

Abstract
Medullary cystic kidney disease (MCKD) is a hereditary disease associated with bilateral medullary polycysts and interstitial fibrosis. MCKD is typically associated with slowly progressive renal dysfunction. We herein report two rare elderly cases with enlarged kidneys and rapidly progressive renal dysfunction without myeloperoxidase anti-neutrophil cytoplasmic antibody (MPO-ANCA), PR3-ANCA, or anti-glomerular basement membrane (GBM) antibodies. Renal biopsies revealed extensive tubular dilatation and atrophy with interstitial fibrosis consistent with MCKD. Both patients began hemodialysis therapy a few months later. Our cases suggest a MCKD subgroup among elderly patients with an undefined genetic background, rapidly progressive renal dysfunction, and enlarged kidneys.
Keywords: kidney enlargement, rapidly progressive renal failure, medullary cystic kidney disease, elderly patients
Introduction
Nephronophthisis (NPH) and medullary cystic kidney disease (MCKD) are renal diseases associated with bilateral medullary polycysts and interstitial fibrosis. These two diseases are thought to represent the same disease process according to similar clinical and pathological features, excluding the age of onset and associated genetic mutations. The concept of juvenile nephronophthisis-medullary cystic disease complex (NPH-MCKD) has been proposed (1).
With the progression of tubular damage, tubular functions, such as sodium reabsorption and urine concentration, become impaired. Inflammation and fibrosis occurs in the tubulointerstitial area of the kidneys, ultimately leading to end-stage kidney disease (ESKD). The progression to ESKD is typically gradual, similar to that observed in polycystic kidney disease. NPH is an autosomal recessive (AR) disease that manifests in early childhood or adolescence and progresses to ESKD in early adolescence. MCKD is an autosomal dominant (AD) disease that develops in adulthood and slowly progresses to ESKD, characterized by multiple cysts, measuring 1-20 mm, in the corticomedullary boundary.
The clinical features of MCKD are nonspecific and include hypertension, polyuria, and sodium wasting due to an impairment in urinary concentration (2). The pathological features include expanded urinary ducts in the corticomedullary boundary areas, diffuse tubulointerstitial nephritis with tubular atrophy, interstitial fibrosis, and an inflammatory cell infiltrate (3). The kidneys are typically atrophic and decreased in size (4,5).
We herein report two cases of MCKD in elderly patients with enlarged kidneys and a rapid progression to ESKD. Both patients had no apparent genetic history of kidney disease, and their clinical courses were not typical for traditional hereditary MCKD.
Case Reports
Case 1
An 84-year-old woman had a 3-year history of hypertension, a femoral neck fracture 1 year previously, and a left radial fracture 1 month previously. She had no family history of renal disease or dialysis. She was noted to have renal dysfunction of unknown etiology 1 year prior to admission. She visited a regional clinic for edema and was observed to have renal dysfunction [serum creatinine (Cr), 2.7 mg/dL] and anemia 2 months prior to admission. Since that time, she had developed marked edema and progressive renal dysfunction (serum Cr, 3.7 mg/dL) and was referred to our hospital for further evaluation and treatment. On physical examination, her temperature was 37.1℃, and her blood pressure was 168/63 mmHg. She showed marked abdominal distention due to ascites and also had lower extremity edema without dyspnea. Chest radiography revealed cardiac enlargement and bilateral pleural effusions, and an abdominal computed tomography (CT) scan revealed massive ascites and bilateral kidney enlargement (Fig. 1). The following laboratory results were obtained: white blood cell (WBC) count, 5,400 WBC/mm3; red blood cells (RBC) count, 250×104 RBC/mm3; hemoglobin (Hb), 8.2 g/dL; hematocrit (Ht), 24.0%; platelet count (plt), 15.8×104 plt/mm3; total protein (TP), 5.6 g/dL; albumin (Alb), 2.8 g/dL; blood urea nitrogen (BUN), 45 mg/dL; serum Cr; 3.9 mg/dL; sodium (Na), 140 mEq/L; potassium (K), 3.2 mEq/L; chloride (Cl), 104 mEq/L; C-reactive protein (CRP), 0.5 mg/dL; plasma glucose, 142 mg/dL; hemoglobin A1c (HbA1c), 4.8%; aspartate aminotransferase (AST), 38 IU/L; alanine aminotransferase (ALT), 18 IU/L; alkaline phosphatase (ALP), 787 IU/L; gamma glutamyl transpeptidase (γGT), 239 IU/L; IgG, 828 mg/dL; IgM, 700 mg/dL; and IgA, 148 mg/dL. Cryoglobulin was negative and an immunoglobulin (Ig) M (kappa) spike was identified on immunoelectrophoresis. The third complement component (C3) level was 59 mg/dL (normal 50-130 mg/dL); the fourth component (C4) level was 10.5 mg/dL (normal 10-50 mg/dL); hemolytic complement activity via the classical pathway (CH50) was 13 U/mL (normal 25.0-48.0 U/mL); and antinuclear antibody (ANA) was negative. Myeloperoxidase antibodies (MPO), proteinase 3 antibodies (PR3), antineutrophil cytoplasmic antibodies (ANCA), and anti-glomerular basement membrane (anti-GBM) antibodies were not detected. A urinalysis showed 1.17 g protein/g creatinine and 30-49 RBC/high-power field (HPF). The urinary β-2 microglobulin (β-2MG) level was 25,150 ng/mL. Bence Jones protein was negative in the urine. A bone marrow aspirate clot showed hypercellular bone marrow with 3% plasma cells.
Figure 1.
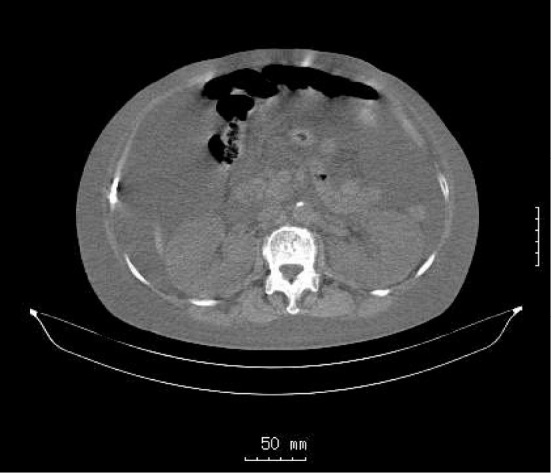
Computed tomography of the abdomen in case 1. Computed tomography of the abdomen shows massive ascites and enlarged kidneys with no evidence of cysts.
At the time of hospitalization, a percutaneous kidney biopsy was not performed because of the patient's massive ascites. A percutaneous kidney biopsy was performed 1 month later after several paracentesis procedures, allowing the patient to lie in a prone position.
Histological findings revealed tubular dilatation, primarily involving the distal tubules, with extensive fibrosis in the interstitium, consistent with nephronophthisis-medullary cystic disease complex(NPH-MCKD). There were no remarkable glomerular changes except for global sclerosis consistent with the patient's age (Fig. 2). Immune deposits of IgG, IgA, IgM, C1q, C3, and fibrinogen were absent, despite the IgM kappa M protein detected in the serum. Electron microscopy revealed irregularities, thinning and disintegration of the tubular basement membrane (TBM). However, it was difficult to explain the patient's massive ascites and rapidly progressive renal dysfunction according to the histological findings.
Figure 2.

Renal biopsy findings in case 1. (a) periodic acid-Schiff (PAS) stain; magnification 10×; (b) PAS stain; 200×. Light microscopy images show tubular dilatation and atrophy. Cystic tubules show irregularities and thinning of the tubular epithelial cells. The glomeruli show global sclerosis and arterial intimal thickening, consistent with the patient's age, without any other remarkable changes such as crescent formation.
Considering her advanced age (84 years of age) and symptomatic relief after paracentesis, no further investigation was done and she was discharged with a serum Cr level of 5.1 mg/dL 2 months after admission. She began hemodialysis therapy 1 month after discharge.
Case 2
A 74-year-old woman was started on antihypertensive therapy 1 year prior to admission when she was noted to have a mild renal impairment with a serum Cr level of 1.1 mg/dL. She was status post-appendectomy at 20 years of age, had a history of ulcerative colitis with rectal cancer at 61 years of age, and a cataract at 68 years of age. She had no family history of renal disease or dialysis. Although her blood pressure was well controlled, her serum Cr level rapidly increased to 1.5 mg/dL at 3 months after the start of antihypertensive therapy and 2.4 mg/dL at 5 months after the start of therapy. She was subsequently referred to our hospital for further evaluation and treatment.
On physical examination, her temperature was 36.5℃, and her blood pressure was 150/79 mmHg. Her conjunctivae were pale and she had lower extremity edema. The following laboratory results were obtained: WBC count, 6,800 WBC/mm3; RBC count, 248×104 RBC/mm3; Hb, 7.3 g/dL; Ht, 21.7%; Plt, 18.2×104/mm3; TP, 7.1 g/dL; Alb, 4.1 g/dL; BUN, 30 mg/dL; Cr, 3.6 mg/dL; Na, 138 mEq/L; K, 5.3 mEq/L; Cl, 107 mEq/L; CRP, 0.21 mg/dL; plasma glucose, 107 mg/dL; HbA1c, 5.1%; IgG, 1,326 mg/dL; IgM, 76 mg/dL; IgA, 239 mg/dL; C3, 91 mg/dL; C4, 24.7 mg/dL; and CH50, 13 U/mL. Cryoglobulin was negative. ANA was negative, and MPO, PR3, ANCA, and anti-GBM were not detected. A urinalysis showed 1.23 g protein/g creatinine and 6 RBC/HPF. Urinary β-2MG was 27,708 ng/mL.
Abdominal CT showed bilateral kidney enlargement that was not seen on an earlier CT exam 1 year previously. Serial CT images are shown in Fig. 3. Magnetic resonance images (MRI) are shown in Fig. 4. MRI, including coronal magnetic resonance images, did not reveal multiple microcysts in the enlarged kidneys, however, a careful observation revealed multiple cysts along the corticomedullary boundary.
Figure 3.

Serial changes of abdominal computed tomography (CT) at (a) 2 years prior to admission, (b) 1 year prior to admission and (c) at admission in case 2. There is no evidence of kidney enlargement at 2 or 1 year prior to admission; however, CT at admission shows kidney enlargement compared with the previous CT scans.
Figure 4.

Magnetic resonance imaging (MRI) of the abdomen: (a) axial section of the abdomen (T1-weighted image) and (b) coronal section of the abdomen (T2-weighted image) in case 2. No apparent microcysts are seen in the enlarged kidneys, including the coronal images, however, a closer observation shows multiple cysts along the corticomedullary boundary in the left kidney in the coronal section (arrows) (b).
A percutaneous renal biopsy was performed on day 2. Of seven glomeruli, two were globally sclerotic, one was segmentally sclerotic, and two showed glomerular cysts. The other glomeruli were essentially normal. The distal tubules were also dilated, consistent with MCKD, and there was mild interstitial fibrosis and inflammatory cell infiltration (Fig. 5). Electron microscopy revealed irregular layering, spheroidizing, thinning, and partial rupture of the TBM. The patient was treated with corticosteroids [prednisolone (PSL) 30 mg/day] in view of the significantly elevated urinary β-2MG level and interstitial cell infiltration. Immediately after PSL administration, a slight reduction in the serum Cr level was noted and her anemia improved. However, the serum Cr level gradually increased and the Hb level gradually decreased 2 months later, when PSL was tapered. Three months after the start of PSL therapy, the patient developed gastrointestinal bleeding and was found to have a serum Cr level of 7 mg/dL. The gastrointestinal bleed may have been an adverse effect of PSL therapy. However, no apparent bleeding source was detected by gastroscopy, colonoscopy, or enteroscopy. Anemia was corrected with blood transfusions, however, the patient's renal function did not improve and she began hemodialysis therapy 5 months after referral.
Figure 5.

Renal biopsy findings in case 2. (a) periodic acid-methenamine-silver (PAM) stain; original magnification 10×, (b) PAS stain original magnification 200×. Light microscopy images show tubular dilatation and atrophy. Cystic tubules show irregularities and thinning of the tubular epithelial cells. Of seven glomeruli, two are globally sclerotic, one is segmentally sclerotic, and two show glomerular cysts. The other glomeruli are intact.
Immunohistochemical findings
We also performed immunohistochemical staining in both cases for CD10, cytokeratin 7 (CK7), epithelial membrane antigen (EMA), E-cadherin, and cytokeratin (34 beta E12) to identify the level of tubular dilatation (Fig. 6). In case 1, the dilated tubules were negative for CD10, a marker of proximal tubules (6). In contrast, immunostaining results for CK7, EMA, and E-cadherin, markers of the distal tubules and collecting ducts, were positive. Immunostaining results for CK (34 beta E12), a marker of collecting ducts, were also positive (7), indicating that the cysts predominantly arose from collecting ducts in case 1. In case 2, the dilated tubules were positive for CK7 and EMA and weakly positive for E-cadherin and CK (34 beta E12).
Figure 6.

Immunohistochemical findings in both cases. Immunohistochemical staining for CD10, cytokeratin 7 (CK7), epithelial membrane antigen (EMA), E-cadherin, and cytokeratin (34 beta E12) in the interstitium of both cases. Original magnification 10×. The tubular basement membrane (TBM) of dilated tubules in both cases is negative for CD10. In case 1, the TBM of dilated tubules is positive for CK7, EMA, E-cadherin, and CK (34 beta E12). In case 2, the TMB of dilated tubules is positive for CK7 and EMA and weakly positive for E-cadherin and CK (34 beta E12).
Discussion
NPH-MCKD is a hereditary disease of the renal tubules, which are important for the reabsorption of sodium and the concentration of urine. Typically, interstitial inflammation and fibrosis occurs, slowly progressing to ESKD, similar to the progression seen in polycystic kidney disease. The genetic mutations associated with NPH-MCKD are shown in Table 1 (8-17).
Table 1.
Gene Mutations Associated with NPH-MCKD.
| Reference | Inheritance | Chromosome | Gene | Protein | ESKD (yrs) |
|---|---|---|---|---|---|
| 8.9 | AR | 2q13 | NPHP1 | Nephrocystin-1 | 13 |
| 10 | AR | 9q22-q31 | NPHP2 | Inversin | <1 |
| 11 | AR | 3q21.1 | NPHP3 | Nephrocystin-3 | 19 |
| 12 | AR | 1p36.22 | NPHP4 | Nephroretinin | 21 |
| 13 | AR | 3q21.1 | IQCB1, NPHP5 | Nephrocystin-5 | 13 |
| 14 | AR | 12q21.33 | CEP290, NPHP5 | Nephrocystin-6 | |
| 15 | AR | 16q13.3 | GLIS2 | Zinc finger protein GLIS2 | |
| 16 | AD | 1q21 | MUC1 | Mucin-1 | 62 |
| 17 | AD | 16q12.3 | UMOD | Uromodulin | 32 |
AR: Autosomal Recessive, AD: Autosomal Dominant
Recently, a human genetic condition termed “ciliopathy” has been proposed. Ciliopathy is thought to be caused by a dysfunction of two functionally and physically associated organelles, the centromere and the cilium. Autosomal dominant polycystic kidney disease (ADPKD) and NPH-MCKD are thought to be related to renal cilia dysfunction (ciliopathy), and more than 30 different genetic mutations have been localized to the cilia and/or centromeres (18,19).
NPH is inherited in an AR manner. The disease onset occurs in early childhood or adolescence and typically progresses to ESKD during adolescence. On the other hand, MCKD is an AD disease. Its onset occurs during adulthood and slowly progresses to ESKD with the formation of multiple cysts, measuring 1-20 mm, in the corticomedullary boundary. The kidneys usually decrease in size and become atrophic. There is currently no effective treatment to prevent progression; sodium supplementation and erythropoietin are used for symptomatic relief.
Two types of MCKD are recognized (MCKD1 and MCKD2) according to the localizing chromosome and the onset of ESKD. MCKD1 progresses more slowly than MCKD2 and has a later onset of ESKD (average age 62 years old; range, 50-70 years). The average age of onset of ESKD in MCKD2 is 32 years (range, 16-54 years).
Genetic mutations are known to affect mucoprotein mucin-1 (MUC1) in MCKD1 and uromodulin (UMOD) in MCKD2 (10). The initial symptoms are polydipsia and polyuria due to decreased urine concentration and sodium loss. The urinalysis is normal early in the disease. Generally, there is a slow progression to ESKD, similar to the progression seen in polycystic kidney disease. The typical CT, MRI, and ultrasound images in MCKD show atrophic kidneys with multiple cysts in the medulla, particularly at the corticomedullary junction (4,5). The differential diagnosis of kidney enlargement includes infiltrative diseases (hematopoietic malignancy, sarcoidosis, and amyloidosis), autosomal dominant polycystic kidney disease, early stage diabetic nephropathy, acute interstitial nephritis, and pyelonephritis (20). In the present cases, an immunohistological investigation revealed numerous microcysts within the dilated distal tubules and collecting ducts, compatible with MCKD. We postulate that these renal microcysts rapidly developed and replaced the renal parenchyma with tubulointerstitial inflammation, a finding rarely seen in MCKD. The cyst formation and subsequent inflammation played a pivotal role in the pathogenesis of the kidney enlargement and dysfunction. We have found similar case reports from Japan (21-23). These cases, including ours, are summarized in Table 2. All five of these cases showed renal enlargement and MCKD-like tubulointerstitial changes with a rapid progression to ESKD. However, there are no previous reports of patients with massive ascites and rapidly progressive renal failure in MCKD. Taneda et al. also reported a similar case in an elderly patient who was not clinically diagnosed, but had been diagnosed at autopsy with ARPKD according to the renal morphology. The case reported by Taneda et al. also showed renal enlargement and a rapid progression to ESKD in the absence of any apparent genetic mutation. The diagnosis of ARPKD was made according to the presence of liver involvement, including hepatic cysts, jaundice, and elevated liver enzyme levels, findings not typically observed in MCKD (24).
Table 2.
Summary of Similar Cases Reported in Japan.
| Reference | Age | Sex | FH | Cr | Hb | CRP | UP | UOB | Uβ2 (µg/L) | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|
| Case1 | 84 | F | (-) | 3.9 | 8.4 | 0.5 | 1+ | 1+ | 25,150 | HD |
| Case2 | 74 | F | (-) | 3.6 | 7.3 | <0.06 | 1+ | - | 27,708 | HD |
| 21 | 70 | M | (-) | 2.3 | 8.3 | 0.37g/gCr | - | 648 | HD | |
| 22 | 66 | F | (-) | 5.87 | 8.9 | 0.24g/gCr | 1+ | 26,123 | HD | |
| 23 | 63 | F | (+)? | 11.6 | 6.2 | 0.06 | 2+ | 1+ | HD |
Because these other cases were reported mainly from Japan, it appears that these diseases are not due to regional or racial differences or a new disease type, unlike traditional hereditary MCKD. A greater accumulation of similar cases with genetic and histopathological analyses is important to further our understanding of this disease entity.
We herein reported two unusual cases that presented with rapidly progressive renal dysfunction, renal enlargement, and acute interstitial injury. The pathological findings were consistent with MCKD, however, the patients had no family history of renal disease. Similar cases, lacking the genetic findings of traditional hereditary MCKD, have been reported in Japan. Further pathological and clinical investigations of future cases are required.
The authors state that they have no Conflict of Interest (COI).
References
- 1.Helen L, Paul W. Cystic disease and developmental kidney defects: Medullary Nephronophithisis-Medullary Cystic Kidney Disease Complex(NPH-MCKD). In: Heptinstall's Pathology of The Kidney. 6th ed Charles J, Jean LO, Melvin MS, Fred GS, Eds. Lippincott Williams & Wilkins, Phialdelphia, 2007: 1273-1278. [Google Scholar]
- 2.Rohatgi R. Clinical manifestations of hereditary cystic kidney disease. Front Biosci 13: 4175-4197, 2008. [DOI] [PubMed] [Google Scholar]
- 3.Bisceglia M, Galliani CA, Senger C, et al. Renal cystic dseases:A review. Adv Anat Pathol 13: 26-56, 2006. [DOI] [PubMed] [Google Scholar]
- 4.Wise SW, Hartman DS, Hardesty LA, et al. Renal medullary cystic disease: Assessment by MRI. Abdom Imaging 23: 649-651, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Scolari F, Ghiggeri GM. Nephronophthisis-medullary cystic kidney disease: From bedside to bench and back again. Saudi J Kidney Dis Transpl 14: 316-327, 2003. [PubMed] [Google Scholar]
- 6.Langner C, Ratschek M, Rehak P, Schips L, Zigeuner R. CD10 is a diagnostic and prognostic marker in renal malignancies. Histopathology 45: 460-467, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Yang XJ, Zhou M, Hes O, et al. Tubulocystic carcinoma of the kidney: Clinicopathologic and molecular characterization. Am J Surg Pathol 32: 177-187, 2008. [DOI] [PubMed] [Google Scholar]
- 8.Hildebrandt F, Otto E, Rensing C, et al. Anovel gene encoding an SH3 domain protein is mutated in nephronophthisis type 1. Net Genet 17: 149-153, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Saunier S, Calado J, Heilig R, et al. A novel gene that encodes a protein with a putative src homology 3 domain is a candidate gene for familial juvenile nephronophthisis. Hum Mol Genet 6: 2317-2323, 1997. [DOI] [PubMed] [Google Scholar]
- 10.Otto EA, Schermer B, Obara T, et al. Mutations in INVS encoding inversion cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet 34: 413-420, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olbrrich H, Fliegauf M, Hoefele J, et al. Mutations in novel gene, NPH3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 34: 455-459, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Otto E, Hoefele J, Ruf R, et al. A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet 71: 1161-1167, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otto EA, Loeys B, Khanna J, et al. Nephrocystin-5, a ciliry IQ domain protein, is mutated in Senior-Loken syndrome are interacts with RPGR and calmodulin. Nat Genet 37: 282-288, 2005. [DOI] [PubMed] [Google Scholar]
- 14.Sayer JA, Otto EA, O'Toole JF, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet 38: 674-681, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Attanasio M, Uhlenhaut NH, Sousa VH, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet 39: 1018-1024, 2007. [DOI] [PubMed] [Google Scholar]
- 16.Kyproula C, Marios T, Christoforos S, et al. Chromosome 1 localization of a gene for autosomal dominant medullary cystic kidney disease (ADMCKD). Hum Molecular Genet 7: 905-911, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Hateboer N, Gumbs C, Teare MD, et al. Confirmation of a gene locus for medullary cystic kidney disease (MCKD2) on chromosome 16p12. Kidney Int 60: 1233-1239, 2001. [DOI] [PubMed] [Google Scholar]
- 18.Hildebrandt F, Attanasio M, Otto E. Nephronophthisis: Disease mechanism of a ciliopathy. J Am Soc Nephrol 20: 23-35, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurd TW, Hildebrandt F. Mechanism of nephronophthisis and related ciliopthies. Nephron Exp Nephrol 118: e9-e14, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storm A, Bhasin B, Rangachari D, et al. Quiz page January 2014: Cachexia, urinary tract infection, nephromegaly, and kidney failure. Am J Kidney Dis 63: A18-A21, 2014. [DOI] [PubMed] [Google Scholar]
- 21.Suzuki T, Iyoda M, Yamaguchi Y, Shibata T. A case of sporadic medullary cystic kidney disease type 1(MCKD1) with kidney enlargement complicated by IgA nephropathy. Pathology Int 65: 379-382, 2015. [DOI] [PubMed] [Google Scholar]
- 22.Matsuura R, Hidaka S, Hasegawa M, et al. A case of MCKD with atrial septal defect. Jpn J Nephrol 55 (Suppl): 1052, 2013. [Google Scholar]
- 23.Amemiya N, Sugiura H, Kamiyama T, et al. A case of medullary cystic kidney disease with rapid progressive renal dysfunction and kidney enlargement. Jpn J Nephrol 58: 660-667, 2016. [PubMed] [Google Scholar]
- 24.Taneda S, Honda K, Aoki A, et al. An autopsy case of clinically un-diagnosed autosomal recessive polycystic kidney disease in 77-year-old male. Pathol Int 62: 811-816, 2012. [DOI] [PubMed] [Google Scholar]