased cell surface expression of the non‐glycosylated Kv1.2N207Q mutant compared to wild‐type Kv1.2 in both cell types by non‐permeabilized immunocytochemistry. This decreased efficiency of surface expression for the glycosylation deficient mutant was further confirmed by whole cell electrophysiological recordings and surface biotinylation studies. Although less current was observed in N207Q mutant channels as a result of decreased surface expression, no change in channel activation and deactivation kinetics was apparent (Table 1). Because the glycosylation deficient mutant channels exhibited normal rate of internalization (see below), the reduced surface expression probably reflects a defect in forward trafficking.
We also monitored the internalization and degradation of Kv1.2 channels expressed on the cell surface. We did not observe a difference in channel endocytosis as visualized by channel internalization immunoassay. For both wild‐type and glycosylation deficient N207Q mutant channels, we measured similar rates of channel internalization and found co‐localization with endosomal markers. However, the internalization immunoassay did detect faster degradation of internalized channels with the N207Q mutation compared to wild‐type. This faster degradation of surface expressed N207Q channels was further validated by surface biotinylation experiments. We also performed surface biotinylation experiments after altering the glycan structure by using a sialidase on live cells. This combination of enzymatic modification of glycan structure along with surface biotinylation allows the study of a particular glycan structure, in this case sialic acid, for surface channel turnover. We observed a decrease in the half‐life of Kv1.2 for sialidase V‐treated cells that was similar to the non‐glycosylated mutant channels. These experiments suggest that removal of the terminal sialic acid residue on the glycan may be sufficient to enhance the degradation of the channels to mimic degradation of non‐glycosylated channels. This faster degradation also supports the observation that, although N207Q mutant channels had internalization rates comparable to those of wild‐type channels (Fig. 4 C), the internalized N207Q channels without glycosylation (also lacking terminal sialic acid residues) were degraded more rapidly. Because a larger fraction of internalized channels without complex glycosylation was degraded rather than recycled back to the cell surface, the N207Q mutant channels, as well as channels without terminal sialic acid residues, exhibited a faster decline of surface biotinylated channel levels (Fig. 6 B).
Taken together, the present study reveals that glycosylation of Kv1.2 is important for channel surface expression, both by promoting forward trafficking and by enhancing the stability of channels on the cell membrane.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
DAT, SBY, YNJ and LYJ conceived and designed the experiments. DAT and SBY collected and analysed the data. DAT, SBY and LYJ interpreted the data. DAT, SBY and LY drafted and revised the manuscript critically for important intellectual content. All authors have read and approved the final copy and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed. All experiments were carried out at the University of California, San Francisco.
Funding
This study was supported by NIMH grant R37MH065334, MOST grant 105‐2320‐B‐001‐003‐MY2 (to SBY) and an NIH NRSA postdoctoral fellowship (to DAT).
Acknowledgements
We thank all members of the Jan laboratory, in particular Drs Woo‐Ping Ge, Wendy Huang, Xiu Ming Wong, Hye Young Lee, Andrew Kim, Jason Tien and David Young, for discussions and suggestions. YNJ and LYJ are Howard Hughes Medical Institute Investigators.
This is an Editor's Choice article from the 15 November 2016 issue.
References
- Baycin‐Hizal D, Gottschalk A, Jacobson E, Mai S, Wolozny D, Zhang H, Krag SS & Betenbaugh MJ (2014). Physiologic and pathophysiologic consequences of altered sialylation and glycosylation on ion channel function. Biochem Biophys Res Commun 453, 243–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campomanes CR, Carroll KI, Manganas LN, Hershberger ME, Gong B, Antonucci DE, Rhodes KJ & Trimmer JS (2002). Kv beta subunit oxidoreductase activity and Kv1 potassium channel trafficking. J Biol Chem 277, 8298–8305. [DOI] [PubMed] [Google Scholar]
- Carchon H, Van Schaftingen E, Matthijs G & Jaeken J (1999). Carbohydrate‐deficient glycoprotein syndrome type IA (phosphomannomutase‐deficiency). Biochim Biophys Acta 1455, 155–165. [DOI] [PubMed] [Google Scholar]
- Chen R and Chung S‐H (2012). Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS ONE 7, e47253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, Chow A, Lau D, McCormack T, Moreno H, Nadal MS, Ozaita A, Pountney D, Saganich M, Vega‐Saenz de Miera E & Rudy B (1999). Molecular diversity of K+ channels. Ann NY Acad Sci 868, 233–285. [DOI] [PubMed] [Google Scholar]
- Ednie AR and Bennett ES (2012). Modulation of voltage‐gated ion channels by sialylation. Compr Physiol 2, 1269–1301. [DOI] [PubMed] [Google Scholar]
- Fozzard HA and Kyle JW (2002). Do defects in ion channel glycosylation set the stage for lethal cardiac arrhythmias? Sci STKE 130, PE19. [DOI] [PubMed] [Google Scholar]
- Glozman R, Okiyoneda T, Mulvihill CM, Rini JM, Barriere H & Lukacs GL (2009). N‐glycans are direct determinants of CFTR folding and stability in secretory and endocytic membrane traffic. J Cell Biol 184, 847–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Anderson CL, January CT & Zhou Z (2002). Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am J Physiol Heart Circ Physiol 283, H77–H84. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Jan YN & Jan LY (2003). A conserved domain in axonal targeting of Kv1 (Shaker) voltage‐gated potassium channels. Science 301, 646–649. [DOI] [PubMed] [Google Scholar]
- Gu C, Zhou W, Puthenveedu MA, Xu M, Jan YN & Jan LY (2006). The microtubule plus‐end tracking protein EB1 is required for Kv1 voltage‐gated K+ channel axonal targeting. Neuron 52, 803–816. [DOI] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W & Wang X (2005). International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage‐gated potassium channels. Pharmacol Rev 57, 473–508. [DOI] [PubMed] [Google Scholar]
- Hall MK, Weidner DA, Edwards MA, Schwalbe RA (2015). Complex N‐glycans influence the spatial arrangement of voltage gated potassium channels in membranes of neuronal‐derived cells. PLoS ONE 10, e0137138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ionic Channels of Excitable Membranes. Sinauer, Sunderland, MA. [Google Scholar]
- Jaeken J, Stibler H & Hagberg B (1991). The carbohydrate‐deficient glycoprotein syndrome. A new inherited multisystemic disease with severe nervous system involvement. Acta Paediatr Scand 375 Suppl, 1–71. [PubMed] [Google Scholar]
- Jan LY and Jan YN (1997). Cloned potassium channels from eukaryotes and prokaryotes. Annu Rev Neurosci 20, 91–123. [DOI] [PubMed] [Google Scholar]
- Kharrat R, Mansuelle P, Sampieri F, Crest M, Oughideni R, Van Rietschoten J, Martin‐Eauclaire MF, Rochat H & El Ayeb M (1997). Maurotoxin, a four disulfide bridge toxin from Scorpio maurus venom, purification, structure and action on potassium channels. FEBS Lett 406, 284–290. [DOI] [PubMed] [Google Scholar]
- Lennarz W (1983). Role of intracellular membrane systems in glycosylation of proteins. Methods Enzymol 98, 91–97. [DOI] [PubMed] [Google Scholar]
- Li M, Tonggu L, Tang L & Wang L (2015). Effects of N‐glycosylation on hyperpolarization‐activated cyclic nucleotide‐gated (HCN) channels. Biochem J 466, 77–84. [DOI] [PubMed] [Google Scholar]
- Martens JR, Kwak YG & Tamkun MM (1999). Modulation of Kv channel alpha/beta subunit interactions. Trends Cardiovasc Med 9, 253–258. [DOI] [PubMed] [Google Scholar]
- Rezazadeh S, Kurata HT, Claydon TW, Kehl SJ & Fedida D (2007). An activation gating switch in Kv1.2 is localized to a threonine residue in the S2‐S3 linker. Biophys J 93, 4173–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Nakahira K, Hammond S, Rhodes KJ, Schechter LE & Trimmer JS (1996). Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 16, 843–852. [DOI] [PubMed] [Google Scholar]
- Shi G and Trimmer JS (1999). Differential asparagine‐linked glycosylation of voltage‐gated K+ channels in mammalian brain and in transfected cells. J Membr Biol 168, 265–273. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY & Jan YN (2003). Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI3‐kinase activity. Cell 112, 63–75. [DOI] [PubMed] [Google Scholar]
- Sutachan JJ, Watanabe I, Zhu J, Gottschalk A, Recio‐Pinto E & Thornhill WB (2005). Effects of Kv1.1 channel glycosylation on C‐type inactivation and simulated action potentials. Brain Res 1058, 30–43. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Zhu J, Recio‐Pinto E & Thornhill WB (2004). Glycosylation affects the protein stability and cell surface expression of Kv1.4 but not Kv1.1 potassium channels. J Biol Chem 279, 8879–8885. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Zhu J, Sutachan JJ, Gottschalk A, Recio‐Pinto E & Thornhill WB (2007). The glycosylation state of Kv1.2 potassium channels affects trafficking, gating, and simulated action potentials. Brain Res 1144, 1–18. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Zhu J, Recio‐Pinto E & Thornhill WB (2015). The degree of N‐glycosylation affects the trafficking and cell surface expression levels of Kv1.4 potassium channels. J Membr Biol 248, 187–196. [DOI] [PubMed] [Google Scholar]
- Woo SK, Kwon MS, Ivanov A, Geng Z, Gerzanich V & Simard JM (2013). Complex N‐glycosylation stabilizes surface expression of transient receptor potential melastatin 4b protein. J Biol Chem 288, 36409–36417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Watanabe I, Poholek A, Koss M, Gomez B, Yan C, Recio‐Pinto E & Thornhill WB (2003). Allowed N‐glycosylation sites on the Kv1.2 potassium channel S1–S2 linker: implications for linker secondary structure and the glycosylation effect on channel function. Biochem J 375, 769–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yan J & Thornhill WB (2012). N‐glycosylation promotes the cell surface expression of Kv1.3 potassium channels. FEBS J 279, 2632–2644. [DOI] [PubMed] [Google Scholar]