

Abstract
Background
Fertility is considered an important economic trait in dairy cattle. Most studies have investigated cow fertility while bull fertility has received much less consideration. The main objective of this study was to perform a comprehensive genomic analysis in order to unravel the genomic architecture underlying sire fertility in Holstein dairy cattle. The analysis included the application of alternative genome-wide association mapping approaches and the subsequent use of diverse gene set enrichment tools.
Results
The association analyses identified at least eight genomic regions strongly associated with bull fertility. Most of these regions harbor genes, such as KAT8, CKB, TDRD9 and IGF1R, with functions related to sperm biology, including sperm development, motility and sperm-egg interaction. Moreover, the gene set analyses revealed many significant functional terms, including fertilization, sperm motility, calcium channel regulation, and SNARE proteins. Most of these terms are directly implicated in sperm physiology and male fertility.
Conclusions
This study contributes to the identification of genetic variants and biological processes underlying sire fertility. These findings can provide opportunities for improving bull fertility via marker-assisted selection.
Electronic supplementary material
The online version of this article (doi:10.1186/s12863-016-0454-6) contains supplementary material, which is available to authorized users.
Keywords: Bovine sperm, Conception rate, Gene set analysis, Whole-genome scan
Background
Improving reproductive efficiency of dairy cattle has become one of the major challenges of the dairy industry worldwide. The intense selection for production traits in the last decades has led to a decrease in fertility [1, 2]. Fertilization failure and early embryonic loss have been identified as the two main factors contributing to this decline [3, 4]. For instance, fertilization rate in high-producing dairy cows is about 75 %, and only 65 % of the fertilized eggs are considered viable at 5–6 days post-fertilization [5]. It is no surprise that conception rates are only 35–45 % [5]. Many reasons may account for this decline in reproductive performance, including physiological, nutritional, environmental, and genetic factors. In this sense, several studies have recognized that there is substantial genetic variation underlying reproductive success in dairy cattle [6, 7].
Reproduction is a very complex process that involves numerous consecutive events, including gametogenesis, fertilization, and early embryo development, that should be accomplished in a well-orchestrated manner in order to achieve a successful pregnancy. The relative importance of the parental effects on the reproductive success, i.e., maternal versus paternal contribution to the zygote, is still largely unknown [8]. Most studies in dairy cattle have focused on female fertility, while male fertility has received much less attention. It is worth noting that the service sire has a direct influence not only in the fertilization process but also on the viability of the preimplantation embryo [9, 10]. In fact, previous studies have reported that the service sire represents an important source of variation for conception rate in dairy cattle [11–13].
Both candidate gene [14–16] and whole-genome scan [17–21] approaches have attempted to identify genomic regions and individual genes responsible for the genetic variation in bull fertility. For instance, two highly conserved spermatogenesis genes, MAP1B and PPP1R11, were significantly associated with male fertility in Holsteins [16]. In addition, genetic markers in BTA2, BTA5, BTA14, and BTAX were associated with testicular development, sperm quality, and hormone levels in young Brahman and Tropical Composite bulls [19, 21]. It should be noted that these association studies detect in general only the most significant markers, and hence, the vast majority of the genetic variants contributing to the trait remains hidden. In this context, gene set or pathway-based analysis offers an alternative strategy based on evaluating modules of functionally related genes, rather than focusing only on the most significant markers [22, 23]. This approach provides unique opportunities to detect the genetic mechanisms underlying complex phenotypes. Indeed, using this pathway-based approach, we have identified some processes, such as small GTPases mediated signal transduction or calcium ion binding, that may explain part of the differences in sire fertility [24].
The main objective of this study was to unravel the genomic architecture underlying sire fertility in dairy cattle. Sire Conception Rate (SCR) was used as a measured of bull fertility. SCR is a new and more accurate phenotypic evaluation of dairy sire fertility calculated using field data. Two complementary genome-wide association approaches plus different gene set analyses were performed in order to identify genomic regions, individual genes, functional gene terms, and biological pathways associated with sire fertility. These findings can contribute to a better understanding of the genetics underlying this complex trait and may point out opportunities for improving bull fertility via selective breeding.
Methods
Phenotypic and genotypic data
The Animal Improvement Programs Laboratory of the United States Department of Agriculture (AIPL-USDA) implemented in 2008 a national phenotypic evaluation of bull fertility called Sire Conception Rate (SCR). The model that is being used in the U.S. bull fertility evaluation includes both factors related to the service sire under evaluation (including age of the bull and AI organization) and also factors (nuisance variables) associated with the cow that receives the unit of semen (including herd-year-season, cow age, parity, and milk yield) [25, 26]. The trait SCR is defined as the expected difference in conception rate of a given bull compared to the mean of all other evaluated bulls; in other words, a bull with an SCR value of +5.0 % is expected to achieve a conception rate of 37 % in a herd that normally averages 32 % and uses average SCR bulls. It is worth noting that the U.S. bull fertility evaluation, in contrast to evaluations for other traits such as production, is intended as a phenotypic rather that a genetic evaluation, because the estimates include not only genetic but also some (permanent) environmental effects.
The entire evaluation of U.S. Holstein bull fertility was used in this study. Specifically, a total of 44,449 SCR records were available from a total of 10,884 Holstein bulls. These SCR records were obtained from 23 consecutive evaluations provided to the U.S. dairy industry between August 2008 and April 2016. These 23 different SCR evaluations are available at the Council of Dairy Cattle Breeding (CDCB) website (https://www.cdcb.us/). Additional file 1 shows (A) the distribution of SCR values per evaluation and (B) the distribution of the number of SCR records per bull, i.e., total number of repeated measurements per sire evaluated. The reliabilities of the SCR records, calculated as a function of the number of breedings, were also available for the analyses.
Genotype data for 60,671 single nucleotide polymorphism (SNP) markers were available for 7447 out of the 10,884 Holstein bulls with SCR evaluation. The SNP data were kindly provided by the Cooperative Dairy DNA Repository (CDDR). Those SNP markers that mapped to the sex chromosomes, or were monomorphic, or had minor allele frequency less than 1 % were removed from our dataset. After data editing, a total of 58,029 SNP markers were retained for subsequent genomic analysis.
Statistical methods for genome-wide association mapping
The association analysis between phenotypes and genotypes using related individuals with repeated measurements can be implemented within the framework of the classical repeatability animal model,
where y is the vector of phenotypic records (SCR values), β is the vector of fixed effects included in the model, u is the vector of random animal effects, pe is the vector of random permanent environmental and non-additive effects, and e is the vector of random residual effects. The matrices X, Z, and W are the incidence matrices relating phenotypic records to fixed, animal, and permanent environmental effects, respectively. In this context, the random effects are assumed to follow a multivariate normal distribution,
where σ 2u, σ 2pe, and σ 2e are the animal additive genetic, permanent environmental, and residual variances respectively; K is a kinship matrix that can be calculated using either pedigree or genotypic information, and R is typically an identity matrix (I) or a diagonal matrix.
In this particular study, two alternative genome-wide association mapping approaches were performed: (1) single-step genomic best linear unbiased prediction (ssGBLUP) and (2) classical genome-wide association study (cGWAS) using regular single-marker regression analysis but with correction for population structure. The ssGBLUP combines all the available phenotypic, pedigree and genotypic information, and fits all the SNP simultaneously, while cGWAS typically uses only animals that have both phenotypic and genotypic data, and fits the SNP markers one at a time.
Genome-wide association mapping using ssGBLUP
The ssGBLUP method is one of a group of statistical methods that were originally developed for genomic prediction and later were extended for performing gene mapping. Indeed, ssGBLUP model is a modification of the classical BLUP model where the pedigree relationship matrix A is replaced by H which combines pedigree and genotypic information [27]. The combined pedigree-genomic relationship matrix H − 1 is calculated as follows,
where G − 11 is the inverse of the genomic relationship matrix and A − 122 is the inverse of the pedigree-based relationship matrix for genotyped animals. In this case, G 1 has dimensions 7,993 × 7,993 and it was created using the 7447 sires with both SCR and SNP data plus 546 genotyped sires with no SCR records. In addition, the A matrix (25,075 × 25,075) was calculated based on a five generation pedigree downloaded from AIPL-USDA website. The random effects were assumed multivariate normal with u ∼ N(0, H σ 2u), pe ∼ N(0, I n σ 2pe), and e ∼ N(0, Q − 1N σ 2e). Note that in this case the original kinship matrix K is replaced by H, and the residual matrix R is the inverse of a diagonal matrix Q with its elements representing the reliabilities of the SCR values. The subscripts n and N indicate the size of the matrices and represent the number of individuals with SCR records (n = 10, 884) and the total number of SCR records (N = 44, 449), respectively.
Candidate regions associated with sire fertility were identified based on the amount of genetic variance explained by 1.5 Mb window of adjacent SNPs evaluated across the entire bovine genome. Given the genomic estimated breeding values (GEBVs), the SNP effects can be estimated as ŝ = DZ′[ZDZ′]− 1 â g, where ŝ is the vector of SNP marker effects, D is a diagonal matrix of weights of SNPs, and â g is the vector of GEBVs [28]. The percentage of genetic variance explained by a given 1.5 Mb genomic region was then calculated as,
where u i is the genetic value of the i th genomic region under consideration, B is the total number of adjacent SNPs within the 1.5 Mb region, and s j is the marker effect of the j th SNP within the i th region. All the ssGBLUP calculations were performed using the BLUPF90 family of programs from Ignacy Misztal and collaborators, University of Georgia.
Genome-wide association mapping using single marker regression (cGWAS)
For the whole genome single marker regression, we extended the repeatability model as,
where X SNP is the design matrix for the SNP under study (coded as 0, 1 or 2) and β SNP is the regression coefficient or SNP effect (also known as the allele substitution effect). In this particular case, the distribution of the random effects were assumed multivariate normal with u ∼ N(0, G 2 σ 2u), pe ∼ N(0, I m σ 2pe), and e ∼ N(0, I M σ 2e). Here the original kinship matrix K is replaced by G 2 that is calculated based on the 7447 sires that had both SCR records and genotypic data. The subscripts m and M indicate the size of the identity matrices and represent the number of individuals with SCR records (n = 7,447) and the total number of SCR records (N = 32, 590) used in this particular analysis.
Note that the extended repeatability model can be written as y = Xβ + X SNP β SNP + ϵ, where ϵ ∼ N(0, V) with V = ZG 2 Z′σ 2u + WW′σ 2pe + I M σ 2e. In this scenario, the significant effect of the SNP marker can be tested using a standard Wald statistics computed from the ratio of the estimate of β SNP and its standard error. However, the application of this test across the whole genome is computationally prohibitive. Alternatively, the association of a given SNP with SCR can be evaluated in a more computationally efficient way using the following test statistic,
which approximates the Wald test, and hence, is asymptotically standard normal. Here, V o is computed as V but from a model where the term X SNP β SNP is removed, and is obtained from the model y = Xβ + X SNP β SNP + e, assuming e ∼ N(0, V oσ2e). These analyses were performed using the R package RepeatABEL [29].
Gene set analysis
The gene set analysis consists basically in three different steps [24, 30]: (i) the assignment of SNPs to genes, (ii) the assignment of genes to functional categories, and finally (iii) the association analysis between each functional category and the phenotype of interest.
The SNPs were assigned to bovine genes based on the UMD3.1 bovine genome sequence assembly [31] using the Bioconductor R package biomaRt [32, 33]. A given SNP was assigned to a particular gene if it was located within the gene or at most 15 kb either upstream or downstream the gene. An arbitrary threshold of P-value ≤ 0.01 was used to define significant SNPs (based on the results of the cGWAS); in this context, significant genes were defined as those genes that contained at least one significant SNP.
The databases Gene Ontology (GO) [34], and Medical Subject Headings (MeSH) [35, 36] were used to define functional categories of genes. The idea is that genes assigned to the same functional category can be considered as members of a group of genes that share some particular properties, typically their involvement in the same biological or molecular process.
-
The significant association of a given term with SCR was analyzed using Fisher’s exact test. The P-value of observing g significant genes in the term was calculated by
where S is the total number of significant genes associated with SCR, N is the total number of genes that were analyzed, and k is the total number of genes in the term considered [24, 37]. The GO gene set enrichment analysis was performed using the R package goseq (using method hypergeometric) [38] while the MeSH enrichment analysis was carried out using the R package meshr [39, 40]. Additionally, the semantic similarities among GO functional terms were calculated based on the GO hierarchy using the R package GOSemSim [41].
Results
Whole genome association analysis
Two complementary genome-wide association approaches, ssGBLUP and cGWAS, were performed in order to identify genomic regions and candidate genes associated with Sire Conception Rate (ĥ 2 = 0.32). These two alternative methods slightly differ in how they identify significant regions or genes associated with the phenotype of interest. On the one hand, ssGLUP allows to identify genomic regions that explain a given amount of genetic variance. On the other hand, using cGWAS, it is possible to formally evaluate the significance of the association (using a statistical test) between each genetic marker and the phenotype of interest. In our study, these two methods yielded very similar results; in fact, the spearman’s rank correlation coefficient between the SNP effects calculated with ssGLUP and cGWAS was equal to 0.943. In addition, the corresponding Manhattan plots showed similar profiles with common significant regions in BTA21 and also BTA25 (Fig. 1). Note that, as expected, ssGBLUP yields less noisy results with well-defined peaks across the entire genome.
Fig. 1.
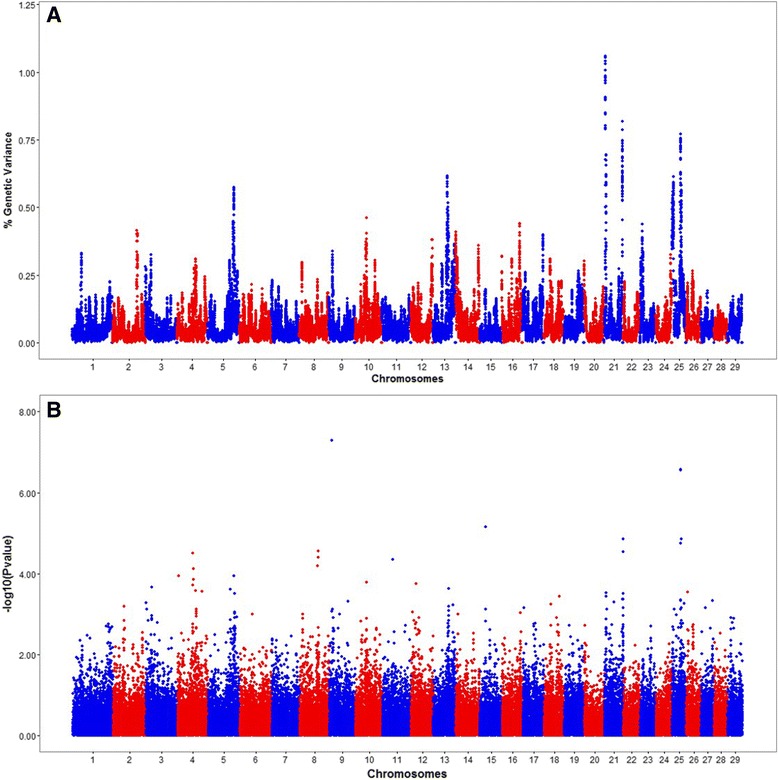
Manhattan plots showing the results of the genome-wide association mapping for Sire Conception Rate: a Percentage of genetic variance explained by 1.5 Mb SNP windows across the genome (ssGBLUP method), and b − log 10(Pvalue) for each of the genetic markers evaluated across the genome (cGWAS method)
Figure 1a displays the results obtained with ssGLUP method in terms of the proportion of genetic variance explained by 1.5 Mb SNP windows across the entire bovine genome. A total of six different genomic regions, distributed on chromosomes BTA5, BTA13, BTA21 and BTA25, explained more than 0.50 % of the genetic variance for sire conception rate. Figure 2 shows the genomic location, the percentage of genetic explained, and the list of genes located in each of these SNP windows. The region that explained the highest percentage of genetic variance (1.06 %) was located on chromosome 21 (21:8031396–9528223). Interestingly, this region harbors IGF1R, an insulin-like growth factor receptor that plays critical roles in different reproductive events, including testis development and spermatogenesis. Another SNP-window on BTA21 (21:68,846,429-70,294,301) explained also a substantial amount of genetic variance (0.82 %); this regions harbors two genes, TDRD9 and CKB, which are implicated in sperm development and sperm quality, respectively. Moreover, two different regions on BTA25 (25:3148958–4647188, and 25:26736589–28233820) explained together almost 1.50 % of the genetic variance. Notably, these regions harbor several putative candidate genes for bull fertility, including MGRN1 and SEPT12, which are directly involved in spermatogenesis, and CCT6A that is implicated in the fertilization process. Finally, two genomic regions on BTA5 and BTA13 were also identified; each of these windows explains roughly 0.60 % of the genetic variance. The region located on BTA5 (5:105357507–106813133) harbors two genes, PARP11 and AKAP3, that are involved in sperm maturation and motility. In addition, at least two putative genes related to male infertility, CTCFL and SPO11, are located in the middle of the region detected on BTA13 (13:58456868–59951247).
Fig. 2.

Genomic regions (1.5 Mb) that explain more than 0.50 % of the genetic variance for Sire Conception Rate: genomic location, percentage of variance explained, and list of genes. Adapted from www.ensembl.org using bovine assembly UMD 3.1
Figure 1b displays the results obtained with cGWAS in terms of − log 10(Pvalue) for each of the SNP markers evaluated across the genome. In addition, Table 1 describes in detail the six most significant SNP markers detected in this analysis (P-value ≤ 1.5 × 10− 5; q-value ≤ 0.15). The most significant SNP (BTB-01438088, P-value = 5.1 × 10− 8) is located in BTA9 in an intron of the gene RIMS1. This gene regulates synaptic vesicle exocytosis and is also involved in the regulation of voltage-gated calcium channels. Unsurprisingly, the RIMS1 allele negatively associated with conception rate is in low frequency in the population (f B = 0.038). Two SNP markers located in chromosome 25, BTA-59768-no-rs and ARS-BFGL-NGS-112660, showed remarkable associations with sire conception rate (P-value = 2.8 × 10− 7). Note that this genomic region (BTA25 26–28 Mb) was also detected using ssGLUP method. The two significant markers were highly correlated (high linkage disequilibrium), and therefore, it is very likely that they represent the same genetic signal. The marker BTA-59768-no-rs is located in an intron of the gene KAT8. This gene encodes a histone acetylase implicated in chromatin modification and gene expression regulation. Finally, like ssGBLUP, the single marker regression also detected the region in BTA21 at 68–71 Mb as significantly associated with sire fertility (P-value = 1.4 × 10− 5). The significant SNP marker ARS-BFGL-NGS-106232 is located within the gene BRF1, which encodes one the subunits of the RNA polymerase III transcription factor complex, and hence, it is directly involved in transcription initiation.
Table 1.
Most significant genetic markers associated with Sire Conception Rate (SCR)
| Marker | Chr | Position | Frequency | β ± se | P-value | q-value | Nearest gene |
|---|---|---|---|---|---|---|---|
| BTB-01438088 | 9 | 11867269 | 0.038 | −0.65 ± 0.12 | 5.1 × 10− 8 | 0.001 | RIMS1 (within) |
| BTB-01138539 | 15 | 26472899 | 0.815 | 0.26 ± 0.06 | 7.0 × 10− 6 | 0.102 | CADM1 (22 kb) |
| ARS-BFGL-NGS-106232 | 21 | 71210609 | 0.670 | 0.20 ± 0.05 | 1.4 × 10− 5 | 0.136 | BRF1 (within) |
| BTA-59768-no-rs | 25 | 27477941 | 0.266 | −0.29 ± 0.06 | 2.7 × 10− 7 | 0.005 | KAT8 (within) |
| ARS-BFGL-NGS-112660 | 25 | 27672891 | 0.266 | −0.29 ± 0.06 | 2.8 × 10− 7 | 0.005 | ITGAM (34 kb) |
| Hapmap8541-BTA-59825 | 25 | 28711626 | 0.150 | −0.30 ± 0.07 | 1.4 × 10− 5 | 0.136 | TYW1 (within) |
Gene set analysis
The whole-genome association analysis was complemented with a gene set enrichment analysis in order to detect potential functional categories and molecular mechanisms associated with sire fertility. Of the 58,029 SNP markers evaluated in the analysis, 27,066 were located within or surrounding annotated genes; this set of SNPs pointed a total of 17,259 annotated genes. A subset of 349 of these 17,259 genes had at least one SNP with P-value ≤ 0.01, and hence, were defined as significantly associated with bull fertility.
Figure 3 displays a set of GO Biological Process terms that were significantly enriched with genes associated with SCR. Noticeably, some of these terms are closely associated with male fertility, such as reproduction process (GO:0022414) and fertilization (GO:0009566). These two categories, highly related in the GO hierarchy, had four significant genes in common, namely BSP3, BSP5, SLC22A16, and ZP2, all of them directly involved in the process of spermatogenesis and subsequent ovum fecundation. Furthermore, many significant GO terms were associated with ion transport and homeostasis, including cation transport (GO:0006812), zinc II ion transport (GO:0006829), regulation of sodium ion transport (GO:0002028), zinc ion homeostasis (GO:0055069), and cellular metal ion homeostasis (GO:0006875). Moreover, terms related to developmental biology (e.g. GO:0048588), small GTPase mediated signal transduction (e.g. GO:0032482), and mRNA processing (e.g. GO:0050685) were also enriched with significant genes.
Fig. 3.
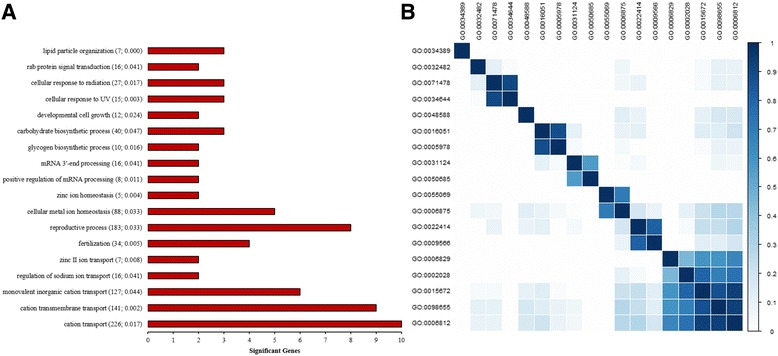
Gene Ontology Biological Process terms significantly enriched with genes associated with Sire Conception Rate: a Name, total number of genes, P-value, and total number of significant genes per functional term, and b Semantic similarity among functional terms
Several GO terms classified into the Molecular Function domain showed an overrepresentation of genes associated with sire fertility (Additional file 2). Especially, functional terms related to channel regulation [e.g., calcium channel regulator activity (GO:0005246, P-value = 0.020) and sodium channel regulator activity (GO:0017080, P-value = 0.010)], and transmembrane transporter activity [e.g., inorganic cation transmembrane transporter activity (GO:0022890, P-value = 0.009) and ion transmembrane transporter activity (GO:0015075, P-value = 0.015)] showed an overrepresentation of significant genes. Of particular interest, two closely related terms, SNARE binding (GO:0000149, P-value = 0.007) and SNAP receptor activity (GO:0005484, P-value = 0.003), which involve a group of membrane-associated proteins that participate in different reproductive events including spermatogenesis and acrosome reaction, were significantly enriched with at least three genes, STX1A, STX1B and STX8, associated with sire conception rate.
Table 2 shows a panel of MeSH terms that were enriched with genes associated with SCR. Many of these terms are closely related to male fertility, such as spermatozoa (D013094), sperm capacitation (D013075), and sperm motility (D013081). Five genes associated with SCR, namely AKAP3, BSP3, BSP5, NTRK2 and ZP2, were part of these terms. Additionally, two other terms related to fertility, follicle stimulating hormone (D005640) and pregnancy rate (D018873), were also enriched with significant genes, including AKT1, CTTNBP2NL, FSHR and IGF1R. Finally, functional categories involving protein kinases (D017868) and GTPases (D020691) were also detected as significant in the MeSH-informed enrichment analysis.
Table 2.
MeSH terms significantly enriched with genes associated with Sire Conception Rate (SCR)
| Mesh term ID | MeSH term name | No. genes | No. significant genes | P-value |
|---|---|---|---|---|
| D005640 | Follicle stimulating hormone | 34 | 4 | 6.4 × 10− 3 |
| D013075 | Sperm capacitation | 9 | 2 | 1.6 × 10− 2 |
| D013081 | Sperm motility | 13 | 4 | 1.4 × 10− 4 |
| D013094 | Spermatozoa | 71 | 5 | 2.0 × 10− 2 |
| D017868 | Cyclic AMP-dependent protein kinases | 75 | 5 | 2.5 × 10− 2 |
| D018698 | Glutamic acid | 35 | 4 | 7.1 × 10− 3 |
| D018873 | Pregnancy rate | 4 | 2 | 2.8 × 10− 3 |
| D020691 | rab GTP-binding proteins | 12 | 3 | 2.0 × 10− 3 |
Discussion
There is growing evidence that bull fertility is influenced by genetic factors. The present study was specifically performed to unravel the genomic architecture underlying sire conception rate, an accurate phenotypic measure of dairy sire fertility. Although previous studies have attempted to identify potential genes and pathways related to SCR [17, 24], this study has some unique features, including the analysis of a large dataset including almost 11 k bulls with about 45 k fertility records, the use of alternative methods for gene mapping, and the application of novel gene set tools, such as MeSH enrichment analysis.
Many methods have been proposed to detect and localize genes underlying complex traits. Given that there is no method that is clearly superior than the others, it is recommended to combine multiple approaches in order to obtain more reliable findings [42]. As such, two alternative whole genome scans were implemented in this study, including a regular single marker regression (cGWAS) and a single-step genomic prediction method (ssGLUP). It is worth noting that these two methods yielded very similar results. In particular, both approaches have identified candidate genomic regions in BTA21 and BTA25 that may be underlying the genetic variation in dairy sire fertility.
The significant region in BTA21 located at 68–71 Mb (see Figs. 1 and 2) harbors at least two candidate genes, namely CKB and TDRD9 that might be directly involved in sire fertility. Gene CKB encodes the enzyme creatine kinase, and previous studies have reported that elevated levels of creatine kinase in the sperm are associated with severe oligospermia and male infertility [43]. In fact, some researchers have proposed that creatine kinase should be used as an indicator of sperm quality and maturity in humans [44]. Similarly, gene TDRD9 encodes an helicase which plays an important role during spermatogenesis by silencing potential transposable elements, and hence, protecting the integrity of the male germline [45]. Hence, our findings provide a foundation for future studies that seek to decipher the specific roles of CKB and TDRD9 in bull fertility. No less important, the results of ssGBLUP in BTA21 at 8–9 Mb strongly suggest IFGF1 as a candidate gene for sire conception rate. This gene belongs to a family of insulin-like growth factors that has important roles in sex determination, testis development, spermatogenesis and steroidogenesis [46]. Interestingly, IGF1R has been implicated in regulating Sertoli cell proliferation and maturation, testis size, and sperm capacitation [47, 48]. Therefore, our findings provide more evidence of the association between IGF1R and male fertility.
Both ssGBLUP and cGWAS identified the region in BTA25 at 26–28 Mb as significantly associated with SCR. This region harbors at least two genes, namely KAT8 and CCT6A, with potential roles in dairy sire fertility. The gene KAT8, a member of the MYST histone acetyltransferase family, is highly expressed during sperm development [49], and it plays essential roles during early embryonic development [50]. In addition, the gene CCT6A encodes a molecular chaperone that mediates the sperm-ooctyte interaction during fertilization [51]. Moreover, the significant region detected in BTA25 but at 3–4 Mb also contains candidate genes for bull fertility, such as SEPT12 and MGRN1. Indeed, SEPT12 is expressed specifically in the testis and encodes a GTP-binding protein that has been implicated in sperm morphogenesis, sperm motility and male infertility [52, 53]. Likewise, the gene MGRN1 is widely expressed in the male reproductive system, and recent studies have shown that MGRN1 knockout in mice results in male infertility, with disruption of hormones secretion and impaired sperm motility [54]. It should be noted that this specific region in BTA25 had been already associated with sire fertility [17]. Overall, our findings provide further evidence for the presence of one or more genes that affect bull fertility in these regions of BTA25. Additional functional studies, including resequencing and fine mapping, are needed to decipher the roles that these genomic regions have in male fertility.
Given that whole-genome scans only detect the most significant regions, and these regions explain only a small fraction of the genetic variance, additional approaches are needed in order to dissect the complex genetic architecture of a quantitative trait. In the present study, different pathway-based approaches, using GO and MeSH databases, were used in order to obtain additional insights regarding the genetic determinants and biological mechanisms underlying sire fertility. Interestingly, some biological processes directly related to male fertility, such as fertilization and sperm motility, were among the most significant functional categories. Further analyses revealed that at least six genes associated with SCR, including AKAP3, BSP3, BSP5, NTRK2, SLC22A16, and ZP2, were part of these functional categories. Interestingly, the gene AKAP3 is expressed in the spermatozoa and is involved in sperm motility, sperm capacitation, and the acrosome reaction [55]. In addition, the genes BSP3 and BSP5 are two binder of sperm proteins implicated in sperm capacitation and fertilization [56]. The gene ZP2 encodes a sperm receptor that mediates gamete recognition during the fertilization [57]. These findings clearly demonstrate that gene set tools can greatly complement genome-wide association studies in order to understand the genetic basis of complex traits.
Of special interest, GO molecular function terms related to SNARE proteins showed an overrepresentation of significant genes. SNARE proteins are implicated in membrane fusion events, including several events that occur during spermatogenesis and also the acrosome reaction [58]. In fact, it was proposed that SNARE proteins are key players involved in controlling the acrosome reaction during fertilization [59]. Therefore, our findings provide further evidence regarding the active role of SNARE proteins in male fertility. On the other hand, several GO terms associated with ion transport and channel regulation also showed a significant enrichment of genes associated with SCR. It is well-documented that ion channels regulate several sperm physiological responses, including maturation, motility, and chemotaxis [60]. Interestingly, most of the significant terms were related to calcium transport and regulation, and several studies have reported that calcium is indeed implicated in the regulation of sperm motility, and it is an essential second messenger for the acrosome reaction [61]. Therefore, our findings provide further evidence of the important association between calcium and sperm physiology. More in general, note that the genetic markers located in genes initially detected in our GO or MeSH-informed enrichment analysis may facilitate the incorporation and implementation of genomic selection in commercial breeding schemes.
Conclusions
In this study, a comprehensive genomic analysis was performed with the purpose of unravelling the genetic architecture underlying sire conception rate in Holstein dairy cattle. Genomic regions in BTA5, BTA9, BTA13, BTA15, BTA21 and BTA25 were associated with sire fertility. Most of these regions harbor genes with known roles in sperm biology, including sperm maturation, motility and fertilization. Moreover, gene set analysis revealed that many of the significant terms, such as reproductive process, calcium ion channels, and SNARE proteins, are implicated in biological processes related to male fertility. Overall, this integrative study sheds light on the genetic variants and mechanisms underlying this complex phenotype in cattle. In addition, these findings can provide opportunities for improving bull fertility via marker-assisted selection.
Acknowledgements
The authors thank the Cooperative Dairy DNA Repository for providing the genotypic data.
Funding
This research was supported by the Florida Agricultural Experiment Station and the Department of Animal Sciences, University of Florida.
Availability of data and materials
The phenotypic data are available at the website of the Council of Dairy Cattle Breeding (https://www.cdcb.us/). Moreover, the genotypic data are available upon reasonable request to the Cooperative Dairy DNA Repository (Columbia, MO).
Authors’ contributions
FP conceived and designed the study. YH and FP performed the experiments and analyzed the data. YH and FP wrote the manuscript. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- GEBV
Genomic estimated breeding value
- GO
Gene Ontology
- GWAS
Genome-wide association studies
- MeSH
Medical Subject Headings
- SCR
Sire Conception Rate
- SNP
Single nucleotide polymorphism
- ssGLUP
Single-step genomic best linear unbiased prediction
Additional files
{kind=link}
Descriptive statistics for Sire Conception Rate (SCR): (A) Distribution of SCR values per evaluation, and (B) Distribution of the total number of SCR records per bull (number of repeated measurements). (JPG 135 kb)
Gene Ontology Molecular Function terms significantly enriched with genes associated with Sire Conception Rate. (DOCX 23 kb)
Contributor Information
Yi Han, Email: yhan2014@ufl.edu.
Francisco Peñagaricano, Phone: (352) 392-1981, Email: fpenagaricano@ufl.edu.
References
- 1.Lucy MC. Reproductive loss in high-producing dairy cattle: where will it end? J Dairy Sci. 2001;84(6):1277–93. doi: 10.3168/jds.S0022-0302(01)70158-0. [DOI] [PubMed] [Google Scholar]
- 2.Royal MD, Darwash AO, Flint APE, Webb R, Woolliams J, Lamming GE. Declining fertility in dairy cattle: changes in traditional and endocrine parameters of fertility. Anim Sci. 2000;70:487–501. doi: 10.1017/S1357729800051845. [DOI] [Google Scholar]
- 3.Morris D, Diskin M. Effect of progesterone on embryo survival. Animal. 2008;2(8):1112–9. doi: 10.1017/S1751731108002474. [DOI] [PubMed] [Google Scholar]
- 4.Diskin MG, Parr MH, Morris DG. Embryo death in cattle: an update. Reprod Fertil Dev. 2012;24(1):244–51. doi: 10.1071/RD11914. [DOI] [PubMed] [Google Scholar]
- 5.Santos JEP, Thatcher WW, Chebel RC, Cerri RLA, Galvao KN. The effect of embryonic death rates in cattle on the efficacy of estrus synchronization programs. Anim Reprod Sci. 2004;82–3:513–35. doi: 10.1016/j.anireprosci.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 6.Shook GE. Major advances in determining appropriate selection goals. J Dairy Sci. 2006;89(4):1349–61. doi: 10.3168/jds.S0022-0302(06)72202-0. [DOI] [PubMed] [Google Scholar]
- 7.Weigel KA. Prospects for improving reproductive performance through genetic selection. Anim Reprod Sci. 2006;96(3–4):323–30. doi: 10.1016/j.anireprosci.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 8.Kropp J, Penagaricano F, Salih SM, Khatib H. Invited review: genetic contributions underlying the development of preimplantation bovine embryos. J Dairy Sci. 2014;97(3):1187–201. doi: 10.3168/jds.2013-7244. [DOI] [PubMed] [Google Scholar]
- 9.Amann RP, DeJarnette JM. Impact of genomic selection of AI dairy sires on their likely utilization and methods to estimate fertility: a paradigm shift. Theriogenology. 2012;77(5):795–817. doi: 10.1016/j.theriogenology.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Stalhammar EM, Janson L, Philipsson J. Genetic studies on fertility in AI bulls. II. Environmental and genetic effects on non-return rates of young bulls. Anim Reprod Sci. 1994;34(3–4):193–207. doi: 10.1016/0378-4320(94)90016-7. [DOI] [Google Scholar]
- 11.Nagamine Y, Sasaki O. Effect of environmental factors on fertility of Holstein-Friesian cattle in Japan. Livest Sci. 2008;115(1):89–93. doi: 10.1016/j.livsci.2008.01.023. [DOI] [Google Scholar]
- 12.Jamrozik J, Fatehi J, Kistemaker GJ, Schaeffer LR. Estimates of genetic parameters for Canadian Holstein female reproduction traits. J Dairy Sci. 2005;88(6):2199–208. doi: 10.3168/jds.S0022-0302(05)72895-2. [DOI] [PubMed] [Google Scholar]
- 13.DeJarnette JM, Marshall CE, Lenz RW, Monke DR, Ayars WH, Sattler CG. Sustaining the fertility of artificially inseminated dairy cattle: the role of the artificial insemination industry. J Dairy Sci. 2004;87(Supplement):E93–104. doi: 10.3168/jds.S0022-0302(04)70065-X. [DOI] [Google Scholar]
- 14.Khatib H, Monson RL, Huang W, Khatib R, Schutzkus V, Khateeb H, Parrish JJ. Validation of in vitro fertility genes in a Holstein bull population. J Dairy Sci. 2010;93(5):2244–9. doi: 10.3168/jds.2009-2805. [DOI] [PubMed] [Google Scholar]
- 15.Lan XY, Peñagaricano F, DeJung L, Weigel KA, Khatib H. A missense mutation in the PROP1 (prophet of Pit 1) gene affects male fertility and milk production traits in the US Holstein population. J Dairy Sci. 2013;96(2):1255–7. doi: 10.3168/jds.2012-6019. [DOI] [PubMed] [Google Scholar]
- 16.Li G, Penagaricano F, Weigel KA, Zhang Y, Rosa G, Khatib H. Comparative genomics between fly, mouse, and cattle identifies genes associated with sire conception rate. J Dairy Sci. 2012;95(10):6122–9. doi: 10.3168/jds.2012-5591. [DOI] [PubMed] [Google Scholar]
- 17.Peñagaricano F, Weigel KA, Khatib H. Genome-wide association study identifies candidate markers for bull fertility in Holstein dairy cattle. Anim Genet. 2012;43:65–71. doi: 10.1111/j.1365-2052.2012.02350.x. [DOI] [PubMed] [Google Scholar]
- 18.Feugang JM, Kaya A, Page GP, Chen L, Mehta T, Hirani K, Nazareth L, Topper E, Gibbs R, Memili E. Two-stage genome-wide association study identifies integrin beta 5 as having potential role in bull fertility. BMC Genomics. 2009;10:176. doi: 10.1186/1471-2164-10-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fortes MRS, Reverter A, Hawken RJ, Bolormaa S, Lehnert SA. Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in Brahman bulls. Biol Reprod. 2012;87(3):58. doi: 10.1095/biolreprod.112.101089. [DOI] [PubMed] [Google Scholar]
- 20.Blaschek M, Kaya A, Zwald N, Memili E, Kirkpatrick BW. A whole-genome association analysis of noncompensatory fertility in Holstein bulls. J Dairy Sci. 2011;94(9):4695–9. doi: 10.3168/jds.2010-3728. [DOI] [PubMed] [Google Scholar]
- 21.Fortes MRS, Reverter A, Kelly M, McCulloch R, Lehnert SA. Genome-wide association study for inhibin, luteinizing hormone, insulin-like growth factor 1, testicular size and semen traits in bovine species. Andrology. 2013;1(4):644–50. doi: 10.1111/j.2047-2927.2013.00101.x. [DOI] [PubMed] [Google Scholar]
- 22.Chasman DI. On the utility of gene set methods in genomewide association studies of quantitative traits. Genet Epidemiol. 2008;32(7):658–68. doi: 10.1002/gepi.20334. [DOI] [PubMed] [Google Scholar]
- 23.Peng G, Luo L, Siu HC, Zhu Y, Hu PF, Hong SJ, Zhao JY, Zhou XD, Reveille JD, Jin L, et al. Gene and pathway-based second-wave analysis of genome-wide association studies. Eur J Hum Genet. 2010;18(1):111–7. doi: 10.1038/ejhg.2009.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peñagaricano F, Weigel KA, Rosa GJM, Khatib H. Inferring quantitative trait pathways associated with bull fertility from a genome-wide association study. Front Genet. 2013;3:307. doi: 10.3389/fgene.2012.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuhn MT, Hutchison JL. Prediction of dairy bull fertility from field data: Use of multiple services and identification and utilization of factors affecting bull fertility. J Dairy Sci. 2008;91(6):2481–92. doi: 10.3168/jds.2007-0743. [DOI] [PubMed] [Google Scholar]
- 26.Kuhn MT, Hutchison JL, Norman HD. Modeling nuisance variables for prediction of service sire fertility. J Dairy Sci. 2008;91(7):2823–35. doi: 10.3168/jds.2007-0946. [DOI] [PubMed] [Google Scholar]
- 27.Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ. Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci. 2010;93(2):743–52. doi: 10.3168/jds.2009-2730. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Misztal I, Aguilar I, Legarra A, Muir WM. Genome-wide association mapping including phenotypes from relatives without genotypes. Genet Res. 2012;94(2):73–83. doi: 10.1017/S0016672312000274. [DOI] [PubMed] [Google Scholar]
- 29.Rönnegård L, McFarlane SE, Husby A, Kawakami T, Ellegren H, Qvarnström A. Increasing the power of genome wide association studies in natural populations using repeated measures – evaluation and implementation. Methods Ecol Evol. 2016;7:792–9. doi: 10.1111/2041-210X.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gambra R, Peñagaricano F, Kropp J, Khateeb K, Weigel KA, Lucey J, Khatib H. Genomic architecture of bovine k-casein and beta-lactoglobulin. J Dairy Sci. 2013;96(8):5333–43. doi: 10.3168/jds.2012-6324. [DOI] [PubMed] [Google Scholar]
- 31.Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, Hanrahan F, Pertea G, Van Tassell CP, Sonstegard TS, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics. 2005;21(16):3439–40. doi: 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
- 33.Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4(8):1184–91. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Jr, Crooker BA, Van Tassell CP, Yang J, Wang S, Matukumalli LK, et al. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary US Holstein cows. BMC Genomics. 2011;12:1. doi: 10.1186/1471-2164-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson SJ, Schopen M, Savage AG, Schulman JL, Arluk N. The MeSH translation maintenance system: structure, interface design, and implementation. Stud Health Technol Inform. 2004;107(Pt 1):67–9. [PubMed] [Google Scholar]
- 37.Abdalla EA, Peñagaricano F, Byrem TM, Weigel KA, Rosa GJM. Genome-wide association mapping and pathway analysis of leukosis incidence in a US Holstein cattle population. Anim Genet. 2016;47:395–407. doi: 10.1111/age.12438. [DOI] [PubMed] [Google Scholar]
- 38.Young M, Wakefield M, Smyth G, Oshlack A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 2010;11(2):R14. doi: 10.1186/gb-2010-11-2-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuyuzaki K, Morota G, Ishii M, Nakazato T, Miyazaki S, Nikaido I. MeSH ORA framework: R/Bioconductor packages to support MeSH over-representation analysis. BMC Bioinformatics. 2015;16(1):1–17. doi: 10.1186/s12859-015-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morota G, Peñagaricano F, Petersen JL, Ciobanu DC, Tsuyuzaki K, Nikaido I. An application of MeSH enrichment analysis in livestock. Anim Genet. 2015;46(4):381–7. doi: 10.1111/age.12307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu G, Li F, Qin Y, Bo X, Wu Y, Wang S. GOSemSim: an R package for measuring semantic similarity among GO terms and gene products. Bioinformatics. 2010;26(7):976–8. doi: 10.1093/bioinformatics/btq064. [DOI] [PubMed] [Google Scholar]
- 42.Legarra A, Croiseau P, Sanchez MP, Teyssedre S, Salle G, Allais S, Fritz S, Moreno CR, Ricard A, Elsen JM. A comparison of methods for whole-genome QTL mapping using dense markers in four livestock species. Genet Sel Evol. 2015;47:6. doi: 10.1186/s12711-015-0087-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gergely A, Szollosi J, Falkai G, Resch B, Kovacs L, Huszar G. Sperm creatine kinase activity in normospermic and oligozospermic Hungarian men. J Assist Reprod Genet. 1999;16(1):35–40. doi: 10.1023/A:1022545612784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hallak J, Sharma RK, Pasqualotto FF, Ranganathan P, Thomas AJ, Agarwal A. Creatine kinase as an indicator of sperm quality and maturity in men with oligospermia. Urology. 2001;58(3):446–51. doi: 10.1016/S0090-4295(01)01224-9. [DOI] [PubMed] [Google Scholar]
- 45.Shoji M, Tanaka T, Hosokawa M, Reuter M, Stark A, Kato Y, Kondoh G, Okawa K, Chujo T, Suzuki T, et al. The TDRD9-MIWI2 complex is essential for piRNA-mediated retrotransposon silencing in the mouse male germline. Dev Cell. 2009;17(6):775–87. doi: 10.1016/j.devcel.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 46.Griffeth RJ, Bianda V, Nef S. The emerging role of insulin-like growth factors in testis development and function. Basic Clin Androl. 2014;24:12. doi: 10.1186/2051-4190-24-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pitetti J-L, Calvel P, Zimmermann C, Conne B, Papaioannou MD, Aubry F, Cederroth CR, Urner F, Fumel B, Crausaz M, et al. An essential role for insulin and IGF1 receptors in regulating sertoli cell proliferation, testis size, and FSH action in mice. Mol Endocrinol. 2013;27(5):814–27. doi: 10.1210/me.2012-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, Qi L, Huang S, Zhou T, Guo Y, Wang G, Guo X, Zhou Z, Sha J. Quantitative phosphoproteomics analysis reveals a key role of insulin growth factor 1 receptor (IGF1R) tyrosine kinase in human sperm capacitation. Mol Cell Proteomics. 2015;14(4):1104–12. doi: 10.1074/mcp.M114.045468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas T, Loveland KL, Voss AK. The genes coding for the MYST family histone acetyltransferases, Tip60 and Mof, are expressed at high levels during sperm development. Gene Expr Patterns. 2007;7(6):657–65. doi: 10.1016/j.modgep.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 50.Thomas T, Dixon MP, Kueh AJ, Voss AK. Mof (MYST1 or KAT8) is essential for progression of embryonic development past the blastocyst stage and required for normal chromatin architecture. Mol Cell Biol. 2008;28(16):5093–105. doi: 10.1128/MCB.02202-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dun MD, Smith ND, Baker MA, Lin M, Aitken RJ, Nixon B. The chaperonin containing TCP1 complex (CCT/TRiC) is involved in mediating sperm-oocyte interaction. J Biol Chem. 2011;286(42):36875–87. doi: 10.1074/jbc.M110.188888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuo Y-C, Shen Y-R, Chen H-I, Lin Y-H, Wang Y-Y, Chen Y-R, Wang C-Y, Kuo P-L. SEPT12 orchestrates the formation of mammalian sperm annulus by organizing core octameric complexes with other SEPT proteins. J Cell Sci. 2015;128(5):923–34. doi: 10.1242/jcs.158998. [DOI] [PubMed] [Google Scholar]
- 53.Lin Y-H, Lin Y-M, Wang Y-Y, Yu IS, Lin Y-W, Wang Y-H, Wu C-M, Pan H-A, Chao S-C, Yen PH, et al. The expression level of septin12 is critical for spermiogenesis. Am J Pathol. 2009;174(5):1857–68. doi: 10.2353/ajpath.2009.080955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cheng D, Xiong C, Li J, Sui C, Wang S, Li H, Jiang X. The effect of mahogunin gene mutant on reproduction in male mice: a new sight for infertility? Andrologia. 2014;46(2):98–105. doi: 10.1111/and.12050. [DOI] [PubMed] [Google Scholar]
- 55.Ficarro S, Chertihin O, Westbrook VA, White F, Jayes F, Kalab P, Marto JA, Shabanowitz J, Herr JC, Hunt DF, et al. Phosphoproteome analysis of capacitated human sperm - evidence of tyrosine phosphorylation of a kinase-anchoring protein 3 and valosin-containing protein/p97 during capacitation. J Biol Chem. 2003;278(13):11579–89. doi: 10.1074/jbc.M202325200. [DOI] [PubMed] [Google Scholar]
- 56.Hung PH, Suarez SS. Alterations to the bull sperm surface proteins that bind sperm to oviductal epithelium. Biol Reprod. 2012;87(4):88. doi: 10.1095/biolreprod.112.099721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Avella MA, Baibakov B, Dean J. A single domain of the ZP2 zona pellucida protein mediates gamete recognition in mice and humans. J Cell Biol. 2014;205(6):801–9. doi: 10.1083/jcb.201404025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gamboa S, Ramalho-Santos J. SNARE proteins and caveolin-1 in stallion spermatozoa: possible implications for fertility. Theriogenology. 2005;64(2):275–91. doi: 10.1016/j.theriogenology.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 59.Ramalho-Santos J, Schatten G, Moreno RD. Control of membrane fusion during spermiogenesis and the acrosome reaction. Biol Reprod. 2002;67(4):1043–51. doi: 10.1095/biolreprod67.4.1043. [DOI] [PubMed] [Google Scholar]
- 60.Lishko PV, Kirichok Y, Ren DJ, Navarro B, Chung JJ, Clapham DE. The control of male fertility by spermatozoan ion channels. In: Julius D, Clapham DE, editors. Annual review of physiology. 2012. pp. 453–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Darszon A, Nishigaki T, Beltran C, Trevino CL. Calcium channels in the development, maturation, and function of spermatozoa. Physiol Rev. 2011;91(4):1305–55. doi: 10.1152/physrev.00028.2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The phenotypic data are available at the website of the Council of Dairy Cattle Breeding (https://www.cdcb.us/). Moreover, the genotypic data are available upon reasonable request to the Cooperative Dairy DNA Repository (Columbia, MO).