

Abstract
The posttranscriptional addition of nucleotides to the 3′ end of RNA regulates the maturation, function, and stability of RNA species in all domains of life. Here, we show that in flies, 3′ terminal RNA uridylation triggers the processive, 3′‐to‐5′ exoribonucleolytic decay via the RNase II/R enzyme CG16940, a homolog of the human Perlman syndrome exoribonuclease Dis3l2. Together with the TUTase Tailor, dmDis3l2 forms the cytoplasmic, terminal RNA uridylation‐mediated processing (TRUMP) complex that functionally cooperates in the degradation of structured RNA. RNA immunoprecipitation and high‐throughput sequencing reveals a variety of TRUMP complex substrates, including abundant non‐coding RNA, such as 5S rRNA, tRNA, snRNA, snoRNA, and the essential RNase MRP. Based on genetic and biochemical evidence, we propose a key function of the TRUMP complex in the cytoplasmic quality control of RNA polymerase III transcripts. Together with high‐throughput biochemical characterization of dmDis3l2 and bacterial RNase R, our results imply a conserved molecular function of RNase II/R enzymes as “readers” of destabilizing posttranscriptional marks—uridylation in eukaryotes and adenylation in prokaryotes—that play important roles in RNA surveillance.
Keywords: Dis3l2, exoribonuclease, RNA decay, RNA surveillance, uridylation
Subject Categories: RNA Biology
Introduction
Upon transcription, RNA species are subjected to a plethora of posttranscriptional modifications that regulate RNA maturation, function, and stability (Machnicka et al, 2013). Among these, the addition of non‐templated nucleotides to the 3′ hydroxyl end of RNA (i.e. tailing) is one of the most frequent marks, exhibiting deep conservation and serving diverse molecular functions.
RNA tailing is catalyzed by a group of template‐independent terminal ribonucleotidyltransferases (TNTases), containing a DNA polymerase β‐like nucleotidyltransferase domain (Aravind & Koonin, 1999). Beyond the well‐established canonical poly(A) polymerases that generate the poly(A) tail of eukaryotic mRNA, many non‐canonical poly(A) polymerases (ncPAPases) have been described in plants, yeast, and animals (Martin & Keller, 2007; Norbury, 2013). Because some ncPAPs catalyze the incorporation of uridine instead of adenine, they are also referred to as terminal uridylyltransferases (TUTases), whereas others exhibit relaxed nucleotide specificity, catalyzing both uridylation and adenylation. The genome of flies and mammals encodes seven ncPAPs, with distinct substrate specificity and subcellular localization.
Depending on organism and cellular compartment, RNA adenylation can have opposing functions: While the addition of short A‐tails triggers mRNA degradation in bacteria and is associated with exosomal decay of aberrant transcripts in eukaryotic nuclei, polyadenylation increases the stability and translatability of mRNA in the cytoplasm of eukaryotes (Dreyfus & Régnier, 2002; Houseley & Tollervey, 2009).
In contrast to adenylation, uridylation only recently emerged as a widespread posttranscriptional mark associated with an expanding set of transcripts including both coding and non‐coding RNA. Messenger RNA uridylation was initially observed at the 3′ ends of small RNA‐directed cleavage products in plants and mammalian cells, perhaps marking these fragments for decay (Shen & Goodman, 2004). But also full‐length mRNA is subjected to 3′ terminal uridylation: Human replication‐dependent histone mRNAs, which lack a poly(A) tail, are uridylated at the end of S phase or upon inhibition of DNA replication (Mullen & Marzluff, 2008; Su et al, 2013). Here, uridylation prompts rapid decay in both 5′‐to‐3′ direction by XRN1, DCP2, and LSM1 and 3′‐to‐5′ direction by the exosome and ERI1 (3′hExo) (Mullen & Marzluff, 2008; Hoefig et al, 2013; Slevin et al, 2014). The addition of uridine nucleotides to the 3′ end of canonical, polyadenylated mRNA was first reported in fission yeast for a small number of transcripts, which were stabilized in the absence of the TUTase Cid1 (Rissland et al, 2007; Rissland & Norbury, 2009). Because uridylation was enhanced in mutants defective of deadenylation and decapping, uridylation and deadenylation were proposed to act redundantly to induce mRNA decapping. Recent high‐throughput methods for the inspection of mRNA 3′ ends at the genomic scale (TailSeq) revealed infrequent uridylation of the majority (> 85%) of protein‐coding transcripts in cultured mammalian cells (Chang et al, 2014). Messenger RNA uridylation in mammals is mediated by the TUTases TUT4/ZCCHC11 and TUT7/ZCCHC6 and contributes to mRNA turnover by targeting deadenylated mRNA for decay (Lim et al, 2014). Like in mammals, mRNAs are also subjected to uridylation in plants, a modification that is predominantly introduced by the TUTase URT1 in Arabidopsis (Sement et al, 2013). However, URT1‐depletion did not impact mRNA turnover, but instead prompted the accumulation of truncated, deadenylated mRNA, implying a role of uridylation in establishing 5′‐to‐3′ directionality of mRNA turnover. In cooperation with PABP, URT1 was subsequently proposed to contribute to the repair of deadenylated mRNA (Zuber et al, 2016). Finally, RNA uridylation also contributes to mRNA expression in kinetoplastid protists, where the insertion and deletion of uridine nucleotides edits cryptogene transcripts to generate otherwise unstable mitochondrial mRNA (Göringer, 2012).
The addition of uridine(s) to the 3′ end of microRNAs (miRNAs) and their precursors recently emerged as a hallmark for the regulation of miRNA biogenesis and turnover in plants and animals (Heo & Kim, 2009; Ameres & Zamore, 2013). Small RNA uridylation was first observed in Arabidopsis, where the small RNA methyltransferase HEN1 protects miRNAs and small interfering RNAs (siRNAs) from HESO1‐directed uridylation and degradation (Li et al, 2005; Ren et al, 2012; Zhao et al, 2012). In animals, more versatile functions of uridylation have been described: In worms and mammals, the RNA‐binding protein Lin28 recruits the TUTase TUT4/ZCCHC11/PUP2 to pre‐let‐7, resulting in its processive uridylation (Hagan et al, 2009; Heo et al, 2009; Lehrbach et al, 2009). Pre‐let‐7 uridylation prevents miRNA maturation and induces degradation by Dis3l2, a 3′ uridylation‐triggered exoribonuclease (Chang et al, 2013; Faehnle et al, 2014). Notably, Dis3l2 is conserved in fission yeast, plants, and animals, and increasing evidence supports a more general role of Dis3l2 in RNA metabolism (Lubas et al, 2013; Malecki et al, 2013; Thomas et al, 2015; Haas et al, 2016; Łabno et al, 2016). In the absence of Lin28, mono‐uridylation of pre‐miRNAs by the TUTases TUT4/ZCCHC11 and TUT7/ZCCHC6 restores the two‐nucleotide 3′ overhangs of selected pre‐miRNAs to enhance maturation, a process that may be conserved in flies (Heo et al, 2012; Reimão‐Pinto et al, 2015). Similar mechanisms may also contribute to the identification of defective pre‐miRNAs that lack intact 3′ overhangs and trigger their destruction via the exosome in mammals (Liu et al, 2014). In flies, uridylation of pre‐miRNAs by the TUTase Tailor prevents the maturation of splicing‐derived mirtrons (Bortolamiol‐Becet et al, 2015; Reimão‐Pinto et al, 2015). Tailor exhibits intrinsic specificity for RNAs ending in 3′‐G (and 3′‐U), facilitating the identification of mirtron hairpin 3′ ends through the conserved splice‐acceptor sequence (AG‐3′). In contrast, conserved Drosophila pre‐miRNAs are significantly depleted in 3′ guanosine, supporting the hypothesis that hairpin uridylation may serve as a barrier to the de novo creation of microRNAs in Drosophila (Bortolamiol‐Becet et al, 2015; Reimão‐Pinto et al, 2015). Finally, destabilization of mature miRNAs upon binding to highly complementary targets is associated with small RNA uridylation (and adenylation) in flies and mammals (Ameres et al, 2010; Xie et al, 2012).
While increasing evidence suggests broad and conserved roles of uridylation in the regulation of RNA fate and function, the molecular mechanisms, cellular targets, and biological functions of RNA uridylation remain largely unexplored. Here, we report that 3′ terminal uridylation prompts 3′‐to‐5′ exoribonucleolytic decay in flies. We describe the molecular basis for uridylation‐triggered RNA decay, revealing a novel complex that plays important roles in cytoplasmic RNA surveillance. Based on RNA immunopurification and high‐throughput sequencing, we uncover putative targets in flies, providing a road map for the functional dissection of uridylation‐triggered RNA decay. In that regard, we uncover an unexpected function of uridylation‐triggered RNA decay in the quality control of RNA polymerase III transcripts. Our work provides novel insights into the targets, functional consequence, and molecular reader of a conserved epitranscriptomic mark with important implications for posttranscriptional gene regulation and RNA surveillance.
Results
Identification of the terminal RNA uridylation‐mediated processing (TRUMP) complex
To identify putative reader(s) of Tailor‐directed RNA uridylation in flies (Fig 1A), we performed immunoprecipitation of FLAG‐tagged Tailor upon expression in Drosophila S2 cells, followed by mass spectrometry analysis (Fig EV1A and B; Table EV1). As the most prominent, statistically significant interaction candidate that withstood high‐salt wash steps emerged the previously uncharacterized, putative 3′‐to‐5′ exoribonuclease CG16940 (Figs 1B and C, and EV1B, D and E). CG16940 is a homolog of the human Perlman syndrome exoribonuclease Dis3l2 (Astuti et al, 2012). Dis3l2 belongs to the RNase II/R enzyme family, is conserved in plants, yeast, and animals, and has previously been implicated in the degradation of uridylated RNA species in fission yeast and mammals (Chang et al, 2013; Lubas et al, 2013; Malecki et al, 2013; Ustianenko et al, 2013). Because of its high overall sequence conservation, we refer to CG16940 as dmDis3l2. To validate the interaction between Tailor and dmDis3l2, we raised a monoclonal antibody against an N‐terminal unstructured fragment of dmDis3l2 (amino acids 97–259) and performed immunoprecipitation of endogenous dmDis3l2 in lysate of Drosophila S2 cells, followed by Western blot analysis. This approach confirmed the interaction between Tailor and dmDis3l2 (Fig EV1C). Because immunodepletion of dmDis3l2 did not result in complete recovery of Tailor from the supernatant, we concluded that Tailor may exist in both a dmDis3l2‐bound and dmDis3l2‐unbound state. To map the interaction between dmDis3l2 and Tailor, we performed co‐immunoprecipitation and Western blot analysis, as well as recombinant protein–protein interaction studies: Immunoprecipitation of FLAG‐dmDis3l2 recovered GFP‐Tailor (but not the unrelated cytoplasmic control protein GFP‐Nibbler), an interaction for which the N‐terminal fragment of Tailor that encompasses the DUF1439 domain was required and sufficient (Figs 1D and EV1F and G). Systematic truncation studies on dmDis3l2 revealed a minimal interaction domain that involves a predicted coiled‐coil motif close to the N‐terminus (Figs 1E and F, and EV1F). Recombinant short protein fragments encompassing Tailor DUF1439 domain (amino acids 63–123) and the N‐terminal coiled‐coil motif in dmDis3l2 (amino acids 2–50) were sufficient to recapitulate this interaction (Figs 1G and EV1G), revealing a minimal protein–protein interaction domain in Tailor and dmDis3l2 underlying complex formation.
Figure 1. Identification of the terminal RNA uridylation‐mediated processing (TRUMP) complex.
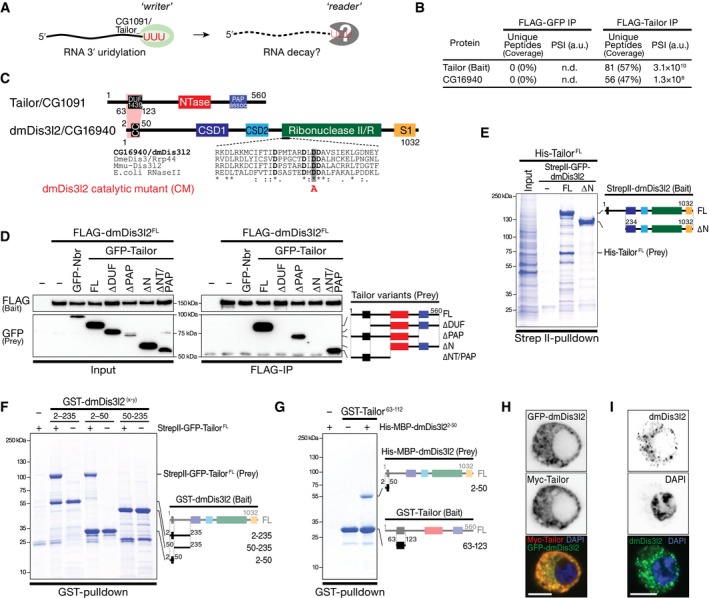
-
AModel for uridylation‐triggered RNA decay. 3′ terminal uridylation by the TUTase Tailor may trigger the degradation of RNA species by unknown reader enzyme(s).
-
BProtein interaction analysis by nano‐LC‐MS. Unique peptide counts and protein sequence coverage, as well as signal intensities (PSI) for Tailor (bait) and CG16940/dmDis3l2 in control IP (FLAG‐GFP) and FLAG‐Tailor IP are indicated. A.u., arbitrary units.
-
CProtein domain architecture of Tailor/CG1091 and dmDis3l2/CG16940. DUF1439, domain of unknown function; NTase, nucleotidyltransferase; PAP assoc, poly(A) polymerase‐associated; CC, predicted coiled‐coil motif; CSD, cold‐shock domain; S1, RNA‐binding domain;
-
DCo‐immunoprecipitation of FLAG‐dmDis3l2 (bait) and GFP‐Tailor full‐length and truncations expressed in Drosophila S2 cells. GFP‐Nibbler (Nbr) served as a negative control. Domain architecture of GFP‐Tailor truncations is indicated.
-
E–GRecombinant protein interaction analysis using the indicated epitope tags and purification methods. Domain architecture of bait and prey are indicated.
-
H, IImmunostaining and imaging of Myc‐Tailor and GFP‐dmDis3l2 (H) or endogenous dmDis3l2 (I) in S2 cells. Single‐color channel images show total inversions. Scale bar = 5 μm.
Source data are available online for this figure.
Figure EV1. Identification of the terminal RNA uridylation‐mediated processing (TRUMP) complex.

-
AExperimental overview of FLAG‐Tailor co‐immunoprecipitation followed by mass spectrometry.
-
BLimma analysis of protein interaction candidates based on three biological replicates of experiment shown in (A). Tailor (bait) is indicated in green. The most prominent interaction candidate showing highest and significant signal accumulation (CG16940) is shown in red.
-
CCo‐immunoprecipitation of endogenous dmDis3l2 and Tailor. dmDis3l2 was immunoprecipitated from S2 cell lysate using an antibody against endogenous dmDis3l2, followed by Western blot analysis of dmDis3l2 and Tailor. Ago1 serves as a control. Asterisk indicates antibody heavy chain signal.
-
DExperimental overview of immunopurification using FLAG‐CG16940 as bait and applying increasing salt concentrations in wash steps, followed by mass spectrometry analysis.
-
EMass spectrometry analysis of the experiment shown in (D). High coverage and signal intensities of Tailor were recovered in FLAG‐CG16940 IP, but not in control IP, irrespective of wash conditions.
-
F, GRecombinant protein interaction studies using the indicated epitope tags and purification methods. Domain architecture of bait and prey are indicated.
-
H, IImmunostaining and imaging of Myc‐Tailor and GFP‐dmDis3l2 (H) or endogenous dmDis3l2 (I) in S2 cells (H and I top panel) or S2 cells depleted of dmDis3l2 by CRISPR/Cas9 (I bottom panel). Single‐color channel images show total inversions. Scale bar = 5 μm.
- J
Source data are available online for this figure.
Co‐expression of epitope‐tagged Tailor and dmDis3l2 in S2 cells revealed predominant—if not exclusive—localization of both proteins dispersed throughout the cytoplasm, without detectable signal in the nucleus (Figs 1H and EV1H). The overall cytoplasmic localization of dmDis3l2 was confirmed by immunofluorescence analyses using the monoclonal antibody against dmDis3l2, resulting in a cytoplasmic signal that was lost upon depletion of dmDis3l2 (Figs 1I and EV1I). Together with the fact that both factors exhibit significant co‐expression across tissues in Drosophila (Pearson's correlation coefficient r > 0.7, P < 10−4; Fig EV1J), we concluded that Tailor and dmDis3l2 form a stable, cytoplasmic, and co‐regulated protein complex in flies, which we refer to as the terminal RNA uridylation‐mediated processing (TRUMP) complex.
CG16940/dmDis3l2 is a uridylation‐triggered 3′‐to‐5′ exoribonuclease
For biochemical characterization, we immunopurified FLAG‐tagged dmDis3l2 upon expression in S2 cells and confirmed enzyme purity by Western blot analysis (Appendix Fig S1A). dmDis3l2 rapidly degraded a 37‐nt, 5′ radiolabeled RNA substrate without detectable decay intermediates (Fig 2A and B). This activity was abolished when one of four highly conserved aspartic acid residues within the RNase II/R domain that constitute the predicted catalytic site was replaced with alanine (Figs 1C and 2B). Together with the conserved domain architecture, we concluded that dmDis3l2 is a bona fide 3′‐to‐5′ exoribonuclease. dmDis3l2‐directed RNA degradation was consistent with the previously proposed molecular mechanism for RNase II/R enzymes, which involves the Mg2+‐dependent nucleophilic attack of phosphodiester bonds (Frazão et al, 2006; Faehnle et al, 2014), because EDTA inhibited, while excess Mg2+ rescued the degradation activity of dmDis3l2 (Appendix Fig S1B).
Figure 2. dmDis3l2/CG16940 is a 3′ terminal uridylation‐triggered, processive 3′‐to‐5′ exoribonuclease.
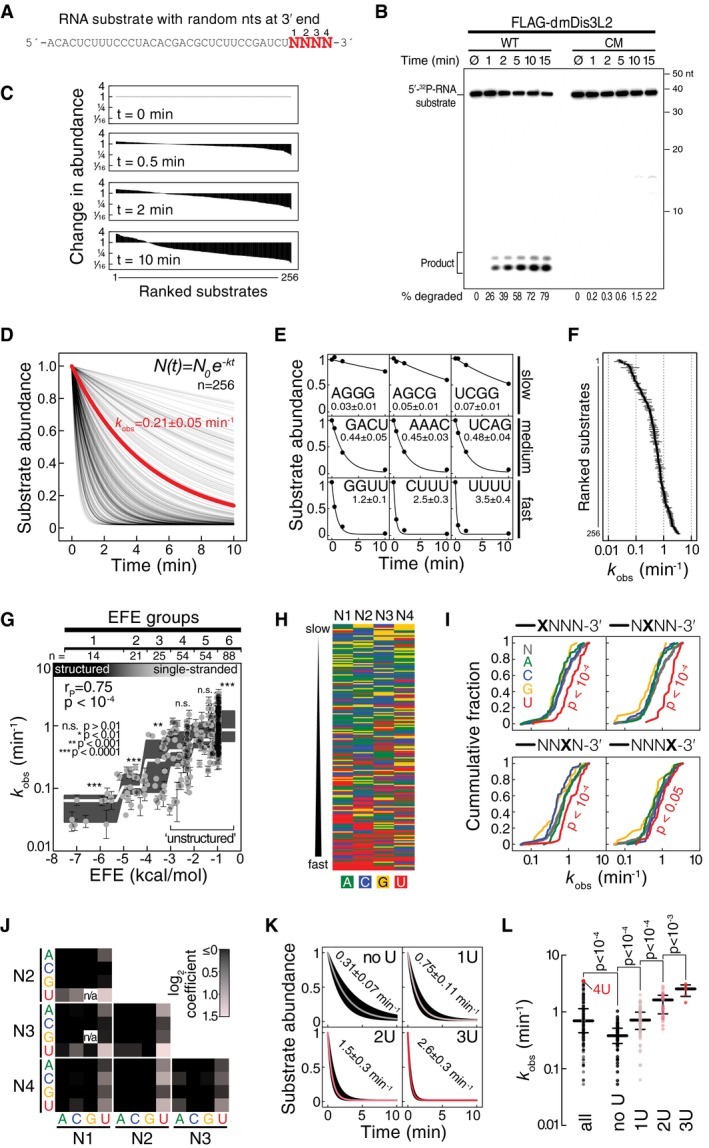
- Sequence of RNA substrate used for in vitro exoribonuclease assays. N, randomized nucleotide.
- In vitro exoribonuclease assay using 5′ radiolabeled RNA substrate and immunopurified wild‐type (WT) or catalytic‐mutant (CM) FLAG‐dmDis3l2 (mutation indicated in Fig 1C), incubated for the indicated time and separated on a 15% polyacrylamide gel followed by phosphorimaging. Quantification of degraded substrate in percent is indicated.
- Change in abundance of 256 different substrate RNAs as determined by high‐throughput sequencing of substrates in experiment shown in (B).
- Decay rates of 256 different substrate RNAs. Data shown in (C) were normalized to overall decrease in substrate abundance as determined by phosphorimaging (as shown in B) and fit to the indicated model for exponential decay. Apparent decay rate of all substrates, as determined by phosphorimaging, is indicated in red.
- Selected examples for individual substrates shown in (D). Values report the observed decay rate (k obs) in min−1.
- Overview of decay rates (k obs) of all 256 different substrate RNAs, as determined in (D). Error bar represents SEM of curve fit.
- Impacts of RNA secondary structures, as determined by RNAfold (Gruber et al, 2008), and reported as effective free energy (EFE, kcal per mol), on decay rates (k obs) are shown. Error bars represent SEM of curve fit. Gray boxes represent Tukey boxplot of individual EFE groups. White line indicates median. P‐value was determined by Mann–Whitney test comparing individual EFE groups against all substrates. Unstructured substrates used for subsequent analysis are indicated (EFE groups 4–6).
- Nucleotide content of single‐stranded RNA substrates, ranked from slow (top) to fast (bottom) decay by dmDis3l2.
- Cumulative distribution of decay rates for RNA substrates with the indicated nucleotide at each one of four randomized positions at the 3′ end of the substrate RNA. P‐value was determined by Mann–Whitney test comparing individual nts against all.
- Functional coupling between two base positions within the randomized 3′ end of substrate RNA. Red squares show average increase in decay rate compared to all substrates. Black squares show no or negative effect.
- Boxplot decay rates of substrates containing no, one, two, or three uridine(s) within the randomized 3′ end of the substrate RNA. Median decay rates ± SEM of fit are indicated.
- Decay rate of all substrates, or substrates containing no, one, two, or three uridine(s) within the randomized 3′ end of the substrate RNA. Median and quartile ranges are indicated. P‐values were determined by Mann–Whitney test. Substrate containing four uridines (4U) is indicated.
Source data are available online for this figure.
To dissect the enzymatic properties of dmDis3l2 in more detail, we developed a high‐throughput biochemical assay for the characterization of 3′‐to‐5′ exoribonucleases. To this end, we employed as a substrate a 37‐nt RNA containing four random 3′ nucleotides (Fig 2A), incubated it with immunopurified dmDis3l2 for 30 s, 2 min, and 10 min, and subjected the remaining substrate to high‐throughput sequencing (Appendix Fig S1C). The randomized 3′ end of the substrate RNA enabled us to simultaneously analyze 256 different substrates, all of which were recovered at each time point of the assay, albeit to varying extent (Fig 2C). Upon normalization to the overall decrease in substrate abundance, as determined by denaturing gel electrophoresis and phosphorimaging (Appendix Fig S1C), analysis of the resulting libraries enabled the calculation of relative degradation rates for all 256 different substrates (Fig 2D), which globally followed a single‐exponential decay model (see examples in Fig 2E). The observed decay rates (k obs) differed between individual substrates by more than two orders of magnitude (Fig 2F). To dissect dmDis3l2 substrate specificities, we first determined the secondary structure stability of each substrate, binned them into effective free energy groups (EFE groups) from highly structured (group 1) to single‐stranded (group 6), and tested each group for significant over‐ or underrepresentation in the observed decay rate (Fig 2G). (Note that the constant region of the substrate was designed to avoid secondary structures not involving the random 3′ nucleotides.) dmDis3l2‐directed decay was strongly affected by secondary structures: Structured RNAs (EFE groups 1–3) were degraded significantly slower (P < 10−3, Mann–Whitney test), whereas single‐stranded substrates were degraded significantly faster (EFE group 6, P < 10−4) compared to all substrates. We concluded that dmDis3l2 is a single‐stranded RNA‐specific exoribonuclease.
But even among single‐stranded substrates (EFE groups 4–6), decay rates varied extensively between individual substrates (Fig 2G). Primary sequence analysis revealed that rapidly degraded single‐stranded substrates were generally enriched in 3′ uridine (Fig 2H). Analysis of each individual position of the randomized four 3′ nucleotides showed that substrates containing uridine at each individual position were degraded significantly faster when compared to any other nucleotide (Fig 2I). Pairwise comparisons between individual positions revealed an additive effect of uridine content on decay activation across all positions (Fig 2J), and grouping substrates according to increasing overall 3′ terminal U‐content confirmed a steady and statistically significant increase in degradation rates (Fig 2K and L). In fact, among all 196 single‐stranded substrates the most efficiently degraded contained four terminal uridines and decayed twelve times faster (k obs = 3.5 ± 0.4 min−1) than the median of all substrates without terminal uridines (k obs = 0.3 ± 0.1 min−1; Fig 2K and L). In summary, we concluded that dmDis3l2 is a 3′ uridylation‐triggered, single‐strand‐specific, processive 3′‐to‐5′ exoribonuclease.
Uridylation by Tailor primes structured RNA for dmDis3l2‐directed decay
The functional characterization of dmDis3l2 implied a model in which 3′ terminal uridylation by Tailor could prime RNA substrates for 3′‐to‐5′ exoribonucleolytic degradation by dmDis3l2. To test this hypothesis, we employed a natural substrate of Tailor, the mirtron hairpin pre‐miR‐1003 (Reimão‐Pinto et al, 2015), and assayed degradation by immunopurified dmDis3l2 in the presence and absence of Tailor (Fig 3A and B). dmDis3l2 alone degraded synthetic, 5′ radiolabeled pre‐miR‐1003 inefficiently (half‐life t 1/2 = 355 min; Fig 3A and B), consistent with the predicted inability of sole dmDis3l2 to attack structured RNA species (Fig 2). The addition of Tailor significantly enhanced pre‐miR‐1003‐degradation, decreasing substrate half‐life by more than one order of magnitude (t 1/2 = 23 min; Fig 3A and B). This decay stimulation followed sigmoidal reaction kinetics, consistent with a model in which Tailor‐directed uridylation precedes dmDis3l2‐directed decay as the rate‐limiting step (Fig 3B). In fact, replacing one of three highly conserved aspartates in the catalytic pol β superfamily motif of Tailor with alanine abolished the enhanced decay of pre‐miR‐1003 by dmDis3l2 entirely, resulting in a substrate half‐life similar to the one observed with dmDis3l2 alone (Fig 3A and B).
Figure 3. Uridylation‐triggered RNA decay of mirtron hairpins by Tailor and dmDis3l2 in vitro .
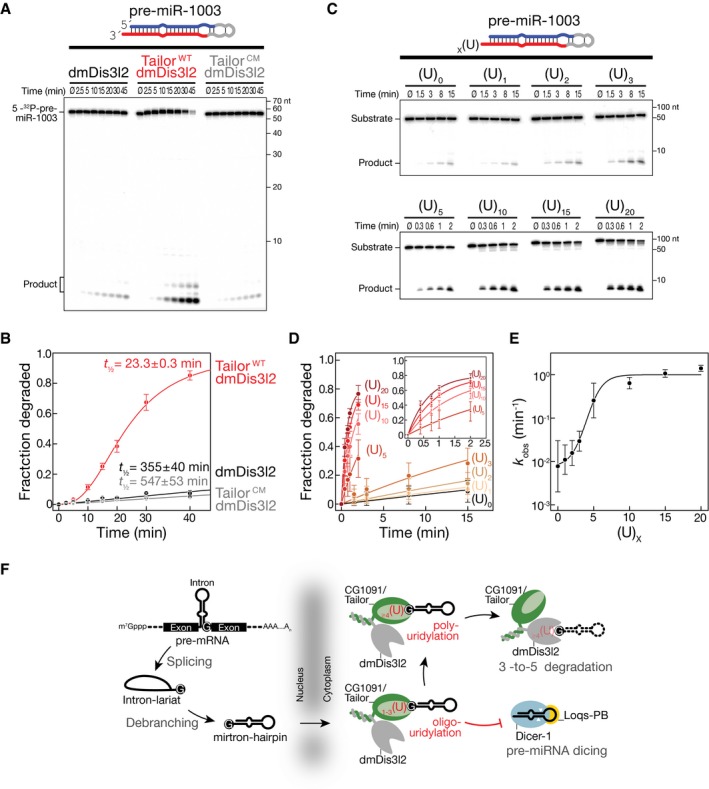
- RNA decay assay using synthetic, 5′ radiolabeled pre‐miR‐1003, immunopurified dmDis3l2, and wild‐type (WT) or catalytic‐mutant (CM) Tailor. Reactions were incubated for the indicated time and separated by 15% denaturing polyacrylamide gel electrophoresis and subjected to phosphorimaging.
- Quantification of at least three independent replicates of experiment shown in (A). Data represent mean ± SD. Half‐life was determined by fitting data to single‐exponential (dmDis3l2 and dmDis3l2/TailorCM) or sigmoidal (dmDis3l2/TailorWT) reaction kinetics.
- RNA decay assay using synthetic, 5′ radiolabeled pre‐miR‐1003 containing the indicated number or uridine residues at the 3′ end and immunopurified dmDis3l2.
- Quantification of three independent replicates of the experiment shown in (C). Data represent mean ± SD. Data were fit to single‐exponential reaction kinetics.
- Reaction kinetics (k obs) determined in (D) plotted against number of 3′ terminal uridine residues. Error of fit is shown as SEM.
- Two‐step model for the regulation of mirtron hairpin maturation in Drosophila. Mirtron hairpins are generated by splicing and lariat debranching. Upon export from the nucleus, Tailor uridylates mirtron hairpins by recognizing the 3′ guanine, a hallmark of splice‐acceptor sequences. Short uridylation masks the 2‐nt 3′ overhang required for Dicer‐1 to recognize pre‐miRNAs. Extended uridine tails eventually trigger processive, 3′‐to‐5′ exoribonucleolytic decay by dmDis3l2, resulting in mirtron clearance.
Source data are available online for this figure.
To directly test the ability of dmDis3l2 to degrade uridylated mirtron hairpins—the products of Tailor activity—we generated synthetic pre‐miR‐1003 variants that contained 3′ terminal uridine extensions of varying length and assayed degradation by sole dmDis3l2 in the absence of Tailor (Fig 3C–E). While the addition of one, two, or three uridine residues only mildly enhanced dmDis3l2‐directed decay of pre‐miR‐1003 by 1.5‐, 2‐, and 3.8‐fold, respectively, penta‐uridylation resulted in 32‐fold and deca‐uridylation in 81‐fold enhancement of degradation when compared to unmodified pre‐miR‐1003 (Fig 3D and E). Although the addition of 15 or 20 uridine residues further accelerated decay, strongest increase in decay activity was observed between 3 and 10 uridine additions (Fig 3E), suggesting that short U‐tails are sufficient for biologically meaningful enhancement of dmDis3l2‐directed decay. Notably, degradation of uridylated mirtron hairpins in vitro by dmDis3l2 proceeded without visible decay intermediates, implying that dmDis3l2 degrades even highly structured RNA species processively once it encounters a uridine‐rich, single‐stranded 3′ extension of sufficient length that can prime decay.
Our data show that uridylation can prime the decay of structured RNA substrates, which—if not uridylated—serve as poor substrates for dmDis3l2‐directed decay (Fig 2), presumably because uridylation provides a single‐stranded entry point for the initiation of degradation. To test whether a single‐stranded extension is sufficient for the initiation of degradation irrespective of its nucleotide composition, we compared decay rates of pre‐miR‐1003 variants that contain an (A)10 or a (U)10 extension (Fig EV2). While deca‐adenylation enhanced the degradation of pre‐miR‐1003 merely by 4.8‐fold, a uridine tail of similar length was 8.3‐fold more efficient in triggering dmDis3l2‐directed decay, resulting in a 40‐fold increase in decay rates compared to untailed pre‐miR‐1003 (Fig EV2). We concluded that primary sequence composition of Tailor‐directed tailing is the major criterion that prompts the degradation of structured RNA by dmDis3l2.
Figure EV2. Uridylation‐triggered RNA decay of mirtron hairpins by Tailor and dmDis3l2 in vitro .
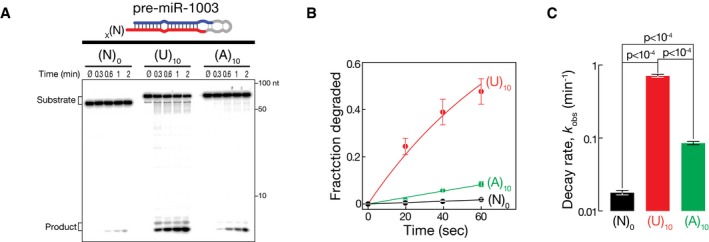
- RNA decay assay using synthetic, 5′ radiolabeled pre‐miR‐1003 containing the indicated number of nucleotide extensions, and immunopurified dmDis3l2.
- Quantification of three independent replicates of experiment shown in (A). Data represent mean ± SD. Data were fit to single‐exponential decay kinetics.
- Decay rates (k obs) determined in (B). Error bars indicate SEM of fit. P‐values were determined using Student's t‐test.
Source data are available online for this figure.
Together, our data are consistent with a two‐step mechanism for the inhibition of mirtron maturation by uridylation in flies (Fig 3F): (i) Initial addition of short uridine tails masks the 2‐nt 3′ overhang, with immediate impairment in Dicer‐mediated processing (Reimão‐Pinto et al, 2015). (ii) Further extension of U‐tails ultimately prompts the processive, 3′‐to‐5′ exoribonucleolytic decay of uridylated mirtron hairpins by dmDis3l2, causing their final clearance. Notably, this model is supported by the fact that in the absence of Tailor, not only mature mirtrons, but also their precursor hairpins accumulate (Reimão‐Pinto et al, 2015).
Tailor and dmDis3l2 cooperate in the degradation of cellular RNA in vitro
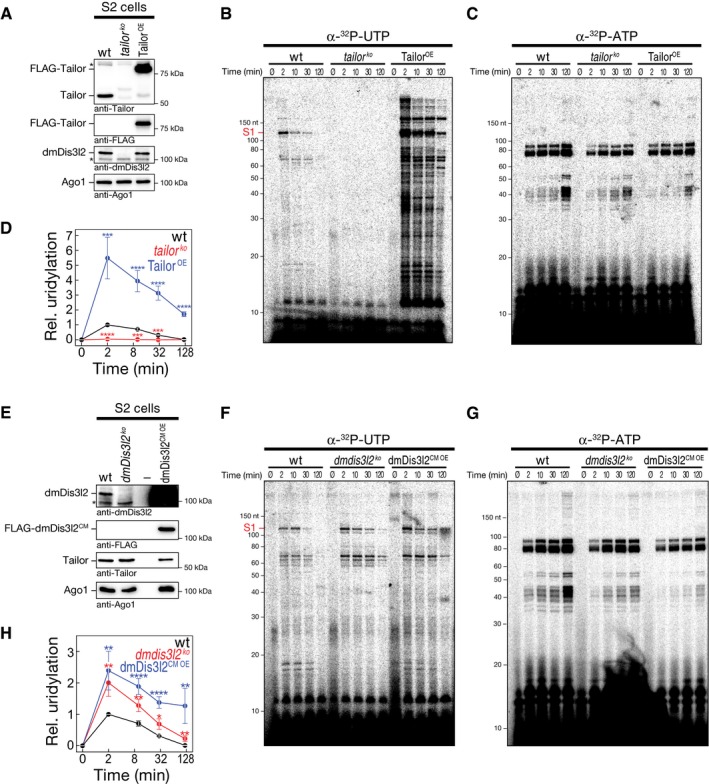
Whether and how 3′ terminal RNA uridylation contributes to general RNA metabolism in flies is unknown. To determine the extent, consequences, and genetic requirements for 3′ terminal RNA uridylation, we established a quantitative uridylation assay in Drosophila S2 cell lysate that monitors the posttranscriptional addition of uridine to cellular RNA over time using α‐32P‐UTP as a cofactor (Fig 4). In wild‐type S2 cell lysate, we observed several RNA species undergoing uridylation as manifested by the rapid appearance of 32P‐labeled RNA species of different molecular weight (wt; Fig 4B and D), an activity that was entirely absent upon depletion of Tailor by CRISPR/Cas9 genome editing (tailor ko; Fig 4A, B and D). (Note that loss of Tailor results in co‐depletion of dmDis3l2, indicating that dmDis3l2 protein stability depends on TRUMP complex formation.) To control for overall quality of the lysate and its general ability to recapitulate posttranscriptional RNA modifications, we simultaneously assayed adenylation, employing α‐32P‐ATP as a cofactor in the same experimental setup, revealing a distinct set of target RNAs (Fig 4C). Adenylation activity was comparable in both wt and tailor ko lysates (Fig 4C and Appendix Fig S2). Together with the fact that RNA uridylation patterns were significantly enhanced in lysate prepared from S2 cells overexpressing an epitope‐tagged version of wild‐type Tailor (TailorOE, P < 0.001, Student's t‐test; Fig 4A, B and D), our data show that multiple endogenous RNA species undergo Tailor‐dependent uridylation in lysate.
Figure 4. In vitro uridylation assay in S2 cell lysate recapitulates uridylation‐triggered RNA decay of cellular RNA transcripts.
-
AWestern blot analysis of genetically modified Drosophila S2 cells. Unmodified S2 cells (wild‐type, wt) or S2 cells depleted of Tailor (tailor ko) by CRISPR/Cas9 or expressing FLAG‐tagged Tailor (TailorOE) were analyzed. Antibodies are indicated. Note that panels were assembled from two technical replicates of identical lysates (see source data).
-
B, CLysates of S2 cells (as characterized in A) were incubated with α‐32P‐UTP (B) or α‐32P‐ATP (C) for the indicated time followed by RNA extraction, denaturing polyacrylamide gel electrophoresis and phosphorimaging.
-
DQuantification of at least five independent replicates of experiment shown in (B, C). Uridylation signal intensity for substrate S1 (indicated in B) was corrected for adenylation activity and normalized to wild‐type 2‐min time point. Data represent mean ± SEM. P‐values were determined using Student's t‐test. ***P < 10−3, ****P < 10−4.
-
EWestern blot analysis of genetically modified Drosophila S2 cells. Unmodified S2 cells (wild‐type, wt) or S2 cells depleted of dmDis3l2 (dmdis3l2 ko) by CRISPR/Cas9 or expressing FLAG‐tagged catalytic‐mutant dmDis3l2 (dmDis3l2CM OE) were analyzed. Antibodies are indicated. Note that panels were assembled from two technical replicates of identical lysates (see source data).
-
F, GLysates of S2 cells (as characterized in E) were incubated with α‐32P‐UTP (F) or α‐32P‐ATP (G) for the indicated time followed by RNA extraction, denaturing polyacrylamide gel electrophoresis and phosphorimaging.
-
HQuantification of at least seven independent replicates of experiment shown in (F, G). Uridylation signal intensity for substrate S1 (indicated in F) was corrected for adenylation activity and normalized to wild‐type 2‐min time point. Data represent mean ± SEM. P‐values were determined using Student's t‐test. *P < 0.05, **P < 10−2, ****P < 10−4.
Source data are available online for this figure.
Quantitative characterization of α‐32P‐UTP‐incorporation signals indicated that RNA uridylation acts as a destabilizing and/or transient mark: Representative quantifications (substrate S1; Fig 4B) revealed highest uridylation signal at early time points (2 min; Fig 4D), followed by a decrease in signal intensity over time (Fig 4D and Appendix Fig S2G). In contrast, adenylation signals were consistent with a persistent, stabilizing posttranscriptional mark, resulting in statistically significant higher stability compared to uridylation (P < 10−4; Fig 4C and G; Appendix Fig S2G).
Because in vitro reconstitution assays indicated that Tailor‐directed uridylation triggers dmDis3l2‐mediated exoribonucleolytic degradation (Fig 3), we next tested whether cellular RNA uridylation in lysate genetically interacts with dmDis3l2. To this end, we generated S2 cells depleted of dmDis3l2 by CRISPR/Cas9 genome editing. Depletion was controlled by Western blot analysis (Fig 4E). (Note that Tailor protein levels stayed unaltered upon depletion of dmDis3l2, consistent with our earlier observation that a fraction of the cellular Tailor pool does not exist in complex with dmDis3l2 [Fig EV1C].) In fact, cellular RNA uridylation signals were significantly elevated across the whole time course in the absence of dmDis3l2 when compared to wild‐type lysate (P < 0.05, Student's t‐test; Fig 4F–H). Similar results were obtained upon overexpression of epitope‐tagged catalytic‐mutant dmDis3l2 (dmDis3l2CM OE, P < 0.05; Fig 4F–H), indicating a dominant‐negative, stabilizing function of dmDis3l2CM. Note that even in the absence of dmDis3l2, an overall steady decrease in uridylation signatures over time indicated that uridylation might also feed into alternative RNA decay pathways (Fig 4H). In summary, we developed an in vitro assay based on Drosophila S2 cell lysate that quantitatively recapitulates posttranscriptional uridylation of cellular RNA by Tailor in whole‐cell lysate, a transient modification that genetically interacts with dmDis3l2.
Identification of TRUMP complex targets
To identify TRUMP complex substrates at the genomic scale, we expressed FLAG‐tagged Tailor or dmDis3l2, as well as their catalytically inactive variants, in Drosophila S2 cells, followed by immunopurification. We reasoned that this strategy might enable co‐purification of endogenous substrates for downstream identification by sequencing. To detect RNA species upon immunopurification, we performed RNA extraction and dephosphorylation by treatment with calf intestinal phosphatase, followed by radiolabeling using γ‐32P‐ATP and T4‐polynucleotide kinase. Labeled RNA was subsequently separated on a denaturing polyacrylamide gel and detected by phosphorimaging (Figs 5A and EV3A). As negative control we included FLAG‐tagged GFP, which did not co‐purify with RNA, as expected (Fig 5A). We likewise did not observe any stable interaction between cellular RNA and wild‐type Tailor or its catalytic‐mutant form, as well as wild‐type dmDis3l2, indicating weak, transient, or intractable interactions between these proteins and their RNA substrates (Fig 5A). However, co‐purified RNA could be readily detected upon immunopurification of a catalytic‐mutant version of dmDis3l2, with strong signal intensities at a size range of 10–20 nt (dmDis3l2CM; Fig 5A). Closer inspection of these RNA species by small RNA cloning and high‐throughput sequencing revealed a length distribution peaking at 14 nts (Fig EV3B). This fragment size is reminiscent of the length covered by the substrate channel in a co‐crystal structure of murine Dis3l2 bound to RNA (Faehnle et al, 2014). While the short length of these RNA species did not permit the identification of their genomic origin, nucleotide‐content analysis revealed a high occurrence of uridine across the five 3′ terminal nucleotides (Fig EV3B). This observation suggests a model in which dmDis3l2CM binds to 3′ uridylated endogenous RNA species that are truncated by 5′‐to‐3′ or endonucleolytic decay—presumably in the course of the purification process—to short fragments that reflect a footprint of dmDis3l2‐substrate interaction. Consistent with this view, a penta‐uridine tail is sufficient for priming degradation of a synthetic substrate by wild‐type dmDis3l2, implying high‐affinity binding (Fig 2). By reducing incubation time and supplementing EDTA (which prevents 5′‐to‐3′ RNA decay by major divalent cation‐dependent cellular RNases, such as Xrn‐1), we optimized dmDis3l2CM immunopurification conditions, prompting the accumulation of higher molecular weight RNA substrates, while reducing the abundance of short, footprint RNA species (Fig EV3C and D). In order to identify full‐length, dmDis3l2CM‐bound substrates, we subjected immunoprecipitated RNA to 3′ adapter ligation [note that products of Tailor‐directed uridylation contain a 3′ end chemistry that is amenable to 3′ adapter ligation (Reimão‐Pinto et al, 2015)], followed by reverse transcription and PCR amplification using random forward priming (Fig 5B; see Materials and Methods for details). The resulting cDNA library was subjected to high‐throughput sequencing in reverse orientation, in order to assess identity and 3′ end modification status of cloned RNA species. Among 17 million analyzed reads, 87% mapped to ribosomal RNA, the vast majority of which (> 90%) stemmed from 5S rRNA loci (Fig 5C). The second most abundant substrate class mapped to non‐coding RNA, consisting mostly of RNase MRP RNA (44%) and the signal recognition particle RNA 7SL (41%). Together with transfer RNA (tRNA, 1.6%), small nuclear RNA (snRNA, 0.9%), and small nucleolar RNA (snoRNA, 0.6%), the vast majority of transcripts bound by dmDis3l2CM therefore consisted of RNA polymerase III transcripts. To a lesser extent, dmDis3l2 also associated with RNA polymerase II transcripts, mapping to introns (2.8%), pseudogenes (1.4%), and mRNA (0.4%).
Figure 5. Identification of TRUMP complex substrates by RNA immunoprecipitation and high‐throughput sequencing.

- Protein/RNA co‐immunopurification experiments. FLAG‐tagged versions of indicated proteins were expressed in S2 cells, followed by immunopurification and RNA isolation. RNA was visualized by CIP/PNK treatment using γ‐32P‐ATP followed by denaturing polyacrylamide gel electrophoresis and phosphorimaging. Asterisk indicates non‐specific contaminant.
- Overview of experimental approach to identify dmDis3l2CM‐associated RNA by high‐throughput sequencing. See text for details.
- Statistics of library generated from dmDis3l2CM‐bound RNA upon mapping to the Drosophila melanogaster genome. rRNA, ribosomal RNA; ncRNA, non‐coding RNA; tRNA, transfer RNA; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; mRNA, messenger RNA; pre‐miRNA, precursor microRNA.
- Posttranscriptional modification status of RNA classes shown in (C) compared to total RNA, subjected to the identical cloning protocol.
- Nucleotide identity of 3′ terminal non‐genome‐matching additions and change in uridine‐tail content for RNA classes shown in (C) compared to total RNA, subjected to the identical cloning protocol. Semitransparent bars correspond to total RNA, and full bars correspond to dmDis3l2CM‐IP. Tail signature reports the three most abundant posttranscriptional modifications detected in the respective RNA class and cloning approach.
Source data are available online for this figure.
Figure EV3. Identification of TRUMP complex substrates by RNA immunoprecipitation and high‐throughput sequencing.

- Western blot analysis associated with experiment shown in Fig 5A. Actin serves as a control. Asterisk indicates non‐specific signal, corresponding to hc‐antibody signal.
- Small RNA sequencing analysis. Strong RNA signal intensities between 10 and 20 nt observed for immunopurified dmDis3l2CM were gel‐purified and subjected to small RNA cloning (see Materials and Methods). Resulting libraries were analyzed for RNA length (top right graph) and nucleotide content (bottom right graph). Small RNAs bound to dmDis3l2CM correspond to 3′ uridine‐rich, RNase‐protected fragments.
- Western blot analysis of FLAG immunopurification in the presence of the indicated concentration of EDTA. Ago1 and actin are controls.
- RNA–protein co‐immunopurification experiments in the presence of the indicated concentration of EDTA. FLAG‐tagged versions of indicated proteins were expressed in S2 cells, followed by immunopurification and RNA isolation. Co‐immunopurified RNA was visualized by CIP/PNK treatment using γ‐32P‐ATP followed by denaturing polyacrylamide gel electrophoresis and phosphorimaging. Asterisk indicates non‐specific contaminant.
- Overview of experimental approach for S2 cell total RNA 3′ end sequencing.
- Statistics of total RNA 3′ end sequencing library generated from S2 cell total RNA following the approach described in (E). rRNA, ribosomal RNA; ncRNA, non‐coding RNA; tRNA, transfer RNA; snRNA, small nuclear RNA; snoRNA, small nucleolar RNA; mRNA, messenger RNA; mitochond., mitochondrial RNA; pre‐miRNA, precursor microRNA.
Source data are available online for this figure.
We next examined the 3′ terminal posttranscriptional modification status of dmDis3l2CM‐bound RNA (Fig 5D and E) and compared it to steady‐state, cellular total RNA, subjected to the same cloning procedure (Figs 5D and E, and EV3E and F). Compared to total RNA, dmDis3l2CM‐bound RNA species exhibited a higher degree of 3′ terminal posttranscriptional modifications across all classes of substrates, with up to 99 and 95% of tailed reads in the case of pseudogenes and rRNA, respectively (Fig 5D). Closer inspection of the tail nucleotide identity in dmDis3l2CM‐bound RNA revealed that posttranscriptional modifications predominantly consisted of uridines across all substrate classes (Fig 5E). The only exception to this was tRNA, which displayed a higher degree of posttranscriptional modification in total RNA (86%; Fig 5D). These modifications predominantly consisted of CCA‐addition events (Fig 5E), an integral feature of bona fide mature tRNAs. But even for tRNA, dmDis3l2CM‐bound substrates were uridine‐tailed, albeit to a lesser extent compared to other substrates, and generally devoid of CCA‐tailing signal (Fig 5E). Not surprisingly given their low expression levels compared to other dmDis3l2CM‐bound non‐coding RNA species, pre‐miRNA hairpins represented only a minor class of dmDis3l2‐substrates (0.003%; Fig 5C). Nevertheless, 13 pre‐miRNAs immunopurified with dmDis3l2CM, half of which stemmed from mirtron loci (miR‐2489, miR‐1006, miR‐3642, miR‐3654, miR‐2501, and miR‐1007), consistent with the proposed function of TRUMP in the degradation of uridylated mirtron hairpins.
Together, our data provide the starting point for a systematic annotation of transcripts subjected to posttranscriptional uridylation and decay via the TRUMP complex in flies.
Molecular basis for TRUMP complex‐mediated quality control of RNA polymerase III transcripts
As a major feature of substrates bound by the TRUMP complex emerged RNA polymerase III products, such as 5S rRNA, RNase MRP, tRNA, snoRNA, and snRNA (Fig 5). In fact, more than 90% of RNA bound by dmDis3l2CM could be assigned to such transcripts. To get further insights into the functional interaction between TRUMP and RNA polymerase III transcripts, we performed Northern hybridization experiments in genetically modified S2 cells and flies. We detected the appearance of a higher molecular weight isoform for 5S rRNA in S2 cells expressing a catalytic‐inactive version of dmDis3l2, implying a role of dmDis3l2 in the decay of aberrant, unprocessed 5S rRNA transcripts (Fig EV4A). The fact that this molecular phenotype is not detectable upon depletion of dmDis3l2 indicated compensatory, alternative routes of decay. Validation of the ncRNA substrate RNase MRP, an essential ribonucleoprotein complex implicated in rRNA processing and initiation of mitochondrial DNA replication (Esakova & Krasilnikov, 2010), revealed a significant accumulation upon depletion of dmDis3l2 (P < 10−2) and expression of a catalytic‐mutant version of dmDis3l2 (P < 10−4), confirming a role of dmDis3l2 in the degradation of RNase MRP in cultured cells (Fig EV4E and G). To confirm these data in vivo, we performed CRISPR/Cas9 genome editing in flies, introducing either a frameshift mutation into the first common exon of the dmdis3l2 locus or an amino acid‐exchange mutation in the predicted catalytic site of the endogenous dmdis3l2 locus (Fig EV4B–D). Similar to our observation in cultured cells, we also detected a significant upregulation of RNase MRP RNA in whole male flies expressing a catalytic‐inactive version of dmDis3l2 (P < 0.002; Fig EV4F and G). Further investigation revealed the accumulation of longer, apparently unprocessed isoforms of RNase MRP upon depletion of dmDis3l2 in S2 cells and in flies (Fig EV4I and J). This was even more apparent upon expression of catalytic‐mutant dmDis3l2 (Fig EV4J). We made similar observations for tRNA, a class of non‐canonical substrates that exhibited lowest level of uridylation among all substrates (Fig 5D and E). Closer investigation of individual tRNA loci, such as tRNAAla(TGC), revealed that dmDis3l2CM‐bound species did not overlap with the annotated, mature tRNA 3′ end, but instead extended ~12 nt downstream, overlapping with a U‐rich region, presumably corresponding to the polymerase III terminator (Fig 6A), similar to the observations of the RNase MRP locus (Fig EV4H). dmDis3l2CM‐bound species therefore corresponded to unprocessed, 3′ trailer‐containing, primary tRNA transcripts. In contrast, the major tRNA 3′ end in total RNA overlapped precisely with the annotated, mature tRNA, containing non‐genome‐matching CCA additions (prefix‐matching reads, PM; Fig 6A), a signal that was absent in dmDis3l2CM (Fig 6A). For a more global view, we performed meta‐analysis of 68 tRNA loci (see Appendix Supplementary Materials and Methods for details), which were abundantly recovered in sequencing data of dmDis3l2CM‐bound RNA. Across all loci, we detected strongest signal downstream of the annotated tRNA 3′ end, overlapping with high genomic thymine content (Fig 6B). This observation supports the hypothesis that TRUMP complex targets unprocessed, 3′ trailer‐containing tRNA transcripts. In fact, Northern hybridization experiments using total RNA prepared from adult male flies depleted of dmDis3l2 by CRISPR/Cas9 genome editing revealed a significant increase in signal using a probe against the 3′ trailer region of tRNAAla(TGC) (P < 0.006, Student's t‐test; Fig 6C and D). An even higher accumulation was observed for the catalytic‐inactive dmDis3l2 (P < 0.002; Fig 6C and D).
Figure EV4. Validation of dmDis3l2CM‐bound RNA reveals unprocessed RNA pol III transcripts as targets for TRUMP‐mediated decay.
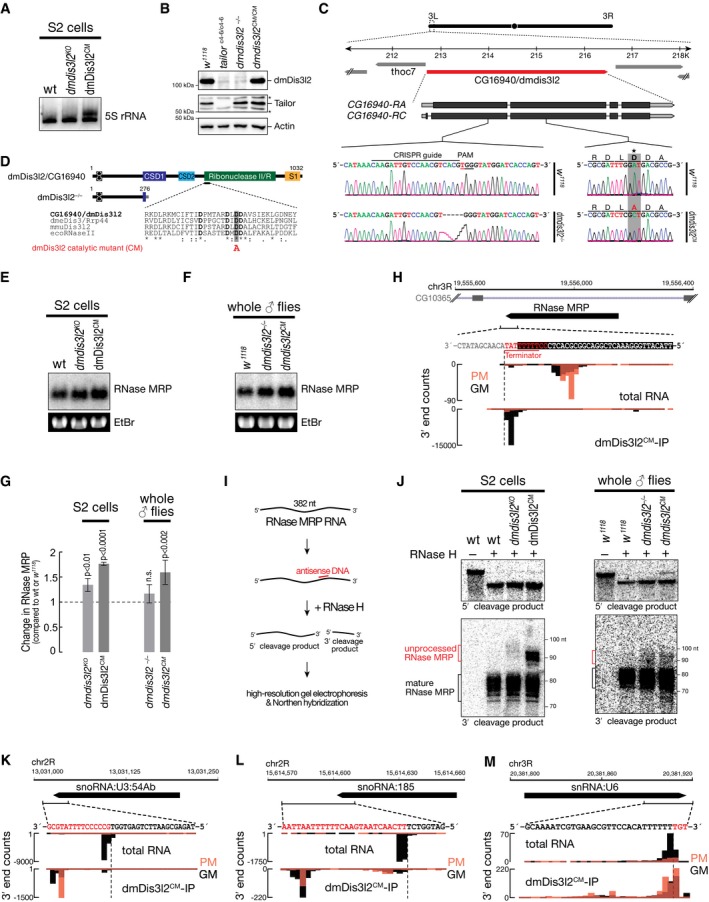
-
ANorthern hybridization experiments using total RNA derived from wild‐type (wt) S2 cells, S2 cells depleted of dmDis3l2 by CRISPR/Cas9 (dmdis3l2 ko), or S2 cells expressing FLAG‐tagged catalytic‐mutant dmDis3l2 (dmDis3l2CM OE). A probe against 5S rRNA was used.
-
BWestern blot analysis of ovary lysate derived from wild‐type flies (w 1118), flies depleted of Tailor (tailor c4/6) or dmDis3l2 (dmdis3l2 −/−), and flies in which the endogenous dmdis3l2 locus was edited by CRISPR/Cas9 to express a catalytic‐inactive version of dmDis3l2 (dmdis3l2 CM/CM). Levels of dmDis3l2 and Tailor were determined using monoclonal antibodies. Actin serves as a loading control.
-
CSchematic representation of the predicted structure of dmdis3l2/cg16940 gene and RNA transcripts. The location of a 5‐bp deletion introduced by CRISPR/Cas9 genome engineering in flies (dmdis3l2 −/−), or a mutation introducing an amino acid exchange (D to A) at the predicted catalytic site of dmDis3l2 is indicated and confirmation by Sanger sequencing of a PCR product prepared from the locus of WT (w 1118) flies and flies homozygous for dmdis3l2 −/− or dmdis3l2 CM/CM is shown.
-
DProtein domain organization of wild‐type dmDis3l2 and a predicted truncation generated by the frameshift mutation shown in (C), as well as the location of the catalytic mutation, is depicted.
-
ENorthern hybridization experiments using total RNA derived from Drosophila S2 cells. Wild‐type S2 cells, cells depleted of dmDis3l2 by CRISPR/Cas9 (dmdis3l2 KO), or cells expressing a catalytic‐mutant version of dmDis3l2 (dmDis3l2CM) were used. A probe against RNase MRP RNA was used; ethidium bromide staining serves as a loading control.
-
FNorthern hybridization experiments using total RNA derived from adult whole male flies. Wild‐type (w 1118) flies, flies depleted of dmDis3l2 by CRISPR/Cas9 (dmdis3l2 −/−), or flies homozygous for a point mutation in the predicted catalytic site of endogenous dmdis3l2 (dmdis3l2 CM) were used. A probe against RNase MRP RNA was used; ethidium bromide staining serves as a loading control.
-
GQuantification of four independent biological replicates of experiment shown in (E) and (F). Abundance of RNase MRP RNA in the respective genetic mutant background relative to wild‐type samples is reported. Values report mean ± SD. P‐values were determined by Student's t‐test.
-
H3′ end counts for the indicated locus in the Drosophila melanogaster genome, corresponding to RNase MRP RNA, in libraries generated from total RNA or dmDis3l2CM‐IP. PM, prefix‐matching (i.e., tailed); GM, genome‐matching.
-
IExperimental approach to test 3′ end heterogeneity of RNase MRP RNA. A DNA oligonucleotide was annealed to RNase MRP RNA in total RNA followed by RNase H treatment, resulting in the endonucleolytic cleavage of the transcript. Shortening of the transcript enables to test 3′ end heterogeneity in high‐resolution Northern hybridization experiments.
-
JNorthern hybridization experiment using the experimental approach described in (I). Total RNA from S2 cells or adult whole male flies of the indicated genotype were used. Probes against the 5′ (top) or 3′ cleavage product (bottom) were used. Signals corresponding to extended, unprocessed 3′ isoforms of RNase MRP RNA are indicated.
-
K–M3′ end counts for the indicated loci in the Drosophila melanogaster genome, in libraries generated from total RNA or dmDis3l2CM‐IP. SnoRNA:U3:54b (K), snoRNA:185 (L), and snRNA:U6 (M) are shown. PM, prefix‐matching (i.e. tailed); GM, genome‐matching.
Source data are available online for this figure.
Figure 6. The TRUMP complex targets unprocessed, 3′ trailer‐containing tRNA for decay.
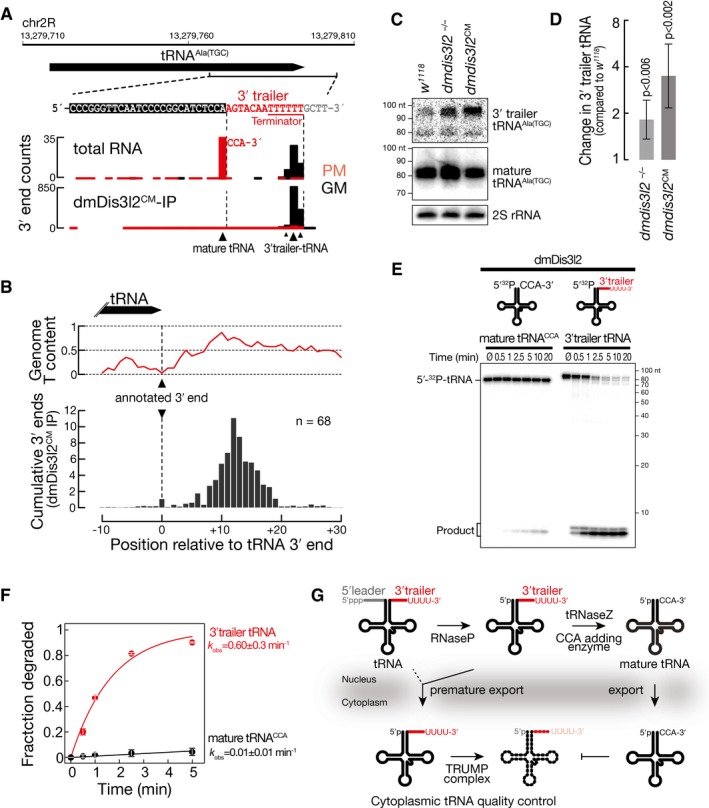
- 3′ end counts for the indicated locus in the Drosophila melanogaster genome, corresponding to tRNA alanine (TGC) in libraries generated from total RNA or dmDis3l2CM‐IP. PM, prefix‐matching (i.e., tailed); GM, genome‐matching.
- Meta‐analysis of 3′ end counts in dmDis3l2CM‐IP mapping to the indicated region around the annotated 3′ end of 68 tRNA loci in the Drosophila melanogaster genome. Cumulative relative distribution of 3′ end signal, relative to the annotated 3′ end, is reported (bottom). The average genome thymine (T)‐content for the same regions is displayed (top).
- Northern hybridization experiment using total RNA from adult whole male flies using probes against 3′ trailer or mature tRNAAla(TGC). Wild‐type flies (w 1118) or flies bearing a homozygous frameshift mutation in the first coding exon (dmDis3l2 −/−) or a homozygous amino acid‐exchange mutation in the catalytic site (dmDis3l2 CM) in the endogenou dmdis3l2 locus in the same genetic background were used. Probes against 2S rRNA served as a loading control.
- Quantification of four independent biological replicates of experiment shown in (C). P‐values determined by Student's t‐test. Data represent mean ± SD.
- Exoribonuclease assay using immunopurified dmDis3l2 and in vitro‐transcribed, 5′ radiolabeled mature tRNA, containing a CCA modification or 3′ trailer‐containing tRNA. Reactions were incubated for the indicated time and subjected to polyacrylamide gel electrophoresis and phosphorimaging.
- Quantification of three independent replicates of experiment shown in (E). Data represent mean ± SD. Decay rates (k obs) ± SEM of fit were determined upon fitting to single‐exponential decay kinetics.
- Model for TRUMP‐mediated quality control of tRNA. In the process of tRNA maturation, RNase P removes the 5′ leader and tRNase Z the 3′ trailer sequence, followed by CCA addition and export. Premature export of unprocessed tRNA results in degradation by dmDis3l2, recognizing the uridine‐rich 3′ end of the 3′ trailer, a hallmark of RNA polymerase III terminators. In contrast, mature CCA‐modified tRNA is resistant against TRUMP‐mediated decay.
Source data are available online for this figure.
Because unprocessed tRNA transcripts contain uridine‐rich 3′ ends—the signature of RNA polymerase III terminators—we predicted that dmDis3l2 would degrade such transcripts independent of the TUTase Tailor. To test this hypothesis, we performed in vitro RNA decay assays, using immunopurified dmDis3l2 and in vitro‐transcribed, 5′ radiolabeled tRNA. In fact, we observed rapid decay of 3′ trailer‐containing tRNA at rates that were 60‐fold faster (k obs = 0.6 min−1), compared to mature, CCA‐modified tRNA, which remained stable throughout the duration of the assay (k obs = 0.01 min−1; Fig 6E and F).
Together, our results support a role of the TRUMP complex in cytoplasmic tRNA quality control (Fig 6G): Upon premature export to the cytoplasm, unprocessed, 3′ trailer‐containing tRNAs are targeted for rapid decay, a process that is stimulated by uridine‐rich 3′ ends, introduced through inherent features of RNA polymerase III terminators.
Together with initial observations on 5S rRNA and RNase MRP RNA, further inspection of dmDis3l2CM‐bound sequences adverts to a more general function of TRUMP in the quality control of RNA polymerase III transcripts beyond tRNA, possibly also contributing to the decay of unprocessed snoRNA (i.e. snoRNA U3 and snoRNA 185; Fig EV4K and L) and mis‐localized LSM‐class snRNA (i.e. snRNA U6; Fig EV4M).
RNase II/R enzymes act as evolutionarily adapted “readers” of posttranscriptional marks
The 3′ terminal posttranscriptional modification of RNA species by tailing is a deeply conserved strategy for priming 3′‐to‐5′ exoribonucleolytic decay. A conceptually similar system to the here described cytoplasmic uridylation‐triggered RNA decay by dmDis3l2 operates in bacteria (Kushner, 2002). In this case, however, 3′ terminal adenylation—instead of uridylation—promotes degradation. For example, the homolog of dmDis3l2 in Escherichia coli—RNase R—processively degrades 3′ adenylated RNA species in one of three major routes for exoribonucleolytic mRNA turnover in bacteria (Regnier & Arraiano, 2000; Andrade et al, 2009). We therefore tested if RNase R—like Dis3l2 in animals—acts as a direct “reader” of destabilizing posttranscriptional marks. To this end, we incubated recombinant E. coli RNase R (ecoRNase R) with 5′ radiolabeled RNA substrate containing four randomized 3′ terminal nucleotides (Fig 2A) for 5, 10, and 30 s and subjected residual substrate to high‐throughput sequencing (Fig 7 and Appendix Fig S3A). Then, we compared the decay rates of 256 different substrates between ecoRNase R and immunopurified dmDis3l2 (Fig 7 and Appendix Fig S3B–D). Similar to dmDis3l2, ecoRNase R exhibited strong susceptibility to RNA secondary structures: Structured RNAs (EFE groups 1–3) were degraded significantly slower (P < 10−4, Mann–Whitney test), whereas single‐stranded substrates were degraded significantly faster (EFE group 6, P < 10−4; Fig 7A). The inability to attack RNA 3′ends that are embedded in secondary structures therefore constitutes a conserved feature of RNase II/R enzymes. Primary sequence analysis of single‐stranded, unstructured substrates revealed that slowly degrading substrates exhibited an enrichment in guanosine for both dmDis3l2 and ecoRNase R (Fig 7B). [Note that the dmDis3l2‐associated terminal uridylyltransferase Tailor exhibits high uridylation activity on substrates ending in 3′ G, indicating a functional cooperativity in the conversion of RNA species into dmDis3l2‐substrates (Bortolamiol‐Becet et al, 2015; Reimão‐Pinto et al, 2015).] In contrast, rapidly degraded substrates were generally enriched in uridine for dmDis3l2, but exhibited strongest enrichment for adenine in the case of ecoRNase R (Fig 7B). Subsequent grouping of substrates according to individual nucleotide content confirmed the statistically significant, faster decay of substrates containing two (P < 0.05, Mann–Whitney test) or three 3′ terminal adenines (P < 0.01) for ecoRNase R, while in the absence of adenine RNA decay was significantly slower (P < 0.01; Fig 7C). In contrast, increasing uridine content did not accelerate decay significantly (Fig 7C). Also among homopolymeric 3′ ends, (A)4‐substrates were degraded fastest. This observation contrasted similar analyses for dmDis3l2: Only increasing U‐content had a statistically significant accelerating effect on RNA decay (P < 10−4 for 3′ ends containing two or three uridine; Fig 7C), and the absence of uridine significantly decreased decay kinetics (P < 10−4; Fig 7C). Taken together, our high‐throughput biochemical characterization provides experimental evidence for an evolutionarily adapted function of RNase II/R enzymes as “readers” of destabilizing posttranscriptional marks—adenylation in prokaryotes and uridylation in eukaryotes (Fig 7D).
Figure 7. High‐throughput biochemical comparison of Escherichia coli RNase R and D. melanogaster Dis3l2.
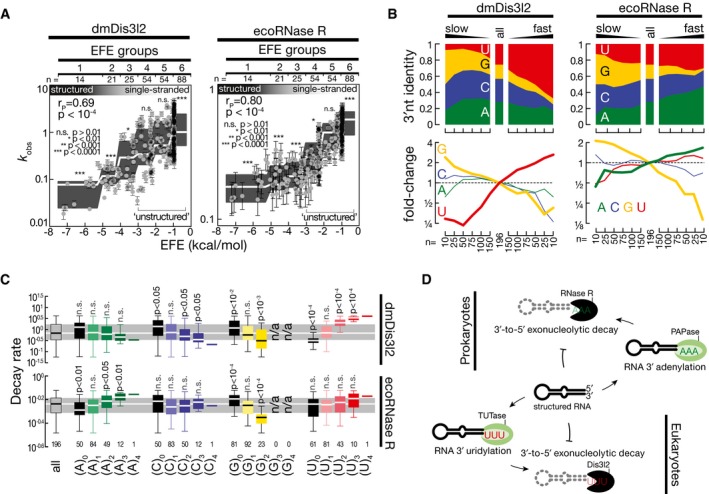
- Impact of RNA secondary structures on RNA decay rates (k obs) of 256 different RNA substrates subjected to E. coli RNase R (ecoRNase R)‐ or Drosophila melanogaster Dis3l2 (dmDis3l2)‐directed RNA decay assay as described in Fig 2. RNA secondary structures were determined by RNAfold (Gruber et al, 2008) and reported as effective free energy (EFE). Error bars represent SEM of curve fit. Gray boxes represent Tukey boxplot of individual EFE groups. White line indicates median. P‐value determined by Mann–Whitney test comparing individual EFE groups against all substrates. Unstructured RNAs used for subsequent analysis are indicated (EFE groups 4–6).
- Nucleotide content of randomized 3′ end sequence among the indicated number of substrate RNAs, ranked according to decay kinetics from slow (left) to fast (right). Fold change in abundance for individual nucleotides is reported (bottom). Only unstructured RNA substrates (see A) were considered.
- Decay rate of the indicated number of sequences, grouped according to individual nucleotide content in the randomized 3′ end sequence. Data are represented as boxplot, outliers are not shown. P‐values report significant differences compared to all substrates as determined by Mann–Whitney test. Gray area represents the inner quartile range and white line the median of all substrates.
- RNase II/R enzymes act as conserved molecular “readers” of destabilizing posttranscriptional modifications. In prokaryotes, for example, E. coli, structured RNA is marked by a poly(A) polymerase (PAPase) for subsequent degradation by RNase R, 3′‐to‐5′ exoribonuclease with intrinsic preference for 3′ adenylated RNA. In eukaryotes, for example, flies or mammals, structured RNA undergoes 3′ terminal uridylation by a terminal uridylyltransferase (TUTase) for subsequent degradation by Dis3l2, a 3′‐to‐5′ exoribonuclease with intrinsic preference for 3′ uridylated RNA.
Discussion
Uridylation of RNA species represents an emerging new layer in the regulation of gene expression. Here, we show that uridylation triggers RNA degradation in Drosophila. Through a series of experiments, we established the molecular basis for uridylation‐triggered RNA decay in flies. We first identified the terminal RNA uridylation‐mediated processing (TRUMP) complex consisting of the TUTase Tailor and the previously undescribed, bona fide 3′‐to‐5′ exoribonuclease CG16940/dmDis3l2 (Fig 1). Together with its ortholog Dis3/Rrp44, the core catalytic subunit of the exosome, Dis3l2 belongs to the RNase II/R family of exoribonucleases and is conserved in fission yeast, plants (aka SOV), and animals (Gallouzi & Wilusz, 2013). In contrast to Dis3, Dis3l2 lacks a N‐terminal PIN domain which mediates the association of Rrp44 with the exosome in yeast (Schneider et al, 2009). An exosome‐independent function of dmDis3l2 is experimentally supported by the absence of any exosome component in our mass spectrometry data (Table EV1). Hence, our data establish a novel and autonomous route to cytoplasmic 3′‐to‐5′ exoribonucleolytic decay in flies.
Tailor and dmDis3l2 stably interact through N‐terminal dimerization modules consisting of a DUF1439 domain in Tailor and a predicted coiled‐coil motif in dmDis3l2 (Figs 1D–G and EV1F and G). To our knowledge, this finding represents the first evidence for a direct coupling of a TUTase to exonucleolytic decay. Tailor and dmDis3l2 interact not only physically but also functionally: dmDis3l2 possesses a preference for single‐stranded, 3′ uridylated RNA as determined in an unbiased assay for the biochemical characterization of 3′‐to‐5′ exoribonucleases (Fig 2). Uridylation‐triggered decay seems to be a conserved feature of Dis3l2 enzymes, because a similar specificity has been reported for its mammalian homolog (Chang et al, 2013; Ustianenko et al, 2013; Faehnle et al, 2014). In fact, a molecular explanation for how uridylation may prompt decay by Dis3l2 has been proposed based on the crystal structure of murine Dis3l2 bound to oligouridine (Faehnle et al, 2014). Three RNA‐binding domains (S1, CSD1, and CSD2) in Dis3l2 form an open funnel on one side of the catalytic domain that navigates RNA substrates to the active site in the RNase II/R domain, a path that is accompanied through multiple uracil‐specific interactions (Faehnle et al, 2014; Lv et al, 2015).
Our data propose a dual function of 3′ terminal uridylation: (i) The addition of uridines to single‐stranded RNA significantly enhances decay kinetics, most certainly because it increases the affinity of substrates to dmDis3l2 (Fig 2) (Faehnle et al, 2014). (ii) Uridylation can also prime the decay of structured RNA substrates, which—if not tailed—serve as poor substrates for dmDis3l2‐directed decay (Figs 2 and 3). In this case, oligo‐uridylation provides a single‐stranded landing site for the initiation of 3′‐to‐5′ exoribonucleolytic decay. Notably, the primary sequence composition of the single‐stranded RNA extension is the major criterion for the initiation of dmDis3l2‐directed decay of structured RNA (Fig EV2). How exactly uridylated RNA species are handed over to dmDis3l2 upon uridylation remains to be established, but the observed sigmoidal reaction kinetics of Tailor‐primed decay (Fig 3A and B) and the strong increase in observed decay rates upon penta‐ and deca‐uridylation (Fig 3C and D) may indicate an underlying shift in relative substrate affinities.
In flies, Tailor‐directed uridylation regulates the maturation of microRNA precursors, particularly mirtrons and evolutionary young miRNAs, possibly serving as a barrier to the de novo generation of miRNAs (Bortolamiol‐Becet et al, 2015; Reimão‐Pinto et al, 2015). Here, we expand the model for pre‐miRNA regulation, by proposing a two‐step model: (i) The addition of short uridine tails (i.e., mono‐ to tri‐uridylation) masks the canonical two‐nucleotide 3′ overhang of miRNA hairpins and prevents the ability of Dicer‐1 to efficiently generate mature miRNAs (Reimão‐Pinto et al, 2015). (ii) Extension of the uridine tail to 5 nts or longer ultimately initiates dmDis3l2‐directed processive 3′‐to‐5′ exonucleolytic decay, prompting the clearance of hairpins. In this regard, the regulation of miRNA maturation in flies is reminiscent of a model proposed for the regulation of let‐7 maturation in mouse embryonic stem cells (Chang et al, 2013; Ustianenko et al, 2013). A major difference however lies in the targeting strategies. While uridylation in mammals is induced through recruitment of TUT4 by its RNA‐binding partner protein Lin28, specificity for mirtron uridylation in flies arises from intrinsic substrate preferences of Tailor that targets RNAs ending in 3′G (or 3′U) (Reimão‐Pinto et al, 2015). Notably, the G‐preference of Tailor complements the inability of dmDis3l2 to degraded even single‐stranded RNA ending in G (Fig 2).
By establishing an in vitro assay for the quantitative analysis of posttranscriptional modification of cellular RNA in lysate of Drosophila S2 cells, we could show that multiple RNA species are subjected to posttranscriptional uridylation (Fig 4). This assay implies that uridylation acts as a transient, destabilizing posttranscriptional mark. In combination with genetic manipulations using CRISPR/Cas9, we find that Tailor is required for uridylation, and depletion of dmDis3l2 results in the stabilization of uridylated RNA. Our data therefore implicate the TRUMP complex as a novel player in general cellular RNA metabolism.
Through RNA immunopurification of dmDis3l2CM, followed by high‐throughput sequencing, we found that a variety of transcripts, including rRNA, ncRNA, tRNA, snoRNA, snRNA, and mRNA, are subjected to uridylation in cultured Drosophila cells, albeit to varying extent (Fig 5). Our data therefore provide a road map for the functional dissection of uridylation‐triggered RNA decay. Notably, germline mutations in DIS3L2 have been shown to cause Perlman syndrome in humans, a congenital overgrowth disorder with high neonatal mortality and predisposition to Wilms’ tumor (Astuti et al, 2012; Higashimoto et al, 2013). But the disease‐causing molecular targets of Dis3l2 remain enigmatic. Future investigations into the substrates and consequences of uridylation‐triggered RNA decay at the organismal level will certainly contribute to better understanding the molecular origin of this disease.
The identification of TRUMP substrates revealed RNA polymerase III transcripts as major targets for uridylation‐triggered decay. Using genetically modified S2 cells and flies, combined with biochemical reconstitution assays, we showed that dmDis3l2 targets aberrant, unprocessed RNA polymerase III transcripts for decay, revealing a novel, unexpected function of uridylation‐triggered RNA decay in cytoplasmic RNA quality control (Fig 6). This is exemplified by tRNAs, a class of non‐coding RNAs that undergo sequential processing in the nucleus, resulting in the removal of short extensions at the 5′ (5′ leader) and 3′ end (3′ trailer) of primary transcripts, followed by the addition of CCA, which ultimately undergoes aminoacylation upon export to the cytoplasm, resulting in mature tRNAs that can participate in protein synthesis (Phizicky & Hopper, 2010). The accumulation of 3′ trailer‐containing transcripts in flies depleted of dmDis3l2, or genetically modified to express a catalytic‐mutant version, indicates a new, cytoplasmic layer of tRNA quality control, targeting unprocessed 3′ trailer‐containing transcripts for decay. A similar layer of quality control may be expanded to other substrates, including RNA polymerase III‐transcribed snRNA and snoRNA (Fig EV4K–M). A key common feature that apparently drives decay in the cytoplasm is uridine‐rich 3′ ends that represent signatures of RNA polymerase III terminators, hence serving as a molecular identifier for decay (Fig 6). In this respect, it is unclear whether transcripts containing the signature of RNA polymerase III terminators actually require the activity of Tailor to initiate dmDis3l2‐directed decay, since 3′ trailer‐containing tRNAs are readily degraded by immunopurified dmDis3l2 in vitro in the absence of Tailor (Fig 6E and F). However, previous results showed that Tailor exhibits enhanced uridylation activity on RNA substrates ending not only in 3′G but also in 3′U (Reimão‐Pinto et al, 2015), indicating that extended uridylation of terminator‐signature‐containing transcripts might further enhance decay rates in vivo. Note that while this manuscript was under review, two studies implicated mammalian Dis3l2 in the quality control of non‐coding RNAs, providing further support to a conserved function of Dis3l2 in cytoplasmic RNA surveillance (Łabno et al, 2016; Pirouz et al, 2016).
While the TRUMP complex provides a molecular explanation for uridylation‐triggered RNA decay, we currently cannot exclude that uridylation in flies may also serve a function other than prompting RNA decay. In fact, immunoprecipitation of endogenous dmDis3l2 revealed that Tailor may exist in a dmDis3l2‐bound and dmDis3l2‐unbound state (Fig EV1C). In contrast, genetic evidence indicates that dmDis3l2 protein stability depends on TRUMP complex formation, because depletion of Tailor results in co‐depletion of dmDis3l2 both in cultured cells and in vivo in flies (Figs 4A and EV4B). Notably, previous gain‐of‐function experiments indicate that Tailor can enhance the biogenesis of selected miRNAs by restoring the 2‐nt 3′ overhang in miRNA precursors, a hallmark for the efficient processing by Dicer (Reimão‐Pinto et al, 2015). Hence, future studies are required to carefully dissect the consequences of Tailor‐directed uridylation in flies, also taking possible functions other than RNA decay into account.
In terms of both function and substrates, the TRUMP complex may act as the cytoplasmic counterpart of the nuclear TRAMP complex, which prompts the exosomal degradation of aberrant transcripts in eukaryotes (Houseley & Tollervey, 2009; Schmidt & Butler, 2013). The TRAMP complex is composed of a non‐canonical poly(A) polymerase Trf4 or Trf5, a zinc knuckle protein Air1 or Air2, and the RNA helicase Mtr4 and tags defective nuclear RNAs with a short poly(A) tail, ultimately resulting their degradation via the nuclear exosome, involving Rrp6 and Dis3/Rrp44 (LaCava et al, 2005; Vanacova et al, 2005; Wyers et al, 2005; Jia et al, 2011; Schmidt & Butler, 2013). However, in contrast to the TRUMP complex, exosomal degradation initiated by the TRAMP complex does not seem to involve the sequence‐specific recognition of the posttranscriptional modification. Instead, the RNA helicase Mtr4 may bridge TRAMP and exosome functions by handing off transcripts directly to the exosome or Rrp6 (Jackson et al, 2010; Weir et al, 2010). Consistent with this hypothesis, TRAMP can trigger the degradation of a subset of target RNAs even in the absence of functional Trf4 and Mtr4, while others require adenylation and RNA unwinding activities, presumably promoting the productive presentation of RNA substrates to Rrp6 or the exosome (Castaño et al, 1996; LaCava et al, 2005; Wyers et al, 2005; Houseley et al, 2007; Rougemaille et al, 2007; San Paolo et al, 2009; Callahan & Butler, 2010). This is different to the situation in bacteria, where adenylation likewise promotes the decay of RNA species, such as tRNA, rRNA and mRNA (Cheng & Deutscher, 2003, 2005; Mohanty & Kushner, 2010). Among the three major 3′‐to‐5′ exoribonucleases in E. coli, RNase R is the only enzyme that is able to efficiently digest through regions of extensive secondary structure, while RNase II cannot and PNPase requires the assistance of the DEAD‐box RNA helicase RhlB (Cheng & Deutscher, 2002, 2005; Khemici & Carpousis, 2004). In this respect, RNase R is very similar to Dis3l2 because both require a single‐stranded 3′ extension as an entry site for initiation of decay that then can proceed through highly structured regions. In E. coli, this entry site is generated by PAP I, a poly(A) polymerase that adenylates mRNA, tRNA, and rRNA (Andrade et al, 2009; Mohanty & Kushner, 2010). Based on high‐throughput biochemical characterization, we provide evidence that RNase R acts as a terminal adenylation‐triggered exoribonuclease (Fig 7). Our data therefore imply a conserved molecular function of RNase II/R enzymes as “readers” of destabilizing posttranscriptional marks—uridylation in eukaryotes and adenylation in prokaryotes—that play important roles in RNA surveillance.
Materials and Methods
General methods
Total RNA from S2‐S cells and whole flies was extracted using TRIzol reagent (Ambion). Stable transgenic and genetically modified Drosophila S2 cells and fly stocks were generated as described (Ameres et al, 2010; Reimão‐Pinto et al, 2015). Northern hybridization experiments were performed as described previously (Han et al, 2011). Protein interaction analysis by mass spectrometry was performed using nano‐LC‐MS. A monoclonal antibody against dmDis3l2/CG16940 was generated upon expression and purification of a His‐tagged N‐terminal sequence of dmDis3l2/CG16940 (‐PC, amino acids 97–259) in E. coli. Flies carrying a 5‐bp frameshift mutation in the first common exon of cg16940/dmdis3l2 or a single amino acid exchange in the catalytic site of the endogenous cg16940/dmdis3l2 locus were generated by CRISPR/Cas9 genome editing and verified by Sanger sequencing and Western blot analysis (see Fig EV4B–D). Detailed information is given in Appendix Supplementary Materials and Methods.
Co‐immunoprecipitation
S2 cells were transiently co‐transfected with pAFMW‐CG16940/dmDis3l2‐PC and pAGW‐Tailor encoding full‐length or truncated versions of Tailor or pAGW‐Nibbler, followed by lysate preparation using 1× lysis/IP buffer (30 mM HEPES–KOH pH 7.4, 100 mM KAc, 2 mM MgAc, 5 mM DTT, 0.5% NP‐40, 5% glycerol). Immunoprecipitation of FLAG‐CG16940/dmDis3l2 was performed using mouse anti‐FLAG M2 (Sigma) coupled to Protein G Dynabeads (Invitrogen), including five wash steps with 1× lysis/IP buffer. Interaction was determined by Western blot analysis using mouse anti‐FLAG M2 (Sigma) and rabbit anti‐GFP (gift from S. Dorner). For immunoprecipitation of endogenous dmDis3l2, mouse‐α‐dmDis3l2 was coupled to GammaBind G Sepharose (GE Healthcare).
Recombinant protein interaction studies
Recombinant proteins for Strep II pull‐down experiments were expressed in insect cells using Baculovirus expression system, bound to Strep‐Tactin beads and co‐purified with His‐tag proteins followed by SDS–PAGE and Coomassie blue staining. Recombinant proteins for GST pull‐down experiments were expressed in E. coli Rosetta II, and interaction was tested using Glutathione Sepharose 4 Fast Flow beads (GE Healthcare), followed by SDS–PAGE and Coomassie blue staining. For more details, see Appendix Supplementary Materials and Methods.
Immunocytochemistry
Immunostaining of S2 cells was performed as previously described (Reimão‐Pinto et al, 2015). For co‐imaging of Tailor and dmDis3l2, S2 cells were transiently transfected with pAFMW‐Tailor and pAGW‐dmDis3l2. Anti‐Myc antibody (M4439; Sigma) was used at 1:2,000 dilution. Anti‐dmDis3l2 was used at 1:150 dilution.
In vitro RNA decay assay
FLAG‐tagged dmDis3l2 and Tailor were immunopurified upon expression in S2 cells as described previously (Reimão‐Pinto et al, 2015). In vitro RNA decay assays employed 10 nM 5′ radiolabeled substrate RNA and FLAG‐dmDis3l2 (and if indicated Tailor) under RNAi assay conditions at 25°C (Ameres et al, 2010), but omitting the ATP‐regeneration system and including 0.6 μM UTP in assays that included Tailor. RNase R (Biozym) was assayed under the same experimental conditions.
CRISPR/Cas9 genome editing in S2 cells
Drosophila S2 cells were transfected with a mixture of two pAc‐sgRNA‐Cas9 plasmids encoding Cas9 and a sgRNA targeting a 46‐bp region in the second exon of tailor/cg1091B or a 48‐bp region in the second exon of dmdis3l2/cg16940. For guide RNA sequences, see Appendix Table S1. Cells were selected for transgene expression using puromycin (5 μg ml−1) for 9 days, followed by serial dilution under non‐selective conditions and expansion of single‐cell clones. Individual clones were tested for successful targeting by Surveyor assay, and positive candidates were verified by Western blot analysis (Fig 4A and E).
RNA uridylation assay in S2 cell lysate
For lysate preparation, S2 cells were pelleted and washed twice in 1× PBS. The pellet was resuspended in 2 cell pellet volumes of 1× lysis/IP buffer (30 mM HEPES–KOH pH 7.4, 100 mM KOAc, 2 mM MgOAc, 0.5% NP‐40, 5 mM DTT, complete ETDA‐free proteinase inhibitor cocktail [Roche]), and the cells were disrupted with ~50 strokes of a Dounce homogenizer using a “B” pestle followed by centrifugation at 18,000 × g for 30 min at 4°C. Cleared lysate was normalized for protein content and assayed under RNAi conditions at 25°C using 625 nM α‐32P‐UTP or α‐32P‐ATP (3,000 Ci mmol−1; Perkin Elmer). Samples were taken at the indicated time points, and reaction was stopped by adding 20 volumes of 2× PK buffer (200 mM Tris–HCl pH 7.5, 25 mM EDTA pH 8, 300 mM NaCl, and 2% w/v SDS). Reactions were treated with proteinase K at 65°C for 15 min, followed by phenol/chloroform extraction and ethanol precipitation. RNA pellets were resuspended in formamide loading buffer and subjected to 15% denaturing polyacrylamide gel electrophoresis. Gels were dried and exposed to storage phosphorimager screens.
RNA library construction
Libraries for high‐throughput biochemical characterization of dmDis3l2 and ecoRNase R and small RNA libraries were generated as previously described (Reimão‐Pinto et al, 2015). For dmDis3l2 substrate identification, RNA co‐immunoprecipitated with FLAG‐tagged dmDis3l2 (or total RNA prepared from S2 cells as control) was subjected to 3′ adapter ligation as described previously (Reimão‐Pinto et al, 2015), followed by library preparation using the QuantSeq‐Flex RNA‐Seq Library preparation Kit (Lexogen), according to instructions of the manufacturer.
Data analysis
Gel images were quantified using ImageQuant TL v7.0 (GE Healthcare). Curve fitting was performed according to the integrated rate law for a first‐order reaction in Prism v7.0a (GraphPad). Statistical analyses were performed in Prism 7 v7.0a (GraphPad) or Excel v15.22 (Microsoft).
Quantitative protein interaction analysis of mass spectrometry data was performed using limma (Smyth, 2005).
Bioinformatics analysis
For high‐throughput characterization of dmDis3l2 and ecoRNase R substrate preferences, 3′ adapter was cut once with cutadapt (v1.2.1). The random 4mers of 3′ adapter were removed with fastx_trimmer of the fastx‐toolkit (v0.0.13). Sequences with exact 4 nt length were counted, and the reads per million for each 4mer were calculated.
For small RNA library analysis (dmDis3l2 footprint RNA; Fig EV3B), adapter sequences were cut once with cutadapt (v1.2.1). The random 4mers on 5′ and 3′ were removed with fastx_trimmer of the fastx‐toolkit (v0.0.13). Summary statistics (length, nt count, nt composition from end) were computed with custom scripts for the range of 7–18 nt.
For analysis of libraries generated from dmDis3l2CM‐bound RNA or total RNA, only the reverse run of PE50 sequencing was used. The random 4mers on the 5′ were removed with fastx_trimmer of the fastx‐toolkit (v0.0.13). The sequences were reverse‐complemented. The reads were aligned with Tailor (v1.1) with a minimal prefix match length of 18 and one mismatch allowed in the middle of the query (Chou et al, 2015). Counts were calculated with htseq‐count (v0.6.1p1). The parameters were stranded = yes and “intersection‐nonempty”. A GTF containing the FlyBase r5.57 annotation was used. Reads with multiple alignments were counted as their fraction to each location/feature. Feature types were summarized by following hierarchy: rRNA, tRNA, pre‐miRNA, transposable_element, pseudogene, ncRNA, snoRNA, snRNA, mRNA, intron.
Accession number
The accession number for high‐throughput sequencing datasets in this manuscript is GEO: GSE84466.
Author contributions
SLA, MMR‐P, and RAM designed and conducted the experiments and wrote the manuscript. TRB, MMR‐P, and SLA performed bioinformatics analysis of high‐throughput sequencing datasets. PS and MJ performed protein interaction experiments on recombinant Tailor and dmDis3l2. KM performed mass spectrometry analysis.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Expanded View
Acknowledgements
We thank P. Duchek for CRISPR/Cas9 genome editing in flies, I. Steinmacher and R. Imre for mass spectrometry analysis, I. Sowemimo for fly husbandry, S. Dorner for rabbit anti‐GFP antibody, and all members of the Ameres Lab for input and discussions. A monoclonal antibody against dmDis3l2 was raised at the MFPL‐mAB facility (S. Schuechner and E. Ogris). HTP sequencing was performed at the VBCF NGS Unit (www.vbcf.ac.at). This work was supported by a Boehringer Ingelheim Fonds PhD Fellowship to M.M.R.‐P., the European Research Council (ERC‐StG‐337284 ANTIVIRNA) to M.J., and the Austrian Academy of Sciences, the Austrian Science Fund FWF (Y‐733‐B22 START, W127‐B09, and F4322), and the European Research Council (ERC‐StG‐338252 miRLIFE) to S.L.A.
The EMBO Journal (2016) 35: 2417–2434
References
- Ameres SL, Horwich MD, Hung J‐H, Xu J, Ghildiyal M, Weng Z, Zamore PD (2010) Target RNA‐directed trimming and tailing of small silencing RNAs. Science 328: 1534–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, Zamore PD (2013) Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol 14: 475–488 [DOI] [PubMed] [Google Scholar]
- Andrade JM, Hajnsdorf E, Regnier P, Arraiano CM (2009) The poly(A)‐dependent degradation pathway of rpsO mRNA is primarily mediated by RNase R. RNA 15: 316–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV (1999) DNA polymerase beta‐like nucleotidyltransferase superfamily: identification of three new families, classification and evolutionary history. Nucleic Acids Res 27: 1609–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astuti D, Morris MR, Cooper WN, Staals RHJ, Wake NC, Fews GA, Gill H, Gentle D, Shuib S, Ricketts CJ, Cole T, van Essen AJ, van Lingen RA, Neri G, Opitz JM, Rump P, Stolte‐Dijkstra I, Müller F, Pruijn GJM, Latif F et al (2012) Germline mutations in DIS3L2 cause the Perlman syndrome of overgrowth and Wilms tumor susceptibility. Nat Genet 44: 277–284 [DOI] [PubMed] [Google Scholar]
- Bortolamiol‐Becet D, Hu F, Jee D, Wen J, Okamura K, Lin C‐J, Ameres SL, Lai EC (2015) Selective suppression of the splicing‐mediated microRNA pathway by the terminal uridyltransferase tailor. Mol Cell 59: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, Wan KH, Yu C, Zhang D, Carlson JW, Cherbas L, Eads BD, Miller D, Mockaitis K, Roberts J, Davis CA et al (2014) Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan KP, Butler JS (2010) TRAMP complex enhances RNA degradation by the nuclear exosome component Rrp6. J Biol Chem 285: 3540–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño IB, Heath‐Pagliuso S, Sadoff BU, Fitzhugh DJ, Christman MF (1996) A novel family of TRF (DNA topoisomerase I‐related function) genes required for proper nuclear segregation. Nucleic Acids Res 24: 2404–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H‐M, Triboulet R, Thornton JE, Gregory RI (2013) A role for the Perlman syndrome exonuclease Dis3l2 in the Lin28–let‐7 pathway. Nature 497: 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang H, Lim J, Ha M, Kim VN (2014) TAIL‐seq: genome‐wide determination of poly(A) tail length and 3′ end modifications. Mol Cell 53: 1044–1052 [DOI] [PubMed] [Google Scholar]
- Cheng Z‐F, Deutscher MP (2002) Purification and characterization of the Escherichia coli exoribonuclease RNase R. Comparison with RNase II. J Biol Chem 277: 21624–21629 [DOI] [PubMed] [Google Scholar]
- Cheng Z‐F, Deutscher MP (2003) Quality control of ribosomal RNA mediated by polynucleotide phosphorylase and RNase R. Proc Natl Acad Sci USA 100: 6388–6393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Z‐F, Deutscher MP (2005) An important role for RNase R in mRNA decay. Mol Cell 17: 313–318 [DOI] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT (2007) Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39: 715–720 [DOI] [PubMed] [Google Scholar]
- Chou M‐T, Han BW, Hsiao C‐P, Zamore PD, Weng Z, Hung J‐H (2015) Tailor: a computational framework for detecting non‐templated tailing of small silencing RNAs. Nucleic Acids Res 43: e109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus M, Régnier P (2002) The poly(A) tail of mRNAs: bodyguard in eukaryotes, scavenger in bacteria. Cell 111: 611–613 [DOI] [PubMed] [Google Scholar]
- Esakova O, Krasilnikov AS (2010) Of proteins and RNA: the RNase P/MRP family. RNA 16: 1725–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faehnle CR, Walleshauser J, Joshua‐Tor L (2014) Mechanism of Dis3l2 substrate recognition in the Lin28‐let‐7 pathway. Nature 514: 252–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazão C, McVey CE, Amblar M, Barbas A, Vonrhein C, Arraiano CM, Carrondo MA (2006) Unravelling the dynamics of RNA degradation by ribonuclease II and its RNA‐bound complex. Nature 443: 110–114 [DOI] [PubMed] [Google Scholar]
- Gallouzi IE, Wilusz J (2013) A DIStinctively novel exoribonuclease that really likes U. EMBO J 32: 1799–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göringer HU (2012) “Gestalt”, composition and function of the Trypanosoma brucei editosome. Annu Rev Microbiol 66: 65–82 [DOI] [PubMed] [Google Scholar]
- Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL (2008) The Vienna RNA websuite. Nucleic Acids Res 36: W70–W74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas G, Cetin S, Messmer M, Chane‐Woon‐Ming B, Terenzi O, Chicher J, Kuhn L, Hammann P, Pfeffer S (2016) Identification of factors involved in target RNA‐directed microRNA degradation. Nucleic Acids Res 44: 2873–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JP, Piskounova E, Gregory RI (2009) Lin28 recruits the TUTase Zcchc11 to inhibit let‐7 maturation in mouse embryonic stem cells. Nat Struct Mol Biol 16: 1021–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han BW, Hung J‐H, Weng Z, Zamore PD, Ameres SL (2011) The 3′‐to‐5′ exoribonuclease nibbler shapes the 3′ ends of microRNAs bound to Drosophila argonaute1. Curr Biol 21: 1878–1887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Kim VN (2009) Regulating the regulators: posttranslational modifications of RNA silencing factors. Cell 139: 28–31 [DOI] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim Y‐K, Ha M, Yoon M‐J, Cho J, Yeom K‐H, Han J, Kim VN (2009) TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre‐microRNA uridylation. Cell 138: 696–708 [DOI] [PubMed] [Google Scholar]
- Heo I, Ha M, Lim J, Yoon M‐J, Park J‐E, Kwon SC, Chang H, Kim VN (2012) Mono‐uridylation of pre‐microRNA as a key step in the biogenesis of group II let‐7 microRNAs. Cell 151: 521–532 [DOI] [PubMed] [Google Scholar]
- Higashimoto K, Maeda T, Okada J, Ohtsuka Y, Sasaki K, Hirose A, Nomiyama M, Takayanagi T, Fukuzawa R, Yatsuki H, Koide K, Nishioka K, Joh K, Watanabe Y, Yoshiura K‐I, Soejima H (2013) Homozygous deletion of DIS3L2 exon 9 due to non‐allelic homologous recombination between LINE‐1s in a Japanese patient with Perlman syndrome. Eur J Hum Genet 21: 1316–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefig KP, Rath N, Heinz GA, Wolf C, Dameris J, Schepers A, Kremmer E, Ansel KM, Heissmeyer V (2013) Eri1 degrades the stem‐loop of oligouridylated histone mRNAs to induce replication‐dependent decay. Nat Struct Mol Biol 20: 73–81 [DOI] [PubMed] [Google Scholar]
- Houseley J, Kotovic K, El Hage A, Tollervey D (2007) Trf4 targets ncRNAs from telomeric and rDNA spacer regions and functions in rDNA copy number control. EMBO J 26: 4996–5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, Tollervey D (2009) The many pathways of RNA degradation. Cell 136: 763–776 [DOI] [PubMed] [Google Scholar]
- Jackson RN, Klauer AA, Hintze BJ, Robinson H, van Hoof A, Johnson SJ (2010) The crystal structure of Mtr4 reveals a novel arch domain required for rRNA processing. EMBO J 29: 2205–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia H, Wang X, Liu F, Guenther U‐P, Srinivasan S, Anderson JT, Jankowsky E (2011) The RNA helicase Mtr4p modulates polyadenylation in the TRAMP complex. Cell 145: 890–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khemici V, Carpousis AJ (2004) The RNA degradosome and poly(A) polymerase of Escherichia coli are required in vivo for the degradation of small mRNA decay intermediates containing REP‐stabilizers. Mol Microbiol 51: 777–790 [DOI] [PubMed] [Google Scholar]
- Kushner SR (2002) mRNA decay in Escherichia coli comes of age. J Bacteriol 184: 4658–4665; discussion 4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Łabno A, Warkocki Z, Kuliński T, Krawczyk PS, Bijata K, Tomecki R, Dziembowski A (2016) Perlman syndrome nuclease DIS3L2 controls cytoplasmic non‐coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res doi:10.1093/nar/gkw649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, Jacquier A, Tollervey D (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121: 713–724 [DOI] [PubMed] [Google Scholar]
- Lehrbach NJ, Armisen J, Lightfoot HL, Murfitt KJ, Bugaut A, Balasubramanian S, Miska EA (2009) LIN‐28 and the poly(U) polymerase PUP‐2 regulate let‐7 microRNA processing in Caenorhabditis elegans . Nat Struct Mol Biol 16: 1016–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Yang Z, Yu B, Liu J, Chen X (2005) Methylation protects miRNAs and siRNAs from a 3′‐end uridylation activity in Arabidopsis . Curr Biol 15: 1501–1507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, Kim VN (2014) Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159: 1365–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zheng Q, Vrettos N, Maragkakis M, Alexiou P, Gregory BD, Mourelatos Z (2014) A microRNA precursor surveillance system in quality control of microRNA synthesis. Mol Cell 55: 868–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubas M, Damgaard CK, Tomecki R, Cysewski D, Jensen TH, Dziembowski A (2013) Exonuclease hDIS3L2 specifies an exosome‐independent 3. EMBO J 32: 1855–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv H, Zhu Y, Qiu Y, Niu L, Teng M, Li X (2015) Structural analysis of Dis3l2, an exosome‐independent exonuclease from Schizosaccharomyces pombe . Acta Crystallogr D Biol Crystallogr 71: 1284–1294 [DOI] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin‐Horkawicz S, Rother KM, Helm M, Bujnicki JM, Grosjean H (2013) MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res 41: D262–D267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki M, Viegas SC, Carneiro T, Golik P, Dressaire C, Ferreira MG, Arraiano CM (2013) The exoribonuclease Dis3L2 defines a novel eukaryotic RNA degradation pathway. EMBO J 32: 1842–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W (2007) RNA‐specific ribonucleotidyl transferases. RNA 13: 1834–1849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty BK, Kushner SR (2010) Bacterial/archaeal/organellar polyadenylation. Wiley Interdiscip Rev RNA 2: 256–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen TE, Marzluff WF (2008) Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5′ to 3′ and 3′ to 5′. Genes Dev 22: 50–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norbury CJ (2013) Cytoplasmic RNA: a case of the tail wagging the dog. Nat Rev Mol Cell Biol 14: 643–653 [DOI] [PubMed] [Google Scholar]
- Phizicky EM, Hopper AK (2010) tRNA biology charges to the front. Genes Dev 24: 1832–1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirouz M, Du P, Munafò M, Gregory RI (2016) Dis3l2‐mediated decay is a quality control pathway for noncoding RNAs. Cell Rep 16: 1861–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regnier P, Arraiano CM (2000) Degradation of mRNA in bacteria: emergence of ubiquitous features. BioEssays 22: 235–244 [DOI] [PubMed] [Google Scholar]
- Reimão‐Pinto MM, Ignatova V, Burkard TR, Hung J‐H, Manzenreither RA, Sowemimo I, Herzog VA, Reichholf B, Fariña‐Lopez S, Ameres SL (2015) Uridylation of RNA hairpins by tailor confines the emergence of microRNAs in Drosophila . Mol Cell 59: 203–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Chen X, Yu B (2012) Uridylation of miRNAs by HEN1 SUPPRESSOR1 in Arabidopsis . Curr Biol 22: 695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Mikulasova A, Norbury CJ (2007) Efficient RNA polyuridylation by noncanonical poly(A) polymerases. Mol Cell Biol 27: 3612–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissland OS, Norbury CJ (2009) Decapping is preceded by 3′ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16: 616–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougemaille M, Gudipati RK, Olesen JR, Thomsen R, Séraphin B, Libri D, Jensen TH (2007) Dissecting mechanisms of nuclear mRNA surveillance in THO/sub2 complex mutants. EMBO J 26: 2317–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Paolo S, Vanacova S, Schenk L, Scherrer T, Blank D, Keller W, Gerber AP (2009) Distinct roles of non‐canonical poly(A) polymerases in RNA metabolism. PLoS Genet 5: e1000555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt K, Butler JS (2013) Nuclear RNA surveillance: role of TRAMP in controlling exosome specificity. Wiley Interdiscip Rev RNA 4: 217–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Leung E, Brown J, Tollervey D (2009) The N‐terminal PIN domain of the exosome subunit Rrp44 harbors endonuclease activity and tethers Rrp44 to the yeast core exosome. Nucleic Acids Res 37: 1127–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sement FM, Ferrier E, Zuber H, Merret R, Alioua M, Deragon J‐M, Bousquet‐Antonelli C, Lange H, Gagliardi D (2013) Uridylation prevents 3′ trimming of oligoadenylated mRNAs. Nucleic Acids Res 41: 7115–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Goodman HM (2004) Uridine addition after microRNA‐directed cleavage. Science 306: 997 [DOI] [PubMed] [Google Scholar]
- Slevin MK, Meaux S, Welch JD, Bigler R, Miliani de Marval PL, Su W, Rhoads RE, Prins JF, Marzluff WF (2014) Deep sequencing shows multiple oligouridylations are required for 3′ to 5′ degradation of histone mRNAs on polyribosomes. Mol Cell 53: 1020–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2005) limma: linear models for microarray data In Bioinformatics and computational biology solutions using R and bioconductor, Gentleman R, Carey VJ, Huber W, Irizarry RA, Dudoit S. (eds), pp 397–420. New York, NY: Springer New York; [Google Scholar]
- Su W, Slepenkov SV, Slevin MK, Lyons SM, Ziemniak M, Kowalska J, Darzynkiewicz E, Jemielity J, Marzluff WF, Rhoads RE (2013) mRNAs containing the histone 3′ stem‐loop are degraded primarily by decapping mediated by oligouridylation of the 3′ end. RNA 19: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MP, Liu X, Whangbo J, McCrossan G, Sanborn KB, Basar E, Walch M, Lieberman J (2015) Apoptosis triggers specific, rapid, and global mRNA decay with 3′ uridylated intermediates degraded by DIS3L2. Cell Rep 11: 1079–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ustianenko D, Hrossova D, Potesil D, Chalupnikova K, Hrazdilova K, Pachernik J, Cetkovska K, Uldrijan S, Zdrahal Z, Vanacova S (2013) Mammalian DIS3L2 exoribonuclease targets the uridylated precursors of let‐7 miRNAs. RNA 19: 1632–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, Friedlein A, Langen H, Keith G, Keller W (2005) A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3: e189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JR, Bonneau F, Hentschel J, Conti E (2010) Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc Natl Acad Sci USA 107: 12139–12144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J‐C, Dufour M‐E, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B, Libri D, Jacquier A (2005) Cryptic Pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737 [DOI] [PubMed] [Google Scholar]
- Xie J, Ameres SL, Friedline R, Hung J‐H, Zhang Y, Xie Q, Zhong L, Su Q, He R, Li M, Li H, Mu X, Zhang H, Broderick JA, Kim JK, Weng Z, Flotte TR, Zamore PD, Gao G (2012) Long‐term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods 9: 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yu Y, Zhai J, Ramachandran V, Dinh TT, Meyers BC, Mo B, Chen X (2012) The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr Biol 22: 689–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber H, Scheer H, Ferrier E, Sement FM, Mercier P, Stupfler B, Gagliardi D (2016) Uridylation and PABP cooperate to repair mRNA deadenylated ends in Arabidopsis . Cell Rep 14: 2707–2717 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Expanded View