

Abstract
Key points
In the vertebrate retina, photoreceptors influence the signalling of neighbouring photoreceptors through lateral‐inhibitory interactions mediated by horizontal cells (HCs). These interactions create antagonistic centre‐surround receptive fields important for detecting edges and generating chromatically opponent responses in colour vision.
The mechanisms responsible for inhibitory feedback from HCs involve changes in synaptic cleft pH that modulate photoreceptor calcium currents. However, the sources of synaptic protons involved in feedback and the mechanisms for their removal from the cleft when HCs hyperpolarize to light remain unknown.
Our results indicate that Na+–H+ exchangers are the principal source of synaptic cleft protons involved in HC feedback but that synaptic cleft alkalization during light‐evoked hyperpolarization of HCs also involves changes in bicarbonate transport across the HC membrane.
In addition to delineating processes that establish lateral inhibition in the retina, these results contribute to other evidence showing the key role for pH in regulating synaptic signalling throughout the nervous system.
Abstract
Lateral‐inhibitory feedback from horizontal cells (HCs) to photoreceptors involves changes in synaptic cleft pH accompanying light‐evoked changes in HC membrane potential. We analysed HC to cone feedback by studying surround‐evoked light responses of cones and by obtaining paired whole cell recordings from cones and HCs in salamander retina. We tested three potential sources for synaptic cleft protons: (1) generation by extracellular carbonic anhydrase (CA), (2) release from acidic synaptic vesicles and (3) Na+/H+ exchangers (NHEs). Neither antagonizing extracellular CA nor blocking loading of protons into synaptic vesicles eliminated feedback. However, feedback was eliminated when extracellular Na+ was replaced with choline and significantly reduced by an NHE inhibitor, cariporide. Depriving NHEs of intracellular protons by buffering HC cytosol with a pH 9.2 pipette solution eliminated feedback, whereas alkalinizing the cone cytosol did not, suggesting that HCs are a major source for protons in feedback. We also examined mechanisms for changing synaptic cleft pH in response to changes in HC membrane potential. Increasing the trans‐membrane proton gradient by lowering the extracellular pH from 7.8 to 7.4 to 7.1 strengthened feedback. While maintaining constant extracellular pH with 1 mm HEPES, removal of bicarbonate abolished feedback. Elevating intracellular bicarbonate levels within HCs prevented this loss of feedback. A bicarbonate transport inhibitor, 4,4′‐diisothiocyano‐2,2′‐stilbenedisulfonic acid (DIDS), also blocked feedback. Together, these results suggest that NHEs are the primary source of extracellular protons in HC feedback but that changes in cleft pH accompanying changes in HC membrane voltage also require bicarbonate flux across the HC membrane.
Keywords: calcium current, horizontal cell, pH regulation, photoreceptor, retina
Key points
In the vertebrate retina, photoreceptors influence the signalling of neighbouring photoreceptors through lateral‐inhibitory interactions mediated by horizontal cells (HCs). These interactions create antagonistic centre‐surround receptive fields important for detecting edges and generating chromatically opponent responses in colour vision.
The mechanisms responsible for inhibitory feedback from HCs involve changes in synaptic cleft pH that modulate photoreceptor calcium currents. However, the sources of synaptic protons involved in feedback and the mechanisms for their removal from the cleft when HCs hyperpolarize to light remain unknown.
Our results indicate that Na+–H+ exchangers are the principal source of synaptic cleft protons involved in HC feedback but that synaptic cleft alkalization during light‐evoked hyperpolarization of HCs also involves changes in bicarbonate transport across the HC membrane.
In addition to delineating processes that establish lateral inhibition in the retina, these results contribute to other evidence showing the key role for pH in regulating synaptic signalling throughout the nervous system.
Abbreviations
- BAF
bafilomycin A1
- CA
carbonic anhydrase
- cariporide
N‐(aminoiminomethyl)‐5‐cyclopropyl‐1‐(5‐quinolinyl)‐1H‐pyrazole‐4‐carboxamide
- DIDS
4,4′‐diisothiocyano‐2,2′‐stilbenedisulfonic acid
- EIPA
5‐(N‐ethyl‐N‐isopropyl)amiloride
- FC5‐207a
benzothiophene‐3‐ylmethylsulfamide
- HCs
horizontal cells
- ICa
Ca2+ current
- Ifeedback
feedback current
- Ipeak
peak current
- NHEs
Na+/H+ exchangers
- V50
voltage when I peak is half‐maximal
- v‐ATPase
vacuolar‐type H+‐ATPase
- zoniporide
N‐(diaminomethylidene)‐3‐methanesulfonyl‐4‐(propan‐2‐yl)benzamide
Introduction
Lateral inhibition of cones by second‐order horizontal cells (HCs) facilitates comparisons between responses of neighbouring photoreceptors that improve the ability of photoreceptors to detect contrast borders and colours (reviewed by Thoreson & Mangel, 2012). Inhibitory feedback from HCs to cones modulates the L‐type Ca2+ current (I Ca) in presynaptic cone terminals and thereby regulates release of the neurotransmitter glutamate onto downstream neurons (Verweij et al. 1996). While other mechanisms may also contribute (Wu, 1991; Kamermans et al. 2001; Kemmler et al. 2014), HC feedback involves a change in extracellular pH at the synapses between HCs and cones (Hirasawa & Kaneko, 2003; Wang et al. 2014). Light‐evoked hyperpolarization of HCs alkalizes the synaptic cleft, relieving proton‐mediated inhibition of I Ca and shifting its activation to more negative potentials. Extracellular protons can rapidly depart the synaptic cleft by diffusion and so the maintenance of HC feedback requires a constant source of extracellular protons in darkness as well as mechanisms for proton removal to produce synaptic cleft alkalization in light. In this study, we set out to identify the major sources of protons involved in feedback and the mechanisms for proton removal from the cleft during light.
The principal mechanism for proton extrusion from neurons is the activity of Na+/H+ exchangers (NHEs) (Koskelainen et al. 1993; Kalamkarov et al. 1996; Obara et al. 2008; Casey et al. 2010; Ruffin et al. 2014). Protons can also be released into the extracellular space by exocytosis of acidic synaptic vesicles (DeVries, 2001; Chesler, 2003) or the activity of vacuolar‐type H+‐ATPases (v‐ATPases) inserted into the plasma membrane during vesicle fusion (Casey et al. 2010; Zhang et al. 2010). By hydrating CO2, extracellular carbonic anhydrase (CA) can also be a source of protons. Several CA isoforms are anchored to the extracellular membrane face by glycosylphosphatidylinositol links (CA IV, XV) while others are transmembrane proteins with an extracellular active site (CA IX, XII and XIV) (Supuran, 2008). We tested the contributions of these mechanisms to inhibitory feedback from HCs to cones by using whole cell recordings in salamander retina. Changes in cone I Ca induced by feedback from HCs were studied by illumination of the receptive field surround or by directly polarizing voltage‐clamped HCs during paired recordings. Using various pharmacological and chemical manipulations, we found that while all three mechanisms appear to contribute extracellular protons, NHEs are the main source of protons involved in HC feedback. Bicarbonate transporters are also important mechanisms for pH regulation (Obara et al. 2008; Casey et al. 2010; Ruffin et al. 2014) and we found that bicarbonate is essential for feedback. Our results suggest that changes in bicarbonate transport activity are likely to be responsible for changes in extracellular pH that occur in response to changes in HC membrane potential. In addition to delineating crucial processes that establish lateral inhibition in the retina, these results contribute to an emerging body of evidence showing the key role of pH changes in regulating signalling at many different synapses throughout the nervous system (Beg et al. 2008; Obara et al. 2008; Casey et al. 2010; Wang et al. 2014; Highstein et al. 2014).
Methods
Preparations
Negative feedback from HCs to cones was studied using two different preparations of salamander retina: flat mount and slice preparations. Neotenic tiger salamanders (Ambystoma tigrinum, 18–25 cm in length, male or female, Charles D. Sullivan, Co., Nashville, TN, USA) were housed on a 12 h light/dark cycle. Experiments were begun 1–2 h into the dark phase. Animals were killed in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center and in compliance with the animal ethics guidelines of the Journal of Physiology (Grundy, 2015). Salamanders were first anaesthetized by immersion in tricaine methanesulfonate (0.25 g l−1) for ≥15 min and then decapitated with heavy shears. The head was then hemisected and the spinal column rapidly pithed. A total of 230 animals were used in these experiments. For each animal, an eye was enucleated and set on a saline‐soaked cotton wad placed on a linoleum block for dissection (Van Hook & Thoreson, 2013). The anterior portion of the eye was removed and the eyecup was cut into three or four sections. For flat mount preparations, tissue was dissected under infrared illumination using GenIII image intensifiers (Nitemate NAV3, Litton Industries, Tempe, AZ, USA) mounted on a dissecting microscope. A section of dark‐adapted retina was isolated and laid ganglion cells down onto a piece of nitrocellulose filter paper (type AAWP, 0.8 μm pores, EMD Millipore, Billerica, MA, USA) with a small hole cut in the centre to allow light to be projected onto the retina. For retinal slice preparations, dissection was done under room lights. A piece of eyecup was placed on a rectangular piece of nitrocellulose filter paper (5×10 mm) and the retina was then isolated by removing the retinal pigment epithelium, choroid and sclera. The filter paper with retina was cut into strips with a width of 125 μm. The retinal slices were rotated 90 deg and placed in the recording chamber where they were viewed through a long‐working distance, water‐immersion objective (40×, 0.7 NA, Olympus or 60×, 1.0 NA, Nikon) on an upright‐fixed stage microscope (BHWI, Olympus or E600FN, Nikon).
Slices were superfused at ∼1 ml min−1 with pH 7.4 bicarbonate‐buffered amphibian saline solution (all in mm: 101 NaCl, 22 NaHCO3, 2.5 KCl, 2.0 CaCl2, 0.5 MgCl2, 11 glucose). Solutions were bubbled continuously with 95% O2/5% CO2. In some experiments, NaHCO3 was changed to 12 or 32 mm to achieve a solution pH of 7.1 or 7.8, respectively. In these solutions, the amount of NaCl was adjusted to maintain an osmolality of 240–244 mosmol kg–1 as assessed by a vapour pressure osmometer (Wescor, Logan, UT, USA). In another set of experiments, we maintained the bicarbonate concentration at 22 mm but altered the pH from 7.4 to 7.9 by bubbling this solution for a few minutes with 100% O2. We bubbled the solution for only a few minutes with 100% O2 to avoid calcium precipitating out of solution. If the solution became too alkaline, we added HCl to attain a final pH of 7.9 just before applying it to the retina. We prepared a nominally Na+‐free solution by replacing Na+ with choline (all in mm: 101 choline chloride, 22 choline‐HCO3, 2.5 KCl, 2.0 CaCl2, 0.5 MgCl2, 11.0 glucose, bubbled for 10 min with 95% O2/5% CO2). For some experiments, we added 10 mm 4‐(2‐hydroxyethyl)‐1‐piperazineethanesulfonic acid (HEPES) to the standard bicarbonate‐buffered solution without adjusting the osmolality and used NaOH to return the pH to 7.4. We used two solutions that included 1 mm HEPES. In one, we added 1 mm HEPES to the standard bicarbonate‐containing superfusate. In the other, we added 1 mm HEPES to a bicarbonate‐free solution created by replacing NaHCO3 with NaCl. We adjusted the pH of both 1 mm HEPES solutions to 7.4 just prior to use on each day. For some experiments on bafilomycin A1 (BAF), we incubated eyecups overnight at 4°C in a bicarbonate‐buffered saline solution supplemented with 1% BSA with or without BAF.
Electrophysiology
Patch pipettes for whole‐cell patch clamp recordings were pulled from borosilicate glass (1.2 mm outer diameter, 0.95 mm inner diameter, with an internal filament, World Precision Instruments, Sarasota, FL, USA) using a PP‐830 micropipette puller (Narishige, Tokyo, Japan). Pipette resistance was 10–15 MΩ. Pipettes were filled with an internal recording solution containing (in mm): 50 caesium gluconate, 40 caesium glutamate, 10 TEA‐Cl, 3.5 NaCl, 1 CaCl2, 1 MgCl2, 9.4 Mg‐ATP, 0.5 Na‐GTP, 5.0 EGTA, 10 HEPES (pH 7.2). The presence of glutamate in cone pipette solution enhances and sustains post‐synaptic currents in HCs evoked by depolarizing steps applied to cones (Bartoletti & Thoreson, 2011). In some experiments, we increased the pipette solution pH to 9.2 by adding CsOH. In other experiments, we elevated intracellular bicarbonate levels in HCs by using a pipette solution containing (in mm): 90 KHCO3, 30 caesium gluconate, 10 TEA‐Cl, 3.5 NaCl, 1 CaCl2, 1 MgCl2, 10 Mg‐ATP, 0.5 Na‐GTP, 5.0 EGTA, 10 Hepes (pH 7.2). Cones and HCs were voltage‐clamped using either an Axopatch 200B patch clamp amplifier (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA) with an Alembic VE‐2 amplifier (Alembic Instruments, Montreal, Quebec, Canada) or with a Multiclamp 700A amplifier (Axon Instruments). Whole‐cell currents were digitized with an Axon Digidata 1440 or 1550 interface and acquired using Clampex 10 software (Molecular Devices). We did not correct for a measured liquid‐junction potential of −9 mV.
Cones and HCs were identified by their morphology and physiological properties (Van Hook & Thoreson, 2013). Charging curves in a sample of cones (n = 15) yielded the following passive parameters: membrane capacitance = 76.1 ± 6.68 pF, access resistance = 25.7 ± 2.86 MΩ, membrane resistance = 229.9 ± 53.3 MΩ and time constant = 1.48 ms ± 0.07. For HCs (n = 15): membrane capacitance = 41.9 ± 4.42 pF, access resistance = 36.0 ± 2.87 MΩ, membrane resistance = 239.4 ± 75.9 MΩ and time constant = 1.03 ± 0.11 ms.
Centre‐surround antagonistic stimulation
With the flat mount preparation, we obtained whole cell voltage‐clamp recordings from cones. Cones were held at −70 mV and then stepped to various test potentials from −60 to −10 mV to activate I Ca to different degrees. A spot plus annulus sequence for light stimulation was created in Microsoft PowerPoint and projected onto the retina from a compact LED projector (MPro110; 3M, Maplewood, MN, USA) through a prism placed in the condenser pathway. The cone was first illuminated with small spot of light (diameter = 45 μm) for 1.5 s and then the surrounding region of its receptive field was illuminated with an annulus (inner diameter = 45 μm; outer diameter = 1 mm) for another 1.5 s to stimulate feedback from nearby HCs. The onset and offset of light were detected with a photodiode. Annular illumination sometimes evoked direct light responses due to light scatter into the receptive field centre of the cone. To subtract these photocurrents, we subtracted the amplitude of currents evoked at annulus onset and offset when the cone was held at −60 mV, below the threshold for I Ca activation.
Paired recording protocol
For slice recordings, we obtained whole cell recordings simultaneously from both a cone and an HC. We tested synaptic connectivity by stimulating the cone with a step pulse (from −70 to −10 mV, 50 ms) to see if the simultaneously voltage‐clamped HC (V m = −60 mV) responded with a post‐synaptic current. To assess feedback strength, the HC was stepped to four different test potentials (−90, −60, −30 or 0 mV) for 1.9 s and, 1.5 s into the step, the cone I Ca was measured using a ramp‐voltage protocol (−90 to +60 mV, 0.5 mV ms−1). We waited 45 s between each HC test step and repeated the entire test step sequence twice, once beginning with the step to −90 mV and the other beginning with 0 mV.
We evaluated the strength of HC to cone feedback by determining the changes in both the peak amplitude (I peak) of the cone I Ca and the voltage at which the I Ca is half maximal (V 50) (Cadetti & Thoreson, 2006). I peak was normalized as a percentage change relative to the cone I peak values obtained when the post‐synaptic HC was voltage‐clamped at −90 mV. Changes in voltage dependence were measured as the difference in V 50 relative to the V 50 measured when the HC was held at −90 mV.
Data were analysed using Clampfit 10 (Molecular Devices) and Prism 4 (GraphPad, LaJolla, CA, USA). Statistical significance was evaluated by Student's t‐test or ANOVA at P < 0.05.
Results
In the first set of experiments on negative feedback from HCs to cones, we obtained whole‐cell, voltage‐clamp recordings from cones in a flat mount retina preparation and measured HC feedback to cones evoked by annular illumination, as illustrated in Fig. 1. For the examples shown in Fig. 1 A and B, the cone was voltage clamped at −30 mV, near the activation midpoint for the L‐type I Ca found in these cells. Application of a bright central spot of light onto this cone evoked an outward photocurrent. With maintained central illumination, we then applied an annulus to illuminate the surrounding region of retina. Illumination of the receptive field surround by the annulus evoked an inward feedback current (I feedback) resulting from release of negative HC feedback (Fig. 1 A, centre). Removal of annular illumination decreased I feedback and thus caused an outward current (Fig. 1 A, bottom). As expected (Hirasawa & Kaneko, 2003; Trenholm & Baldridge, 2010), increasing pH buffering by addition of 10 mm HEPES to the bicarbonate‐buffered superfusate abolished inward and outward feedback currents at both onset and offset of annular illumination (Fig. 1 B). In this example, the response to central illumination was also increased by HEPES (Fig. 1 B, top). However, this increase in the outward photocurrent was seen only at potentials above −50 mV, suggesting that it was due to inhibition of countervailing inward feedback currents. In both control and HEPES conditions, we repeated spot and annular stimulation at different cone holding potentials from −60 to −10 mV. We plotted the amplitude of feedback currents evoked at annulus onset and offset in Fig. 1 C. Previous studies (Verweij et al. 1996; Hirasawa & Kaneko, 2003) have shown that the inward feedback current at onset of the annulus is due to an increase in I Ca (and the outward current at annulus offset due to a decrease in I Ca). Changes in I feedback observed at onset and offset of annular illumination showed a voltage dependence consistent with I Ca (Fig. 1 C). Application of HEPES, which blocks feedback‐induced changes in I Ca voltage dependence and amplitude (Hirasawa & Kaneko, 2003; Cadetti & Thoreson, 2006), also blocked I feedback over this voltage range (Fig. 1 C). Light scattered back into the receptive field centre of the voltage‐clamped cone during annular illumination sometimes stimulated cone photocurrents. For the graph in Fig. 1 C, the amplitude of these photocurrents was subtracted from changes in I feedback by subtracting the amplitude of currents evoked at annulus onset or offset when the cone was voltage‐clamped at −60 mV, below the activation voltage for I Ca.
Figure 1. Centre‐surround antagonism assessed by light stimulation .
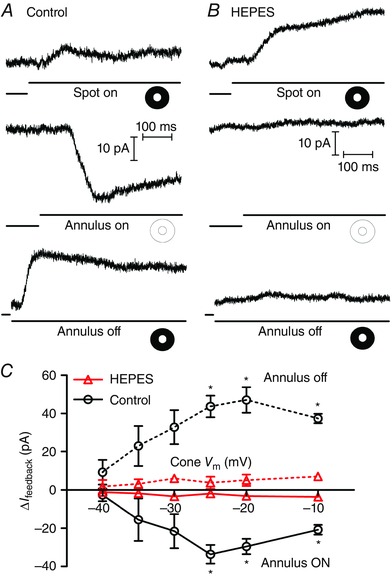
A, representative set of traces from a voltage‐clamped cone in a flat mount retina. Illumination of the cone with a spot of light evoked an outward current (top trace). Superposition of annular illumination evoked an inward feedback current (I feedback; middle trace). Removal of the annulus caused an outward current reflecting loss of I feedback (bottom trace). For the bottom traces, acquisition of the record was triggered at annulus offset by a photodiode. B, same protocol with 10 mm HEPES buffer added to the bath solution. Application of HEPES caused a slight increase in the outward light‐evoked current due to inhibition of inward feedback currents (top trace). No currents were observed at the onset (middle trace) or offset (bottom trace) of annular illumination showing that I feedback was eliminated by buffering synaptic pH changes with 10 mm HEPES. C, graph of the changes in feedback amplitude (ΔI feedback) upon onset (solid lines) and offset (dashed lines) of annular illumination plotted against the cone membrane potential (V m, n = 5). The differences in ΔI feedback between control (circles) and 10 mm HEPES (triangles, n = 5) achieved statistical significance at cone potentials of −25, −20 and −10 mV (annulus ON: cone V m = −25 mV P = 0.0063, −20 mV P = 0.0092, −10 mV P = 0.0077; annulus OFF: cone V m = −25 mV P = 0.0417, −20 mV P = 0.0367, −10 mV P = 0.0151). [Colour figure can be viewed at wileyonlinelibrary.com]
Extracellular carbonic anhydrase
One potential source of extracellular protons in the synaptic cleft is the activity of extracellular CA that catalyses the conversion of CO2 and H2O into H2CO3, which in turn spontaneously dissociates into H+ and HCO3 −. CA can be expressed both intra‐ and extracellularly (Haugh‐Scheidt & Ripps, 1998; Sarthy & Ripps, 2002; Supuran, 2008) and extracellular CA XIV has been observed in the outer plexiform layer of goldfish and mouse retina (Nagelhus et al. 2005; Fahrenfort et al. 2009). The CA inhibitors methazolamide and benzolamide have both been reported to inhibit feedback (Vessey et al. 2005; Fahrenfort et al. 2009). Benzolamide is often described as being membrane‐impermeant but is actually membrane‐permeant (Supuran & Scozzafava, 2004). Using the protocol described above, we tested an improved membrane‐impermeant CA inhibitor, benzothiophene‐3‐ylmethylsulfamide (FC5‐207a; 1 μm; Winum et al. 2007; Fiaschi et al. 2013). FC5‐207a caused a negative shift in the activation midpoint (V 50) for I Ca of −4.6 ± 0.84 mV and increase in the peak amplitude of I Ca (I peak) of 25.3 ± 3.96% (Fig. 2 A; n = 7). These changes are consistent with extracellular alkalization (Barnes et al. 1993) as expected from inhibition of extracellular CA activity. This suggests that CA activity can generate protons that have access to the extracellular face of Ca2+ channels within the cone synapse. However, as shown in Fig. 2 B, FC5‐207a did not significantly inhibit I feedback evoked at onset or offset of surround illumination. This indicates that while CA activity contributes extracellular protons to the synaptic cleft, it is not a major source for protons involved in feedback.
Figure 2. Extracellular carbonic anhydrase (CA) does not contribute significantly to the protons required for feedback .
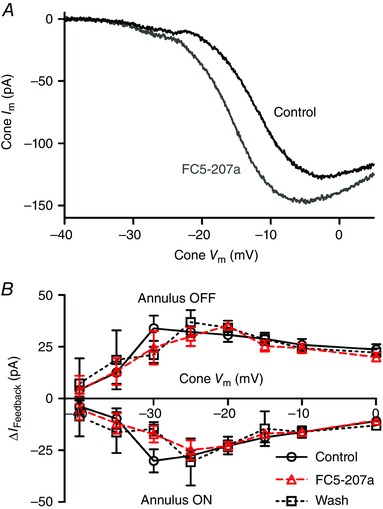
A, effect of the membrane‐impermeant CA antagonist FC5‐207a (1 μm) on I Ca. Application of FC5‐207a (grey trace) shifted the activation threshold of I Ca to more negative potentials and increased the peak current relative to control (black trace), consistent with alkalization of the synaptic cleft. B, changes in I feedback at onset and offset of the annulus were unchanged by application of 1 μm FC5‐207a (triangles) over the entire range of cone test potentials compared to prior control recordings (circles) or recordings obtained after washout (squares; n = 7 experiments). [Colour figure can be viewed at wileyonlinelibrary.com]
As illustrated in Fig. 2 A, we often saw inward current inflections near the threshold for I Ca activation. Similar inflections can also be seen in Figs 3 and 5. These are not due to a T‐type I Ca which is absent from cones (Wilkinson & Barnes, 1996). While the mechanisms responsible for these inflections are not yet clear, our observations suggest that they are due to protons released presynaptically from cones. In the many paired recordings conducted during the course of these experiments, the presence of these inflections was correlated with the presence of post‐synaptic currents evoked by cone depolarization in simultaneously voltage‐clamped HCs. However, we have not observed these current inflections previously in many cone recordings conducted in the presence of strong pH buffering with 10 mm HEPES (our unpublished observations) suggesting that they are pH‐dependent. While these cone current inflections were absent in 10 mm HEPES, post‐synaptic currents evoked in HCs by depolarizing stimulation of cones remain equally large (e.g. Bartoletti & Thoreson, 2011) indicating that inflections are a consequence, not a cause, of glutamate release from cones. Consistent with this conclusion, these inflections were also absent in recordings where cones were pre‐treated with BAF (3.5 μm; n = 7) for 15–90 min, which, as described later, blocks synaptic vesicle acidification. We blocked I h with 3 mm CsCl to test one specific possible mechanism whereby inward inflections might involve activation of I h in cones by vesicular protons (Stevens et al. 2001) but found that Cs+ did not eliminate the inflection (n = 3). This suggests that inward inflections may instead reflect proton influx through proton‐permeable channels in the cone membrane.
Figure 3. Protocol for measuring feedback with paired recordings from a cone and HC .
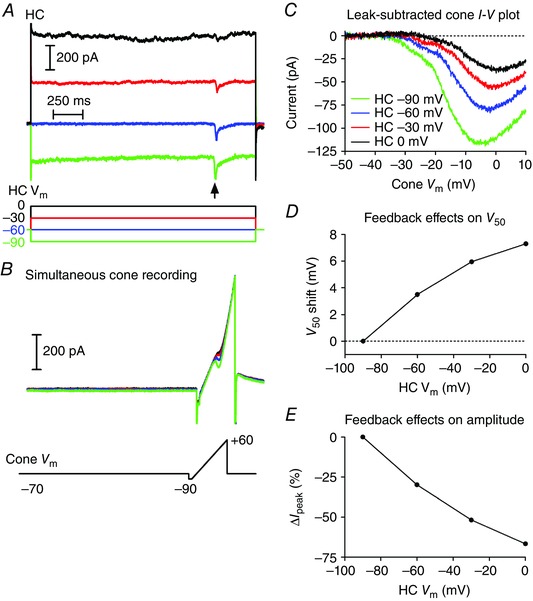
A, currents in an HC during steps from −60 to −90 (green), −60 (blue), −30 (red) and 0 mV (black). Note the inward synaptic currents evoked in the HC by activation of I Ca in the cone by a ramp voltage protocol (arrow). B, simultaneously recorded currents in the cone evoked by a ramp voltage protocol applied while the HC was stepped to different test potentials. C, leak‐subtracted I Ca plotted against the cone holding potential shows the increase in peak amplitude of the I Ca and shift in activation of the I Ca to more negative potentials as the HC was hyperpolarized. D, feedback‐induced changes in I Ca activation (V 50) as a function of HC holding potential. V 50 was measured as the cone V m at which the I Ca attained its half‐maximal amplitude. Values were normalized to the V 50 measured when the HC V m = −90 mV. E, change in peak amplitude of the cone I Ca as a function of changes in HC holding potential. Data are plotted as the percentage decrease in amplitude from that measured when HC V m = −90 mV. All data in this figure are from the same cone/HC pair.
Figure 5. Effects of sodium removal, the NHE antagonist cariporide and alkalizing intracellular pH on feedback .
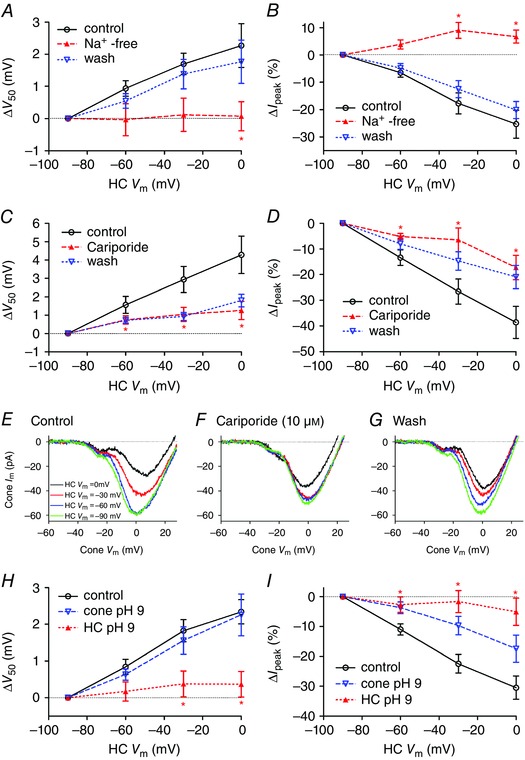
A and B, when Na+ in the extracellular solution was replaced with choline (filled triangles), feedback‐induced shifts in V 50 and changes in peak amplitude of I Ca (I peak) were eliminated (paired t‐test between test and control, n = 5; V 50: HC V m = −30 mV, P = 0.052; 0 mV, P = 0.043; I peak: HC V m = −60 mV, P = 0.059; −30 mV, P = 0.020; 0 mV, P = 0.020). Both measures recovered after washout (open triangles). C and D, cariporide (10 μm, n = 12, open triangles) caused a significant reduction in feedback‐induced changes in V 50 (C) and I peak (D) (paired t‐tests; V 50: HC V m = −60 mV, P = 0.0413; −30 mV, P = 0.0067; 0 mV, P = 0.0008; I peak: HC V m = −60 mV, P = 0.0024; −30 mV, P = 0.0004; 0 mV, P = 0.0026). E–G, this example shows I Ca recorded in a cone using the ramp voltage protocol when a synaptically coupled HC was voltage‐clamped at −90 (green trace), −60 (blue trace), −30 (red trace) and 0 mV (black trace). In control conditions, depolarization of the post‐synaptic HC was accompanied by a reduction in the peak amplitude of the presynaptic cone I Ca and a positive shift in activation voltage (E). Cariporide (10 μm) inhibited HC feedback effects on V 50 and I peak (F). Feedback effects recovered after washout (G). H and I, cone or HC pipette solutions were buffered to pH 9.2 and feedback was assessed using the paired recording protocol. H, alkalizing HC cytosol (filled triangles, n = 7) eliminated the feedback‐induced shift in V 50 with respect to control (n = 22, unpaired t‐test, ΔV 50: HC V m = −60 mV, P = 0.0403; −30 mV, P = 0.0072; 0 mV, P = 0.0035). Alkalizing cone cytosol (open triangles, n = 4) did not alter feedback‐induced shifts in V 50. I, alkalizing HC cytosol caused a significant reduction in feedback‐induced effects on I peak compared to control (unpaired t‐tests; HC alkalization: HC V m = −60 mV, P = 0.0134; −30 mV, P = 0.0005; 0 mV, P = 0.0007).
Paired recording protocol
Many of the drugs we tested could potentially inhibit feedforward glutamate release from cones and thus inhibit feedback simply by blocking HC light responses. To avoid this complication, we also tested feedback directly using an approach in which we manipulated the membrane potential of a voltage‐clamped HC while recording I Ca from a simultaneously voltage‐clamped cone. As illustrated in Fig. 3, we stepped the HC to one of four test potentials (−90, −60, −30 and 0 mV) for 1.9 s. At 1.5 s into the step, we measured I Ca in the presynaptic cone by applying a ramp voltage protocol (−90 to +60 mV, 0.5 mV ms−1). We confirmed synaptic connectivity between the HC and cone from the presence of a post‐synaptic current in the HC evoked by depolarizing stimulation of the cone. For example, Fig. 3 A shows inward synaptic currents (arrow) evoked by activation of I Ca during the voltage ramp applied to the cone (Fig. 3 B). The ramp‐evoked cone currents were leak subtracted and plotted against the cone membrane potential (Fig. 3 C). As illustrated in this example, progressive depolarization of the HC membrane potential caused the cone I Ca to activate at progressively more positive potentials and attain smaller peak currents. To assess the strength of HC feedback, we plotted the change in membrane potential at which the cone I Ca was half maximal (V 50, Fig. 3 D) and the change in I Ca peak amplitude (Fig. 3 E) against the HC holding potential. We normalized the data from different cells by comparing the changes in I Ca amplitude and V 50 relative to values measured when the HC was voltage‐clamped at −90 mV.
Vesicular protons
Synaptic vesicles utilize a v‐ATPase to take up protons and then use this proton gradient to load neurotransmitters such as glutamate and GABA (Poudel & Bai, 2014). v‐ATPase activity produces a luminal vesicle pH of 5–6 (Liu & Edwards, 1997). Fusion of glutamate‐filled vesicles in cone terminals releases protons into the synaptic cleft where they can exert an inhibitory effect on presynaptic I Ca (DeVries, 2001; Palmer et al. 2003; Hosoi et al. 2005; Cho & von Gersdorff, 2014). V‐ATPases inserted into the plasma membrane by synaptic vesicle fusion can also extrude protons directly into the cleft (Zhang et al. 2010). Consistent with earlier findings (Cadetti & Thoreson, 2006), we found that HCs exerted negative feedback effects on cone I Ca only in cell pairs that were synaptically connected but that continued presynaptic release of glutamate was not required for feedback as feedback persisted even after rundown of post‐synaptic currents in paired recordings. This suggests that release of vesicular protons by cones at a given synapse is not required to sustain feedback at that synapse. However, it is possible that GABA release might contribute protons because HCs are known to contain acidic synaptic vesicles at their terminals (Lee & Brecha, 2010). To test whether acidic synaptic vesicles in cones or HCs are a major source of protons for feedback, we inhibited v‐ATPase activity by bath application of BAF (3.5 μm) for 15–90 min. The ability of vesicular protons to inhibit I Ca presynaptically has been shown previously in cones, retinal bipolar cells and hair cells (DeVries, 2001; Palmer et al. 2003; Hosoi et al. 2005; Cho & von Gersdorff, 2014). We used proton inhibition of I Ca to assess the ability of BAF to inhibit v‐ATPase activity. As found in these earlier studies, in control cones without BAF, I Ca grew in amplitude as the vesicular release of protons subsided during a 100 ms depolarizing step (Fig. 4 A, black trace). This increase in I Ca was inhibited by blocking v‐ATPase activity with BAF (Fig. 4 A, grey trace). We quantified the impact of BAF on proton inhibition of presynaptic cone I Ca in two ways: (1) by comparing the ratio of I Ca measured at 20 ms to that measured at 90 ms (I 20ms/I 90ms, Fig. 4 B) and (2) by measuring total charge transfer during the first 25 ms of I Ca (Fig. 4 B). Both measurements showed that BAF significantly increased the initial I Ca amplitude, consistent with reduced proton inhibition. Post‐synaptic currents evoked in HCs by depolarizing steps applied to cones were also rapidly inhibited by BAF treatment and virtually absent from slices treated for >30 min (data not shown). The effects of HC GABA release on cones or HCs are not sufficiently reliable or predictable to test for BAF effects but we assume that BAF also blocked HC GABA release. Treatment with BAF (n = 7) caused a significant negative shift in V 50 for cone I Ca (−7.3 ± 2.1 mV, P = 0.0014, unpaired t‐test) relative to an untreated control group (n = 28) consistent with extracellular alkalization. To look at effects on HC to cone feedback, we used the paired recording protocol and measured changes in V 50 and peak amplitude of cone I Ca induced by changes in HC holding potential. We found no statistically significant differences in the strength of feedback between the two groups (unpaired t‐tests, Fig. 4 C and D). To ensure that we had thoroughly blocked the loading of protons into synaptic vesicles, we also incubated retinas overnight with BAF along with 1% BSA. Even after treatment with BAF for more than 12 h (n = 5), we found no significant change in the strength of feedback assessed by changes in V 50 or I peak compared to HC/cone pairs (n = 4) recorded from control slices incubated overnight in 1% BSA solution without BAF (Fig. 4 E and F). These results, which are consistent with findings from Vroman et al. (2014), show that while protons released into the cleft by vesicular ATPase activity can influence presynaptic cone I Ca, they are not the primary source of protons involved in lateral inhibitory feedback from HCs to cones.
Figure 4. v‐ATPase inhibitor, bafilomycin (BAF), prevents the loading of protons into synaptic vesicles but did not eliminate feedback .

A, I Ca recorded from a cone in control solution (black) and from another cone in the presence of 3.5 μm BAF (grey). I Ca was evoked by a voltage step from −70 to −10 mV for 100 ms. Passive currents were subtracted by a P/8 protocol. BAF treatment increased the initial portion of I Ca relative to control, showing loss of proton inhibition. B, differences in the inhibition of the initial portion of I Ca were statistically significant by two different measures: (1) comparing the amplitude of I Ca measured 20 and 90 ms after initiation of the step (I 20ms/I 90ms; n = 6; unpaired t‐test, P < 0.00001) and (2) comparing the total charge transfer (Q Ca) that entered the cone during the first 25 ms (grey‐shaded region; n = 6; unpaired t‐test, P < 0.00002). C and D, application of BAF (triangles) for 15–60 min produced no statistically significant effects on feedback assessed by changes in the peak amplitude of I Ca (C) and V 50 (D) that were induced by changes in HC holding potential (n = 7) when compared to control pairs (n = 28; unpaired t‐tests). E and F, effects of BAF (triangles) on feedback assessed by changes in V 50 (E) or peak amplitude of I Ca (F) were also not statistically significant in retinas treated for at least 12 h with BAF plus 1% BSA (n = 5) relative to control tissue incubated overnight in 1% BSA without BAF (n = 4). [Colour figure can be viewed at wileyonlinelibrary.com]
Sodium–hydrogen exchangers (NHEs)
The extrusion of protons by NHEs is the primary mechanism for extrusion of protons by most neurons (Koskelainen et al. 1993; Kalamkarov et al. 1996; Saarikoski et al. 1997; Haugh‐Scheidt & Ripps, 1998). To test for a contribution from NHEs, we began by testing whether feedback requires an inward Na+ driving force by replacing extracellular Na+ with choline. Na+ replacement eliminated feedback‐induced changes in both I peak and V 50 of cone I Ca caused by changes in HC holding potential (Fig. 5 A and B). Feedback‐induced changes in I peak and V 50 both recovered after returning to the control superfusate (Fig. 5 A and B). The finding that feedback requires an inward gradient for Na+ is consistent with a major role for NHE activity although it could also reflect involvement of other sodium‐dependent processes such as Na+/bicarbonate co‐transport.
Vessey et al. (2005) showed previously that an NHE antagonist, amiloride, inhibited HC to cone feedback. However, in addition to blocking NHE, amiloride can interact with other mechanisms such as epithelial sodium channels (ENaCs), acid‐sensing ion channels (ASICs) and transient receptor potential (TRP) channels. As a further test of the idea that NHE may be involved, we tested the NHE antagonist (Masereel et al. 2003) cariporide [N‐(aminoiminomethyl)‐5‐cyclopropyl‐1‐(5‐quinolinyl)‐1H‐pyrazole‐4‐carboxamide] for its effects on feedback. This compound has strong inhibitory effects on NHE1 and NHE2 at micromolar concentrations (Masereel et al. 2003). We found that cariporide (10 μm) caused a significant reduction in feedback‐induced changes in both I peak (n = 12 cone/HC pairs, paired comparisons at −60 mV, P = 0.0024; −30 mV, P = 0.0004; and 0 mV, P = 0.0026) and V 50 (paired comparison at −60 mV, P = 0.0413; −30 mV, P = 0.0067; and 0 mV, P = 0.0080; Fig. 5 C and D). Recovery during washout was slow but as illustrated by an example from one cell shown in Fig. 5 E–G, we saw recovery of feedback after lengthy washout in four cell pairs (Fig. 5 E–G). This example shows I Ca recorded in a cone using the ramp voltage protocol when a synaptically coupled HC was voltage‐clamped at −90 (green trace), −60 (blue trace), −30 (red trace) and 0 mV (black trace). As shown in Fig. 3, depolarization of the post‐synaptic HC in control conditions was accompanied by a reduction in the peak amplitude in the presynaptic cone I Ca and a positive shift in the activation voltage for I Ca. These changes were inhibited by cariporide and recovered after washout. A related compound, zoniporide [N‐(diaminomethylidene)‐3‐methanesulfonyl‐4‐(propan‐2‐yl)benzamide], showed a trend towards reduced feedback (25 μm; n = 5), but effects on I peak and V 50 did not attain statistical significance (data not shown). We also tested an amiloride derivative that acts as a CA inhibitor, 5‐(N‐ethyl‐N‐isopropyl)amiloride (EIPA, 25 μm), but it rapidly inhibited cone I Ca, preventing its use in assessing changes in feedback strength.
To assess whether HCs or cones are a major source for synaptic cleft protons in feedback, we tested effects of reducing the intracellular proton concentration to restrict the availability of protons needed for NHE activity. To do so, we obtained paired recordings and introduced an alkaline patch pipette solution with pH 9.2 into either the cone or the HC. We compared the changes in V 50 and amplitude with control recordings obtained using the normal pH 7.2 pipette solution. When we introduced the pH 9.2 solution into HCs, feedback‐induced differences in both V 50 and peak amplitude of cone I Ca were significantly reduced (n = 7) at HC holding potentials of −60, −30 and 0 mV (Fig. 5 H and I). HCs retained their viability as evidenced by the fact that holding currents were unchanged during 12–16 min of recording with pH 9.2 pipette solution (0.0 ± 10.2 pA, n = 7). In contrast to the profound reduction in feedback caused by alkalization of the HC cytosol, feedback‐induced differences in V 50 were not reduced by alkalization of the cone cytosol (Fig. 5 H; n = 4). Feedback effects of HC membrane potential changes on the peak amplitude of cone I Ca were, however, somewhat reduced by use of pH 9.2 pipette solutions in the cone (Fig. 5 I). To explain this result, we postulated that alkalization of the cone interior may prevent changes in extracellular pH from altering protonation of amino acid residues located within the channel pore that are critical for mediating proton effects on conductance (Chen et al. 1996; Chen & Tsien, 1997). To test this idea, we compared the changes in peak amplitude of cone I Ca caused simply by changing extracellular superfusate pH while recording with either control or pH 9.2 pipette solutions. When using the control pipette solution, alkalization of the superfusate from pH 7.4 to 7.8 doubled the amplitude of I Ca (213.4 ± 25.5% relative to pH 7.4, n = 6) but when using pH 9.2 pipette solution, the same alkalization did not increase I Ca amplitude (94.4 ± 9.8% relative to pH 7.4, n = 4; difference compared to alkalization‐induced changes with control pipette solution, unpaired t‐test: P = 0.0057). This is consistent with the idea that the Ca2+ channel site regulating pH‐dependent amplitude changes is accessible from the interior of the cell (Chen et al. 1996; Chen & Tsien, 1997) and that alkalization of the cone interior with pH 9.2 solution limits the changes in amplitude that would normally be produced by changes in extracellular pH. While it is possible that alkalization of the HC interior may disrupt feedback by acting on some other mechanism, the finding that use of pH 9.2 pipette solutions in HCs but not cones prevents feedback‐induced changes in V 50 is consistent with the idea that protons involved in feedback are derived largely from HC cytosol.
Effects of extracellular pH on feedback
The data above point to NHE activity in HCs as being a major source for protons involved in HC feedback to cones. However, NHEs are not known to be voltage‐sensitive and so while NHEs may provide a tonic source of protons, we hypothesized that a separate mechanism is probably responsible for rapid changes in synaptic cleft pH caused by HC membrane potential changes. There are two general possibilities: changes in proton flux through ion channels in the HC membrane or changes in buffering of free extracellular protons (e.g. by phosphate or bicarbonate). To explore these possibilities, we began by testing whether feedback was sensitive to the proton driving force. We measured feedback strength from paired cone/HC recordings in control pH 7.4 solution and then switched to a solution with either pH 7.1 or 7.8. As expected, acidification decreased cone I Ca and shifted its activation to more positive values (Barnes et al. 1993). Reducing the pH from 7.8 to 7.4 caused a +10.5 ± 1.7 mV shift in V 50 (n = 5) and reducing it further from pH 7.4 to 7.1 caused an additional +5.5 ± 1.2 mV shift (n = 6; Fig. 6 A). Although I Ca amplitude was reduced by extracellular acidification, feedback‐induced changes in both V 50 (Fig. 6 A; P < 0.05, one‐way ANOVA at HC V m = −30 and 0 mV) and I Ca amplitude (Fig. 6 B; P < 0.015, one‐way ANOVA at HC V m = −60, −30 and 0 mV) increased significantly as the pH was reduced from 7.8 to 7.4 to 7.1.
Figure 6. Lowering extracellular pH increases feedback strength .
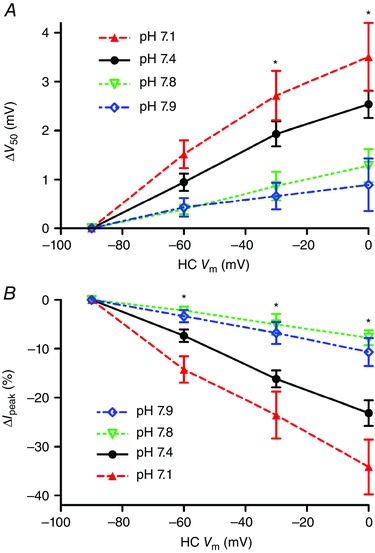
Using the paired recording protocol, feedback was tested at pH 7.8 (open triangles, n = 5), 7.4 (filled circles, n = 23) or 7.1 (open triangles, n = 7). There were significant increases in both the V 50 shift (A) and change in I peak (B) as external pH was lowered from 7.8 to 7.4 to 7.1 by changing bicarbonate from 12 to 22 to 32 mm (one‐way ANOVA, V 50: HC V m = −60 mV, P = 0.0571; −30 mV, P = 0.0143; 0 mV, P = 0.0093; I peak: HC V m = −60 mV, P = 0.0043; −30 mV, P = 0.0018; 0 mV, P = 0.0016). Similar decreases in both V 50 shift (A) and change in I peak (B) were seen as pH was increased from 7.4 to 7.9 (diamonds) in the presence of 22 mm bicarbonate. [Colour figure can be viewed at wileyonlinelibrary.com]
In these experiments, the pH 7.1 and 7.8 solutions were prepared by using 12 or 32 mm NaHCO3, respectively, rather than 22 mm used for the control pH 7.4 solution. To test whether the stronger feedback observed in more acidic extracellular environments might be due to differences in bicarbonate levels rather than protons, we tested a solution in which we maintained a constant bicarbonate concentration of 22 mm but altered the pH from 7.4 to 7.9. To do so, we bubbled the standard bicarbonate‐buffered solution for a few minutes with 100% O2 and then, if necessary, added HCl to attain a final pH of 7.9 just before applying it to the retina. As illustrated in Fig. 6 (open circles), feedback strength assessed by both changes in V 50 and amplitude was weakened significantly by increasing the pH to 7.9 in this way, attaining levels similar to those observed when the pH was changed to 7.8 by altering bicarbonate levels. These data indicate that extracellular pH changes by themselves can have a major impact on the strength of feedback.
Effects of bicarbonate on feedback
The finding that feedback strength is enhanced by extracellular acidification is consistent with the possibility that light‐evoked alkalization of the cleft during feedback may be due to an influx of protons into hyperpolarized HCs but could also be explained by an increase in extracellular pH buffering. We save consideration of specific possible mechanisms for the Discussion, but note there are a variety of proton‐permeable ion channels that might mediate proton entry into HCs when they hyperpolarize. Alternatively, at physiological pH, a small change in bicarbonate concentration can cause a large change in pH, suggesting that increased bicarbonate efflux (or reduced bicarbonate influx) might also be responsible for cleft alkalization in light. We therefore tested whether bicarbonate was required to maintain feedback. HEPES at 10 mm blocks feedback entirely (Hirasawa & Kaneko, 2003; Cadetti & Thoreson, 2006; Trenholm & Baldridge, 2010) and so we first tested whether feedback could be maintained after supplementing the standard pH 7.4 bicarbonate solution with a lower concentration of 1 mm HEPES. Addition of 1 mm HEPES weakened feedback but did not abolish it (Fig. 7 A and B). In the presence of 1 mm HEPES to stabilize the extracellular pH at 7.4, we then applied a bicarbonate‐free solution. Bicarbonate removal eliminated the remaining feedback (Fig. 7 A and B) showing that feedback‐induced changes in cleft pH require the presence of bicarbonate.
Figure 7. Bicarbonate is required for feedback and feedback is blocked by the anion transport inhibitor DIDS .
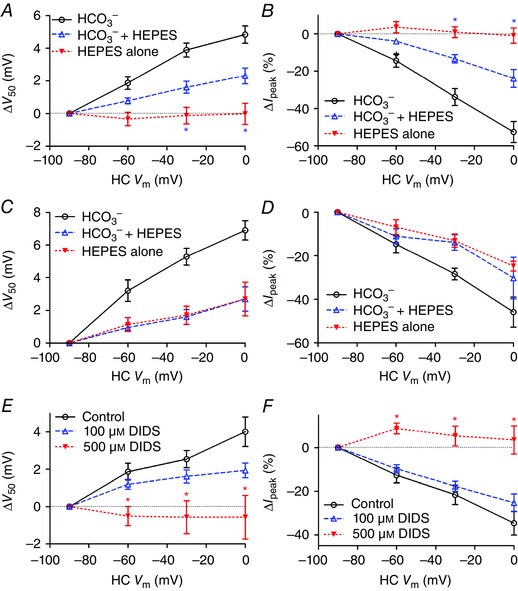
A, a slight increase in buffering capacity of the external solution by addition of 1 mm HEPES decreased the magnitude of the activation shift in V 50 [unpaired Student's t‐test, control (open circles; n = 9) vs. NaHCO3 + 1 mM HEPES (open triangles; n = 6), ΔV 50: HC V m = −30 mV, P = 0.0406; 0 mV, P = 0.0458], while removal of bicarbonate (filled triangles; n = 12) eliminated the shift (NaHCO3 + 1 mm Hepes vs. 1 mm HEPES only, 0 mV, P = 0.0315; −30 mV, P = 0.0379; −60 mV, P = 0.0836). B, feedback‐induced changes in I peak were reduced when 1 mm Hepes was added to the normal solution (open triangles; unpaired Student's t‐test, control vs. NaHCO3 + 1 mm HEPES, I peak: HC V m = 0 mV, P = 0.0034; −30 mV, P = 0.0073; −60 mV, P = 0.034) and abolished when bicarbonate was removed entirely (filled triangles; unpaired Student's t‐test, NaHCO3 + 1 mm Hepes vs. 1 mm HEPES only, HC V m = 0 mV, P = 0.0034; −30 mV, P = 0.0054; −60 mV, P = 0.1034). In paired recordings where the HC pipette contained 90 mm HCO3 −, removal of extracellular HCO3 − did not abolish feedback‐induced changes in V 50 (C) or I peak (D). Application of the anion transport inhibitor DIDS (500 μm, n = 5; filled triangles) significantly inhibited feedback‐induced changes in V 50 (E) and I peak (F) (unpaired Student's t‐test, d.f. = 15; V 50: HC V m = −60 mV, P = 0.0080; −30 mV, P = 0.0039; 0 mV, P = 0.0063; I peak: HC V m = −60 mV, P = 0.0019; −30 mV, P = 0.0029; 0 mV, P = 0.0011). A lower concentration of DIDS (100 μm; n = 7; open triangles) had a weaker effect that did not attain statistical significance. [Colour figure can be viewed at wileyonlinelibrary.com]
Does feedback depend only on the extracellular presence of bicarbonate or was feedback eliminated because of a secondary depletion of intracellular bicarbonate from HCs? To answer this question, we obtained paired recordings in which the HC recording pipette contained high levels of HCO3 − (90 mm). After elevation of HCO3 − in the HC, removal of extracellular HCO3 − from the superfusate did not abolish feedback as assessed by changes in V 50 or amplitude (Fig. 7 C and D). Together, these results indicate that extracellular pH changes during feedback involve an efflux of bicarbonate from HCs.
As a further test for possible involvement of bicarbonate transporters, we used the inhibitor, 4,4′‐diisothiocyano‐2,2′‐stilbene‐disulfonic acid (DIDS) (Romero et al. 2013; Virkki et al. 2002; Shmukler et al. 2014). At a concentration of 500 μm (n = 5), DIDS significantly inhibited HC to cone feedback as assessed by shifts in V 50 and changes in peak amplitude of cone I Ca caused by changes in HC membrane potential (Fig. 7 E and F). We did not observe recovery from 500 μm DIDS treatment after washout. However, feedback was stable for equally long periods in control recordings and as illustrated in Fig. 5 we observed recovery after washout from sodium replacement experiments. Further evidence that the reduction observed in DIDS was not simply due to response rundown comes from the finding that a lower concentration of DIDS (100 μm, n = 7; Fig. 7 E and F) did not produce significant inhibition. In addition to reducing effects of HC feedback on activation voltage and amplitude of cone I Ca, DIDS also caused an overall negative activation shift in I Ca V 50 (−14.6 ± 2.63 mV, P = 0.0052) and an increase in I Ca peak amplitude (+53.4 ± 31.3%; P = 0.0081) when the HC membrane potential was held at a constant value of −90 mV. These feedback‐independent changes in I Ca are consistent with alkalization of the synaptic cleft (Barnes et al. 1993). In addition to blocking bicarbonate transport, DIDS can also inhibit a number of anion channels. DIDS did not cause a significant change in HC membrane conductance (n = 5). In cones, tail currents activated by strong depolarizing steps are due largely to Ca2+‐activated Cl− currents (Mercer et al. 2011). Inward tail currents observed after voltage ramps (−90 to +60 mV, 300 ms) were actually larger following DIDS treatment (P = 0.03). This is probably an indirect effect of the DIDS‐induced increase in I Ca. Ca2+‐activated Cl− currents can reduce the strength of feedback (Endeman et al. 2012). However, it is unlikely that this contributed significantly to inhibitory effects of DIDS on feedback in our experiments as we assessed feedback by using voltage ramp protocols in which Ca2+‐activated Cl− currents are not significantly active until after the peak of I Ca has been attained. While effects of DIDS include a number of possible targets, the ability of this compound to inhibit feedback supports bicarbonate removal experiments in suggesting that bicarbonate transport is required to maintain lateral‐inhibitory feedback from HCs to cones.
Discussion
The mechanisms of inhibitory feedback from HCs to cones remain a puzzle despite many years of study, but a consensus has emerged that regulation of cone Ca2+ channels by extracellular protons within the synaptic cleft plays an important role in this process (Liu et al. 2013; Wang et al. 2014; Vroman et al. 2014; Kemmler et al. 2014). The present data show that NHE activity in the HC membrane is a major source of protons participating in HC feedback. However, the presence of intracellular bicarbonate within HCs is also essential, suggesting that HC voltage changes may modify extracellular pH by regulating the transmembrane flux of bicarbonate.
CA can either generate protons and bicarbonate (by hydrating CO2) or convert them into CO2 and water (bicarbonate dehydration reaction). CA enzymatic activity is coupled with other proteins involved in ion transport and metabolism. Extracellular CO2 is processed by CA isoforms with extracellular active sites (CA IX, XII, XIV or IV) that work in concert with anion exchangers and sodium/bicarbonate co‐transporters to recycle bicarbonate. This dynamic process can produce extracellular acidification and concomitant intracellular alkalization (Neri & Supuran, 2011). The CA inhibitors, benzolamide and methazolamide, have been shown to interfere with feedback (Vessey et al. 2005; Fahrenfort et al. 2009). While often considered to be membrane‐impermeant, benzolamide, like methazolamide, can actually cross the membrane (Supuran & Scozzafava, 2004). We therefore tested an improved membrane‐impermeant CA inhibitor, FC5‐207a. The effects of this compound on the V 50 and amplitude of cone I Ca are consistent with alkalization of the synaptic cleft (Barnes et al. 1993). While this suggests that extracellular CA activity generates extracellular protons, it does not appear to be the major source of extracellular protons involved in feedback as FC5‐207a did not significantly reduce the strength of HC feedback (Fig. 2 B). The ability of benzolamide and methazolamide to inhibit feedback is thus more likely to be due to effects on intracellular CA activity. Consistent with this idea, Vessey et al. (2005) found that methazolamide only eliminated feedback effects induced by hyperpolarization of HCs and suggested that intracellular acidification by methazolamide perhaps reduced the inward driving force for protons upon hyperpolarization.
Consistent with release of protons during fusion of glutamatergic synaptic vesicles (DeVries, 2001; Hosoi et al. 2005), blocking v‐ATPase activity with BAF eliminated the initial inhibition of I Ca seen during depolarizing stimulation of cones. In addition, we observed that BAF treatment caused a negative activation shift in cone I Ca consistent with cleft alkalization. Using a pH‐sensitive dye, 5‐hexadecanoylaminofluorescein, Jouhou et al. (2007) reported that BAF reduced extracellular acidification stimulated by depolarization of isolated fish HCs, suggesting that v‐ATPase activity in HCs may also provide a source of extracellular protons. However, a subsequent study concluded that this dye reports near‐membrane intracellular pH, not extracellular pH (Jacoby et al. 2014). Wang et al. (2014) found that BAF blocked the ability of HC depolarization to acidify the cone synaptic cleft by using transgenic zebrafish with a pH‐sensitive dye attached to the extracellular surface of cone Ca2+ channels (caliphluorin). Our results showed that although v‐ATPase activity contributes protons to the extracellular environment, BAF did not eliminate HC feedback effects on cone I Ca (Fig. 4). This suggests that synaptic vesicle protons are not a major steady‐state source of protons involved in feedback. However, note that because we measured feedback in cone/HC pairs by applying cone voltage ramps 1.5 s after a voltage step was applied to the HC, we could not detect rapid effects of vesicular protons on feedback that might have occurred immediately after changes in HC membrane potential. Thus, while our results showed that vesicular protons are not a major steady‐state source of protons in feedback, dynamic changes in vesicle release might be capable of modifying the kinetics of feedback.
The major source of protons in HC feedback appears to derive from NHE activity. This conclusion is supported by experiments showing that removal of Na+ from the extracellular medium and application of the NHE antagonist cariporide both significantly inhibited feedback‐induced changes in I Ca. It is also supported by the finding that another NHE antagonist, amiloride, also blocks HC feedback to cones (Vessey et al. 2005). A major role for NHE activity in regulating extracellular pH at the cone/HC synapse is consistent with other studies showing that NHEs serve as the primary mechanism for proton extrusion by neurons (Koskelainen et al. 1993; Kalamkarov et al. 1996; Saarikoski et al. 1997; Haugh‐Scheidt & Ripps, 1998). As discussed above, extracellular CA and v‐ATPase activity both appear to contribute protons to the cleft and so it is not entirely surprising that cariporide did not completely block feedback. Another potential source for synaptic cleft protons is efflux through voltage‐gated proton channels (DeCoursey, 2013). From the retinal gene expression database of Siegert et al. (2012), these channels appear to be strongly expressed in Müller glia. Voltage‐gated proton channels are blocked by low concentrations of zinc that have also been shown to block feedback (Vroman et al. 2015). However, zinc can also interfere with numerous other mechanisms including CLC chloride channels, AMPA receptors, calcium channels and GABA receptors (Ripps & Chappell, 2014; Stauber et al. 2012).
Reducing the free proton concentration in HCs by using a pipette solution of pH 9.2 eliminated feedback‐induced changes in cone I Ca. By contrast, introducing pH 9.2 solution into cones did not alter feedback‐induced shifts in V 50, suggesting that feedback remained intact. However, alkalization of the cone intracellular milieu did reduce changes in peak amplitude of cone I Ca caused by changes in HC membrane potential. Changes in pH influence the amplitude of L‐type I Ca by altering the protonation state of four key glutamate residues lying within the Ca2+ channel pore (Chen et al. 1996). One of these residues is exposed to the cytosol and this particular residue stabilizes the protonation states of the other three glutamate residues (Chen et al. 1996; Chen & Tsien, 1997). Strongly alkalizing the cone cytosol by use of pH 9.2 pipette solution may act on this exposed cytosolic residue to stabilize protonation of the other residues and thereby diminish the impact of extracellular pH changes on I Ca amplitude. Consistent with this, changes in cone I Ca amplitude caused by changes in superfusate pH were also inhibited by use of a pH 9.2 pipette solution in cones. We cannot exclude the possibility that alkalizing the HC cytosol might block feedback by interfering with other mechanisms such as bicarbonate transport, but renal bicarbonate transport is not altered by changes in intracellular proton levels (Zhou et al. 2006). While proton extrusion by NHEs in other nearby cells including Müller glia might also contribute, the finding that lowering the free proton concentration in the HC cytosol inhibited feedback suggests that many of the protons supplied by NHE activity derive from HCs.
As NHEs are not voltage‐dependent, it seems unlikely that voltage‐dependent changes in NHE activity are directly responsible for the cleft alkalization caused by HC hyperpolarization (Demaurex et al. 1995; Fuster et al. 2004). We instead hypothesized that NHEs provide a steady background source of protons and that alkalization of the synaptic cleft during HC hyperpolarization arises from some other mechanism. We considered three other ways that HC hyperpolarization might reduce the free proton concentration in the cleft: (1) influx of protons into HCs through ion channels (e.g. through TRP channels, gap junction hemichannels or other proton‐permeable channels; Vessey et al. 2005; Wang et al. 2014); (2) increase in bicarbonate levels in the cleft (e.g. through bicarbonate transporters or anion channels; Liu et al. 2013); and (3) efflux of phosphate buffer into the cleft (e.g. by hydrolysis of ATP exiting HCs through pannexin channels; Vroman et al. 2014). Vroman et al. (2014) suggested that the phosphate buffer created by hydrolysis of ATP has a pKa ≈ 7.2, and so feedback should be weakened at this pH. This appears contrary to our observations showing that feedback was stronger at pH 7.1 and weakened progressively as the pH was increased to 7.4 and 7.8. Additionally, we found that the presence of bicarbonate is required for feedback. A key role for bicarbonate can account for the finding that feedback strength was increased by extracellular acidification and weakened by alkalization. For example, alkalization of the superfusate from pH 7.4 to 7.9 diminished the strength of feedback even when superfusate bicarbonate levels were not changed (Fig. 6). With fewer free protons available for buffering, the impact of local cleft changes in bicarbonate (e.g. following bicarbonate extrusion from HCs) will diminish in a more alkaline solution. More significantly, we found that removing bicarbonate from the superfusate abolished feedback and boosting intracellular bicarbonate levels within HCs prevented this loss of feedback. These results show that bicarbonate is essential for feedback and suggests that removal of bicarbonate from the superfusate blocks feedback by depriving HCs of intracellular bicarbonate. The ability of DIDS to inhibit feedback provides further support for the involvement of HCO3 − transporters although DIDS can also act on a variety of anion transport mechanisms. Among the DIDS‐sensitive Slc4 family of bicarbonate transporters, the retinal gene expression database from Siegert et al. (2012) indicates that murine HCs express Slc4a3 and Slc4a5. Slc4a5 is a sodium‐bicarbonate cotransporter that shows punctate expression in the outer plexiform layer (Kao et al. 2011) but low expression levels in photoreceptors (Siegert et al. 2012), suggesting that expression is probably concentrated in HC dendrites. Slc4a5 knockout mice show a loss of photoreceptors and diminished electroretinogram a‐ and b‐waves but feedback from HCs was not studied in these animals (Kao et al. 2011). Involvement of a sodium‐bicarbonate cotransporter would accommodate the findings that sodium replacement inhibited feedback. Interestingly, there is evidence that Slc4a5 can be voltage‐dependent, increasing HCO3 − entry into cells upon depolarization (Virkki et al. 2002). However, our finding that high levels of bicarbonate in the HC recording pipette prevented loss of feedback after removal of extracellular bicarbonate does not support the idea that HC hyperpolarization alkalizes the synaptic cleft by causing a voltage‐dependent reduction in the influx of bicarbonate into HCs. Slc4a3 is an Na+‐independent Cl−/HCO3 − exchanger expressed throughout the entire HC (Kobayashi et al. 1994). While there is no evidence that Slc4a3 activity is voltage‐sensitive (Halligan et al. 1991), voltage‐sensitivity might be conferred indirectly by voltage‐dependent changes in chloride efflux through nearby channels. Such a mechanism could explain why feedback strength can be reduced by increases in cone Cl− conductance (e.g. by activating GABA receptors or Ca2+‐activated Cl− channels; Endeman et al. 2012). Voltage‐dependent changes in bicarbonate flux through anion channels might also confer voltage‐dependence. Liu et al. (2013) postulated a mechanism by which depolarization of HCs stimulated release of GABA, which then acts in an autocrine fashion to activate HC GABA receptors. Reduced influx of bicarbonate through GABA receptor channels into the HC when it hyperpolarizes causes the cleft to alkalinize (Liu et al. 2013). Studies in a number of species have shown that GABA receptor antagonists do not block HC feedback to cones (Thoreson & Burkhardt, 1990; Verweij et al. 1996, 2003), suggesting that while GABA receptors may modify feedback under certain conditions (e.g. during certain circadian or illumination conditions), they are not the principal mechanism for feedback.
In summary, our results identify two principal mechanisms that work together to produce the extracellular pH changes that mediate inhibitory feedback from HCs to cones. The steady extrusion of protons by NHEs is necessary to maintain a sufficient concentration of free protons in the synaptic cleft. However, HC to cone feedback also requires the presence of bicarbonate in HCs, suggesting that voltage‐dependent changes in bicarbonate transport across the HC membrane, rather than voltage‐dependent changes in NHE activity, are primarily responsible for the changes in synaptic cleft pH produced by HC voltage changes.
Additional information
Competing interests
The authors declare no competing interests.
Author contributions
WT, TW and MVH designed experiments, performed recordings and analysed data. CS provided novel reagents and critical interpretation. TW drafted the article and WT, MVH and CS revised it. All authors approved the final version. Experiments were conducted at the University of Nebraska Medical Center in Omaha.
Funding
This study was upported by NIH Grants R01EY10542 (WBT) and F32EY023864 (MJVH) and a Senior Scientific Investigator Award from Research to Prevent Blindness (WBT).
References
- Barnes S, Merchant V & Mahmud F (1993). Modulation of transmission gain by protons at the photoreceptor output synapse. Proc Natl Acad Sci USA 90, 10081–10085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoletti TM & Thoreson WB (2011). Quantal amplitude at the cone ribbon synapse can be adjusted by changes in cytosolic glutamate. Mol Vis 17, 920–931. [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Ernstrom GG, Nix P, Davis MW & Jorgensen EM (2008). Protons act as a transmitter for muscle contraction in C. elegans . Cell 132, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadetti L & Thoreson WB (2006). Feedback effects of horizontal cell membrane potential on cone calcium currents studied with simultaneous recordings. J Neurophysiol 95, 1992–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey JR, Grinstein S & Orlowski J (2010). Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol 11, 50–61. [DOI] [PubMed] [Google Scholar]
- Chen XH, Bezprozvanny I & Tsien RW (1996). Molecular basis of proton block of L‐type Ca2+ channels. J Gen Physiol 108, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XH & Tsien RW (1997). Aspartate substitutions establish the concerted action of P‐region glutamates in repeats I and III in forming the protonation site of L‐type Ca2+ channels. J Biol Chem 272, 30002–30008. [DOI] [PubMed] [Google Scholar]
- Chesler M (2003). Regulation and modulation of pH in the brain. Physiol Rev 83, 1183–1221. [DOI] [PubMed] [Google Scholar]
- Cho S & von Gersdorff H (2014). Proton‐mediated block of Ca2+ channels during multivesicular release regulates short‐term plasticity at an auditory hair cell synapse. J Neurosci 34, 15877–15887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCoursey TE (2013). Voltage‐gated proton channels: molecular biology, physiology, and pathophysiology of the HV family. Physiol Rev. 93, 599–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaurex N, Orlowski J, Brisseau G, Woodside M & Grinstein S (1995). The mammalian Na+/H+ antiporters NHE‐1, NHE‐2, and NHE‐3 are electroneutral and voltage independent, but can couple to an H+ conductance. J Gen Physiol 106, 85–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVries SH (2001). Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron 32, 1107–1117. [DOI] [PubMed] [Google Scholar]
- Endeman D, Fahrenfort I, Sjoerdsma T, Steijaert M, ten Eikelder H & Kamermans M (2012). Chloride currents in cones modify feedback from horizontal cells to cones in goldfish retina: chloride currents in cones. J Physiol 590, 5581–5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahrenfort I, Steijaert M, Sjoerdsma T, Vickers E, Ripps H, van Asselt J, Endeman D, Klooster J, Numan R, ten Eikelder H, von Gersdorff H & Kamermans M (2009). Hemichannel‐mediated and pH‐based feedback from horizontal cells to cones in the vertebrate retina. PLoS One 4, e6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiaschi T, Giannoni E, Taddei ML, Cirri P, Marini A, Pintus G, Nativi C, Richichi B, Scozzafava A, Carta F, Torre E, Supuran CT & Chiarugi P (2013). Carbonic anhydrase IX from cancer‐associated fibroblasts drives epithelial–mesenchymal transition in prostate carcinoma cells. Cell Cycle 12, 1791–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster D, Moe OW & Hilgemann DW (2004). Lipid‐ and mechanosensitivities of sodium/hydrogen exchangers analyzed by electrical methods. Proc Natl Acad Sci USA 101, 10482–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology . Exp Physiol 100, 755–758. [DOI] [PubMed] [Google Scholar]
- Halligan RD, Shelat H & Kahn AM (1991). Na+‐independent Cl––HCO3 – exchange in sarcolemmal vesicles from vascular smooth muscle. Am J Physiol 260, C347–C354. [DOI] [PubMed] [Google Scholar]
- Haugh‐Scheidt L & Ripps H (1998). pH regulation in horizontal cells of the skate retina. Exp Eye Res 66, 449–463. [DOI] [PubMed] [Google Scholar]
- Highstein SM, Holstein GR, Mann MA & Rabbitt RD (2014). Evidence that protons act as neurotransmitters at vestibular hair cell‐calyx afferent synapses. Proc Natl Acad Sci USA 111, 5421–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa H & Kaneko A (2003). pH changes in the invaginating synaptic cleft mediate feedback from horizontal cells to cone photoreceptors by modulating Ca2+ channels. J Gen Physiol 122, 657–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi N, Arai I & Tachibana M (2005). Group III metabotropic glutamate receptors and exocytosed protons inhibit L‐type calcium currents in cones but not in rods. J Neurosci 25, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby J, Kreitzer MA, Alford S & Malchow RP (2014). Fluorescent imaging reports an extracellular alkalinization induced by glutamatergic activation of isolated retinal horizontal cells. J Neurophysiol 111, 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouhou H, Yamamoto K, Homma A, Hara M, Kaneko A & Yamada M (2007). Depolarization of isolated horizontal cells of fish acidifies their immediate surrounding by activating V‐ATPase. J Physiol 585, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamkarov G, Pogozheva I, Shevchenko T, Koskelainen A, Hemila S & Donner K (1996). pH changes in frog rods upon manipulation of putative pH‐regulating transport mechanisms. Vision Res 36, 3029–3036. [DOI] [PubMed] [Google Scholar]
- Kamermans M, Fahrenfort I, Schultz K, Janssen‐Bienhold U, Sjoerdsma T & Weiler R (2001). Hemichannel‐mediated inhibition in the outer retina. Science 292, 1178–1180. [DOI] [PubMed] [Google Scholar]
- Kao L, Kurtz LM, Shao X, Papadopoulos MC, Liu L, Bok D, Nusinowitz S, Chen B, Stella SL, Andre M, Weinreb J, Luong SS, Piri N, Kwong JMK, Newman D & Kurtz I (2011). Severe neurologic impairment in mice with targeted disruption of the electrogenic sodium bicarbonate cotransporter NBCe2 (Slc4a5 gene). J Biol Chem 286, 32563–32574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemmler R, Schultz K, Dedek K, Euler T & Schubert T (2014). Differential regulation of cone calcium signals by different horizontal cell feedback mechanisms in the mouse retina. J Neurosci 34, 11826–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Morgans CW, Casey JR & Kopito RR (1994). AE3 anion exchanger isoforms in the vertebrate retina: developmental regulation and differential expression in neurons and glia. J Neurosci 14, 6266–6279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskelainen A, Donner K, Lerber T & Hemilä S (1993). pH regulation in frog cones studied by mass receptor photoresponses from the isolated retina. Vision Res 33, 2181–2188. [DOI] [PubMed] [Google Scholar]
- Lee H & Brecha NC (2010). Immunocytochemical evidence for SNARE protein‐dependent transmitter release from guinea pig horizontal cells. Eur J Neurosci 31, 1388–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hirano AA, Sun X, Brecha NC & Barnes S (2013). Calcium channels in rat horizontal cells regulate feedback inhibition of photoreceptors through an unconventional GABA‐ and pH‐sensitive mechanism. J Physiol 591, 3309–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y & Edwards RH (1997). The role of vesicular transport proteins in synaptic transmission and neural degeneration. Annu Rev Neurosci 20, 125–156. [DOI] [PubMed] [Google Scholar]
- Masereel B, Pochet L & Laeckmann D (2003). An overview of inhibitors of Na+/H+ exchanger. Eur J Med Chem 38, 547–554. [DOI] [PubMed] [Google Scholar]
- Mercer AJ, Rabl K, Riccardi GE, Brecha NC, Stella SL Jr, & Thoreson WB (2011). Location of release sites and calcium‐activated chloride channels relative to calcium channels at the photoreceptor ribbon synapse. J Neurophysiol. 105, 3213–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Bateman AC, Haug F‐M, Ottersen OP, Grubb JH, Waheed A & Sly WS (2005). Carbonic anhydrase XIV is enriched in specific membrane domains of retinal pigment epithelium, Muller cells, and astrocytes. Proc Natl Acad Sci USA 102, 8030–8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri D, Supuran CT (2011). Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov. 10, 767–777. [DOI] [PubMed] [Google Scholar]
- Obara M, Szeliga M & Albrecht J (2008). Regulation of pH in the mammalian central nervous system under normal and pathological conditions: facts and hypotheses. Neurochem Int 52, 905–919. [DOI] [PubMed] [Google Scholar]
- Palmer MJ, Hull C, Vigh J & von Gersdorff H (2003). Synaptic cleft acidification and modulation of short‐term depression by exocytosed protons in retinal bipolar cells. J Neurosci 23, 11332–11341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudel KR & Bai J (2014). Synaptic vesicle morphology: a case of protein sorting? Curr Opin Cell Biol 26, 28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripps H & Chappell RL (2014). Zinc's functional significance in the vertebrate retina. Mol Vis 20, 1067–1074. [PMC free article] [PubMed] [Google Scholar]
- Romero MF, Chen AP, Parker MD & Boron WF (2013). The SLC4 family of bicarbonate (HCO3 –) transporters. Mol Aspects Med 34, 159–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffin VA, Salameh AI, Boron WF & Parker MD (2014). Intracellular pH regulation by acid–base transporters in mammalian neurons. Front Physiol 5, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikoski J, Ruusuvuori E, Koskelainen A & Donner K (1997). Regulation of intracellular pH in salamander retinal rods. J Physiol 498, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarthy V & Ripps H (2002). The Retinal Müller Cell Structure and Function. Kluwer Academic Publishers, New York. [Google Scholar]
- Shmukler BE, Reimold FR, Heneghan JF, Chen C, Zhao T, Paw BH & Alper SL (2014). Molecular cloning and functional characterization of zebrafish Slc4a3/Ae3 anion exchanger. Pflügers Arch 466, 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegert S, Cabuy E, Scherf BG, Kohler H, Panda S, Le Y‐Z, Fehling HJ, Gaidatzis D, Stadler MB & Roska B (2012). Transcriptional code and disease map for adult retinal cell types. Nat Neurosci 15, 487–495, S1–S2. [DOI] [PubMed] [Google Scholar]
- Stauber T, Weinert S & Jentsch TJ (2012). Cell biology and physiology of CLC chloride channels and transporters. Compr Physiol 2, 1701–1744. [DOI] [PubMed] [Google Scholar]
- Stevens DR, Seifert R, Bufe B, Müller F, Kremmer E, Gauss R, Meyerhof W, Kaupp UB, Lindemann B (2001). Hyperpolarization‐activated channels HCN1 and HCN4 mediate responses to sour stimuli. Nature 413, 631–635. [DOI] [PubMed] [Google Scholar]
- Supuran CT (2008). Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 7, 168–181. [DOI] [PubMed] [Google Scholar]
- Supuran CT & Scozzafava A (2004). Benzolamide is not a membrane‐impermeant carbonic anhydrase inhibitor. J Enzyme Inhib Med Chem 19, 269–273. [DOI] [PubMed] [Google Scholar]
- Thoreson WB & Burkhardt DA (1990). Effects of synaptic blocking agents on the depolarizing responses of turtle cones evoked by surround illumination. Vis Neurosci 5, 571–583. [DOI] [PubMed] [Google Scholar]
- Thoreson WB & Mangel SC (2012). Lateral interactions in the outer retina. Prog Retin Eye Res 31, 407–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trenholm S & Baldridge WH (2010). The effect of aminosulfonate buffers on the light responses and intracellular pH of goldfish retinal horizontal cells. J Neurochem 115, 102–111. [DOI] [PubMed] [Google Scholar]
- Van Hook MJ & Thoreson WB (2013). Simultaneous whole‐cell recordings from photoreceptors and second‐order neurons in an amphibian retinal slice preparation. J Vis Exp e50007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij J, Hornstein EP & Schnapf JL (2003). Surround antagonism in macaque cone photoreceptors. J Neurosci 23, 10249–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij J, Kamermans M & Spekreijse H (1996). Horizontal cells feed back to cones by shifting the cone calcium‐current activation range. Vision Res 36, 3943–3953. [DOI] [PubMed] [Google Scholar]
- Vessey JP, Stratis AK, Daniels BA, Da Silva N, Jonz MG, Lalonde MR, Baldridge WH & Barnes S (2005). Proton‐mediated feedback inhibition of presynaptic calcium channels at the cone photoreceptor synapse. J Neurosci 25, 4108–4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virkki LV, Wilson DA, Vaughan‐Jones RD & Boron WF (2002). Functional characterization of human NBC4 as an electrogenic Na+‐HCO3 – cotransporter (NBCe2). Am J Physiol Cell Physiol 282, C1278–C1289. [DOI] [PubMed] [Google Scholar]
- Vroman R, Klaassen LJ, Howlett MHC, Cenedese V, Klooster J, Sjoerdsma T & Kamermans M (2014). Extracellular ATP hydrolysis inhibits synaptic transmission by increasing ph buffering in the synaptic cleft. PLoS Biol 12, e1001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vroman R & Kamermans M (2015). Feedback‐induced glutamate spillover enhances negative feedback from horizontal cells to cones. J Physiol 593, 2927–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T‐M, Holzhausen LC & Kramer RH (2014). Imaging an optogenetic pH sensor reveals that protons mediate lateral inhibition in the retina. Nat Neurosci 17, 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson MF & Barnes S (1996). The dihydropyridine‐sensitive calcium channel subtype in cone photoreceptors. J Gen Physiol 107, 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winum J‐Y, Thiry A, Cheikh KE, J‐M Dogné, Montero J‐L, Vullo D, Scozzafava A, Masereel B & Supuran CT (2007). Carbonic anhydrase inhibitors. Inhibition of isoforms I, II, IV, VA, VII, IX, and XIV with sulfonamides incorporating fructopyranose‐thioureido tails. Bioorg Med Chem Lett 17, 2685–2691. [DOI] [PubMed] [Google Scholar]
- Wu SM (1991). Input–output relations of the feedback synapse between horizontal cells and cones in the tiger salamander retina. J Neurophysiol 65, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Nguyen KT, Barrett EF & David G (2010). Vesicular ATPase inserted into the plasma membrane of motor terminals by exocytosis alkalinizes cytosolic pH and facilitates endocytosis. Neuron 68, 1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Bouyer P & Boron WF (2006). Effects of angiotensin II on the CO2 dependence of HCO3 – reabsorption by the rabbit S2 renal proximal tubule. Am J Physiol Renal Physiol 290, F666–F673. [DOI] [PubMed] [Google Scholar]