

Abstract
Background
Neurosarcoidosis is a rare variant of sarcoidosis and is only described in small cohort studies. We define clinical features, treatment and outcome of patients with neurosarcoidosis over the last 35 years.
Methods
We performed a systematic review and meta-analysis of studies on neurosarcoidosis published between 1980 and 2016. Studies were included if they reported at least 5 cases. Studies describing one specific neurological presentation were excluded.
Results
We identified 29 articles describing 1088 patients diagnosed between 1965 and 2015. Neurosarcoidosis occurred in 5% of patients with systemic sarcoidosis. Mean age at presentation was 43 years and neurological symptoms were the first clinical manifestation of sarcoidosis in 52%. The most commonly reported feature of neurosarcoidosis was cranial neuropathy in 55%, with the facial and optic nerve most commonly affected, followed by headache in 32%. Pleiocytosis and elevated CSF protein were found in 58 and 63%. MRI of the brain showed abnormalities in 70%. Chest X-ray, chest CT, or gallium-67-scintigraphy showed findings consistent with sarcoidosis in 60%, 70% and 69%, respectively. First line therapy with corticosteroids was initiated in 434 of 539 patients (81%). Second and third line therapy was started in 27 and 9%. Outcome consisted of complete remission in 27%, incomplete remission in 32%, stable disease in 24%, deterioration in 6% and death in 5%.
Conclusion
Neurosarcoidosis has a heterogeneous clinical presentation and the diagnosis can be difficult because of low sensitivity of ancillary investigations. New treatments have emerged, but nevertheless one third of patients do not respond to treatment. Prospective cohort studies and RCTs on treatment are urgently needed.
Electronic supplementary material
The online version of this article (doi:10.1186/s12883-016-0741-x) contains supplementary material, which is available to authorized users.
Keywords: Neurosarcoidosis, Clinical neurology, Auto-immune disease, Systemic disease, Systematic review, Meta-analysis
Background
Sarcoidosis is a multisystem granulomatous inflammatory disease of unknown aetiology, that typically affects young adults [1]. The incidence varies throughout the world, but is estimated to be between 10 and 20 per 100.000 population [2]. It mainly affects lung, skin and eyes, and has been reported to involve the nervous system in 5–20% [3, 4]. Neurologic manifestations described are cranial nerve palsy, aseptic meningitis, peripheral neuropathy and myopathy [5]. Data on neurosarcoidosis are mostly derived from single-centre retrospective studies and vary considerably between studies [6]. Treatment is based on expert opinion, and no randomized controlled trials have been done comparing treatment in patients with neurosarcoidosis [7].
Diagnostic criteria for neurosarcoidosis have been proposed based on a clinical presentation suggestive of neurosarcoidosis, results of ancillary investigation and exclusion of other diagnoses [5, 8–10]. A definite diagnosis of neurosarcoidosis is met in only a minority of patients because this needs histologic confirmation of non-caseating granulomas of affected nervous system tissue. A probable diagnosis is defined as evidence of nervous system inflammation on magnetic resonance imaging (MRI) or cerebrospinal fluid (CSF; elevated protein, cells, immunoglobin G indices, or presence of oligoclonal bands) in combination with evidence of systemic sarcoidosis with histological confirmation and/or at least two of the indirect indicators consisting of fluorodeoxyglucose positron emission tomography (FDG-PET), gallium scan, chest imaging, and serum angiotensin-converting enzyme. Possible neurosarcoidosis is defined a clinical suspicion and exclusion of other diagnoses, but above mentioned criteria are not met.
We performed a systematic review and meta-analysis to determine clinical features, treatment, and outcome of patients with neurosarcoidosis over the last 35 years.
Methods
PubMed and Embase were searched using the search terms “neurosarcoidosis”, “sarcoidosis” and “nervous system”. Studies written in Dutch, English, French, German, or Spanish published between 1980 and March 2016 were considered for inclusion. Studies were included in the meta-analysis if they reported at least 5 cases of neurosarcoidosis, involving at least intracranial manifestations other than isolated hypothalamo-pituitary neurosarcoidosis. Studies were excluded if they: 1) reported only on a specific subset of neurosarcoidosis manifestations (e.g., neuro-ophthalmic or spinal cord neurosarcoidosis); or 2) were duplicate publications. Clinical features described in at least five case studies are reported. Data on study characteristics, demographic features, clinical manifestations, ancillary investigations, treatment, and outcome were systematically scored by DF and MB. Therapy was classified as first line, second line, or third line therapy. First line therapy consists of corticosteroid treatment, second line treatment consists of immunosuppressive therapy with methotrexate, azathioprine, mycophenolate mofetil, cyclosporine A, or (hydroxyl) chloroquine, and third line therapy either consists of cyclophosphamide or immunomodulatory medication (tumor necrosis factor-alpha inhibitors (TNF-alpha) or B-cell targeted therapy) [3]. Remission was defined as complete improvement, without residual symptoms. Favourable outcome was defined as remission, either complete or incomplete, and no need for alternative immunosuppressants.
We performed a pooled reanalysis of all published data in the included studies. Because of heterogeneity between studies, all data is presented as a number for which a certain characteristic is present out of the total number of patients for which it was described (n/N [%]) and a 95% confidence interval (95% CI) was used. For continuous data the standard deviation (SD) was used. Heterogeneity between studies was calculated for all reported variables using the Cochrane Q and I2 statistical tests.
Results
Our search identified 2994 articles of which 29 studies remained after screening (Fig. 1), including 1088 patients diagnosed from 1965 to 2015 [6, 8, 9, 11–36]. The number of patients per study ranged from 5 to 305 patients (median 27, interquartile range [IQR] 13–37, Additional file 1: Table S1). The individual study periods varied between 3 and 31 years (median 13 years, IQR 9–15). Only one of 29 studies was performed prospectively (3%); [6] 24 of 29 (83%) were single-centre studies [6, 9, 11–19, 21–27, 29–31, 34–36]. Inclusion criteria were described in 27 of 29 studies (93%) and were based on the Zajicek criteria in 11 studies [8, 9, 25, 27–31, 33, 35, 36]. Four studies included patients with definite or probable neurosarcoidosis according to these criteria, and seven included definite, probable, or possible neurosarcoidosis [8, 9, 25, 27–31, 33, 35, 36]. Eleven studies included biopsy-proven sarcoidosis [6, 11, 14, 16, 18, 20–23, 32, 34]. Five studies included patients with the diagnosis based on either histological evidence of non-caseating granulomas or clinical and radiologic findings consistent with the diagnosis sarcoidosis [13, 15, 17, 19, 24]. Twelve studies considered only central nervous system involvement of neurosarcoidosis [8, 9, 12, 17, 18, 24, 26–28, 30, 33, 34]. Calculation of the heterogeneity of data reported in the pooled study results showed significant heterogeneity (I 2 > 50%) occurred for 48 of 110 studied parameters (Additional file 1: Table S2 A-C).
Fig. 1.
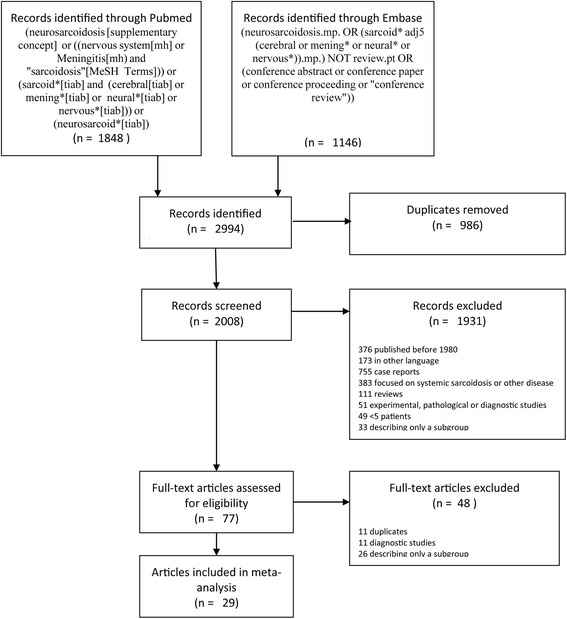
Flow chart of inclusion of studies
Patient characteristics
Neurosarcoidosis was diagnosed in 246 cases of 5263 reported cases of systemic sarcoidosis (5%, range 2–26%) [6, 13–15, 19, 22, 31]. Mean age at presentation was 43 years (range 11–84 years, SD 11.57, recalculated from 27 studies including 967 patients; Table 1). Overall, 602 of 1088 patients (55%, 95% CI 52–58%) were female. Ethnicity was reported in 739 patients of which 461 patients (62%) were Caucasian and 218 patients (29%) were of African descent. In total 652 patients were diagnosed according to Zajicek diagnostic criteria: [8, 9] 159 patients (25%) were classified as having definite neurosarcoidosis, 371 (59%) as probable neurosarcoidosis and 95 (15%) as possible neurosarcoidosis.
Table 1.
Patient Characteristics
| Characteristic | n/Na (%) | Characteristic | n/Na (%) |
|---|---|---|---|
| Age, yr., meanb(SD) | 43.1 (11.57) | Neurological symptoms | |
| Sex, male | 486/1088 (45) | Headache | 271/860 (32) |
| Ethnicity | Sensory abnormalities | 138/484 (29) | |
| Caucasian | 461/739 (62) | Hypaesthesia | 39/174 (22) |
| African | 218/739 (29) | Paraesthesia | 69/334 (21) |
| Other | 27/739 (4) | Neuropathic pain | 25/216 (12) |
| Unknown | 33/739 (4) | Gait abnormalities | 128/466 (28) |
| Presentation of disease | Visual impairment | 105/450 (23) | |
| History of sarcoidosis | 229/728 (31) | Fatigue | 47/202 (23) |
| Systemic sarcoidosis at admission | 256/440 (58) | Motor abnormalities | 103/552 (19) |
| Primary neurological presentation | 344/662 (52) | Hemiparesis | 41/454 (9) |
| Isolated neurosarcoidosis | 169/770 (22) | Paraparesis | 37/348 (11) |
| Systemic involvement | Ataxia | 64/387 (17) | |
| Pulmonary | 316/469 (67) | Vertigo | 39/277 (14) |
| Eye | 117/469 (25) | Hearing impairment | 97/716 (14) |
| Dermatological | 99/469 (21) | Seizures | 132/965 (14) |
| Rheumatological | 99/469 (21) | Nausea | 11/84 (13) |
| Otorhinolaryngeal | 43/469 (9) | Diplopia | 41/361 (11) |
| Hepatic | 37/469 (8) | Micturition abnormalities | 23/206 (11) |
| Cardial | 26/469 (6) | Dysarthria | 36/413 (9) |
| Constitutional symptoms | 106/469 (23) | Dysphagia | 44/481 (9) |
| Cranial nerve palsy | 572/1047 (55) | Psychiatric symptoms | 30/390 (8) |
| n. VII palsy | 227/937 (24) | Nystagmus | 11/172 (6) |
| n. II palsy | 190/910 (21) | Papilledema | 23/354 (6) |
| n. V palsy | 111/942 (12) | Site of neurological involvement | |
| n. VIII palsy | 102/914 (11) | Spinal cord disease | 98/543 (18) |
| n. VI palsy | 63/906 (7) | Peripheral neuropathy | 97/558 (17) |
| n. III palsy | 46/869 (5) | Polyneuropathy | 56/487 (11) |
| n. IX–X palsy | 32/882 (4) | (Multiple Mononeuropathies) | 16/137 (12) |
| n. I palsy | 17/889 (2) | Radiculopathy | 7/140 (5) |
| n. IV palsy | 17/869 (2) | Meningitis | 105/648 (16) |
| n. XI palsy | 5/882 (1) | Myopathy | 43/288 (15) |
| n. XII palsy | 12/882 (1) | Neuro-endocrinec | 90/1034 (9) |
| >1 cranial nerve involved | 206/724 (28) | Hydrocephalus | 52/570 (9) |
a n/N: number for which a certain characteristic is present out of the total number of patients for which it was described
b The mean age was recalculated from averages presented in 27 studies (967 patients)
c Panhypopituirism (n = 34), Diabetes insipidus (n = 21), amenorrhea (n = 5), hypothermia/hypotension (n = 1), galactorrhoea (n = 1), not further specified (n = 28)
Clinical characteristics
Neurological symptoms were the first clinical manifestation of sarcoidosis in 344 of 662 patients (52%; 95% CI 48–56%). Patients diagnosed with neurosarcoidosis had a known diagnosis of sarcoidosis outside the nervous system in 229 of 728 patients (31%, 95% CI 28–35%). Systemic manifestations of sarcoidosis outside the nervous system at any time during disease course occurred in 284 of 339 patients (84%; 95% CI 80–88%), consisting of pulmonary involvement in 338 of 589 patients (57%), ocular sarcoidosis in 117 of 589 patients (20%), lymphadenopathy in 105 of 589 patients (18%), dermatological manifestations in 104 of 589 patients (18%) and rheumatological manifestations in 104 out of 589 patients (18%).
The most commonly reported presenting feature of neurosarcoidosis was cranial neuropathy, which occurred in 572 of 1047 patients (55%, 95% CI 52–58%). Of all cranial nerves, the facial nerve (24%, 95% CI 21–27%) and optic nerve (21%, 95% CI 18–24%) were involved most frequently. Other common presenting features were headache (32%, 95% CI 28–35%), sensory abnormalities (29%, 95% CI 24–33%), and motor symptoms (19%; 95% CI 15–22%) consisting of hemiparesis (9%; 95% CI 6–12%) and paraparesis (11%; 95% CI 7–14%). Meningitis was reported in 105 of 648 patients (16%, 95% CI 13–19%). Spinal cord abnormalities were reported in 18% (95% CI 15–21%) and involvement of the peripheral nervous system in 17% of patients (95% CI 14–21%), mainly consisting of polyneuropathy; myopathy was reported in 43 of 288 evaluated patients (15%; 95% CI 11–9%).
Ancillary investigations
Results of CSF analysis were described in 22 studies (Table 2); a lumbar puncture was performed in the majority of patients (78%). Pleiocytosis was found in 424 of 730 patients (58%; 95% CI 54–62%), and CSF white cell counts ranged from 5 to 1571 cells per mm3. CSF total protein was elevated in 457 of 729 patients (63%, 95% CI 59–66%). The IgG-index was elevated in 36 of 89 evaluated cases (40%, 95% CI 30–51%) with CSF oligoclonal bands present in 77 of 184 patients (46%, 95% CI 35–49%). CSF angiotensin-converting enzyme (ACE) levels were elevated in 189 of 410 patients (46%, 95% CI 41–51%).
Table 2.
Ancillary investigations on presentation
| Characteristic | n/Na (%) | Characteristic | n/Na (%) |
|---|---|---|---|
| Blood chemical tests | Abnormal ancillary investigation | ||
| Serum ACE increasedb | 238/674 (35) | Chest X-ray | 277/461 (60) |
| Serum calcium increasedb | 19/222 (9) | Chest CT | 93/132 (70) |
| ESR >20 mm/h | 64/202 (32) | Gallium-67 scintigraphy | 81/123 (66) |
| Cranial CT | 83/168 (49) | ||
| Cerebrospinal fluid analysis | Cranial MRI | 283/362 (78) | |
| Lumbar puncture performed | 774/988 (78) | Parenchymal lesions | 191/378 (51) |
| White cell count (cells/mm3) | 5–1571 | Meningeal enhancement | 282/610 (46) |
| >5 cells/mm3 | 424/730 (58) | Mass lesions | 82/501 (16) |
| Protein (g/L) | 0.45–22.4 | Cranial nerve enhancement | 123/478 (26) |
| >0.45 g/Lc | 457/729 (63) | Spinal MRI | 89/185 (48) |
| Hypoglycorrhachiac | 43/312 (14) | Diagnosis | |
| Increased IgG-indexc | 36/89 (40) | Histopathological confirmationd | 455/550 (81) |
| Oligoclonal bands present | 77/184 (42) | Definite neurosarcoidosis | 159/625 (25) |
| Increased CSF ACEc | 189/410 (46) | Probable neurosarcoidosis | 371/625 (59) |
| Normal | 90/332 (27) | Possible neurosarcoidosis | 95/625 (15) |
a n/N: number for which a certain characteristic is present out of the total number of patients for which it was described
b As reported in articles. The normal values of serum calcium were not reported in studies. The normal values of serum ACE varied between > 52 IU/L and >100 IU/L
c As reported in the articles. The normal values of CSF glucose, CSF IgG index and CSF ACE varied between studies (CSF glucose between <35 mg/dL and <0.50 mg/dL, IgG index between >0.6 and >0.7 and CSF ACE between > 1U/L and 3 U/L)
d Total histopathological confirmation, either in the CNS or systemic disease
Cranial MRI showed abnormalities in 283 of 362 evaluated patients (79%, 95% CI 74–83%), consisting of parenchymal lesions in 51% (95% CI 45–56%), and contrast enhancement of the meninges in 46% (95% CI 42–50%) and of cranial nerves in 26% (95% CI 22–30%). Spine MRI showed abnormalities in 89 of 185 evaluated patients (48%; 95% CI 41–55%). Chest X-ray showed findings consistent with pulmonary or lymph node sarcoidosis in 277 of 461 patients (60%, 95% CI 56–65%) and chest CT in 93 of 132 patients (70%, 95% CI 63–78%). Gallium-67-scintigraphy findings were consistent with sarcoidosis in 96 of 140 patients (69%, 95% CI 61–76%). Whole body 18FDG-PET CT, reported in only one study, was performed in 19 of 52 patients and showed abnormalities consistent with sarcoidosis in 15 patients (78%, 95% CI 57–93%).
Serum ACE level was elevated in 238 of 674 patients (35%, 95% CI 32–39%), erythrocyte sedimentation rate was elevated in 64 of 202 patients (32%, 95% CI 25–38%), and hypercalcemia was present in 19 of 222 evaluated patients (9%, 95% CI 5–12%). A Kveim test was performed in eight studies, and was reported positive in 93 of 117 patients (79%, 95% CI 72–87%).
Treatment
The treatment strategies are reported in Table 3. No immunosuppressive treatment was indicated in 99 of 655 patients (15%, 95% CI 12–18%). First line therapy, consisting of corticosteroids, was initiated in 434 of 539 patients (81%, 95% CI 77–84%); subsequently, 24% of these patients were switched from first line to second or third line therapy. Second line therapy, consisting of methotrexate, azathioprine, (hydroxyl) chloroquine, mycophenolate mofetil, and cyclosporine A, was initiated in 144 of 539 patients (27%, 95% CI 23–31%). Third line therapy, consisting of cyclophosphamide and TNF-alpha antagonists, was initiated in 49 of 539 patients (9%, 95% CI 7–12%). Overall, 546 of 655 patients (83%, 95% CI 80–86%) were treated with corticosteroids, either as monotherapy or combination therapy with a second or third line agent; methotrexate was administered in 16% (CI 13–19%), azathioprine in 8% (95% CI 6–10%), cyclophosphamide in 6% (95% CI 4–8%), and TNF-alpha antagonists in 4% (95% CI 2–5%). The use of TNF-alpha antagonists increased over time (Fig. 2). Other reported treatments consisted of neurosurgical intervention in 30 of 230 patients (13%, 95% CI 9–17%), hormonal substitution therapy in 18 of 227 patients (8%, 95% CI 4–12%), and anti-epileptic medication in 14 of 99 patients (14%, 95% CI 7–21%).
Table 3.
Treatment and outcome
| Characteristic | n/Na (%) | Characteristic | n/Na (%) |
|---|---|---|---|
| No treatment | 99/655 (15) | Other treatment modalities | |
| First line therapy | 434/539 (81) | Neurosurgical intervention | 30/230 (13) |
| Second line therapy | 144/539 (27) | Anti-epileptic medication | 14/106 (13) |
| Third line therapy | 49/539 (9) | Hormonal substitution | 18/227 (8) |
| Overall treatment | Follow-up, yr., mean (SD) | 4.4 (SD 3.15) | |
| Corticosteroids | 546/655 (83) | Outcome | |
| Methotrexate | 105/655 (16) | Remission | 126/415 (27) |
| Azathioprine | 54/655 (8) | Improvement | 147/415 (32) |
| (Hydroxy) chloroquine | 24/655 (4) | Stable disease | 100/415 (22) |
| Mycophenolate mofetil | 12/655 (2) | Deterioration | 19/415 (4) |
| Cyclosporine A | 10/655 (2) | Mortality | 42/826 (5) |
| Cyclophosphamide | 40/655 (6) | ||
| TNF-alpha antagonists | 24/655 (4) | Favourable outcome per treatment group | |
| Treatment switches | First line therapy | 161/227 (71) | |
| First to second or third line | 88/363 (24) | Second line therapy | 47/85 (55) |
| Second to third line | 14/104 (13) | Third line therapy | 7/18 (39) |
| Between third line | 7/23 (30) | ||
a n/N: number for which a certain characteristic is present out of the total number of patients for which it was described
Fig. 2.
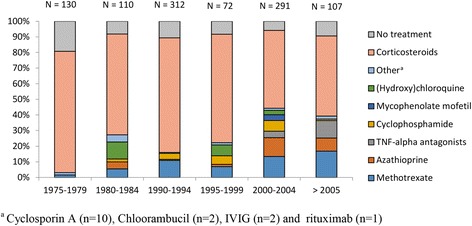
Proportion of treatment used according to timeframe
Outcome
Treatment response is reported in Table 3. The average time of follow-up was 4 years (IQR 2–7, SD 4.4). Mortality was reported in 23 of 29 studies (79%), ranging from 0 to 33%. Overall, 42 of 826 patients died (5%; 95% CI 4–7%). The temporal trend of patients with neurosarcoidosis with reported substantial improvement is noted in Fig. 3. Total remission was achieved in 126 of 465 patients (27%, 95% CI 23–31%), incomplete remission in 147 of 465 (32%, 95% CI 27–36%), stable disease in 111 of 465 (24%; 95% CI 20–28%), and deterioration in 28 of 465 patients (6%; 95% CI 4–8%). Favourable outcome was reported in 161 out of 227 patients (71%; 95% CI 65–77%) who received only first line treatment. Favourable outcome was reported in 47 of 85 patients (55%; 95% CI 45–66%) who received second line therapy and were not switched to third line therapy. Seven out of 18 patients (39%; 95% CI 16–62%) who received third line therapy had a favourable outcome.
Fig. 3.
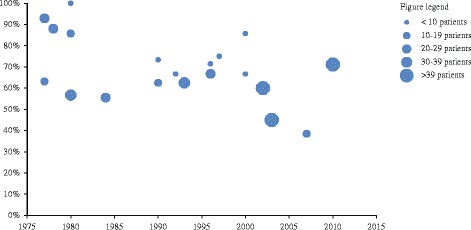
Temporal trend of patients with neurosarcoidosis with reported substantial improvement
Discussion
Our data show that neurosarcoidosis is a diverse illness, with heterogeneous clinical presentation, varying results of ancillary investigation, and considerable inter-individual differences in treatment response. Four clinical presentations could be distilled from our meta-analysis: those presenting with cranial neuropathy, spinal cord inflammation, peripheral neuropathy or myopathy, or (chronic) meningitis. Neurological symptoms were the first clinical manifestation of sarcoidosis in about half of patients, and of those known with sarcoidosis about 5% eventually develop neurosarcoidosis. Although new treatments, such as TNF-alpha antagonists, are increasingly used, the mortality rate among patients with neurosarcoidosis remains 5% and about one third does not have a substantial clinical improvement on treatment.
Our systematic review and meta-analysis has limitations. First, all but one study were retrospective, introducing selection bias. This limits the external validity of our results and may overestimate effect of treatment in this disease. Second, most studies were performed in tertiary referral centres in European countries or the USA, which is also reflected in the ethnicity of included patients, again introducing selection bias. Tertiary care patients might be more severely affected and perhaps less responsive to treatment. Third, studies used heterogeneous inclusion criteria and not all items were reported for all patients. Fourth, patients were included over a period of 30 years. Within this time period, alternative diagnoses mimicking neurosarcoidosis have been described which previously would be classified as probable or even definite neurosarcoidosis. Examples of such mimics are IgG4-related disease or the POEMS syndrome [37–39]. These rare diseases may present with similar clinical features and histopathology. It may well be that different aetiologies may exist for a disease that we now call neurosarcoidosis. Finally, when performing the meta-analyses we found there frequently was significant heterogeneity between studies on the reported data. This confirms the influence of the abovementioned biases.
Making a diagnosis of neurosarcoidosis can be difficult. A definite diagnosis was made only in one out of four patients. The far majority of patients were classified as probable neurosarcoidosis. Diagnostic classification for neurosarcoidosis includes MRI, CSF and serum inflammatory markers, FDG-PET, gallium scan, and chest imaging [10]. Our study shows that none of these diagnostic markers seems to have a good accuracy for the diagnosis. Multicentre prospective diagnostic studies in patients with suspected neurosarcoidosis are needed to test diagnostic accuracy of these tests in the diagnostic process. These studies should determine the value of markers noted in the diagnostic criteria as well as other promising new markers, such as soluble interleukin-2-receptor [35, 40].
Clear guidance for the treatment of patients with neurosarcoidosis is lacking. Most treatment strategies are extrapolated from few studies on pulmonary sarcoidosis. In our meta-analysis 83% of patients received corticosteroids, which can be considered the mainstay of treatment in neurosarcoidosis. However, 24% of the patients initially treated with first line therapy were switched to second or third line therapy. In addition, corticosteroid-associated side effects occur frequently and can be severe [8, 20]. Over time, second and third line therapy emerged into the treatment of neurosarcoidosis, most recently TNF-alpha antagonists. Value of TNF-alpha antagonists has been suggested only in a phase II randomised controlled trial (RCT) in pulmonary sarcoidosis [41]. This trial including 138 patients showed that infliximab therapy resulted in a statistically significant improvement in percentage of predicted forced vital capacity at week 24. The value of TNF-alpha antagonists is based on sporadic case series, and it is unclear if patients benefit from a combined therapy of TNF-alpha antagonists and first and second line therapy. It is also unclear for how long TNF-alpha antagonists should be continued and whether this therapy is associated with side effects. RCTs on treatments or treatment strategies in this disease are urgently needed.
Conclusions
Neurosarcoidosis develops in 5% of the sarcoidosis patients, and in more than half of these patients neurological symptoms are the primary presentation of sarcoidosis. The diagnosis is difficult due to the heterogeneous clinical presentation and low sensitivity of ancillary investigations. Despite the increasing use of second and third line medication, still one-third of patients do not improve or deteriorate. These data stress the need for prospective cohort studies and treatment trials.
Acknowledgements
We would like to thank René Spijker (Medical information specialist Dutch Cochrane Centre and Medical Library AMC) for his help with the literature search.
Funding
Matthijs Brouwer is supported by a grant from the Netherlands Organization for Health Research and Development (ZonMw; NWO-Veni grant 2012 [916.13.078]), D.v.d.B. is supported by grants from the Netherlands Organization for Health Research and Development (ZonMw; NWO-Vidi grant 2010 [016.116.358]) and the European Research Council (ERC Starting Grant 281156). This study was not industry-sponsored.
Availability of data and materials
This study included articles which are available via PubMed. All information analysed in this study was collected in a dataset and this is available from the corresponding author on reasonable request.
Authors’ contributions
D.F. designed the study, conducted the literature search, collected, analysed and interpreted the data and drafted the first manuscript. D.vd.B. interpretation of data and revision of manuscript for intellectual content. M.B. study design, literature search, analysis and interpretation of data and revision of the manuscript for intellectual content. All authors have read and approved the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- 95% CI
95% confidence interval
- ACE
Angiotensin-converting enzyme
- CSF
Cerebrospinal fluid
- CT
Computer tomography
- FDG-PET
Fludeoxyglucose-positron emission tomography
- IQR
Interquartile range
- MRI
Magnetic resonance imaging
- RCT
Randomised controlled trial
- SD
Standard deviation
- TNF-alpha
Tumor necrosis factor-alpha
Additional file
Study descriptives. This includes a summary of all studies included concerning the first author, date of publication, country of study, study design, whether it is a single or multi center study, number of included patients and inclusion period. Table S2 A-C. Heterogeneity of variables between studies. This table shows the heterogeneity between studies concerning the various variables. This includes the Q statistic and I2. Table S3. List of scored variables per study. This is a comprehensive list of all variables we scored per article. (DOC 155 kb)
Contributor Information
Daan Fritz, Email: d.fritz@amc.uva.nl.
Diederik van de Beek, Email: d.vandebeek@amc.uva.nl.
Matthijs C. Brouwer, Phone: +31205664042, Email: m.c.brouwer@amc.uva.nl
References
- 1.Iannuzzi MC, Rybicki BA, Teirstein AS. Sarcoidosis. N Engl J Med. 2007;357(21):2153–2165. doi: 10.1056/NEJMra071714. [DOI] [PubMed] [Google Scholar]
- 2.Thomas KW, Hunninghake GW. Sarcoidosis. JAMA. 2003;289(24):3300–3303. doi: 10.1001/jama.289.24.3300. [DOI] [PubMed] [Google Scholar]
- 3.Nozaki K, Judson MA. Neurosarcoidosis. Curr Treat Options Neurol. 2013;15(4):492–504. doi: 10.1007/s11940-013-0242-9. [DOI] [PubMed] [Google Scholar]
- 4.Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol. 2013;26(3):307–313. doi: 10.1097/WCO.0b013e3283608459. [DOI] [PubMed] [Google Scholar]
- 5.Hoitsma E, Faber CG, Drent M, Sharma OP. Neurosarcoidosis: a clinical dilemma. Lancet Neurol. 2004;3(7):397–407. doi: 10.1016/S1474-4422(04)00805-1. [DOI] [PubMed] [Google Scholar]
- 6.Allen RK, Sellars RE, Sandstrom PA. A prospective study of 32 patients with neurosarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 2003;20(2):118–125. [PubMed] [Google Scholar]
- 7.Agnihotri SP, Singhal T, Stern BJ, Cho TA. Neurosarcoidosis. Semin Neurol. 2014;34(4):386–394. doi: 10.1055/s-0034-1390387. [DOI] [PubMed] [Google Scholar]
- 8.Zajicek JP, Scolding NJ, Foster O, Rovaris M, Evanson J, Moseley IF, Scadding JW, Thompson EJ, Chamoun V, Miller DH, et al. Central nervous system sarcoidosis-diagnosis and management. QJM. 1999;92(2):103–117. doi: 10.1093/qjmed/92.2.103. [DOI] [PubMed] [Google Scholar]
- 9.Marangoni S, Argentiero V, Tavolato B. Neurosarcoidosis. Clinical description of 7 cases with a proposal for a new diagnostic strategy. J Neurol. 2006;253(4):488–495. doi: 10.1007/s00415-005-0043-5. [DOI] [PubMed] [Google Scholar]
- 10.Tavee JO, Stern BJ. Neurosarcoidosis. Continuum. Neurol Systemic Dis. 2014;20(3):545–559. doi: 10.1212/01.CON.0000450965.30710.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elkin R, Willcox PA. Neurosarcoidosis. A report of 5 cases. S Afr Med J. 1985;67(23):943–946. [PubMed] [Google Scholar]
- 12.Pentland B, Mitchell JD, Cull RE, Ford MJ. Central nervous system sarcoidosis. Q J Med. 1985;56(220):457–465. [PubMed] [Google Scholar]
- 13.Stern BJ, Krumholz A, Johns C, Scott P, Nissim J. Sarcoidosis and its neurological manifestations. Arch Neurol. 1985;42(9):909–917. doi: 10.1001/archneur.1985.04060080095022. [DOI] [PubMed] [Google Scholar]
- 14.Oksanen V. Neurosarcoidosis: clinical presentations and course in 50 patients. Acta Neurol Scand. 1986;73(3):283–290. doi: 10.1111/j.1600-0404.1986.tb03277.x. [DOI] [PubMed] [Google Scholar]
- 15.Chen RC, McLeod JG. Neurological complications of sarcoidosis. Clin Exp Neurol. 1989;26:99–112. [PubMed] [Google Scholar]
- 16.Chapelon C, Ziza JM, Piette JC, Levy Y, Raguin G, Wechsler B, Bitker MO, Bletry O, Laplane D, Bousser MG, et al. Neurosarcoidosis: signs, course and treatment in 35 confirmed cases. Medicine (Baltimore) 1990;69(5):261–276. doi: 10.1097/00005792-199009000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Zouaoui A, Maillard JC, Dormont D, Chiras J, Marsault C. MRI in neurosarcoidosis. J Neuroradiol. 1992;19(4):271–284. [PubMed] [Google Scholar]
- 18.Radziwill A, Weder B. Neurosarcoidosis of the central nervous system. Analysis of the long-term follow-up of 8 cases. Nervenarzt. 1995;66(12):915–922. [PubMed] [Google Scholar]
- 19.Lower EE, Broderick JP, Brott TG, Baughman RP. Diagnosis and management of neurological sarcoidosis. Arch Intern Med. 1997;157(16):1864–1868. doi: 10.1001/archinte.1997.00440370104011. [DOI] [PubMed] [Google Scholar]
- 20.Sharma OP. Neurosarcoidosis: a personal perspective based on the study of 37 patients. Chest. 1997;112(1):220–228. doi: 10.1378/chest.112.1.220. [DOI] [PubMed] [Google Scholar]
- 21.Briner VA, Muller A, Gebbers JO. Neurosarcoidosis. Schweiz Med Wochenschr. 1998;128(21):799–810. [PubMed] [Google Scholar]
- 22.Pavese P, Brion JP, Chabre O, Fauconnier J, Pasquier B. Neurologic involvement in sarcoidosis. Federation of Systemic Diseases of Grenoble. Presse Med. 1999;28(4):168–172. [PubMed] [Google Scholar]
- 23.Ferriby D, De Seze J, Stojkovic T, Hachulla E, Wallaert B, Destee A, Hatron PY, Vermersch P. Long-term follow-up of neurosarcoidosis. Neurology. 2001;57(5):927–929. doi: 10.1212/WNL.57.5.927. [DOI] [PubMed] [Google Scholar]
- 24.Heuser K, Kerty E. Neuro-ophthalmological findings in sarcoidosis. Acta Ophthalmol Scand. 2004;82(6):723–729. doi: 10.1111/j.1600-0420.2004.00348.x. [DOI] [PubMed] [Google Scholar]
- 25.Kellinghaus C, Schilling M, Ludemann P. Neurosarcoidosis: clinical experience and diagnostic pitfalls. Eur Neurol. 2004;51(2):84–88. doi: 10.1159/000076534. [DOI] [PubMed] [Google Scholar]
- 26.Karouache A, Mounach J, Aziz N, Bouraza A, Satte A, Ouhabi H, Rafiq R, Boutaleb N, Mossadaq R. A report of 9 cases of neurosarcoidosis. Rev Neurol. 2005;161(11):1091–1101. doi: 10.1016/S0035-3787(05)85176-8. [DOI] [PubMed] [Google Scholar]
- 27.Spencer TS, Campellone JV, Maldonado I, Huang N, Usmani Q, Reginato AJ. Clinical and magnetic resonance imaging manifestations of neurosarcoidosis. Semin Arthritis Rheum. 2005;34(4):649–661. doi: 10.1016/j.semarthrit.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 28.Joseph FG, Scolding NJ. Neurosarcoidosis: a study of 30 new cases. J Neurol Neurosurg Psychiatry. 2009;80(3):297–304. doi: 10.1136/jnnp.2008.151977. [DOI] [PubMed] [Google Scholar]
- 29.Pawate S, Moses H, Sriram S. Presentations and outcomes of neurosarcoidosis: a study of 54 cases. QJM. 2009;102(7):449–460. doi: 10.1093/qjmed/hcp042. [DOI] [PubMed] [Google Scholar]
- 30.Shah R, Roberson GH, Cure JK. Correlation of MR imaging findings and clinical manifestations in neurosarcoidosis. AJNR Am J Neuroradiol. 2009;30(5):953–961. doi: 10.3174/ajnr.A1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gascon-Bayarri J, Mana J, Martinez-Yelamos S, Murillo O, Rene R, Rubio F. Neurosarcoidosis: report of 30 cases and a literature survey. Eur J Intern Med. 2011;22(6):e125–e132. doi: 10.1016/j.ejim.2011.08.019. [DOI] [PubMed] [Google Scholar]
- 32.Nozaki K, Scott TF, Sohn M, Judson MA. Isolated neurosarcoidosis: case series in 2 sarcoidosis centers. Neurologist. 2012;18(6):373–377. doi: 10.1097/NRL.0b013e3182704d04. [DOI] [PubMed] [Google Scholar]
- 33.Carlson ML, White JR, Jr, Espahbodi M, Haynes DS, Driscoll CL, Aksamit AJ, Pawate S, Lane JI, Link MJ. Cranial base manifestations of neurosarcoidosis: a review of 305 patients. Otol Neurotol. 2015;36(1):156–166. doi: 10.1097/MAO.0000000000000501. [DOI] [PubMed] [Google Scholar]
- 34.Gozubatik-Celik G, Uygunoglu U, Uluduz D, Atahan E, Musellim B, Saip S, Siva A. Diagnosis and treatment in neurosarcoidosis. Noropsikiyatri Arsivi. 2015;52(1):102–106. doi: 10.5152/npa.2015.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wegener S, Linnebank M, Martin R, Valavanis A, Weller M. Clinically isolated neurosarcoidosis: a recommended diagnostic path. Eur Neurol. 2015;73(1–2):71–77. doi: 10.1159/000366199. [DOI] [PubMed] [Google Scholar]
- 36.Leonhard SE, Fritz D, Van De Beek D, Brouwer MC. Neurosarcoidosis in a tertiary referral centre: a cross-sectional cohort study. Medicine (Baltimore) 2016;95(14) doi: 10.1097/MD.0000000000003277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dispenzieri A, Buadi FK. A review of POEMS syndrome. Oncology (Williston Park) 2013;27(12):1242–1250. [PubMed] [Google Scholar]
- 38.Kamisawa T, Zen Y, Pillai S, Stone JH. IgG4-related disease. Lancet. 2015;385(9976):1460–1471. doi: 10.1016/S0140-6736(14)60720-0. [DOI] [PubMed] [Google Scholar]
- 39.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366(6):539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 40.Petereit HF, Reske D, Tumani H, Jarius S, Markus Leweke F, Woitalla D, Pfister HW, Rubbert A. Soluble CSF interleukin 2 receptor as indicator of neurosarcoidosis. J Neurol. 2010;257(11):1855–1863. doi: 10.1007/s00415-010-5623-3. [DOI] [PubMed] [Google Scholar]
- 41.Baughman RP, Drent M, Kavuru M, Judson MA, Costabel U, Du Bois R, Albera C, Brutsche M, Davis G, Donohue JF, et al. Infliximab therapy in patients with chronic sarcoidosis and pulmonary involvement. Am J Respir Crit Care Med. 2006;174(7):795–802. doi: 10.1164/rccm.200603-402OC. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study included articles which are available via PubMed. All information analysed in this study was collected in a dataset and this is available from the corresponding author on reasonable request.