

Abstract
Background
Sulfate-reducing bacteria (SRB) are key players of the carbon- and sulfur-cycles in the sediments of the world’s oceans. Habitat relevant SRBs are often members of the Desulfosarcina-Desulfococcus clade belonging to the deltaproteobacterial family of Desulfobacteraceae. Despite this environmental recognition, their molecular (genome-based) physiology and their potential to contribute to organic carbon mineralization as well as to adapt to changing environmental conditions have been scarcely investigated. A metabolically versatile representative of this family is Desulfococcus multivorans that is able to completely oxidize (to CO2) a variety of organic acids, including fatty acids up to C14, as well as aromatic compounds.
Results
In this study the complete 4.46 Mbp and manually annotated genome of metabolically versatile Desulfococcus multivorans DSM 2059 is presented with particular emphasis on a proteomics-driven metabolic reconstruction. Proteomic profiling covered 17 substrate adaptation conditions (6 aromatic and 11 aliphatic compounds) and comprised 2D DIGE, shotgun proteomics and analysis of the membrane protein-enriched fractions. This comprehensive proteogenomic dataset allowed for reconstructing a metabolic network of degradation pathways and energy metabolism that consists of 170 proteins (154 detected; ~91 % coverage). Peripheral degradation routes feed via central benzoyl-CoA, (modified) β-oxidation or methylmalonyl-CoA pathways into the Wood-Ljungdahl pathway for complete oxidation of acetyl-CoA to CO2. Dissimilatory sulfate reduction is fueled by a complex electron transfer network composed of cytoplasmic components (e.g., electron transfer flavoproteins) and diverse membrane redox complexes (Dsr, Qmo, Hmc, Tmc, Qrc, Nuo and Rnf). Overall, a high degree of substrate-specific formation of catabolic enzymes was observed, while most complexes involved in electron transfer appeared to be constitutively formed.
Conclusions
A highly dynamic genome structure in combination with substrate-specifically formed catabolic subproteomes and a constitutive subproteome for energy metabolism and electron transfer appears to be a common trait of Desulfobacteraceae members.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3236-7) contains supplementary material, which is available to authorized users.
Keywords: Genome, Proteome, Metabolic reconstruction, Degradation pathways, Energy metabolism, Membrane proteins, Sulfate-reducing bacteria, Desulfococcus multivorans, Desulfobacteraceae
Background
The decomposition of organic matter by sulfate-reducing bacteria (SRB) in anoxic (O2-depleted) marine sediments plays a major role in the global cycles of carbon and sulfur. This process accounts for more than 50 % of the total organic carbon (Corg) mineralization in marine shelf sediments [1, 2] which are characterized by shallow waters and highest Corg input. The observed high process rates were early on proposed to demand complete substrate oxidation to CO2 [3]. The intensively studied deltaproteobacterial Desulfovibrio spp., however, oxidize organic substrates only incompletely to acetyl-CoA and possess only a rather limited substrate range and may, therefore, not be responsible for these rates. In contrast, members of the likewise deltaproteobacterial family Desulfobacteraceae are capable of complete oxidation and are nutritionally versatile [4]. Their substrate spectra range from readily degradable simple fermentation endproducts via long-chain fatty acids to more challenging molecules such as aromatic compounds and hydrocarbons [4]. Biogeographic investigations of various marine sediments revealed members of the Desulfosarcina-Desulfococcus clade (DSS) within Desulfobacteraceae to dominate the SRB community [5, 6]. Members of the family Desulfobacteraceae have long been known to dominate bacterial populations in marine shelf sediments (e.g., [7–10]) and were recently also detected in a sedimental sulfate methane transition zone [11] as well as an oxygen minimum zone off the coast of Namibia [12]. Next to their ecophysiological relevance for the biogeochemistry of marine environments, interest in SRB also arises from their long evolutionary history and their energy metabolism operating at the thermodynamic limit [13]. The first members of the Desulfobacteraceae to have their genomes sequenced are facultatively chemolithoautotrophic Desulfobacterium autotrophicum HRM2 [14], aromatic compound degradation specialist Desulfobacula toluolica Tol2 [15] and the two n-alkane degraders Desulfococcus oleovorans Hxd3 (unpublished) and Desulfatibacillum alkenivorans AK-01 [16]. Studies on the differential proteomic level have been performed with D. autotrophicum HRM2 [17, 18] and D. toluolica Tol2 [15].
The present study extends our current knowledge on Desulfobacteraceae by reporting the first complete genome of a Desulfosarcina-Desulfococcus clade member, the nutritionally versatile Desulfococcus multivorans (Table 1). Moreover, we advance the genome-based metabolic reconstruction of D. multivorans by differential proteomic analysis of cells adapted to 17 different substrate conditions.
Table 1.
Properties of genome-sequenced representatives of completely oxidizing SRB
| Members of Desulfobacteraceae | ||||||
|---|---|---|---|---|---|---|
| Property | Desulfococcus multivorans 1be1 | Desulfococcus biacutus KMRActS | Desulfococcus oleovorans Hxd3 | Desulfatibacillum alkenivorans AK-01 | Desulfobacula toluolica Tol2 | Desulfobacterium autotrophicum HRM2 |
| Physiological characteristics | ||||||
| Organic substrates | ||||||
| Aliphatic hydrocarbons | - | - | C12 - C20 | C13 - C18 | - | - |
| Aromatic hydrocarbons | - | - | - | - | 1 | - |
| Fatty acids | < C14 a, b | < C7 a | C4 - C18 | < C16 | < C4 | < C16 a |
| Other aliphatic compounds | 6 | 8 | - | 3 c | 8 | 8 |
| Polar aromatic compounds | 7 | - | - | - | 7 | - |
| Electron acceptors | ||||||
| Sulfate | + | + | + | + | + | + |
| Sulfite | + | + | + | + | + | - |
| Thiosulfate | + | + | + | + | + | + |
| Autotrophy | - | - | - | + | - | + |
| Syntrophy | - | n.d. | n.d. | + | n.d. | n.d. |
| Genome features | ||||||
| Size (bp) | 4 455 399 | 5 242 029 | 3 944 167 | 6 517 073 | 5 197 905 | 5 589 073 |
| Sequencing status | complete | completed | complete | complete | complete | complete |
| G + C content (mol%) | 57 | 58 | 57 | 56 | 42 | 49 |
| rRNA operons | 3 | 1 | 1 | 2 | 4 | 6 |
| tRNAs | 54 | 51 | 47 | 56 | 48 | 50 |
| Coding sequences (CDS) | 3942 | 4708 | 3267 | 5296 | 4375 | 4943 |
| Coding (%) | 87 | 89 | 88 | 87 | 87 | 88 |
| Average size (bp) | 985 | 987 | 1066 | 1080 | 1039 | 1017 |
| Assigned function | 2648 | 3520 | 2602 | 4053 | 3188 | 3477 |
| Conserved unknown | 689 | 1162e | 665e | 1243e | 681 | 1466e |
| Unknown | 605 | 512 | ||||
| Plasmids | - | - | - | - | - | 1 |
| Phage | - | n.d. | - | - | 1 | 1 |
| CRISPR | 6 | 8 | 2 | 1 | 3 | 1 |
| Accession number | CP015381 | 125602g | CP000859 | CP001322 | FO203503 | CP001087 |
| References | ([31]; this study) | ([75, 76]; IMGf) | ([77]; IMGf) | [16, 78] | [15, 79] | [14, 80] |
n.d., no data available
aalso branched-chain fatty acids
balso cyclohexane carboxylate
cpoorly utilized
ddraft sequence
ewithout predicted function (https://img.jgi.doe.gov/)
f IMG, https://img.jgi.doe.gov/
gNCBI taxon ID
Results and discussion
General genome features
The complete genome of D. multivorans consists of a single 4,455,399 bp circular chromosome containing 3,942 ORFs with an average length of 985 bp. The genome size of D. multivorans lies in between those of other SRBs such as Desulfotalea psychrophila (3.5 Mbp) [19], Desulfovibrio vulgaris Hildenborough (3.6 Mbp) [20], and closely related Desulfococcus oleovorans strain Hxd3 (3.9 Mbp; GenBank accession: CP000859) on the lower side, and those of Desulfobacterium autotrophicum HRM2 (5.6 Mbp) [14] and Desulfobacula toluolica Tol2 (5.2 Mbp) [15] on the upper side. General overviews of genomic features of D. multivorans are illustrated in Fig. 1 and compared to other genome-sequenced members of the Desulfobacteraceae in Table 1.
Fig. 1.
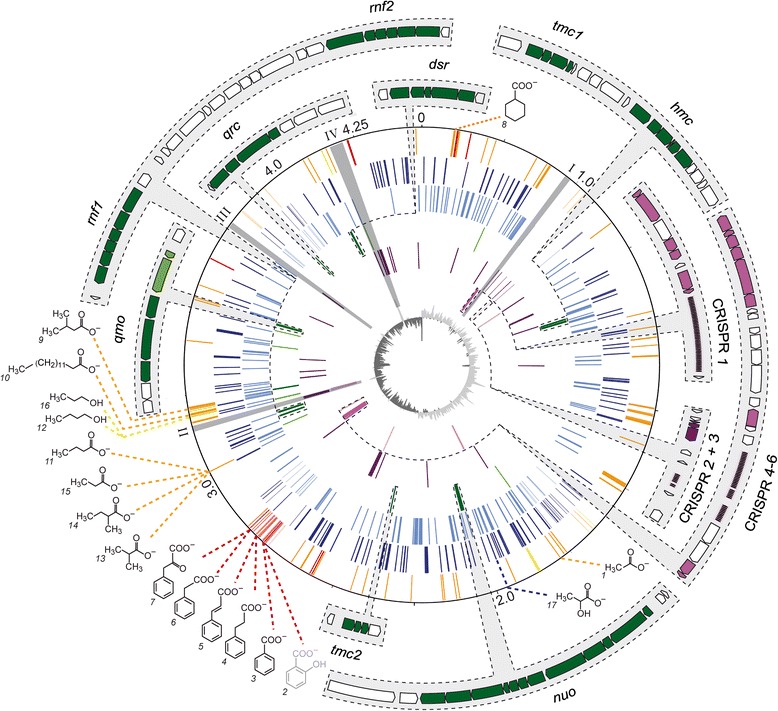
Structural representation of the chromosome of Desulfococcus multivorans. Location of gene clusters for aromatic and aliphatic compound degradation pathways are indicated by the corresponding chemical structure, with the respective names described in legend to Fig. 2 (grey: genome prediction; black: genome prediction confirmed by proteomics). The chromosomal location of redox complexes (dark green) and CRISPR-related and CRISPR-associated proteins (pink) are enlarged in the peripheral panel. Genomic islands I-IV are indicated by grey shading. Selected gene categories are represented by circles (from outside to inside): (i) aromatic compound degradation (red), fatty acid metabolism (orange) and alcohol dehydrogenases (yellow); (ii) other carbon metabolic functions (dark blue); (iii) signal transduction and transcriptional regulation (light blue); (iv) membrane-associated redox complexes (dark green) and sulfate reduction (light green); (v) transposases and mobile elements (purple); (vi) CRISPR-related and CRISPR-associated proteins (pink) and phage-related (light pink); (vii) GC skew (dark grey, below average; light grey, above average). The scale (Mbp) is indicated by the outer rim
Three rRNA operons were detected, all of which contain genes for tRNAIle and tRNAAla located between the 16S and 23S rRNA genes. Overall, genes of 54 tRNAs for the regular proteinogenic amino acids were identified in the genome. Additionally, genes (selA, selB and selD, respectively) for L-seryl-tRNA selenium transferase, Sec-specific elongation factor and selenophosphate synthase were identified. Five genes harbour the selenocysteine codon UGA upstream of a selenocysteine insertion sequence (SECIS) instructing the ribosome to recognize UGA as Sec codon [21]: heterodisulfide reductase subunit A encoding dmul_C02770/80, the δ-subunit of methyl-viologen-reducing hydrogenase (mvhD) encoding dmul_C28840/50, the α-subunit of formate dehydrogenase (cytoplasmic) encoding dmul_C16320/30, selenocysteine-containing peroxiredoxin encoding dmul_C20060/70, and BamE encoding dmul_C24500/10.
In total, 45 genes related to mobile genetic elements and transposases were discovered in the genome, 11 of which locate in four regions (I, 0.47–0.49 Mbp; II, 3.14–3.16 Mbp; III, 3.78–3.81 Mbp; IV, 4.17–4.21 Mbp) predicted to represent genomic islands (indicated by grey shading in Fig. 1). Most ORFs (56 out of 92) within these islands could not be functionally assigned and were annotated to code for uncharacterized proteins. No prophage was detected, however, the presence of 14 genetic elements related to phage assembly (tail and capsid morphogenesis), phage genome replication and transcription regulation indicates phage impact on the genome of D. multivorans.
Six loci containing clustered regularly interspaced palindromic repeats (CRISPR) and 15 genes for CRISPR-associated proteins (Cas) were found in the genome of D. multivorans (Fig. 1; purple colored gene clusters). CRISPR loci were recently reported to be present in 40 % of bacterial genomes and > 66 % of the investigated 45 deltaproteobacterial genomes [22]. CRISPR and Cas are considered to constitute an adaptive nucleic acid-based antiviral defense mechanism affiliated to spacer-phage sequence similarity [23, 24] that provides resistance against a particular phage based on a RNA interference mechanism [25]. The CRISPR locus 1 at 0.45 Mbp is closely neighboring a DNA section predicted to represent the genomic island I and thereby resembles the structural relationship observed for other prokaryotic genomes. It contains 83 spacers and genes related to cas1 and cas2 (dmul_C04070/80) which are required for the integration of new invader-derived spacers [26], cas6 (dmul_C04010) involved in crRNA processing [27, 28], cas5 and cas7 (dmul_C04050/40) responsible for assembly and surveillance of CRISPR ribonucleoprotein complex (crRNP) as well as cas3 (dmul_C04020) mediating the degradation of DNA targets [29, 30]. The two CRISPR loci (2 and 3) at 0.48 Mbp are considerably shorter and contain 9 and 3 spacers, respectively. No cas-related genes were identified in these regions but three genes predicted to code for transposases were found in close proximity. The fourth CRISPR locus at 2.92 Mbp harbours a cluster of cas1-6 homologs (dmul_C25810-60) and a second copy of cas2 (dmul_C25940) upstream to 40 spacers and the fifth CRISPR locus with 10 spacers. The sixth CRISPR locus comprises 18 spacers and homologs of cas1-2 (dmul_C26060/70). The numerous CRISPR systems along with the highly diverse CRISPR spacer content and phage-related elements in the genome is indicative of D. multivorans’ exposure to repeated viral impact in its evolutionary history.
Concept of metabolic reconstruction by proteogenomics
The present study focused on the metabolic reconstruction of D. multivorans since it was originally described as a nutritionally versatile, completely oxidizing SRB [31]. Particular emphasis was on the anaerobic degradation of 6 aromatic (benzoate, 2-hydroxybenzoate, 3-phenylpropanoate, cinnamate, phenylacetate, phenylpyruvate) and 11 aliphatic growth substrates (acetate, 1-propanol, propanoate, n-butanol, butanoate, isobutanoate, 2-methylbutanoate, 3-methylbutanoate, lactate, myristinate and cyclohexane carboxylate) ultimately feeding into the Wood-Ljungdahl pathway for terminal oxidation to CO2 as well as on the energy metabolism associated with dissimilatory sulfate reduction. Genome-based functional predictions were verified and refined by differential proteome profiling of cells adapted to each of the 17 different substrates. To achieve optimal proteome coverage and consider subcellular localization, 2D DIGE-based profiling of soluble proteins was supplemented by shotgun proteomics and analysis of the membrane protein-enriched fractions. For overview on the proteomic data set and complementarity of the applied proteomics methods refer to Additional file 1: Figure S1. The reconstructed metabolic network is illustrated in Fig. 2, with the underlying proteomic data compiled in Figs. 3 and 4 (see also Additional file 1: Table S1), and selected gene clusters are presented in Fig. 5. The network consists of 170 proteins, 154 of which have been identified (Figs. 3 and 4). Notably, most of the degradation capacities are encoded in a concise genomic region of ~500 kbp surrounding the 3.0 Mbp position, while genes for cytoplasmic and transmembrane electron transfer are dispersed across the genome (Fig. 1). The proteomic coverage of the pathways and protein complexes predicted from the genome and shown in Fig. 2 is as follows: (i) aromatic compound degradation, 27/24 proteins predicted/identified (89 % coverage); (ii) aliphatic compound degradation, 46/39 proteins predicted/identified (85 % coverage); (iii) energy metabolism, 67/67 proteins predicted/identified (100 % coverage).
Fig. 2.
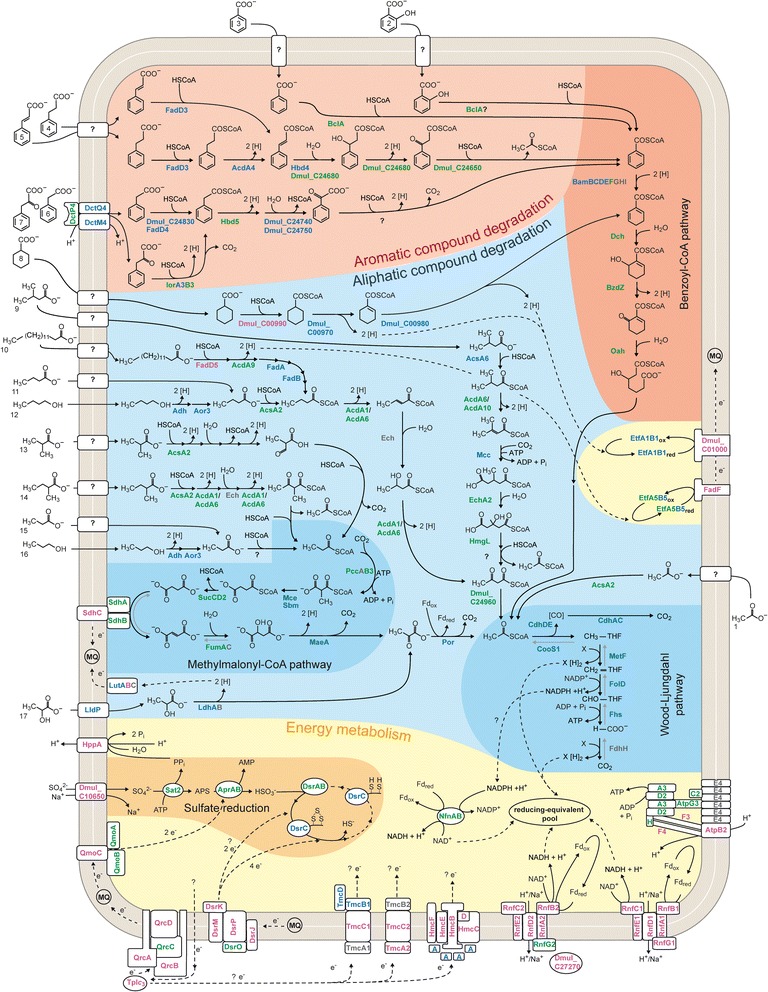
Metabolic reconstruction of Desulfococcus multivorans for 17 different growth substrates based on combined genomic and differential proteomic data. Assigned proteins are colour coded as follows: grey, genome predicted only; coloured, identified by proteomics (green, 2D DIGE; blue, shotgun analysis; pink, membrane protein-enriched fraction). In case of multiple protein identifications, only one method is indicated hierarchically from 2D DIGE via shotgun analysis down to membrane protein-enriched fraction. Enzyme names and their predicted functions are provided in Additional file 1: Table S1. Putative and assumed routes of electron flow are indicated by dashed lines. Compound names: (1) acetate, (2) 2-hydroxybenzoate, (3) benzoate, (4) 3-phenylpropanoate (hydrocinnamate), (5) cinnamate, (6) phenylacetate, (7) phenylpyruvate, (8) cyclohexane carboxylate, (9) 3-methylbutanoate, (10) myristinate, (11) butanoate, (12) n-butanol, (13) isobutanoate, (14) 2-methylbutanoate, (15) propanoate, (16) 1-propanol, and (17) lactate
Fig. 3.

Subproteomes for the degradation of aromatic and aliphatic compounds across different substrate adaptation conditions in Desulfococcus multivorans. Compound names are as detailed in legend to Fig. 2. Identified proteins are ordered according to pathways/functional categories and ascending locus tags. Fold changes in protein abundance were determined by 2D DIGE using lactate-adapted cells as reference state. In case of multiple protein identifications, only one method is indicated hierarchically from 2D DIGE (green) via shotgun analysis (blue) down to membrane protein-enriched fraction (pink). Not detected proteins are indicated in grey. Enzyme abbreviations and functional predictions are according to Additional file 1: Table S1
Fig. 4.

Proteins involved in the energy metabolism of Desulfococcus multivorans adapted to aromatic and aliphatic substrate conditions. Compound names are as detailed in legend to Fig. 2. Identified proteins are ordered according to pathways/functional categories and ascending locus tags. Fold changes in protein abundance were determined by 2D DIGE using lactate-adapted cells as reference state in all cases. In case of multiple protein identifications, only one method is indicated hierarchically from 2D DIGE (green) via shotgun analysis (blue) down to membrane protein-enriched fraction (pink). Not detected proteins are indicated in grey. Enzyme abbreviations and functional predictions are according to Additional file 1: Table S1
Fig. 5.
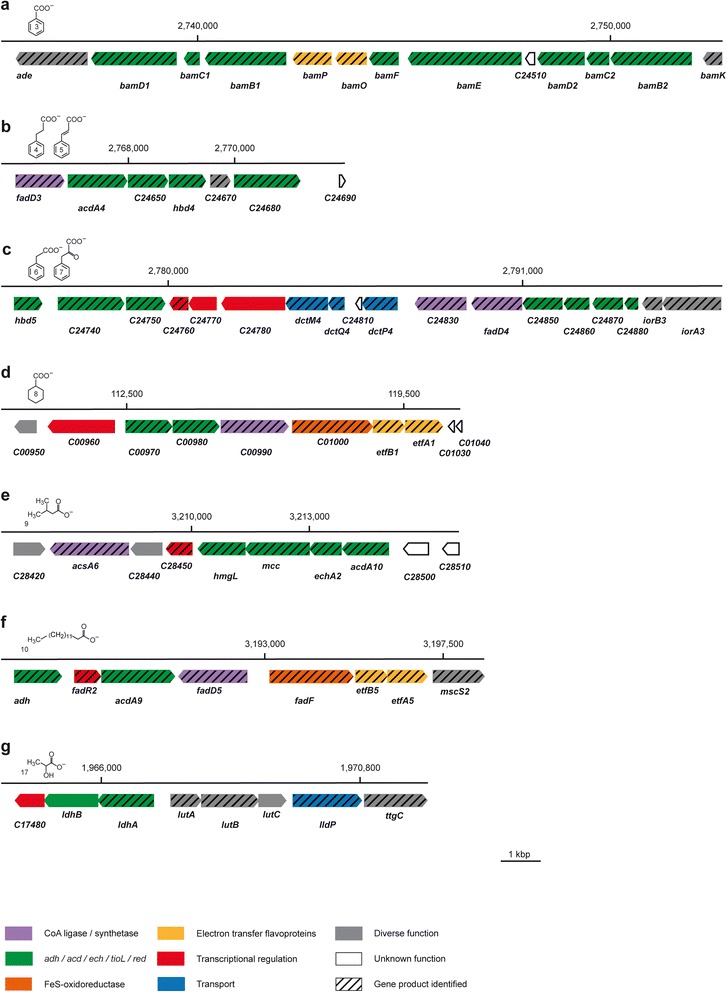
Organization of genes proposed to encode proteins involved in the catabolism of a benzoate, b 3-phenylpropanoate and cinnamate, c phenylacetate and phenylpyruvate, d cyclohexane carboxylate, e 3-methylbutanoate, f myristinate, and g lactate. Genes are color coded according their predicted function, as indicated in the insert. Locus tags without gene names are abbreviated dmul_Cxxxxx
Anaerobic degradation of aromatic compounds
Genes assigned to the conversion of 3-phenylpropanoate, cinnamate, phenylacetate and phenylpyruvate to benzoyl-CoA are framed on the upstream side by bam genes for reductive dearomatization of benzoyl-CoA and downstream by genes involved in β-oxidation and ring opening, together forming a “catabolic hotspot” on the genome (Fig. 1).
3-Phenylpropanoate and cinnamate
Anaerobic degradation of 3-phenylpropanoate (hydrocinnamate) by D. multivorans was demonstrated by Widdel [31] and resembles the pathways reported for “Aromatoleum aromaticum” EbN1 [32] and Rhodopseudomonas palustris [33]. All five enzymes proposed to be involved in 3-phenylpropanoate degradation, are encoded in an operon-like structure (Fig. 5b) and were specifically identified in cinnamate- and 3-phenylpropanoate-adapted cells of D. multivorans (Fig. 3). Initial activation of 3-phenylpropanoate (compound 4 in Fig. 2) to 3-phenylpropionyl-CoA (CoA ligase, FadD3) is followed by oxidation to 3-phenyl-2-propenoyl-CoA (acyl-CoA dehydrogenase, AcdA4). Further β-oxidation of the propenoyl-CoA sidechain yielding 3-oxo-3-phenylpropionyl-CoA is then mediated by 3-hydroxybutyryl-CoA dehydrogenase (Hbd4) and 3-hydroxyacyl-CoA dehydrogenase (Dmul_C24680). Finally, thiolytic cleavage forms acetyl-CoA and benzoyl-CoA (thiolase, Dmul_C24650).
Cinnamate (compound 5 in Fig. 2) is most likely directly CoA-activated to 3-phenyl-2-propenoyl-CoA (FadD3) prior to further degradation via the above described pathway for 3-phenylpropanoate. Contrasting the fatty acid CoA-ligase (EbA5317) in “A. aromaticum” EbN1, which is also proposed to activate p- and o-hydroxylated 3-phenylpropanoids to their corresponding CoA-thioesters, FadD3 (22 % sequence identity with EbA5317) may be restricted to non-hydroxylated substrates since growth tests revealed that D. multivorans does not utilize p-hydroxyphenylpropanoate.
Phenylacetate and phenylpyruvate
The anaerobic peripheral degradation of phenylacetate (compound 6 in Fig. 2) to benzoyl-CoA was previously described for the denitrifying betaproteobacteria Thauera aromatica K172, Azoarcus evansii [34, 35] and “A. aromaticum” EbN1 [36]. The Pad pathway demonstrated with these three strains involves initial activation to phenylacetyl-CoA (PadJ), subsequent α-oxidation forming phenylglyoxylate (PadBCD) and decarboxylation yielding the central intermediate benzoyl-CoA (PadEFGHI). Genome analysis revealed D. multivorans to only possess two genes for PadJ (dmul_C22990/23060; 54 and 35 % sequence identity with EbA5402) and two paralogs of padCD (dmul_C32580/70; 45 and 27 % sequence identity with EbA5395/6, respectively), but no padEFGHI genes. The absence of genes for PadB and PadEFGHI indicates these genes not to be required in phenylacetate and phenylpyruvate catabolism of D. multivorans which was recently also demonstrated for D. toluolica Tol2 [15].
Proteins encoded in 2 adjacent gene clusters at 2.77 Mbp are specifically formed during anaerobic growth with phenylacetate and phenylpyruvate: (i) a TRAP uptake system (DctM4Q4P4) highly similar to the supposedly phenylalanine/phenylacetate importing DctM9Q9P9 system of D. toluolica Tol2 (74–90 % identity), (ii) one of two alternative CoA-ligases for phenylacetyl-CoA formation (Dmul_C24830, FadD4), and (iii) a dehydrogenase (Hbd5), a hydratase (Dmul_C24740) as well as a thiolase (Dmul_C24750) for the formation of phenylglyoxylate. Subsequent conversion of phenylglyoxylate to benzoyl-CoA is unclear at present, since the protein abundance profiles do not provide appropriate enzyme candidates.
In case of anaerobic degradation of phenylpyruvate (compound 7 in Fig. 2), substrate-specific formation of a predicted indolepyruvate:ferredoxin oxidoreductase (IorA3B3) may indicate CoA-activation followed by oxidative decarboxylation to phenylacetyl-CoA. Such an activity, converting arylpyruvates to the corresponding arylacetyl-CoA, was described for the archaeon Pyrococcus furiosus [37] and postulated for phenylpyruvate degradation in D. toluolica Tol2 [15]. In case of the latter, the chromosomal colocalization of iorA2B2 with atoAD and the phenylalanine/phenylacetate-specifically increased abundance in D. toluolica Tol2 suggested an involvement of AtoAD in the subsequent reaction from phenylglyoxylate to benzoyl-CoA [15]. However, no atoAD genes are present in the genome of D. multivorans. Lower level formation of IorA3B3 in phenylacetate-adapted cells may indicate gratuitous induction which was also observed for the orthologs in D. toluolica Tol2 [15].
Notably, the second gene cluster contains genes encoding enzymes for the conversion of phenyllactate to phenylpyruvate (Dmul_C24850-70) (Fig. 5c). Indeed, phenyllactate (4 mM) was positively tested (data not shown) to serve as growth substrate for D. multivorans. This gene cluster also contains two sensor histidine kinases (Dmul_C24770/80) and a CheY-like transcriptional regulator (Dmul_C24760) of a two-component regulatory system. Therefore, the cluster likely represents a rather complete catabolic module differing from that of denitrifiers and also from the organization as two separate gene clusters in D. toluolica Tol2.
Benzoate and the central benzoyl-CoA pathway
The anaerobic degradation of benzoate (compound 3 in Fig. 2) and most monoaromatic compounds yields the central intermediate benzoyl-CoA [38]. Benzoate is initially activated to benzoyl-CoA by benzoate CoA-ligase (BclA). The BclA protein was identified in D. multivorans with all tested aromatic growth substrates (and cyclohexane carboxylate) (Fig. 3), contrasting substrate-specific formation in D. toluolica Tol2 [15].
Subsequent anaerobic degradation of benzoyl-CoA proceeds via reductive dearomatization which involves either the ATP-dependent class I benzoyl-CoA reductase (BcrABCD) in facultative anaerobes (e.g., T. aromatica K172) [39] or the ATP-independent class II benzoyl-CoA reductase (BamB-I) in obligate anaerobes (e.g., the FeIII-reducing deltaproteobacterium Geobacter metallireducens GS-15). The BamB subunit is considered to contain the active site of this enzyme complex, forming a heterodimeric structure with BamC, whereas BamDEFGHI are predicted to be involved in ATP-independent electron transfer [40]. As expected, the genome of D. multivorans harbours no bcrABCD homologues, but one bam-cluster at 2.74 Mbp. Interestingly, two copies of each bamB-D are present in this cluster separated by bamOP, encoding subunits of an electron transfer flavoprotein (Fig. 5a). The two paralogous BamB proteins share 66 % sequence identity with BamB2 being more similar (79 % identity) to the catalytic subunit of G. metallireducens GS-15 (BamB1 only 64 %). Accordingly, phylogenetic analysis clusters BamB2 with BamB homologs of G. metallireducens GS-15, D. toluolica Tol2 (BamB1) and strain NaphS2 (NPH7171) (Additional file 1: Figure S2), suggesting BamB2 to be the active enzyme subunit in ATP-independent reduction of benzoyl-CoA in D. multivorans. However, BamB2 was only detected during anaerobic growth with benzoate and cinnamate while BamB1 was detected with all aromatic growth substrates and also cyclohexane carboxylate. Thus, one may speculate that BamB1/2 jointly function in benzoyl-CoA reduction in D. multivorans.
Genes for the β-oxidation of the formed cyclohex-1,5-diene-1-carboxyl-CoA, i.e., dch, bzdZ and oah (bamQ), are located downstream of the bam-cluster at 2.80 Mbp. Their respective products were detected under all six analyzed aromatic substrate conditions as well as in case of cyclohexane carboxylate and 3-methylbutanoate (Fig. 3).
Aliphatic compounds
Short chain alcohols
The genome of D. multivorans harbours five genes (dmul_C14490, 16490, 16540, 36750 and adh) encoding predicted alcohol dehydrogenases. Since the product of only one of them (Adh) was found to be specifically increased in abundance during anaerobic growth with n-butanol and 1-propanol (compounds 12 and 16 in Fig. 2), it is likely to be responsible for converting these alcohols to their respective aldehydes. Four aldehyde dehydrogenases (Aor1-3, aldehyde:ferredoxin oxidoreductase; Dmul_14960, NAD-dependent aldehyde dehydrogenase) are encoded in the genome, but only the product of aor3 was detected under all tested growth conditions (Fig. 3), pointing towards its involvement in aldehyde catabolism in D. multivorans.
β-Oxidation of unbranched aliphatic carboxylates and cyclohexane carboxylate
D. multivorans utilizes fatty acids up to a chain length of C14 via classic β-oxidation and cyclohexane carboxylate via the modified β-oxidation route of the central benzoyl-CoA pathway (Fig. 2). Accordingly, the genome contains a wide range of genes associated with β-oxidation-like functions: (i) Seven (3 identified) predicted paralogs of long-chain-fatty-acid-CoA-ligases (FadD) as well as 10 (7 identified) predicted AMP-dependent synthetases and ligases for CoA-activation of carboxylates; (ii) 15 (6 identified) predicted acyl-CoA dehydrogenase (Acd), (iii) 5 (2 identified) predicted enoyl-CoA hydratases (EchA) and (iv) one (identified) predicted 3-hydroxyacyl-CoA dehydrogenase (FadB). Out of these 19 proteins, seven could be assigned to specific pathways as discussed below.
The anaerobic degradation of cyclohexane carboxylate (compound 8 in Fig. 2) was recently elucidated for G. metallireducens GS-15. Initial activation to cyclohexanoyl-CoA is followed by 1,2-dehydrogenation to cyclohex-1-ene-1-carboxyl-CoA and subsequent 1,4-dehydrogenation to cyclohex-1,5-diene-1-carboxyl-CoA [41]. Genes (>80 % protein sequence identity to orthologs of G. metallireducens GS-15) encoding putative cyclohexanoyl-CoA dehydrogenase (dmul_C00970) and cyclohex-1-ene-1-carboxyl-CoA dehydrogenase (dmul_C00980) were detected in the genome of D. multivorans, directly adjacent to an AMP-dependent synthetase (dmul_C00990), a membrane-bound FeS oxidoreductase (dmul_C01000) and an electron transfer flavoprotein (etfA1B1) (Fig. 5d). All corresponding proteins were specifically detected during anaerobic growth with cyclohexane carboxylate supporting their involvement in this pathway also in case of D. multivorans. The formed cyclohex-1,5-diene-1-carboxyl-CoA enters the β-oxidation-like branch of the benzoyl-CoA pathway as supported by specifically increased abundances of respective proteins (Dch, Oah, BzdZ) (Fig. 3).
Myristinate (tetradecanoic acid; compound 10 in Fig. 2) conversion to five acetyl-CoA and one butyryl-CoA is assumed to involve CoA-activation by FadD5, AcdA9-catalyzed dehydrogenation, acetyl-CoA acyltransfer by FadA and oxidation of 3-hydroxyacyl-CoA by FadB, inferred from myristinate-specific protein formation. Respective encoding genes colocalize on a stretch of 10.3 kbp at 3.19 Mbp of the chromosome (Fig. 5f), except for fadAB (at 1.03 Mbp). Notably, this genomic locus also includes genes for a transcriptional regulator FadR2 (specifically detected in myristinate-adapted cells) as well as genes (etfA5B5) for an electron transfer flavoprotein (ETF) and a constitutively formed membrane-bound FeS oxidoreductase (fadF) (Figs. 2 and 3). The latter may be involved in myristinate-specific electron transfer (see below section “Electron transfer flavoproteins”). The TetR-type regulator FadR2 may mediate the observed myristinate-specific protein formation.
Butanoate (compound 11 in Fig. 2) is commonly degraded via crotonyl-CoA to acetoacetyl-CoA which is thiolytically cleaved into two acetyl-CoA. An increased abundance of acetyl-CoA dehydrogenases AcdA1/6 in butanoate- as well as n-butanol- and myristinate-adapted cells indicates an involvement of these proteins in conversion of butanoate and unbranched fatty acids in general (Fig. 3). However, none of the many β-oxidation-related genes could be assigned unequivocally to butanoate catabolism based on genomic co-localization and/or substrate-specific product formation, indicating a rather constitutive formation of these enzymes.
Propanoate (compound 15 in Fig. 2) and odd-numbered fatty acids are commonly degraded also via β-oxidation which however yields one propionyl-CoA in addition to the acetyl-CoA moieties. Further degradation of propionyl-CoA proceeds via the methylmalonyl-CoA pathway. Initially, propionyl-CoA is carboxylated to methylmalonyl-CoA, by ATP-dependent propionyl-CoA carboxylase (PccAB3). Methylmalonyl-CoA is transformed from D- to L-conformation by methylmalonyl-CoA epimerase (methylmalonyl-CoA racemase, Mce) prior to being intramolecularly rearranged to succinyl-CoA by methylmalonyl-CoA mutase (Sbm). Succinyl-CoA is further degraded employing enzymes of the tricarboxylic acid cycle (succinyl-CoA ligase, SucCD2; succinate dehydrogenase, SdhABC; fumarate hydratase, FumAC) followed by an oxidative decarboxylation (malic enzyme, MaeA) yielding pyruvate. Genes for all required enzymes are rather scattered across the genome of D. multivorans contrasting the clustered organization in D. autotrophicum HRM2 [14]. Respective gene products of D. multivorans were detected under all aliphatic substrate conditions (except for PccA and FumC) pointing to a constitutive presence of the methylmalonyl-CoA pathway (Figs. 2 and 3).
Branched-chain fatty acids
Isobutanoate (compound 13 in Fig. 2) is probably degraded according to the pathway suggested by Stieb and Schink [42] including initial CoA-activation followed by two oxidation and one hydrolysis step(s) yielding methylmalonic semialdehyde. The latter could be subsequently oxidized and decarboxylated forming propionyl-CoA (Fig. 2). Initial CoA-activation of branched-chain fatty acids in D. multivorans is probably mediated by AcsA2 (Fig. 3). The unambiguous assignment of genes involved in further conversion was, however, not possible and also no isobutanoate-specific formation of respective proteins was observed.
In case of 2-methylbutanoate (compound 14 in Fig. 2), the methyl-branching is compatible with classical β-oxidation (see short chain alcohols), but thiolytic cleavage of intermediary 2-methylacetoacetyl-CoA yields acetyl-CoA and propionyl-CoA (Fig. 2), with the latter feeding into the methylmalonyl-CoA pathway.
3-Methylbutanoate (isovalerate; compound 9 in Fig. 2) is probably degraded in analogy to the pathway previously proposed for a mixed anaerobic marine co-culture [43]. Initially formed isovaleryl-CoA is oxidized to methylcrotonyl-CoA, which is subsequently carboxylated to methylglutaconyl-CoA, following hydration to 3-hydroxy-3-methylglutaconyl-CoA, and final cleavage into acetoacetyl-CoA and acetyl-CoA. Genome analysis revealed a gene cluster at 3.20 Mbp potentially encoding enzymes for isovalerate degradation as well as a transcriptional regulator (Fig. 5e). Differential proteomics corroborated this prediction by demonstrating exclusive formation of acyl-CoA synthase (AcsA6), enoyl-CoA hydratase (EchA2), methylcrotonyl-CoA carboxylase (Mcc) and 3-hydroxy-3-methylglutaconyl lyase (HmgL) in 3-methylbutanoate-adapted cells of D. multivorans (Fig. 3).
Lactate and pyruvate
Lactate (compound 17 in Fig. 2) is probably taken up by L-lactate permease (LldP). The latter is encoded in an operon-like structure together with genes for pyruvate-forming lactate dehydrogenase (ldhAB), L-lactate utilization proteins (lutABC) predicted to transfer electrons from lactate dehydrogenase to membrane-embedded redox partners in Bacillus subtilis [44], and a GntR-type transcriptional regulator (dmul_C17480) (Fig. 5g). These lactate-related proteins were substrate-specifically formed, supporting their predicted function in D. multivorans.
Pyruvate, which is also generated from the C4-dicarboxylate malate by malic enzyme (Mae) as the final step of the methylmalonyl-CoA pathway (see section above), is oxidatively decarboxylated to acetyl-CoA by the pyruvate:ferredoxin oxidoreductase (Por). Agreeing with its central metabolic role, Por was found to be constitutively formed.
Complete oxidation of acetyl-CoA (to CO2) via the Wood-Ljungdahl pathway
A catabolic property of D. multivorans shared with many members of the Desulfobacteraceae is the complete oxidation of acetyl-CoA to CO2 via the Wood-Ljungdahl pathway [13]. Genes for this pathway are present in, but dispersed across the genome of D. multivorans, contrasting the clustered organization observed for D. oleovorans Hxd3, D. toluolica Tol2 and D. autotrophicum HRM2. Except for formate dehydrogenase (FdhH, solely genome-encoded Fdh) all other protein constituents of the pathway were identified across all tested 17 substrate adaptation conditions, pointing to a constitutive formation. This agrees with proteogenomic data available for D. autotrophicum HRM2 [14, 17, 18] and D. toluolica Tol2 [15].
Although, homologs of all enzymes of the TCA cycle are encoded in the genome, the cycle is apparently only partially operative within the framework of the methylmalonyl-CoA pathway due to non-detection of aconitase (AcnA), isocitrate dehydrogenase (Icd) and malate dehydrogenase (Mdh) under any of the tested substrate conditions (Figs. 2 and 3).
Electron transfer flavoproteins
Electron transfer flavoproteins (ETF) are soluble αβ-heterodimeric proteins which serve in accepting electrons from dehydrogenases and transferring them to the membrane (mena)-quinone pool mediated by membrane-bound Etf:quinone oxidoreductases [45]. ETFs can be functionally classified in (i) constitutively formed proteins accepting electrons from a variety of acyl-CoA dehydrogenases and (ii) those being substrate-specifically formed receiving electrons from specific oxidation reactions [46]. EtfAB are predicted in Syntrophus aciditrophicus [47], Syntrophomonas wolfei [48] and D. alkenivorans AK-01 [16] to funnel β-oxidation-derived electrons to the menaquinone-pool. Likewise, electrons liberated during successive dehydrogenation reactions during anaerobic degradation of toluene and p-cresol in D. toluolica Tol2 are suggested to be conveyed by specific ETF proteins encoded directly adjacent to respective catabolic genes [15].
Seven (five identified) ETF proteins (including BamOP) are encoded in the genome of D. multivorans. Three ETF proteins (EtfA2B2, EtfA4B4 and EtfA5B5) were abundantly formed under all analyzed substrate conditions, suggesting a constitutive formation. In case of EtfA5B5 the chromosomally colocalizing membrane-bound FeS-oxidoreductase (FadF) is also present under all tested substrate conditions, suggesting transfer of β-oxidation-generated electrons via EtfA5B5 to the membrane menaquinone pool. The genomes of D. autotrophicum HRM2 and D. oleovorans Hxd3 also harbour genes for ETFs highly similar (66–80 %) to EtfA5B5 of D. multivorans, albeit the respective genomic contexts do not provide any hints on the specific functional roles in these SRB.
The ETF-proteins BamOP were identified under all aromatic substrate conditions, suggesting a specific function in transfer of electrons during reductive dearomatization of benzoyl-CoA. The exclusive detection of EtfA1B1 as well as the associated membrane-bound FeS oxidoreductase Dmul_C01000 in cyclohexane carboxylate-adapted cells indicates their involvement in anaerobic degradation of cyclohexane carboxylate in D. multivorans (Figs. 2 and 3).
Overall, the obtained proteogenomic evidence specific for D. multivorans as well as D. toluolica Tol2 [15] point to an emerging principle of specific ETF-proteins (and associated membrane-bound FeS oxidoreductases) to transfer electrons generated by distinct catabolic cytoplasmic redox reactions to the membrane menaquinone-pool in SRB.
Dissimilatory sulfate reduction
The energy metabolism of D. multivorans centers on dissimilatory sulfate reduction, typical of all SRB. D. multivorans is well equipped for the uptake of sulfate, as the genome contains six genes for putative sodium/sulfate symporters and two predicted proton/sulfate symporters (SulP). Products of four of the sodium/sulfate symporters were identified, and two (Dmul_C10650/18630) were found to be constitutively formed. The latter are reminiscent of the sulfate uptake systems recently dissected by means of BN-PAGE in D. toluolica Tol2 [49] (sequence identities 25–75 %). The product of SulP1 was identified in the membrane protein-enriched fraction of 12 out of the 17 tested substrate conditions, whereas low score detection of SulP2 (Mascot Score 45.8 and 41.4) could indicate the latter to be less relevant for sulfate uptake in D. multivorans.
Dissimilatory reduction of sulfate to sulfide proceeds via initial ATP-dependent activation of sulfate to adenylylsulfate (APS; ATP sulfurylase encoded by sat), followed by successive reduction of APS to sulfite (APS reductase encoded by aprAB) and sulfite to sulfide (dissimilatory sulfite reductase encoded by dsrAB and associated proteins dsrCDE). The genome of D. multivorans contains most of these genes in a region at ~3.50 Mbp: two paralogous sat genes at 3.05 and 3.51 Mbp, aprAB at 3.54 Mbp, dsrABD at 3.17 Mbp, dsrC at 3.43 Mbp and dsrE at 1.77 Mbp. The non-clustered, but rather scattered distribution of these sulfate reduction genes also occurs in the genomes of D. vulgaris Hildenborough, D. autotrophicum HRM2 and D. toluolica Tol2. Differential proteomics revealed the constitutive and abundant formation of Sat2, AprAB and DsrABCDE of D. multivorans under all analyzed substrate conditions (Fig. 4), which was also observed for D. toluolica Tol2 under eight different substrate conditions [15], underpinning the essential function of these proteins in SRB energy metabolism.
Transmembrane redox complexes and cytochromes
QmoABC and DsrMKJOP complexes
Among the various described transmembrane complexes in SRB, only the three-subunit, quinone-interacting, membrane-bound oxidoreductase complex QmoABC and the five-subunit DsrMKJOP complex are strictly conserved in currently genome-sequenced SRB [50]. In D. multivorans the qmoABC and aprAB genes colocalize while dsrMKJOP and dsrABC genes do not. In analogy to other genome sequenced SRB, the QmoABC complex is proposed to channel electrons from the membrane-localized menaquinone pool to AprAB, while the DsrMKJOP performs the analogous function towards DsrAB via soluble DsrC [13, 51]. All QmoABC and DsrMKJOP subunits could be identified in rather high abundance under all tested substrate conditions (Fig. 4), corroborating their predicted essential function in the energy metabolism.
HmcABCDEF, TmcABC(D) and QrcABCD complexes
The six-subunit HmcABCDEF [52] complex and the potentially Hmc-derived four-subunit TmcABCD [53] complex often occur in parallel in SRB and are functionally associated with electron transfer between the cytoplasm and the periplasm. While the Tmc complex is assumed to channel electrons from periplasmic hydrogen oxidation to cytoplasmic dissimilatory sulfate reduction, the Hmc complex operates probably in reverse direction to build up reduced conditions in the periplasm [13]. Notably, the genome of D. multivorans lacks genes encoding periplasmic hydrogenases (e.g., hyn, hys, hyd), agreeing with the original finding of the strain not being able to utilize hydrogen as an electron donor [31].
The genome of D. multivorans encodes a complete Hmc complex at 0.90 Mbp, and formation of several subunits was observed in phenylacetate- (HmcABCEF) as well as phenylpyruvate-adapted (HmcABEF) cells. The comparatively selective and apparently lower-level formation of the Hmc complex indicates an only minor (non-essential) role in general electron transfer, while a potentially specific function in phenylacetate and phenylpyruvate catabolism remains to be demonstrated.
Two paralogous gene clusters for the Tmc complex (tmcA1B1C1D at 0.89 Mbp and tmcA2B2C2 at 2.38 Mbp) are present in the genome of D. multivorans sharing 35–64 % (Tmc1) and 27–35 % (Tmc2) amino acid identity with the orthologs from D. vulgaris Hildenborough [53]. Hmc complex encoding genes are located downstream of tmcA1 (Additional file 1: Figure S3) directly adjacent to genes encoding three DNA-binding response regulator proteins (Dmul_C08090/180/210), a DNA-binding universal stress protein (Dmul_C08170) and four signal transduction histidine kinases (Dmul_C08190/200/220/230), resembling the operon-like structure previously described for D. vulgaris Hildenborough [53]. In contrast to the above described Hmc complex, most constituents of the two encoded Tmc complexes were identified in the membrane protein-enriched fraction under all 17 substrate adaptation conditions, displaying, however, rather low abundances (Fig. 4). Notably, the periplasmic TmcA1 subunit and cytoplasmic TmcB2 were not detected. While the overall low Tmc complex abundance may render the small (15 kDa) and periplasmic TmcA1 not detectable, the cytoplasmic TmcB2 (50 kDa) is apparently not formed. However, the absence of genes encoding periplasmic formate dehydrogenases and hydrogenases challenges a function of the D. multivorans Tmc complexes in the respective electron transfer as described for D. vulgaris Hildenborough [53].
The Qrc complex contains three periplasmic (ABC) and one integral membrane (D) subunit(s) and has been associated with connecting periplasmic components, TpIc 3, and non-membrane-anchored hydrogenase or formate dehydrogenase (for overview see [13, 54, 55]). The genome of D. multivorans harbours a qrcABCD gene cluster at 3.91 Mbp colocalizing with cytochrome biogenesis genes (ccsAB); they share high sequence similarities (33–75 % on amino acid level) with their orthologs from D. vulgaris Hildenborough [54], D. autotrophicum HRM2 [14] and D. alkenivorans AK-01 [16]. All four Qrc subunits of D. multivorans were abundantly detected in the membrane protein-enriched fraction of all 17 substrate adaptation conditions. The inability of D. multivorans to utilize hydrogen and formate, both of which are predicted to serve as periplasmic electron donor for the Qrc complex in D. vulgaris Hildenborough [54], suggests a different, though essential function of the Qrc complex in D. multivorans energy metabolism. The predicted physiological partner may be the periplasmic TpIc 3 cytochrome (Dmul_C11180).
Nuo complex
Genes for the multi-subunit NADH:quinone oxidoreductase complex (Nuo, respiratory complex I; nuoA-N) were identified at 2.03 Mbp of the chromosome. Among Deltaproteobacteria, complete nuo gene sets seem to be characteristic for Desulfobulbaceae and have so far been reported only for seven representatives of Desulfovibrionaceae (for overview see [13, 50]). Thus, D. multivorans is the first member of Desulfobacteraceae possessing the complete nuo gene set in a single cluster (Additional file 1: Figure S3). However, only the cytoplasmic fusion protein NuoBCD and the integral membrane subunit NuoL could be detected under most of the tested substrate conditions at rather low abundance (Fig. 4). Hence, the Nuo complex in D. multivorans may play only a minor role in energy metabolism with its function still to be resolved.
RnfABCDEG complex and Na+-based bioenergetics
The Rnf complex is an electron transporting, membrane-localized ferredoxin:NAD+-oxidoreductase widespread among Deltaproteobacteria. The six subunit RnfABCDEG complex consists of three integral membrane proteins (RnfADE), two membrane associated (RnfBG) and one soluble subunit(s) (RnfC). It is assumed to link the cellular NADH and ferredoxin pools via bidirectional electron transfer, i.e., (i) oxidation of reduced ferredoxin by RnfB and transfer via RnfDG to RnfC for NAD+ reduction coupled to the generation of a Na+-gradient, or (ii), vice versa, from NADH to oxidized ferredoxin driven by the membrane potential [50, 56]. Notably, a different type of the Rnf complex was recently reported to be present in D. toluolica Tol2 [49]. Here, the RnfB subunit involved in ferredoxin oxidation/reduction is larger (~700 amino acids) as compared to characterized RnfB of Clostridium ljungdahlii or Acetobacterium woodii (~300 amino acids) and contains NAD+-binding sites in addition to the 4Fe-4S cluster and ferredoxin-type iron-sulfur binding domain present in all RnfBs. Furthermore, also the RnfC subunit of D. toluolica Tol2 differs, lacking the NADH-ubiquinone oxidoreductase and soluble ligand-binding domains necessitating an RnfC electron acceptor/donor other than NAD+/NADH, which has to be identified. Due to these differences, a mode of action different to the classical Rnf complex was suggested [49] that awaits experimental verification.
The genome of D. multivorans contains two colocalized rnf gene cluster (rnf1 at 3.09 Mbp and rnf2 at 3.11 Mbp) encoding an A. woodii-type Rnf complex with a small RnfB subunit (Rnf1; 35–55 % amino acid identity) as well as a D. toluolica Tol2-type Rnf complex with a large RnfB subunit (Rnf2; 41–84 % amino acid identity), a genetic repertoir similar to D. autotrophicum HRM2 [14]. Differential proteomics revealed the formation of all six subunits of Rnf1 and five subunits of Rnf2 under all tested substrate conditions in the membrane protein-enriched fraction; the only exception is the integral membrane protein RnfA2 being only detected under 6 substrate conditions (Fig. 4). The latter may be due to its size and detectability of the resulting tryptic peptides. The cytochrome encoded in the rnf2 cluster (Dmul_C27670) was only detected in lactate-adapted cells. However, Dmul_C27670 is a small (15 kDa) acidic (pI 5.3) protein, with only few detectable peptides, rendering its identification difficult and implicating its presence may not be completely excluded under the other tested growth substrates.
In contrast to D. toluolica Tol2 [15], the genome of D. multivorans does not contain genes for a Na+-potential-forming NADH:quinone oxidoreductase (NqrA-F).
Electron bifurcation
Flavin-based electron bifurcation (FBEB) was originally discovered with the butyryl-CoA dehydrogenase complex in Clostridium kluyveri and shown to couple the endergonic reduction of ferredoxin with NADH to the exergonic reduction of butyryl-CoA with NADH [57]. Several protein candidates predicted to perform FBEB have been detected also in other anaerobic archaea and bacteria, suggesting this mechanism to be widespread and to play a significant role in cytoplasmic electron transfer of these organisms [50].
Hdr/Mvh complex
A protein complex involved in electron bifurcation is the HdrABC/MvhADG complex, which couples the oxidation of H2 to the reduction of ferredoxin and heterodisulfide (CoM-S-S-CoB) in methanogens [58]. HdrA contains a FAD-binding site and is therefore predicted to be involved in electron bifurcation/confurcation and to represent the site of ferredoxin reduction. In contrast, the Mvh subunits probably catalyze the H2 oxidation (MvhA) and serve in electron transfer (MvhDG) to the FAD centre of HdrA. HdrB catalyzes the reduction of heterodisulfide and is supplied with electrons by HdrC [59]. The hdrA gene is highly conserved in methanogenic archaea and orthologs are present in the genomes of a broad range of anaerobic organisms including SRB. Indeed, some SRB genomes harbour a rather large number of hdrA orthologs, e.g., D. autotrophicum HRM2 (7 orthologs) [14] and D. toluolica Tol2 (13 orthologs) [15].
The genome of D. multivorans harbours only two hdrA homologs (hdrA1 at 0.32 Mbp and hdrA2 at 3.25 Mb). The hdrA1 gene colocalizes with mvhD1 and pyridine nucleotide-disulphide oxidoreductase (Dmul_C02800), whereas hdrA2 is located adjacent to hdrBC and mvhD2 similar to the archaean gene organization. Analysis of the membrane protein enriched-fraction of D. multivorans revealed rather substrate-specific protein formation: HdrA1 was only detected in cell extracts of cells adapted to anaerobic growth with n-butanol, 1-propanol and butanoate, whereas all HdrA2BC subunits were found in cyclohexane carboxylate-adapted cells and HdrA2 was only sporadically observed with other substrate conditions (Fig. 4). These findings agree with the non-detection of HdlA5BC subunits in D. toluolica Tol2 and preclude those protein complexes from serving a general function in cytoplasmic electron transfer. They are more likely involved in specifically linking individual redox reactions to the reducing-equivalent pool.
NfnAB transhydrogenase
Another protein complex involved in FBEB is the transhydrogenase NfnAB (NADH-dependent reduced ferredoxin:NADP+ oxidoreductase). This complex was originally discovered in Clostridium kluyveri and reported to catalyze the reduction of ferredoxin with NADPH in the presence of NAD+ which was also reduced to NADH [60, 61]. Comparative genome analysis revealed the nfnAB genes to be present in many SRB [50]. Deletion of the nfnAB genes slightly affected growth of Desulfovibrio alaskensis G20 with fumarate and malate, suggesting NfnAB to oxidize NADPH (derived from fumarate and malate conversion) and to reduce NAD+ and ferredoxin potentially coupled to energy conservation via Rnf [56].
In D. multivorans the nfnAB genes are located at 0.69 Mbp of the genome. Both protein subunits were identified in all tested substrate-specific subproteomes, with only NfnA revealing slightly fluctuating abundances (Fig. 4). This constitutive formation indicates a rather central role in the energy metabolism of D. multivorans and other Deltaproteobacteria (e.g., D. alaskensis G20 [56]). In D. multivorans, NfnAB may oxidize NADPH derived from acetyl-CoA oxidation by the Wood-Ljungdahl pathway yielding NADH and reduced ferredoxin (Fig. 2).
Response to oxygen stress
Besides their frequent detection in anoxic, marine sediments, Desulfococcus phylotypes were also reported to be the most abundant among SRB in the photooxic zone of cyanobacterial mats reducing sulfate to sulfide while being exposed to oxygen [62]. Accordingly, the genome of D. multivorans contains a plurality of genes coding for oxygen detoxifying enzymes: 4 (1 identified) thioredoxins, 4 (1 identified) peroxidases, 3 (1 identified) superoxide dismutases, 1 (identified) catalase as well as cytochrome oxidase. Additionally, rubredoxin oxidoreductase (Roo1 and Roo2), ruberythrin (Rbr1-5) and desulfoferredoxin (Dfx) may increase the tolerance to oxygen exposure of D. multivorans. Detection of at least one protein of each enzyme class (except for cytochrome oxidase) in the subproteomes of all substrate conditions (i.e., constitutive formation) indicates a permanent readiness to cope with sudden oxygen exposure.
Regulatory potential
The presence of 215 (49 products identified) genes in the genome of D. multivorans coding for predicted sensory/regulatory systems indicate the capacity to regulate the metabolic network in a diversified and fine-tuned manner. In comparison, the genomes of D. autotrophicum HRM2 and D. toluolica Tol2 contain >250 and >430 such genes, respectively [14, 15], providing together first genome-wide evidences for members of the Desulfobacteraceae to have the means for comprehensive sensory capacities and regulatory adaptation.
Conclusions
The genome of D. multivorans is rather similar to several other sulfate-reducing Deltaproteobacteria with respect to size, GC content and encoded proteins. The presence of a large number of encoded proteins involved in signal transduction allows D. multivorans to adapt to changing environmental conditions as evident from formation of substrate-specific catabolic subproteomes. This flexibility is contrasted by the constitutive and abundant formation of multiple membrane-embedded redox complexes involved in electron transfer to cytoplasmic partners (most importantly sulfate reduction). Notably, specifically formed proteins of electron generating catabolic pathways are connected to the membrane-bound electron transfer system via pathway-specific electron-transfer proteins. The latter are encoded in gene clusters with corresponding catabolic proteins, thus constituting complete degradation modules similar to earlier observations with D. toluolica Tol2 [15]. The presence of such modules seems to be a principle to efficiently connect electron generating and consuming processes in completely oxidizing SRB affiliating with Desulfobacteraceae, possibly to optimize energy metabolism at the thermodynamic limit.
Abundant and constitutive formation of membrane redox complexes (e.g., QrcABCD), are described in Desulfovibrio sp. to connect periplasmic hydrogen and formate oxidation. Despite the genetic and apparent physiological inability of D. multivorans to utilize such electron donors, respective membrane redox complexes are abundantly and constitutively formed by this bacterium. This indicates a different, though essential, function of these complexes in its energy metabolism. Furthermore, abundant formation of two most likely functionally different Rnf complexes supposes a prominent role of Na+-based bioenergetics for D. multivorans. Hence, this bacterium appears to be a catabolic generalist employing a multifunctional and highly interconnected membrane redox complex network.
Furthermore, constitutive formation of a large number of oxygen counter-acting proteins will allow D. multivorans to survive oxygen pulses in dynamic marine environments (e.g., tidal systems). The interplay of these characteristics may contribute to its habitat success.
Overall, the study underpins the value of proteomic analysis to enhance functional genomic predictions and represents another puzzle piece in understanding the proteogenomic basis of the habitat-relevance and -success of the deltaproteobacterial SRB family Desulfobacteraceae.
Methods
Media and cultivation
Desulfococcus multivorans DSM 2059 (strain 1be1; originally isolated from a sewage digester (Göttingen, Germany) [31]) was obtained from the Leibniz-Institute DSMZ-German Collection of Microorganisms and Cell Cultures and cultivated under sulfate-reducing conditions in 400 ml flat glass bottles containing defined bicarbonate-buffered brackish water medium [63]. Cultures were incubated at 28 °C with the following growth substrates (in alphabetic order; concentration in mM is given in parenthesis): acetate (30), benzoate (3), n-butanol (5), butanoate (5), cinnamate (3), cyclohexane carboxylate (3), 2-hydroxybenzoate (2), isobutanoate (5), lactate (10), 2-methylbutanoate (5), 3-methylbutanoate (5), myristinate (3), phenylacetate (4), 3-phenylpropanoate (3), phenylpyruvate (3), 1-propanol (7) and propanoate (15). Adaptation of D. multivorans over at least five passages to the individual substrate conditions and cell harvest were performed as described recently [15].
DNA sequencing, assembly and annotation
DNA was isolated with the Genomic DNA kit (Qiagen, Hildesheim, Germany) according to the manufacturer’s instructions. Recombinant plasmid and fosmid shotgun libraries were constructed. Plasmid libraries with average insert sizes of 1.5 and 2.5 kbp were generated from sonified DNA [64]. Additionally, a fosmid library was constructed (84-fold physical coverage) for data finishing and assembly confirmation (Epicentre Technologies, Madison, WI, USA). Templates for sequencing were obtained by insert amplification via PCR or by plasmid isolation. Sequencing was carried out using ABI3730XL capillary systems (ABI). In total, 107,083 sequencing reads were generated and resulted in 15-fold sequencing coverage. Sequence quality assessment and assembly were performed with a quality of less than 1 error in 100,000 bases using PHRAP (Phragment assembly program 1999 (http://www.phrap.org/phredphrapconsed.html)) and Consed [65]. Structural rRNAs and tRNAs were determined using RNAmmer [66] and tRNAscan-SE [67]. Protein-coding sequences (CDS) were predicted by the ORF-finding program Glimmer3 [68] and manually revised and curated using Artemis (v.12.0) [69]. The generated ORF dataset was screened against nonredundant protein databases (SWISSPROT, TREMBL) and the genome was manually annotated applying the annotation platform HTGA (High Throughput Gene Annotation; [70]). The genome sequence of Desulfococcus multivorans DSM 2059 has been submitted to GenBank under the BioProject PRJNA310394 with accession number CP015381.
Profiling of soluble proteins by 2D DIGE and protein identification by MALDI-TOF-MS/MS
Extracts of soluble proteins of D. multivorans were prepared and 2D DIGE performed essentially as reported previously [71]. Cell pellets (approx. 100 mg wet weight) from 4 biological replicates per substrate condition were suspended in lysis buffer (7 M urea, 2 M thiourea, 30 mM Tris/HCl, 4 % CHAPS, pH 8.5), cell breakage was achieved with the PlusOne sample grinding kit (GE Healthcare, Munich, Germany) and the protein concentration was determined according to the method of Bradford [72]. For minimal labelling, 200 picomoles of CyDye DIGE fluors (GE Healthcare) were used to label 50 μg of protein sample. Protein extracts of lactate-adapted cells served as reference state and were labelled with Cy5. Protein extracts from the other 16 substrate adaptation conditions represented the test states and were each labelled with Cy3. The internal standard contained equal amounts of all test and the reference state(s) and was labelled with Cy2. Per gel, 50 μg each of the labelled reference state, test state and internal standard were applied. To account for biological variation [73], four parallel gels were run with labelled protein extracts from four individual cultures (i.e., biological replicates) for each test state. First dimension separation by isoelectric focusing (IEF) was conducted with 24 cm-long IPG strips (pH 3–11 NL; GE Healthcare) run in an IPGphor system (GE Healthcare). The IEF program used was as follows: 30 V for 7 h, 60 V for 6 h, 200 V for 1 h, 1,000 V for 1 h, gradual gradient to 8,000 V within 0.5 h and 8,000 V for 60,000 Vhs. Second dimension separation of proteins according to molecular size was done by SDS-PAGE (12.5 % gels, v/v) using an EttanDalttwelve system (GE Healthcare).
2D DIGE gels were scanned directly after completion of electrophoresis with a Typhoon 9400 scanner (GE Healthcare). Cropped gel images were analyzed with the DeCyder software (version 7.0; GE Healthcare) in two different work packages: one for aromatic (incl. cyclohexane carboxylate) and the other for aliphatic substrates. Parameters for spot detection were as described previously [36]. Three of the four biological replicates were included for the reference and each test state. Changes in the protein abundance of ≥ |1.5|-fold were regarded as significant [73]. Separate preparative colloidal Coomassie Brilliant Blue (cCBB)-stained gels were run (300 μg protein load) to obtain sufficient amounts of protein for reliable mass spectrometric identification [36]. Spots of interest were excised using the EXQuest spot cutter (Bio-Rad) from two cCBB-stained gels per analysed substrate state, and subsequently washed and tryptically digested as described recently [74].
Sample digests were spotted onto Anchorchip steel targets (Bruker Daltonik GmbH, Bremen, Germany) and analysed with an UltrafleXtreme MALDI-TOF/TOF mass spectrometer (Bruker Daltonik GmbH) as recently described [74]. Peptide mass fingerprint (PMF) searches were performed with a Mascot server (version 2.3) against the translated genome of D. multivorans with a mass tolerance of 25 ppm. Five lift spectra were collected to confirm PMF identification and three additional spectra were acquired of unassigned peaks applying feedback by the ProteinScape platform (version 3.1; Bruker Daltonik GmbH). In case of failed PMF identification, eight lift spectra of suitable precursors were acquired. MS/MS searches were performed with a mass tolerance of 100 ppm. For both, MS and MS/MS searches, Mascot scores not meeting the 95 % certainty criterion were not considered significant. A single miscleavage was allowed (enzyme trypsin) and carbamidomethyl (C) and oxidation (M) were set as fixed and variable modifications, respectively.
Analysis of membrane protein-enriched fraction
Total membrane protein fractions were prepared from 2 biological replicates per substrate condition and analyzed as recently reported [74]. Essentially, cell extracts generated by French Press® (Sim-Aminco Ltd, Rochester, NY, USA) were treated with ice-cold carbonate and hot SDS to solubilize the membrane proteins. Protein content was determined with the RC-DC kit (Bio-Rad), and protein separation was achieved using 12.5 % SDS mini gels (10 × 7 cm; Bio-Rad). Each sample lane (10 μg protein load) was divided into 4 gel slices, and each slice cut into smaller pieces (about 1 mm3) prior to washing, reduction, alkylation and tryptic digest [74]. Separation and mass spectrometric analysis of peptides was performed with a nano-LC system (UltiMate 3000; Dionex GmbH, Germering, Germany) equipped with a 25 cm analytical column (C18, 2 μm bead size, 75 μm inner diameter; ThermoFisher Scientific) operated in a trap-column mode (C18, 5 μm bead size, 2 cm length, 75 μm inner diameter) using a 120 min linear gradient [74]. The nano-LC eluent was continuously analyzed by an online-coupled electrospray (captive spray; Bruker Daltonik GmbH) ion-trap mass spectrometer (amaZon speed ETD; Bruker Daltonik GmbH). The instrument was operated in positive mode with a capillary current of 1.3 kV and drygas flow of 3 l/min at 150 °C. Active precursor exclusion was set for 0.2 min. Per full scan MS, 20 MS/MS spectra of the most intense masses were acquired. Protein identification was performed with ProteinScape as described above, allowing for a mass difference of 0.4 Da and applying a target decoy strategy (false discovery rate < 1 %).
Shotgun proteomic analysis
For shotgun analysis, cell pellets from 3 biological replicates per substrate condition were suspended in lysis buffer (7 M urea, 2 M thiourea, 30 mM Tris/HCl, pH 8.5). Cell breakage, removal of cell debris, reduction with dithiotreitol, alkylation with iodoacetamide, and tryptic in-solution digest were performed as previously described [74]. Separation of total peptide mixtures per sample was performed by nano-LC-ESI-MS/MS (see above section) applying a linear 240 min gradient. Protein identification was performed via the ProteinScape platform (see above section).
Acknowledgements
We would like to thank C. Hinrichs (Oldenburg) and D. Thies (Bremen) for technical assistance. Bioinformatic advice of J. Tomaszewski (Siegen) and support of A. Beck (Berlin) is greatly appreciated.
Funding
Thus study was supported by the University of Oldenburg and the Max Planck Society.
Availability of data and materials
The genome sequence of Desulfococcus multivorans DSM 2059 has been deposited at GenBank under the BioProject PRJNA310394 with accession number CP015381. The other datasets supporting the conclusions of this article are in included within the article and its additional files.
Author’s contributions
MD, LW and RRa designed the experiments. MK and RRe performed the genome sequencing. MK provided bioinformatic expertise. MD performed the manual genome annotation. MD and LW performed the proteomic experiments and analyzed the data. MD, LW and RRa prepared the manuscript. The final manuscript was read and approved by all authors.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- FBEB
Flavin-based electron bifurcation
- SRB
Sulfate-reducing bacteria
Additional files
Distribution of the 1,307 detected proteins by 2D DIGE, whole cell shotgun analysis and preparation of the membrane protein-enriched fraction of D. multivorans grown with 17 different substrates. Figure S2 Phylogenetic relationship of the class II benzoyl-CoA reductase catalytic subunit BamB and other uncharacterized aldehyde: ferredoxin oxidoreductases (AFOR) of selected Deltaproteobacteria. Figure S3 Scale model and chromosomal localization of transmembrane redox complex containing genes of D. multivorans. Table S1 Listing of locus tags of genes manually assigned to metabolic pathways and energy conservation as displayed in Figs. 2 and 3. (PDF 433 kb)
Contributor Information
Marvin Dörries, Email: marvin.doerries@uni-oldenburg.de.
Lars Wöhlbrand, Email: lars.woehlbrand@uni-oldenburg.de.
Michael Kube, Email: michael.kube@thuenen.de.
Richard Reinhardt, Email: reinhardt@mpipz.mpg.de.
Ralf Rabus, Email: rabus@icbm.de.
References
- 1.Jørgensen BB. Mineralization of organic matter in the sea bed - the role of sulfate reduction. Nature. 1982;296(5858):643–5. doi: 10.1038/296643a0. [DOI] [Google Scholar]
- 2.Canfield DE, Jørgensen BB, Fossing H, Glud R, Gundersen J, Ramsing NB, et al. Pathways of organic carbon oxidation in 3 continental-margin sediments. Mar Geol. 1993;113(1–2):27–40. doi: 10.1016/0025-3227(93)90147-N. [DOI] [PubMed] [Google Scholar]
- 3.Fenchel TM, Jørgensen BB. Detritus foodchains in aquatic ecosystems: the role of bacteria. In: Alexander M, editor. Adv Microb Ecol. New York: Plenum Press; 1977. pp. 1–58. [Google Scholar]
- 4.Rabus R, Hansen T, Widdel F. Dissimilatory sulfate- and sulfur-reducing prokaryotes. In: Dworkin MF S, Rosenberg E, Schleifer KH, Stackebrandt E, editors. The Prokaryotes: An Evolving Electronic Ressource for the Microbiological Community. 3. New York: Springer; 2000. pp. 659–768. [Google Scholar]
- 5.Ravenschlag K, Sahm K, Knoblauch C, Jørgensen BB, Amann R. Community structure, cellular rRNA content, and activity of sulfate-reducing bacteria in marine arctic sediments. Appl Environ Microb. 2000;66(8):3592–602. doi: 10.1128/AEM.66.8.3592-3602.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llobet-Brossa E, Rabus R, Böttcher ME, Könneke M, Finke N, Schramm A, et al. Community structure and activity of sulfate-reducing bacteria in an intertidal surface sediment: a multi-method approach. Aquat Microb Ecol. 2002;29(3):211–26. doi: 10.3354/ame029211. [DOI] [Google Scholar]
- 7.Na H, Lever MA, Kjeldsen KU, Schulz F, Jørgensen BB. Uncultured Desulfobacteraceae and crenarchaeotal group C3 incorporate 13C-acetate in coastal marine sediment. Environ Microbiol Rep. 2015;7(4):614–22. doi: 10.1111/1758-2229.12296. [DOI] [PubMed] [Google Scholar]
- 8.Mussmann M, Ishii K, Rabus R, Amann R. Diversity and vertical distribution of cultured and uncultured Deltaproteobacteria in an intertidal mud flat of the Wadden Sea. Environ Microbiol. 2005;7(3):405–18. doi: 10.1111/j.1462-2920.2005.00708.x. [DOI] [PubMed] [Google Scholar]
- 9.Gittel A, Mussmann M, Sass H, Cypionka H, Könneke M. Identity and abundance of active sulfate-reducing bacteria in deep tidal flat sediments determined by directed cultivation and CARD-FISH analysis. Environ Microbiol. 2008;10(10):2645–58. doi: 10.1111/j.1462-2920.2008.01686.x. [DOI] [PubMed] [Google Scholar]
- 10.Leloup J, Loy A, Knab NJ, Borowski C, Wagner M, Jørgensen BB. Diversity and abundance of sulfate-reducing microorganisms in the sulfate and methane zones of a marine sediment, Black Sea. Environ Microbiol. 2007;9(1):131–42. doi: 10.1111/j.1462-2920.2006.01122.x. [DOI] [PubMed] [Google Scholar]
- 11.Oni O, Miyatake T, Kasten S, Richter-Heitmann T, Fischer D, Wagenknecht L, et al. Distinct microbial populations are tightly linked to the profile of dissolved iron in the methanic sediments of the helgoland mud area, north sea. Front Microbiol. 2015;6. [DOI] [PMC free article] [PubMed]
- 12.Lavik G, Stuehrmann T, Bruechert V, Van der Plas A, Mohrholz V, Lam P, et al. Detoxification of sulphidic African shelf waters by blooming chemolithotrophs. Nature. 2009;457(7229):581–U86. doi: 10.1038/nature07588. [DOI] [PubMed] [Google Scholar]
- 13.Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IAC. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol. 2015;66:55–321. doi: 10.1016/bs.ampbs.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Strittmatter AW, Liesegang H, Rabus R, Decker I, Amann J, Andres S, et al. Genome sequence of Desulfobacterium autotrophicum HRM2, a marine sulfate reducer oxidizing organic carbon completely to carbon dioxide. Environ Microbiol. 2009;11(5):1038–55. doi: 10.1111/j.1462-2920.2008.01825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wöhlbrand L, Jacob JH, Kube M, Mussmann M, Jarling R, Beck A, et al. Complete genome, catabolic sub-proteomes and key-metabolites of Desulfobacula toluolica Tol2, a marine, aromatic compound-degrading, sulfate-reducing bacterium. Environ Microbiol. 2013;15(5):1334–55. doi: 10.1111/j.1462-2920.2012.02885.x. [DOI] [PubMed] [Google Scholar]
- 16.Callaghan AV, Morris BEL, Pereira IAC, McInerney MJ, Austin RN, Groves JT, et al. The genome sequence of Desulfatibacillum alkenivorans AK-01: a blueprint for anaerobic alkane oxidation. Environ Microbiol. 2012;14(1):101–13. doi: 10.1111/j.1462-2920.2011.02516.x. [DOI] [PubMed] [Google Scholar]
- 17.Amann J, Lange D, Schüler M, Rabus R. Substrate-dependent regulation of carbon catabolism in marine sulfate-reducing Desulfobacterium autotrophicum HRM2. J Mol Microbiol Biotechnol. 2010;18(2):74–84. doi: 10.1159/000277655. [DOI] [PubMed] [Google Scholar]
- 18.Dörries M, Wöhlbrand L, Rabus R. Differential proteomic analysis of the metabolic network of the marine sulfate-reducer Desulfobacterium autotrophicum HRM2. PROTEOMICS 2016;16:2878-93. [DOI] [PubMed]
- 19.Rabus R, Ruepp A, Frickey T, Rattei T, Fartmann B, Stark M, et al. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold arctic sediments. Environ Microbiol. 2004;6(9):887–902. doi: 10.1111/j.1462-2920.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- 20.Heidelberg JF, Seshadri R, Haveman SA, Hemme CL, Paulsen IT, Kolonay JF, et al. The genome sequence of the anaerobic, sulfate-reducing bacterium Desulfovibrio vulgaris Hildenborough. Nat Biotechnol. 2004;22(5):554–9. doi: 10.1038/nbt959. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Gladyshev VN. An algorithm for identification of bacterial selenocysteine insertion sequence elements and selenoprotein genes. Bioinformatics. 2005;21(11):2580–9. doi: 10.1093/bioinformatics/bti400. [DOI] [PubMed] [Google Scholar]
- 22.Staals RHJ, Brouns SJJ. Distribution and mechanism of the Type I CRISPR-Cas Systems. In: Barrangou R, Van der Oost J, editors. CRISPR-Cas Systems. Berlin Heidelberg: Springer; 2013. pp. 145–69. [Google Scholar]
- 23.Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315(5819):1709–12. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- 24.Marraffini LA, Sontheimer EJ. CRISPR Interference limits horizontal gene transfer in Staphylococci by targeting DNA. Science. 2008;322(5909):1843–5. doi: 10.1126/science.1165771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makarova KS, Grishin NV, Shabalina SA, Wolf YI, Koonin EV. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1. [DOI] [PMC free article] [PubMed]
- 26.Yosef I, Goren MG, Qimron U. Proteins and DNA elements essential for the CRISPR adaptation process in Escherichia coli. Nucleic Acids Res. 2012;40(12):5569–76. doi: 10.1093/nar/gks216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321(5891):960–4. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lintner NG, Kerou M, Brumfield SK, Graham S, Liu H, Naismith JH, et al. Structural and functional characterization of an archaeal clustered regularly interspaced short palindromic repeat (CRISPR)-associated complex for antiviral defense (CASCADE) J Biol Chem. 2011;286(24):21643–56. doi: 10.1074/jbc.M111.238485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beloglazova N, Petit P, Flick R, Brown G, Savchenko A, Yakunin AF. Structure and activity of the Cas3 HD nuclease MJ0384, an effector enzyme of the CRISPR interference. Embo J. 2011;30(22):4616–27. doi: 10.1038/emboj.2011.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mulepati S, Bailey S. Structural and biochemical analysis of nuclease domain of clustered regularly interspaced short palindromic repeat (CRISPR)-associated Protein 3 (Cas3) J Biol Chem. 2011;286(36):31896–903. doi: 10.1074/jbc.M111.270017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Widdel F. Anaerober Abbau von Fettsäuren und Benzoesäure durch neu isolierte Arten Sulfat-reduzierender Bakterien [Dissertation] Göttingen: Georg-August Universität zu Göttingen; 1980. [Google Scholar]
- 32.Trautwein K, Wilkes H, Rabus R. Proteogenomic evidence for β-oxidation of plant-derived 3-phenylpropanoids in “Aromatoleum aromaticum” EbN1. Proteomics. 2012;12(9):1402–13. doi: 10.1002/pmic.201100279. [DOI] [PubMed] [Google Scholar]
- 33.Elder DJE, Morgan P, Kelly DJ. Anaerobic degradation of trans-cinnamate and ω-phenylalkane carboxylic-acids by the photosynthetic bacterium Rhodopseudomonas palustris - Evidence for a β-oxidation mechanism. Arch Microbiol. 1992;157(2):148–54. doi: 10.1007/BF00245283. [DOI] [PubMed] [Google Scholar]
- 34.Schneider S, Mohamed ME, Fuchs G. Anaerobic metabolism of L-phenylalanine via benzoyl-CoA in the denitrifying bacterium Thauera aromatica. Arch Microbiol. 1997;168(4):310–20. doi: 10.1007/s002030050504. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch W, Schägger H, Fuchs G. Phenylglyoxylate : NAD(+) oxidoreductase (CoA benzoylating), a new enzyme of anaerobic phenylalanine metabolism in the denitrifying bacterium Azoarcus evansii. Eur J Biochem. 1998;251(3):907–15. doi: 10.1046/j.1432-1327.1998.2510907.x. [DOI] [PubMed] [Google Scholar]
- 36.Wöhlbrand L, Kallerhoff B, Lange D, Hufnagel P, Thiermann J, Reinhardt R, et al. Functional proteomic view of metabolic regulation in “Aromatoleum aromaticum” strain EbN1. Proteomics. 2007;7(13):2222–39. doi: 10.1002/pmic.200600987. [DOI] [PubMed] [Google Scholar]
- 37.Mai XH, Adams MWW. Indolepyruvate ferredoxin oxidoreductase from the hyperthermophilic archaeon Pyrococcus furiosus - a new enzyme involved in peptide fermentation. J Biol Chem. 1994;269(24):16726–32. [PubMed] [Google Scholar]
- 38.Fuchs G, Boll M, Heider J. Microbial degradation of aromatic compounds - from one strategy to four. Nat Rev Microbiol. 2011;9(11):803–16. doi: 10.1038/nrmicro2652. [DOI] [PubMed] [Google Scholar]
- 39.Buckel W, Kung JW, Boll M. The benzoyl-coenzyme A reductase and 2-hydroxyacyl-coenzyme A dehydratase radical enzyme family. Chembiochem. 2014;15(15):2188–94. doi: 10.1002/cbic.201402270. [DOI] [PubMed] [Google Scholar]
- 40.Boll M, Loeffler C, Morris BEL, Kung JW. Anaerobic degradation of homocyclic aromatic compounds via arylcarboxyl-coenzyme A esters: organisms, strategies and key enzymes. Environ Microbiol. 2014;16(3):612–27. doi: 10.1111/1462-2920.12328. [DOI] [PubMed] [Google Scholar]
- 41.Kung JW, Meier A-K, Mergelsberg M, Boll M. Enzymes involved in a novel anaerobic cyclohexane carboxylic acid degradation pathway. J Bacteriol. 2014;196(20):3667–74. doi: 10.1128/JB.02071-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stieb M, Schink B. Anaerobic degradation of isobutyrate by methanogenic enrichment cultures and by a Desulfococcus multivorans strain. Arch Microbiol. 1989;151(2):126–32. doi: 10.1007/BF00414426. [DOI] [Google Scholar]
- 43.Stieb M, Schink B. Anaerobic degradation of isovalerate by a defined methanogenic coculture. Arch Microbiol. 1986;144(3):291–5. doi: 10.1007/BF00410965. [DOI] [Google Scholar]
- 44.Chai Y, Kolter R, Losick R. A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. J Bacteriol. 2009;191(8):2423–30. doi: 10.1128/JB.01464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herrmann G, Jayamani E, Mai G, Buckel W. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J Bacteriol. 2008;190(3):784–91. doi: 10.1128/JB.01422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Weidenhaupt M, Rossi P, Beck C, Fischer HM, Hennecke H. Bradyrhizobium japonicum possesses two discrete sets of electron transfer flavoprotein genes: fixA, fixB and etfS, etfL. Arch Microbiol. 1996;165(3):169–78. doi: 10.1007/BF01692858. [DOI] [PubMed] [Google Scholar]
- 47.McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L, et al. The genome of Syntrophus aciditrophicus: Life at the thermodynamic limit of microbial growth. Proc Natl Acad Sci U S A. 2007;104(18):7600–5. doi: 10.1073/pnas.0610456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sieber JR, Sims DR, Han C, Kim E, Lykidis A, Lapidus AL, et al. The genome of syntrophomonas wolfei: New insights into syntrophic metabolism and biohydrogen production. Environ Microbiol. 2010;12(8):2289–301. doi: 10.1111/j.1462-2920.2010.02237.x. [DOI] [PubMed] [Google Scholar]
- 49.Wöhlbrand L, Ruppersberg HS, Feenders C, Blasius B, Braun HP, Rabus R. Analysis of membrane protein complexes of the marine sulfate reducer Desulfobacula toluolica Tol2 by 1D Blue Native-PAGE complexome profiling and 2D Blue Native-/SDS-PAGE. Proteomics. 2016;16(2):973–88. doi: 10.1002/pmic.201500360. [DOI] [PubMed] [Google Scholar]
- 50.Pereira IAC, Ramos AR, Grein F, Marques MC, da Silva SM, Venceslau SS. A comparative genomic analysis of energy metabolism in sulfate-reducing bacteria and archaea. Front Microbiol. 2011;2:69. doi: 10.3389/fmicb.2011.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santos AA, Venceslau SS, Grein F, Leavitt WD, Dahl C, Johnston DT, et al. A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science. 2015;350(6267):1541–5. doi: 10.1126/science.aad3558. [DOI] [PubMed] [Google Scholar]
- 52.Rossi M, Pollock WBR, Reij MW, Keon RG, Fu RD, Voordouw G. The Hmc operon of Desulfovibrio vulgaris subsp. vulgaris Hildenborough encodes a potential transmembrane redox protein complex. J Bacteriol. 1993;175(15):4699–711. doi: 10.1128/jb.175.15.4699-4711.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pereira PM, Teixeira M, Xavier AV, Louro RO, Pereira IAC. the Tmc complex from Desulfovibrio vulgaris Hildenborough is involved in transmembrane electron transfer from periplasmic hydrogen oxidation. Biochem. 2006;45(34):10359–67. doi: 10.1021/bi0610294. [DOI] [PubMed] [Google Scholar]
- 54.Venceslau SS, Lino RR, Pereira IAC. The Qrc membrane complex, related to the alternative complex III, is a menaquinone reductase involved in sulfate respiration. J Biol Chem. 2010;285(30):22772–81. doi: 10.1074/jbc.M110.124305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venceslau SS, Matos D, Pereira IAC. EPR characterization of the new Qrc complex from sulfate reducing bacteria and its ability to form a supercomplex with hydrogenase and TpIc3. FEBS Lett. 2011;585(14):2177–81. doi: 10.1016/j.febslet.2011.05.054. [DOI] [PubMed] [Google Scholar]
- 56.Price MN, Ray J, Wetmore KM, Kuehl JV, Bauer S, Deutschbauer AM, et al. The genetic basis of energy conservation in the sulfate-reducing bacterium Desulfovibrio alaskensis G20. Front Microbiol. 2014;5:577. doi: 10.3389/fmicb.2014.00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, Thauer RK. Coupled ferredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J Bacteriol. 2008;190(3):843–50. doi: 10.1128/JB.01417-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thauer RK, Kaster A-K, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008;6(8):579–91. doi: 10.1038/nrmicro1931. [DOI] [PubMed] [Google Scholar]
- 59.Buckel W, Thauer RK. Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochimica et Biophysica Acta-Bioenergetics. 2013;1827(2):94–113. doi: 10.1016/j.bbabio.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 60.Thauer RK, Rupprech E, Ohrloff C, Jungerma K, Decker K. Regulation of reduced nicotinamide adenine dinucleotide phosphate-ferredoxin reductase system in Clostridium kluyveri. J Biol Chem. 1971;246(4):954–9. [PubMed] [Google Scholar]
- 61.Wang S, Huang H, Moll J, Thauer RK. NADP(+) reduction with reduced ferredoxin and NADP(+) reduction with NADH are coupled via an electron-bifurcating enzyme complex in Clostridium kluyveri. J Bacteriol. 2010;192(19):5115–23. doi: 10.1128/JB.00612-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Risatti JB, Capman WC, Stahl DA. Community structure of a microbial mat - The phylogenetic dimension. Proc Natl Acad Sci U S A. 1994;91(21):10173–7. doi: 10.1073/pnas.91.21.10173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Widdel F, Pfennig N. Studies on dissimilatory sulfate-reducing bacteria that decompose fatty acids. I. Isolation of new sulfate-reducing bacteria enriched with acetate from saline environments. Description of Desulfobacter postgatei gen. nov., sp. nov. Arch Microbiol. 1981;129(5):395–400. doi: 10.1007/BF00406470. [DOI] [PubMed] [Google Scholar]
- 64.Rabus R, Kube M, Heider J, Beck A, Heitmann K, Widdel F, et al. The genome sequence of an anaerobic aromatic-degrading denitrifying bacterium, strain EbN1. Arch Microbiol. 2005;183(1):27–36. doi: 10.1007/s00203-004-0742-9. [DOI] [PubMed] [Google Scholar]
- 65.Gordon D. Viewing and editing assembled sequences using Consed. Curr protoc Bioinformatics. 2003;2:11.2:11.2.1–11.2.43. [DOI] [PubMed]
- 66.Lagesen K, Hallin P, Rodland EA, Staerfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Delcher AL, Harmon D, Kasif S, White O, Salzberg SL. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999;27(23):4636–41. doi: 10.1093/nar/27.23.4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, et al. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–5. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 70.Rabus R, Kube M, Beck A, Widdel F, Reinhardt R. Genes involved in the anaerobic degradation of ethylbenzene in a denitrifying bacterium, strain EbN1. Arch Microbiol. 2002;178(6):506–16. doi: 10.1007/s00203-002-0487-2. [DOI] [PubMed] [Google Scholar]
- 71.Gade D, Thiermann J, Markowsky D, Rabus R. Evaluation of two-dimensional difference gel electrophoresis for protein profiling. J Mol Microbiol Biotechnol. 2003;5(4):240–51. doi: 10.1159/000071076. [DOI] [PubMed] [Google Scholar]
- 72.Bradford MM. rapid and sensitive method for quantitation of microgram quantities of protein utilizing principle of protein-dye binding. Anal Biochem. 1976;72(1–2):248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 73.Zech H, Echtermeyer C, Wöhlbrand L, Blasius B, Rabus R. Biological versus technical variability in 2-D DIGE experiments with environmental bacteria. Proteomics. 2011;11(16):3380–9. doi: 10.1002/pmic.201100071. [DOI] [PubMed] [Google Scholar]
- 74.Zech H, Hensler M, Kossmehl S, Drüppel K, Wöhlbrand L, Trautwein K, et al. Adaptation of Phaeobacter inhibens DSM 17395 to growth with complex nutrients. Proteomics. 2013;13(18–19):2851–68. doi: 10.1002/pmic.201200513. [DOI] [PubMed] [Google Scholar]
- 75.Platen H, Temmes A, Schink B. Anaerobic degradation of acetone by Desulfococcus biacutus spec. nov. Arch Microbiol. 1990;154(4):355–61. doi: 10.1007/BF00276531. [DOI] [PubMed] [Google Scholar]
- 76.Acosta OBG, Schleheck D, Schink B. Acetone utilization by sulfate-reducing bacteria: draft genome sequence of Desulfococcus biacutus and a proteomic survey of acetone-inducible proteins. BMC Genomics. 2014;15. [DOI] [PMC free article] [PubMed]
- 77.Aeckersberg F, Bak F, Widdel F. Anaerobic oxidation of saturated hydrocarbons to CO2 by a new type of sulfate-reducing bacterium. Arch Microbiol. 1991;156(1):5–14. doi: 10.1007/BF00418180. [DOI] [Google Scholar]
- 78.So CM, Young LY. Isolation and characterization of a sulfate-reducing bacterium that anaerobically degrades alkanes. Appl Environ Microb. 1999;65(7):2969–76. doi: 10.1128/aem.65.7.2969-2976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rabus R, Nordhaus R, Ludwig W, Widdel F. Complete oxidation of toluene under strictly anoxic conditions by a new sulfate-reducing bacterium. Appl Environ Microb. 1993;59(5):1444–51. doi: 10.1128/aem.59.5.1444-1451.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Brysch K, Schneider C, Fuchs G, Widdel F. Lithoautotrophic growth of sulfate-reducing bacteria, and description of Desulfobacterium autotrophicum gen. nov., sp. nov. Arch Microbiol. 1987;148(4):264–74. doi: 10.1007/BF00456703. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The genome sequence of Desulfococcus multivorans DSM 2059 has been deposited at GenBank under the BioProject PRJNA310394 with accession number CP015381. The other datasets supporting the conclusions of this article are in included within the article and its additional files.