

Abstract
We herein present a rare case of an autosomal dominant polycystic kidney disease (ADPKD) patient with Caroli's disease, a congenital embryonic biliary tree ductal plate abnormality often associated with autosomal recessive polycystic kidney disease. A 76-year-old woman with ADPKD on hemodialysis was admitted to our hospital with recurrent cholangitis and hepatobiliary stones. Caroli's disease was diagnosed according to typical imaging findings of cystic intrahepatic bile duct dilatation and the central dot sign. Hepatobiliary system abnormalities such as Caroli's disease should be considered in febrile ADPKD patients, even in the absence of typical clinical signs or symptoms.
Keywords: Caroli's disease, autosomal dominant polycystic kidney disease, recurrent cholangitis, ductal plate malformations
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited disorder mainly associated with renal cyst formation and renal function deterioration. Intrahepatic cysts are the most common hepatic complication of ADPKD (1).
Caroli's disease was first described in 1958 (2). As originally reported, the entity is characterized by: 1) segmental cystic dilatation of the intrahepatic bile ducts; 2) high incidence of intrahepatic cholelithiasis, cholangitis, and hepatic abscesses; 3) absence of cirrhosis and portal hypertension; and 4) association with renal tubular ectasia or medullary sponge kidney and, in some instances, pancreatic cysts. The combination of Caroli's disease and congenital hepatic fibrosis is commonly known as Caroli's syndrome. Caroli's disease or syndrome is often associated with autosomal recessive polycystic kidney disease (ARPKD), but it is a rare complication of ADPKD. Only four case reports describing ADPKD associated with Caroli's disease or syndrome have previously been published (3-6).
Renal and hepatic cyst infections are the major causes of a fever in ADPKD patients (7). Infection of the hepatobiliary tract has also been reported to occur in ADPKD patients with hepatobiliary morphologic abnormalities. We herein report a rare case of ADPKD with Caroli's disease and recurrent hepatobiliary infection. The study of this case was approved by the institutional review board of Toranomon Hospital.
Case Report
A 76-year-old woman was admitted to our hospital for the evaluation of an intractable fever with chills in August 2012. ADPKD had been diagnosed in 1982, and hemodialysis treatment was started in 1998. Transcutaneous renal arterial embolization was performed to reduce abdominal distension related to enlarged kidneys in 2007 (8). A clipping operation was carried out for left anterior cerebral artery aneurysm in 2008. One of her two daughters was also diagnosed with ADPKD. The patient had presented with a high fever and bacteremia once or twice per year since 2009, and renal or hepatic cyst infection was considered to be causative on each occasion.
On this admission, the patient was 150.3 cm tall and weighed 54.7 kg, with a blood pressure of 138/61 mmHg and a temperature of 38.3℃. A physical examination revealed no jaundice or abdominal tenderness. The results of the laboratory examination are shown in Table. Regarding enzymes related to hepatobiliary tract diseases, only alkaline phosphatase (ALP) was increased slightly. The C-reactive protein concentration was 2.7 mg/dL. A blood culture was positive for extended-spectrum β-lactamase-producing Escherichia coli (ESBL-E. coli). Magnetic resonance imaging (MRI) indicated multiple large renal cysts consistent with ADPKD and multiple hepatic cysts located mainly in peribiliary regions (Fig. 1a). There were no signs of cyst infections on MRI, such as an increased intensity on diffusion-weighted imaging, niveau, wall thickening, or gas in the cysts, which we previously reported to be typical findings of infected cysts (9). MRI revealed a common bile duct stone and cystic dilatation of the intrahepatic bile ducts in the left lobe (Fig. 1b). No gallbladder stones were observed. Contrast-enhanced abdominal computed tomography (CT) showed the central dot sign representing the fibrovascular bundle within the dilated cystic intrahepatic ducts (Fig. 2), which was considered to be characteristic feature of Caroli's disease (10). Endoscopic retrograde cholangiography (ERC) also demonstrated cystic dilated biliary ducts with stones (Fig. 3). One black stone suggesting bilirubin origin was removed under ERC, but unremovable stones existed. ESBL-E. coli was also detected in the bile culture. According to these findings, recurrent cholangitis related to Caroli's disease with bile duct stones was diagnosed.
Table.
Laboratory Data on Admission to the Hospital.
| hemoglobin | 8.4 | (11.3-15.0) | g/dL |
| White blood cells | 5,200 | (3.2-7.9) | /µL |
| platelets | 18.5×104 | (15.5-35.0) | /µL |
| total protein | 6.9 | (6.9-8.4) | g/dL |
| albumin | 2.7 | (3.9-5.2) | g/dL |
| urea nitrogen | 29 | (8-21) | mg/dL |
| creatinine | 5.4 | (0.46-0.78) | mg/dL |
| alkaline phosphatase | 492 | (117-350) | IU/L |
| gamma glutamyl transferase | 31 | (9-109) | IU/L |
| alanine aminotransferase | 4 | (8-40) | IU/L |
| aspartate aminotransferase | 15 | (13-33) | IU/L |
| total bilirubin | 0.5 | (0.3-1.1) | mg/dL |
| C-reactive protein | 2.7 | (<0.3) | mg/dL |
Figure 1.

(a) Magnetic resonance imaging (MRI) indicated multiple large renal cysts consistent with ADPKD and multiple hepatic cysts located mainly in peribiliary regions. (b) MRI revealed cystic dilatation of the intrahepatic bile ducts in the left lobe and peribiliary cysts.
Figure 2.
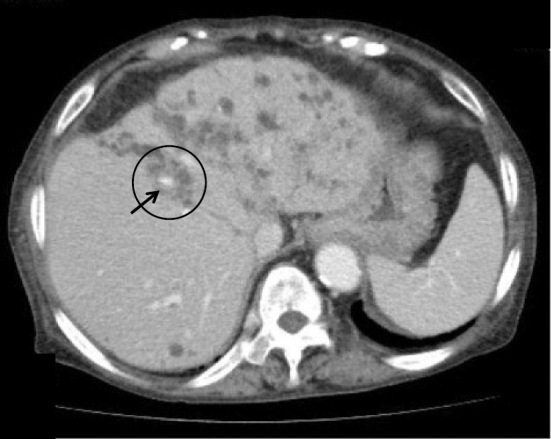
Contrast-enhanced abdominal CT showed the central dot sign (circle), representing the fibrovascular bundle (arrow) within the dilated cystic intrahepatic ducts, which is considered to be a characteristic feature of Caroli's disease.
Figure 3.
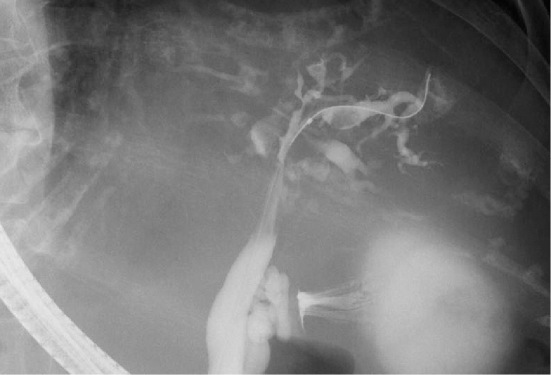
Endoscopic retrograde cholangiography (ERC) demonstrated cystic dilated biliary ducts with stones.
Sepsis was controlled with intravenous meropenem, however, the fever recurred when the treatment was changed to oral antibiotics, as shown in Fig. 4. The patient exhibited febrile episodes five times within a year due to cholangitis, liver abscess, or hepatic cyst infection. Since September 2013, the patient has received intravenous meropenem treatment at every hemodialysis session and cholangitis has been controlled since that time.
Figure 4.

Clinical course of the present case.
Discussion
The presence of Caroli's disease or syndrome has previously been described in conjunction with ARPKD, which usually has a childhood onset. The association of Caroli's disease with ADPKD is rare (3-6). Both Caroli's disease and hepatic cysts in ADPKD are regarded as fibrocystic liver diseases, which comprise a group of congenital disorders resulting from ductal plate malformation, i.e., abnormal embryogenesis of the biliary ductal system (11). A ductal plate is defined as a cylindrical layer of cells that surrounds a branch of the portal vein and develops during the first week of gestation (12). The clinicopathological abnormality of fibrocystic liver disease is dependent on the level of the biliary tree affected during embryogenesis. The involvement of large intra-hepatic ducts results in Caroli's disease. ADPKD represents ductal plate malformation of medium-sized intrahepatic ducts. Small-sized intra-hepatic duct involvement results in congenital hepatic fibrosis. The disease entities can exist as individual conditions or in combination (11,13).
A mutation in PKD1 encoding polycystin-1 or PKD2 encoding polycystin-2 gives rise to ADPKD. The identified gene underling ARPKD is PKHD1 encoding fibrocystin (14). These proteins localize to primary cilia of renal tubule cells and cholangiocytes (15). Although the hepatic phenotype of Caroli's disease generally appears in ARPKD, the phenotype of Caroli's disease was observed in ADPKD patient in the present case.
Hepatic phenotype variants have been previously reported in ADPKD patients. Dranssart et al. observed intrahepatic bile duct abnormalities in 25 of 90 ADPKD patients (27.8%) with magnetic resonance cholangiography, which were correlated with hepatobiliary infection (16). Ishikawa et al. reported that common bile duct dilatation occurred more frequently in ADPKD patients than in non-ADPKD patients (17). Terada et al. examined specimens from autopsied livers and biliary tracts of three patients with ADPKD using intrahepatic cholangiography and found nonobstructive dilatation of the intrahepatic bile ducts in all cases (18). Jordon et al. reported the 17-year history of an ADPKD patient with a recurrent fever of unknown origin that was later attributed to Caroli's disease with multiple episodes of cholangitis (4). These phenotypic variants might be due to differences in the type or locus of the mutation of responsive genes, epigenetic mechanisms, and/or environmental factors (19).
Our case presented with a recurrent fever and septicemia for several years. At first, renal or hepatic cyst infections were thought to be the cause of the fevers, because they are the most common causes of a fever in ADPKD (7). However, diagnostic imaging findings revealed the characteristic hepatobiliary abnormalities of Caroli's disease that lead to recurrent cholangitis with hepatobiliary stones. The patient did not present with abdominal pain, and her laboratory findings did not indicate liver function abnormality, except for a slight elevation of ALP. The treatment of Caroli's disease is largely supportive and should be individualized (20). Cholangitis and sepsis should be treated with appropriate antibiotics and biliary stone extraction whenever feasible. Partial hepatectomy may be curative in rare patients in whom the disease is confined to a single lobe of the liver (21). Patients who have recurrent bouts of biliary infection, particularly those who also have complications related to portal hypertension, may require liver transplantation (22). In the present case, it was difficult to choose surgical intervention because of the patient's old age and general condition. The prognosis is variable depending upon the severity of disease and the presence of coexisting renal dysfunction. Recurrent infections and other complications related to biliary lithiasis can be associated with significant morbidity.
We herein experienced the rare case of an ADPKD patient with Caroli's disease. In conclusion, when we encounter intractable bacterial infection in ADPKD patients, we have to consider cholangitis related to abnormalities of hepatobiliary systems, such as Caroli's disease, as well as cyst infection.
The authors state that they have no Conflict of Interest (COI).
References
- 1.Comfort MW, Gary HK, Dahlin DC, Whitesell FB Jr. Polycystic disease of the liver: a study of 24 cases. Gastroenterology 20: 60-78, 1952. [PubMed] [Google Scholar]
- 2.Caroli J, Soupault R, Kossakowski J, Plocker L, Paradowska M. La dilatation polykystique congénitale des voies bilaires intra-hepatiques. Essai de classification. Sem Hop Paris 34: 128-135, 1958. (in French). [PubMed] [Google Scholar]
- 3.Berenuger J, Olaso V, Rayón M, et al. Dilatacion congenital no obstructive de los condoctos biliares intrahepaticos segmentarios (enfermedad de Caroli) Presentacion de una observacion y revision de la literature. Rev Clin Esp 140: 567-577, 1976. (in Spanish). [PubMed] [Google Scholar]
- 4.Jordon D, Harpaz N, Thung S. Caroli's disease and adult polycystic kidney disease: a rarely recognized association. Liver 9: 30-35, 1989. [DOI] [PubMed] [Google Scholar]
- 5.Mousson C, Rabec M, Cercueil JP, Virot JS, Hillon P, Rifle G. Caroli's disease and autosomal dominant polycystic kidney disease: a rare association? Nephrol Dial Transplant 12: 1481-1483, 1997. [DOI] [PubMed] [Google Scholar]
- 6.Shedda S, Robertson A. Caroli's syndrome and adult polycystic kidney disease. ANZ J Surg 77: 292-294, 2007. [DOI] [PubMed] [Google Scholar]
- 7.Schwab SJ, Bander SJ, Klahr S. Renal infection in autosomal dominant polycystic kidney disease. Am J Med 82: 714-718, 1987. [DOI] [PubMed] [Google Scholar]
- 8.Takei R, Ubara Y, Hoshino J, et al. Percutaneous transcatheter hepatic artery embolization for liver cysts in autosomal dominant polycystic kidney disease. Am J Kidney Dis 49: 744-752, 2007. [DOI] [PubMed] [Google Scholar]
- 9.Suwabe T, Ubara Y, Sumida K, et al. Clinical features of cyst infection and hemorrhage in ADPKD: new diagnostic criteria. Clin Exp Nephrol 16: 892-902, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Choi BI, Yeon KM, Kim SH, Han MC. Caroli disease: central dot sign in CT. Radiology 174: 161-163, 1990. [DOI] [PubMed] [Google Scholar]
- 11.Desmet VJ. Congenital diseases of intrahepatic bile ducts: variations on the theme “ductal plate malformation”. Hepatology 16: 1069-1083, 1992. [DOI] [PubMed] [Google Scholar]
- 12.Brancatelli G, Federle M, Vilgrain V, Vullierme MP, Marin D, Lagalla R. Fibropolycystic liver disease: CT and MR Imaging findings. Radiographics 25: 659-670, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Venkatanarasimha N, Thomas R, Armstrong EM, Shirly JF, Fox BM, Jackson SA. Imaging features of ductal plate malformations in adults. Clin Radiol 66: 1086-1093, 2011. [DOI] [PubMed] [Google Scholar]
- 14.Ward CJ, Hogan MC, Rossetti S, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet 30: 259-269, 2002. [DOI] [PubMed] [Google Scholar]
- 15.Hildebrandt F. Genetic kidney diseases. Lancet 375: 1287-1295, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dranssart M, Cognet F, Mousson C, Cercueil JP, Rifle G, Krause D. MR cholangiography in the evaluation of hepatic and biliary abnormalities in autosomal dominant polycystic kidney disease: study of 93 patients. J Comput Assist Tomogr 26: 237-242, 2002. [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa I, Chikamoto E, Nakamura M, Asaka M, Tomosugi N, Yuri T. High incidence of common bile duct dilatation in autosomal dominant polycystic kidney disease patients. Am J Kidney Dis 27: 321-326, 1996. [DOI] [PubMed] [Google Scholar]
- 18.Terada T, Nakamura Y. Congenital biliary dilatation in autosomal dominant adult polycystic disease of the liver and kidneys. Arch Pathol Lab Med 112: 1113-1116, 1988. [PubMed] [Google Scholar]
- 19.Bergmann C. ARPKD and manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr Nephrol 30: 15-30, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taylor AC, Palmer KR. Caroli's disease: 1977-1995 experiences. Eur J Gastroenterol Hepatol 10: 105-108, 1998.9581983 [Google Scholar]
- 21.Kassahun WT, Kahn T, Wittekind C, et al. Caroli's disease: liver resection and liver transplantation. Experience in 33 patients. Surgery 138: 888-898, 2005. [DOI] [PubMed] [Google Scholar]
- 22.Harring TR, Nguyen NT, Liu H, Goss JA, O'Mahoney CA. Caroli disease patients have excellent survival after liver transplant. J Surg Res 177: 365-372, 2012. [DOI] [PubMed] [Google Scholar]