

Abstract
In a large clinical trial, finasteride reduced the rate of low-grade prostate cancer (PCa) while increasing the incidence of high-grade cancer. Whether finasteride promotes the development of high-grade tumors remains controversial. We demonstrated the role of fibroblasts and c-Jun in chemopreventive and therapeutic effect of finasteride on xenograft models of PCa. LNCaP (PC3) cells or recombinants of cancer cells and fibroblasts were implanted in male athymic nude mice treated with finasteride. Tumor growth, cell proliferation, apoptosis, p-Akt, and p-ERK1/2 were evaluated. In LNCaP (PC3) mono-grafted models, finasteride did not change the tumor growth. In recombinant-grafted models, fibroblasts and c-Jun promoted tumor growth; finasteride induced proliferation of LNCaP cells and repressed PC3 cell apoptosis. When c-Jun was knocked out, fibroblasts and/or finasteride did not promote the tumor growth. Finasteride inhibited p-Akt and p-ERK1/2 in mono-culture cancer cells while stimulating the same signaling molecules in the presence of fibroblasts. Reduced p-Akt and p-ERK1/2 were noted in the presence of c-Jun−/− fibroblasts. Fibroblasts and c-Jun promote PCa growth; finasteride further stimulates tumor growth with promoted proliferation, repressed apoptosis, and up-regulated pro-proliferative molecular pathway in the presence of fibroblasts and c-Jun. Stromal-epithelial interactions play critical roles in finasteride's therapeutic effects on PCa. Our findings have preliminary implications in using finasteride as a chemopreventive or therapeutic agent for PCa patients.
Keywords: chemoprevention, c-Jun, fibroblasts, finasteride, mouse model, prostate cancer
INTRODUCTION
Prostate cancer (PCa) is the most common cancer and is the second leading cause of cancer death in men in the Western world.1 The role of androgen and androgen receptor (AR) in carcinogenesis and progression of PCa are the most investigated mechanisms. However, in recent years, more other pathways such as paracrine signaling between epithelial cells and stromal cells were reported to be involved in the process of tumorigenesis and progression of PCa, which will probably shed some light into the mechanism in investigation on prevention and management of PCa in the near future.2
Finasteride, a 5-alpha reductase (5-AR) inhibitor, was ever investigated as a chemopreventive agent for PCa.3,4 In a PCa Prevention Trial (PCPT), finasteride reduced the rate of low-grade PCa; however, the percentage of patients diagnosed with high-grade disease (Gleason grades 7-10) increased.3 This result led to controversy regarding whether finasteride facilitates the detection of high-grade cancers or it promotes the development of high-grade tumors.
Several articles were published to explain that finasteride facilitates the detection of existing high-grade cancers through increasing the sensitivity of PSA5 and digital rectum examination;6 the effects of finasteride on reducing the prostate volume and selective inhibition of low-grade cancer may be contributed to the increase in high-grade cancers. Also, the finasteride-associated increase in high-grade disease at biopsy was diminished in patients who had radical prostatectomy.7
However, the possibility that finasteride may induce high-grade cancers cannot be excluded. The American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guidelines8 stated that although the majority of the panel judged that the observed higher incidence of high-grade cancers in the finasteride group is most likely to be due to confounding factors, the result of induction by finasteride cannot be excluded with certainty. 2015 EAU guideline9 stated that although it seems that 5-AR inhibitors have a potential benefit in preventing or delaying the development of PCa (~25%, only of Gleason 6 cancer), this must be weighed against treatment-related side effects as well as the potential increased risk of high-grade PCa. It was recently reported that finasteride upregulates expression of androgen receptor in LNCaP cells,10 induces neuroendocrine differentiation and aggressive PCa,11 promotes high-grade PCa by inducing the expression of NF-related factor-2 in androgen refractory cancer cells,12 and down-regulates the apoptotic factors caspase-7 and IGFBP-3 in cancer cells upon finasteride treatment for 30 days before surgery.13
To verify whether finasteride induces the progression of high-grade PCa, a xenograft mouse model was established using human PCa cells and stromal fibroblasts. We demonstrate that in a grafted tumor model, finasteride promotes tumor growth in the presence of stromal cells; furthermore, molecular pathways associated with cancer progression were promoted in cancer cells upon finasteride treatment. To the best of our knowledge, this article is the first to investigate the effects of finasteride on chemoprevention and therapy for PCa in an epithelial-stroma recombinant grafted mouse model. The role of fibroblasts and c-Jun in the chemoprevention and therapy for PCa are discussed.
MATERIALS AND METHODS
Animals
Male athymic Balb/c-nu mice, 4–6 weeks old, were housed in laminar flow racks and provided with sterilized food and drinking water. Sterilized gloves, clean gowns, facemasks, and caps were used when handling the animals. All animals were purchased and used for experiments at Cancer Institute, Chinese Academy of Medical Sciences. All the experiments were approved by the Institutional Animal Care and Use Committee at Cancer Institute, Chinese Academy of Medical Sciences and were performed at Cancer Institute in accordance with ethical guidelines.
Establishment of the LNCaP (PC3) grafted mouse model
Eight mice in each group, 2.5 × 107 LNCaP cells, 5 × 106 PC3 cells, or recombinants of LNCaP (PC3) and fibroblasts (c-Jun+/+ or c-Jun−/−) in a ratio of 1:2.5, suspended in 250 μl of PBS, were mixed with 250 μl of Matrigel (Becton Dickinson labware, Bedford, MA, USA) and were then implanted subcutaneously in male athymic nude mice using a 25-gauge needle. The tumor volume was calculated by the modified ellipsoid formula: length × width2 × 0.52. When tumors grew to approximately 100 mm3, animals were treated with finasteride (intra-gastric feeding, 100 mg kg−1 per day) for 5–6 weeks or with the same amount of coin oil was feed to mice as control group. The sizes of the tumors were measured twice a week. The final weight and volume were determined after the xenografts were harvested.
Immunohistochemistry staining of Ki-67, CK, and vimentin
The tumor tissues were evaluated for cell proliferation using Rabbit anti-human Ki-67 monoclonal antibody (Maixin Biotech, Fuzhou, China). Rabbit Anti-P-CK/Cytokeratin AE1 + AE3 (Bioss, Beijing, China) (CK) and Rabbit Anti-Vimentin (Bioss, Beijing, China) were used to determine the origin of cells in the tumor tissue, CK-positive epithelial cells, or Vimentin-positive stromal cells. Formalin-fixed, paraffin-embedded tissue blocks were cut into 5-μm sections and mounted on positively charged slides. Tissue sections were deparaffinized with xylene and rehydrated with graded alcohol solutions; they were then incubated in citrate buffer (pH 6.0) at 210°C for 10 min and at 160°C for 10 min in a pressure cooker. After incubation in 3.0% hydrogen peroxide for 10 min and PBS wash, the tissue sections were immersed in working solution of anti-Ki-67, anti-cytokeratin, or anti-vimentin for 1 h at 37°C. Tissue sections were exposed with a second antibody (MaxVision™ HRP-Polymer anti-rabbit IHC kit, Maixin Bio, Fuzhou, China) for 15 min at room temperature. Finally, the sections were incubated in DAB chromogen and then counterstained with hematoxylin. Positive and negative controls were used throughout all immunostaining protocols. The ratio of Ki-67-positive cells was defined as the percentage of positive cancer cells by counting 2000 cancer cells at ×200 microscopically.
Tunel
For the xenografts, apoptotic cancer cells were identified using the ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore Corporation, Billerica, MA, USA). Paraffin-embedded specimens were deparaffinized, rehydrated, and incubated in Proteinase K (20 μg ml−1) for 15 min and in 3.00% hydrogen peroxide in PBS for 5 min at room temperature. After being incubated in equilibration buffer for at least 10 s, tissue sections were then incubated in working strength TdT enzyme buffer for 1 h at 37°C and incubated in stop/wash buffer for 10 min at room temperature. Then, apoptotic bodies were labeled using anti-digoxigenin conjugate for 30 min at room temperature. Specimens were incubated in DAB chromogen and, then, counterstained with hematoxylin. The apoptotic index was defined as the percentage of apoptotic cancer cells by counting 2000 cancer cells at ×200 microscopically.
Cells and cell culture conditions
Androgen-sensitive LNCaP and androgen-insensitive PC3 cells were obtained from the cell resource center, the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences, cultured in RPMI 1640 medium (Gibco, Rockville, MD, USA) supplemented with 2 mmol l−1 L-glutamine, 10% fetal bovine serum (FBS) (Gibco, Melbourne, Australia), and 1% penicillin-streptomycin (Hyclone, Logan, Utah, USA) at 37°C with 5% CO2. Human primary prostate fibroblasts (HPF), c-Jun wild-type (c-Jun+/+), and c-Jun knockout (c-Jun−/−) mouse embryonic fibroblasts were obtained from lab of Dr. Aria F. Olumi (Massachusetts General Hospital, School of Medicine, Harvard University). The fibroblasts were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 2 mmol l−1 L-glutamine, 10% FBS, and 1% penicillin-streptomycin at 37°C with 5% CO2. In co-culture experiments, LNCaP or PC3 cells were cultured in 0.4 μm-pore size permeable membrane Transwell inserts (Corning Inc., Corning, NY, USA), and fibroblasts were cultured in 6-well plates. When 50% confluent, LNCaP or PC3 cells were starved in FBS-free DMEM for 24 h and then moved to the 6-well plate with 80% confluent fibroblasts. The stromal-epithelial co-cultures were maintained in 1% FBS DMEM medium containing 100 μmol l−1 Finasteride (LKT Laboratories, Inc., St. Paul, USA); the medium was replaced every 24 h for 72 h.
Cell extracts and Western blot analysis
Cells were harvested for total cell lysates with M-PER Mammalian Protein Extraction Reagent (Pierce Biotechnology, Rockford, IL, USA) containing a mixture of protease inhibitors (Halt Protease Inhibitor Cocktail Kit, Pierce Biotechnology, Rockford, IL, USA) according to the instructions. The protein concentration was determined by the BCA protein assay reagent (Pierce, Rockford, IL, USA). Western blot analysis was performed as previously described.14 The antibodies, including Akt, phospho-Akt (Ser473), extracellular signal-regulated kinase (ERK), and phospho-ERK (Thr202/Tyr204), were purchased from Cell Signaling Technology (Boston, MA, USA); the horseradish peroxidase-conjugated secondary antibody (goat anti-mouse, goat anti-rabbit) from Santa Cruz Biotechnology, Inc., (Santa Cruz, CA, USA); and the GAPDH antibody from Abcam, Inc., (Cambridge, MA, USA).
Statistics
Relevance among growth curves of grafted tumors was analyzed using repeated measures by ANOVA test. Associations between c-Jun, fibroblasts, finasteride, and tumor growth were evaluated by the ANOVA and LSD test using SPSS version 16 (SPSS Inc., Chicago, IL, USA).
RESULTS
Finasteride had no impact on tumor growth in the LNCaP (PC3) mono-grafted mouse model without fibroblast recombination
No difference was found between two growth curves for LNCaP (P = 0.272, Figure 1a) or PC3 (P = 0.210 Figure 1b) mono-grafted mouse groups with or without finasteride treatment. At the endpoint of the curves, finasteride did not change the growth of the LNCaP mono-grafted tumors based on size (Figure 2a) and weight (Figure 2b); similar results were observed in PC3 mono-grafted tumors based on size (Figure 2c) and weight (Figure 2d). The ratio of Ki-67-positive cells was not different in LNCaP tumors (Figure 3a and 3b) or in PC3 tumors (Figure 4a and 4b) between the groups with or without finasteride feeding. Also, the apoptotic index was not different in LNCaP tumors (Figure 3a and 3c) or PC3 tumors (Figure 4a and 4c) between the two groups.
Figure 1.
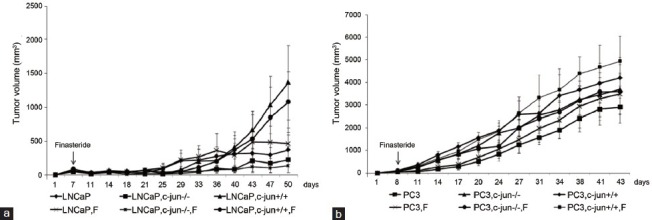
The growth curves of the LNCaP (a) and PC3 (b) mono-grafted and cancer cell-fibroblast recombinant-grafted tumors in the xenograft PCa mouse model. Finasteride did not change the tumor growth of LNCaP or PC3 mono-grafted tumors or in the presence of fibroblasts.
Figure 2.
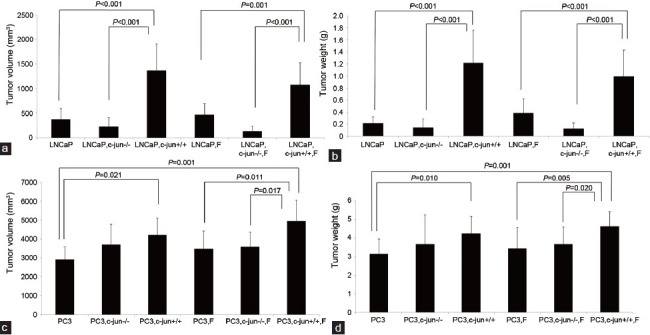
Comparisons of the final tumor volumes and weights after grafted tumors were removed from mice. (a) and (b) Compared the LNCaP tumor volumes and weights among groups on the 50th day from implantation of tumor cells. Finasteride stimulated the LNCaP tumor growth in the presence of wild fibroblasts. (c) and (d) Compared the PC3 tumor volumes and weights among groups on the 43th day from implantation of tumor cells. Finasteride stimulated the PC3 tumor growth in the presence of wild fibroblasts. c-Jun is important in mediating the pro-proliferative effects of fibroblasts and Finasteride for both LNCaP and PC3 grafted tumors.
Figure 3.
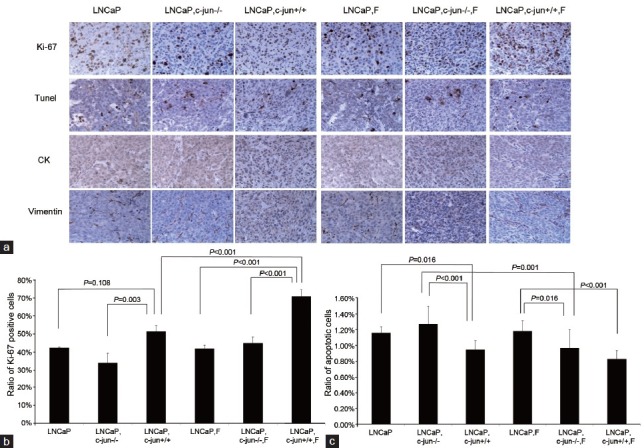
(a) Immunoreactive staining of Ki-67 and apoptotic cancer cells by Tunel in LNCaP tumors treated with finasteride. (b) The ratio of Ki-67-positive cancer cells in LNCaP tumors. Fibroblasts induced the expression of Ki-67 in cancer cells, and finasteride further promoted the expression of Ki-67 in the presence of fibroblasts. c-Jun is important in mediating the pro-proliferative effects of fibroblasts and finasteride. (c) The apoptotic index in LNCaP tumors. Fibroblasts induced the LNCaP cell apoptosis, and finasteride did not further promoted the LNCaP cell apoptosis in the presence of fibroblasts.
Figure 4.
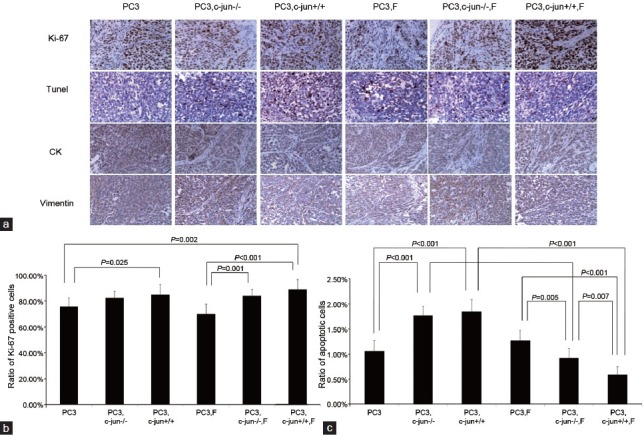
(a) Immunoreactive staining of Ki-67 and apoptotic cancer cells by Tunel in PC3 tumors (b) The ratio of Ki-67-positive cancer cells in PC3 tumors. Fibroblasts induced the expression of Ki-67; finasteride did not further promoted the expression of Ki-67 in the presence of wild fibroblasts. (c) The apoptotic index in PC3 tumors. Fibroblasts induced the PC3 cell apoptosis while finasteride repressed PC3 cell apoptosis in the presence of fibroblasts. c-Jun played a critical role in mediating the apoptosis-repressing effect of finasteride.
Fibroblasts stimulated tumor growth in LNCaP (PC3)-fibroblasts recombinant-grafted mouse model without finasteride treatment
The fibroblasts significantly promoted the LNCaP tumor growth in the recombinant-grafted group compared with the mono-grafted group between two growth curves (P = 0.001, Figure 1a) or based on the final size (1364.3 mm3 vs 372.2 mm3, P < 0.001) (Figure 2a) and final weight (1.216 g vs 0.215 g, P < 0.001) (Figure 2b); similar results were found for the PC3 tumors between two growth curves (P = 0.000, Figure 1b) or based on the final size (4210.3 mm3 vs 2906.7 mm3, P = 0.021) (Figure 2c) and final weight (4.238 g vs 3.150 g, P = 0.010) (Figure 2d). Accordingly, in the LNCaP tumor, fibroblasts reduced the apoptotic index (1.16% vs 0.95%, P = 0.016) (Figure 3a and 3c) and had the tendency to increase the ratio of Ki-67-positive cancer cells (42.50% vs 51.50%, P = 0.108) (Figure 3a and 3b); in PC3 tumors, fibroblasts promoted the ratio of Ki-67-positive cancer cells (75.83% vs 85.00%, P = 0.025) (Figure 4b), while also increasing the apoptotic index of cancer cells (1.06% vs 1.85%, P < 0.001) (Figure 4c).
Finasteride promoted cancer cell proliferation and repressed cell apoptosis in the LNCaP (PC3)-fibroblast recombinant-grafted mouse model
In the presence of fibroblasts, finasteride feeding did not have a significant impact on LNCaP (Figures 1a, 2a, and 2b) and PC3 (Figures 1b, 2c, and 2d) tumor growth. However, microscopically, the ratio of Ki-67-positive cells was up-regulated by finasteride in LNCaP tumors (51.50% vs 71.00%, P = 0.001) (Figure 3a and 3b), and the apoptotic index was repressed in PC3 tumors (1.85% vs 0.59%, P < 0.001) (Figure 4a and 4c).
The role of c-Jun in the fibroblasts’ pro-proliferative function and for finasteride's therapeutic effects on the xenograft PCa tumors
The pro-proliferative effects of c-Jun−/− fibroblasts were not observed in LNCaP tumors (Figures 1a, 2a, and 2b); the Ki-67 inducing effects and the apoptosis-repressing effects of c-Jun−/− fibroblasts were not found in the LNCaP tumor tissue (Figure 3a, 3b and 3c). Compared with the obvious pro-proliferative effect of finasteride on tumor growth in the presence of c-Jun+/+ fibroblasts, finasteride did not stimulate LNCaP tumor growth in the presence of c-Jun−/− fibroblasts (Figures 1a, 2a, and 2b); the apparent Ki-67 stimulating effects of finasteride was no longer observed (Figure 3a and 3b), while finasteride still showed the apoptosis-repressing effect in LNCaP tumors in the presence of c-Jun−/− fibroblasts (1.27% vs 0.97%, P = 0.001) (Figure 3a and 3c).
The pro-proliferative function of c-Jun−/− fibroblasts disappeared in PC3 grafted tumors (Figures 1b, 2c, and 2d), and the Ki-67 inducing effects of c-Jun−/− fibroblasts were no longer noted (Figure 4a and 4b), while the pro-apoptotic effects of c-Jun−/− fibroblasts were still observed (1.06% vs 1.77%, P < 0.001) (Figure 4a and 4c). In the presence of c-Jun−/− fibroblasts in PC3 tumors, the finasteride feeding did not stimulate the tumor growth (Figures 1b, 2c, and 2d) compared with the significant pro-proliferative effect of finasteride in the presence of wild fibroblasts. Finasteride did not stimulate the Ki-67 expression (Figure 4a and 4b), the significant apoptosis-repressing effect was still observed (1.77% vs 0.93%, P < 0.001) (Figure 4a and 4c), although a much stronger apoptosis-repressing effect was noted in the presence of wild fibroblasts (0.93% vs 0.59%, P = 0.007) (Figure 4a and 4c).
Finasteride promoted the expression of p-Akt and p-ERK1/2 in LNCaP (PC3) cells when co-cultured with fibroblasts in vitro
Finasteride inhibited p-Akt and p-ERK1/2 in mono-culture of LNCaP (PC3) cells (Figure 5), but when LNCaP (PC3) cells were grown with mouse fibroblasts or HPF in co-culture transwells, finasteride up-regulated p-Akt and p-ERK1/2 in LNCaP and PC3 cells (Figure 5). When cells were co-cultured with c-Jun−/− fibroblasts, the pro-proliferative effect of finasteride for cancer cells was attenuated, especially for p-ERK1/2 (Figure 5).
Figure 5.

The expression of p-Akt and p-ERK1/2 in LNCaP (a) and PC3 (b) cells in mono-culture or in co-culture with fibroblasts or HPF in co-culture transwells. Finasteride repressed the expression of p-Akt and p-ERK1/2 in mono-culture of LNCaP (PC3) cells. However, when LNCaP (PC3) cells were grown with fibroblasts or HPF in co-culture transwell, Finasteride upregulated the expression of p-Akt and p-ERK1/2 in cancer cells. When c-Jun in fibroblasts was knocked out, the pro-proliferative effect of finasteride for LNCaP (PC3) cells was attenuated, especially for p-ERK1/2 in the same co-culture transwells.
DISCUSSION
Finasteride, a 4-azasteroid and analog of testosterone, works by acting as a potent and specific competitive inhibitor of the 5-AR2. For humans, finasteride suppresses DHT by 70.80% at 24 weeks. The intra-prostatic DHT level changes with differential 5-AR isoform expression in different prostatic disease states. Finasteride is therapeutically effective for BPH patients because 5-AR2 is the predominant form in BPH.15,16 When finasteride was investigated as a chemopreventive agent for PCa, the cases diagnosed as high-grade tumors increased, and it remains controversial whether finasteride promotes the development of high-grade PCa.
PC3 and LNCaP grafted nude mouse models were established and fed with finasteride. Finasteride did not induce tumor growth in the absence of fibroblasts; when fibroblasts were involved, finasteride significantly promoted cancer cell proliferation and inhibited cell apoptosis. We can postulate that with time, oral finasteride may stimulate the progression of high-grade PCa by its synergistic effect of promoting cell proliferation and repressing cell apoptosis. The preventive and therapeutic efficacy of finasteride and dutasteride were determined in TRAMP mice, dutasteride inhibited PIN and/or PCa progression, supporting the therapeutic use of dutasteride for PCa, but finasteride increased the incidence of high-grade PCa,17 suggesting that the increased incidence of high-grade PCa observed in PCPT may be an adverse effect of finasteride treatment.
The 5-AR isozymes’ differential expression in the prostate may be relevant when examining the chemopreventive and therapeutic effects of 5-AR inhibitors in BPH and PCa. Decreased expression of 5-AR2 and increased expression of 5-AR1 in primary PCa were observed compared within BPH (unpublished).16,18 The differential expression of 5-AR isozymes in PCa implies that dual inhibitors of 5-AR1 and 5-AR2 may be more effective in reducing the need for aggressive treatment of low-risk PCa in men under active surveillance19 or delaying PSA progression in patients with biochemical failure after RP or RT for PCa.20,21 The mechanisms driving the differential expression of 5-AR isozymes have not been adequately explored. Methylated CpG islands in the 5-AR2 promoter region could account for the reduced expression in some adult prostates.22 We are currently investigating whether methylation of the 5-AR2 promoter region is involved in the differential expression of 5-AR isozymes in PCa. Second, both 5-AR1 and 5-AR2 are present in epithelial and stromal cells in the human prostate, while 5-AR2 is the predominant isozyme in stromal cells.23 Fibroblast-secreted, soluble factor also induces the transcription of 5-AR2 mRNA in long-term primary cultures of prostate epithelial cells that can no longer transcribe 5-AR2 mRNA in the absence of fibroblasts.24 Fibroblasts and fibroblast-secreted factors are very important for the transcription and translation of 5-AR2 through stroma-epithelia crosstalk in prostatic tissue. Third, androgens, especially DHT, and the androgen receptor are vitally involved in the growth and progression of PCa. Koivisto et al.25 reported that PCa developing during finasteride therapy might have distinct biological properties, such as Xq gains and 6q losses as well as Arg726Leu mutation of the AR gene. One tumor with Xq gain had a 3-fold amplification of the AR gene, suggesting that tumor development in finasteride-treated patients may require an increased copy number and expression of AR.
The Jun-family of proteins is critical transcription factors that act as co-activators of the AR or form activator protein 1 with Fos to regulate the transcription of androgen-regulating genes. Activated c-Jun in the stromal cells plays key roles in stromal-epithelial interactions. Thakur et al. identified a novel binding site for activated c-Jun in the promoter of the Snail1 gene, which triggered TGFβ-induced invasion of human prostate cancer cells.26 We also observed the obvious pro-proliferative effects of c-Jun in fibroblasts for cancer cells, which is in agreement with Li's description that stromally expressed c-Jun promotes prostatic epithelial proliferation through the IGF-1 paracrine signal pathway.14 When c-Jun was knocked out, the pro-proliferative function of fibroblasts was compromised or absent. The stimulating effects of finasteride for cancer growth were attenuated to a reduced level. What we can infer is that the c-Jun mediated response plays critical roles in the pro-proliferative function of fibroblasts and the stimulating effects of finasteride for tumor growth. Chen et al.27 reported that the c-Jun is an AR coactivator that stimulates AR transactivation by mediating receptor dimerization and subsequent DNA binding; siRNA-mediated repression of endogenous c-Jun expression resulted in markedly reduced growth of PCa cells, strongly suggesting an important biological role for c-Jun in PCa, especially for CRCP. The DNA enzyme, Dz13, targeted against the oncogene c-Jun was also able to inhibit both loco-regional and distal metastasis of PCa, which further highlights the growing potential of Dz13 as an anti-neoplastic agent.28,29
The activated p-Akt and p-ERK1/2 pathways are closely related to cancer growth and progression. Although finasteride inhibited p-Akt and p-ERK1/2 in mono-culture cancer cells, finasteride induced p-Akt and p-ERK1/2 to a very high level in cancer cells when co-cultured with fibroblasts. Fibroblasts play critical roles in the chemopreventive or therapeutic effects of finasteride in prostatic diseases. Finasteride up-regulates pro-proliferative signals in cancer cells through its impact on stroma-epithelia crosstalk, stimulating tumor growth by promoting cell proliferation and repressing cell apoptosis. When PCa cells were grown alone or in combination with c-Jun−/− fibroblasts, the pro-proliferative function was repressed, which underlines the importance of c-Jun in epithelia-fibroblast interaction and finasteride's chemopreventive or therapeutic function. Another group observed deteriorating effects of androgen deprivation in a mouse model of high-grade prostatic intraepithelial neoplasia (HG-PIN) induced by PTEN loss in which surgical castration accelerated the disease progression of the stable HG-PIN to invasive CRPC. Targeting the PI3K signaling pathway via either genetic ablation of PI3K components or pharmacological inhibition of the PI3K pathway reversed the PTEN loss-induced HG-PIN phenotype. Concurrent inhibition of the PI3K and MAPK pathways was effective in blocking the growth of PTEN-null CRPC.30 These data indicate the potential adverse effects of anti-androgen chemoprevention in certain modified microenvironments (cancer reactive stroma or CAF) or genetic contexts (PTEN loss or continuously activated c-Jun) while demonstrating the potential promise of paracrine and stromal targeted therapy for the management of PCa, especially for CRPC.
More interestingly, when androgen-sensitive, AR-positive LNCaP and androgen-resistant, AR-negative PC-3 were used to determine the role of fibroblast and c-Jun in the therapeutic effects of finasteride for prostate cancer cells, we obtained very similar results for these two distinctly different cell-lines. Normal growth of these cancer cells and response to stress of finasteride may partly account for the similar results; more importantly, these results underscores the role of fibroblasts and c-Jun for finasteride's effects on both cancer cells through the epithelial-stroma interaction, apart from the AR signaling pathway. However, we also find subtle difference between these two cell-lines that finasteride induced proliferation of LNCaP cells while repressed PC3 cell apoptosis in the presence of fibroblasts. The final results were probably the synergistic effects among different signaling pathways. The paracrine factors between epithelial cells and stromal cells, such as IGF-1, were more and more emphasized, which is our direction in the following investigation.
Here, we need to mention some limitations in this investigation. First, although fibroblasts were reported to induce the growth of prostate epithelial cells,2,14 the data (Figures 1 and 2) in our article are not perfect to explain the inducing effect of fibroblasts for cancer cells, because in recombinant-grafted group, we implanted fibroblasts in addition to cancer cells compared with the cancer cells mono-grafted group, but we still found the pro-proliferative function of fibroblasts by increased Ki-67 expression and repressed apoptosis in cancer cells. In this article, we focused on the role of fibroblasts and c-Jun for the stimulating effects of finasteride for prostatic cancer cells, as above discussed. Second, the animal experiment period is 6–7 weeks, much shorter than the therapeutic time of months or years for men taking finasteride. During the limited period, it is hard to show the effect of finasteride fully for cancer cells in the presence of fibroblasts, although we found some significant differences in proliferation and apoptosis between groups with or without taking finasteride. Third, the application of matrigel is helpful for the LNCaP xenograft tumor growth, especially for the groups of mono-grafted tumors, while it was probably a confounding factor for evaluate the role of fibroblasts for finasteride's effects on cancer cells. We applied matrigel to all the groups, including mono-grafted and recombinant-grafted groups, to eliminate the confounding impact by maintaining the same volume and same concentration of matrigel.
CONCLUSIONS
Stromal cells and c-Jun promote tumor growth; finasteride further stimulates tumor growth through the synergistic effect of promoting cell proliferation and repressing cell apoptosis. Finasteride promotes certain molecular pathways that are associated with tumorigenesis and cancer progression in cancer cells in the presence of stromal cells and c-Jun. Stromal-epithelial interactions play critical roles in finasteride's chemopreventive and therapeutic effects on PCa. Our findings have preliminary implications in using finasteride as a chemopreventive or therapeutic agent for PCa patients.
AUTHOR CONTRIBUTIONS
YNN conceived the project, participated in its design and coordination, and helped to draft the manuscript. KW and SJ carried out animal experiment, data analysis and drafted the manuscript; DDF and MSW completed in vitro experiments and performed the statistical analysis; NZX and SJX helped to conceive of the study and participated coordination. All authors read and approved the final manuscript.
COMPETING INTERESTS
All authors declared no competing interests.
ACKNOWLEDGMENTS
We would like to thank Doctor Aria. F. Olumi for providing research training at Massachusetts General Hospital, the research work in his lab is very critical for us to form the primary idea and execute the experimental design for this investigation. Funding support: the National Natural Science Foundation of China (No. 30973015) and the Beijing Natural Science Foundation (No. 7122074) at Beijing Chaoyang Hospital, Capital Medical University to YNN.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Niu YN, Xia SJ. Stroma-epithelium crosstalk in prostate cancer. Asian J Androl. 2009;11:28–35. doi: 10.1038/aja.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 4.Thompson IJ, Goodman PJ, Tangen CM, Parnes HL, Minasian LM, et al. Long-term survival of participants in the prostate cancer prevention trial. N Engl J Med. 2013;369:603–10. doi: 10.1056/NEJMoa1215932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thompson IM, Chi C, Ankerst DP, Goodman PJ, Tangen CM, et al. Effect of finasteride on the sensitivity of PSA for detecting prostate cancer. J Natl Cancer Inst. 2006;98:1128–33. doi: 10.1093/jnci/djj307. [DOI] [PubMed] [Google Scholar]
- 6.Thompson IM, Tangen CM, Goodman PJ, Lucia MS, Parnes HL, et al. Finasteride improves the sensitivity of digital rectal examination for prostate cancer detection. J Urol. 2007;177:1749–52. doi: 10.1016/j.juro.2007.01.071. [DOI] [PubMed] [Google Scholar]
- 7.Lucia MS, Epstein JI, Goodman PJ, Darke AK, Reuter VE, et al. Finasteride and high-grade prostate cancer in the prostate cancer prevention trial. J Natl Cancer Inst. 2007;99:1375–83. doi: 10.1093/jnci/djm117. [DOI] [PubMed] [Google Scholar]
- 8.Kramer BS, Hagerty KL, Justman S, Somerfield MR, Albertsen PC, et al. Use of 5 alpha-reductase inhibitors for prostate cancer chemoprevention: American Society of Clinical Oncology/American Urological Association 2008 Clinical Practice Guideline. J Urol. 2009;181:1642–57. doi: 10.1016/j.juro.2009.01.071. [DOI] [PubMed] [Google Scholar]
- 9.Mottet N, Bellmunt J, Briers E, van den Bergh RC, Bolla M, et al. Guidelines on prostate cancer: risk factors and chemoprevention. Eur Assoc Urol. 2015:17. [Google Scholar]
- 10.Hsieh JT, Chen SC, Yu HJ, Chang HC. Finasteride upregulates expression of androgen receptor in hyperplastic prostate and LNCaP cells: implications for chemoprevention of prostate cancer. Prostate. 2011;71:1115–21. doi: 10.1002/pros.21325. [DOI] [PubMed] [Google Scholar]
- 11.Tarle M, Spajic B, Kraljic I, Kusic Z. Continuous finasteride therapy for benign prostate hypertrophy upgrades both neuroendorcine differentiation and aggressive prostate cancer. Anticancer Res. 2009;29:1797–801. [PubMed] [Google Scholar]
- 12.Yun DK, Lee J, Keum YS. Finasteride increases the expression of hemoxygenase-1 (HO-1) and NF-E2-related factor-2 (Nrf2) proteins in PC-3 Cells: implication of finasteride-mediated high-grade prostate tumor occurrence. Biomol Ther (Seoul) 2013;21:49–53. doi: 10.4062/biomolther.2012.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bass R, Perry B, Langenstroer P, Thrasher JB, Dennis KL, et al. Effects of short-term finasteride on apoptotic factors and androgen receptors in prostate cancer cells. J Urol. 2009;181:615–9. doi: 10.1016/j.juro.2008.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Wu CL, Febbo PG, Olumi AF. Stromally expressed c-Jun regulates proliferation of prostate epithelial cells. Am J Pathol. 2007;171:1189–98. doi: 10.2353/ajpath.2007.070285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gormley GJ, Stoner E, Bruskewitz RC, Imperato-McGinley J, Walsh PC, et al. The effect of finasteride in men with benign prostatic hyperplasia. The Finasteride Study Group. N Engl J Med. 1992;327:1185–91. doi: 10.1056/NEJM199210223271701. [DOI] [PubMed] [Google Scholar]
- 16.Wang K, Fan DD, Jin S, Xing NZ, Niu YN. Differential expression of 5-alpha reductase isozymes in the prostate and its clinical implications. Asian J Androl. 2014;16:274–9. doi: 10.4103/1008-682X.123664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Opoku-Acheampong AB, Unis D, Henningson JN, Beck AP, Lindshield BL. Preventive and therapeutic efficacy of finasteride and dutasteride in TRAMP mice. PLoS One. 2013;8:e77738. doi: 10.1371/journal.pone.0077738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas LN, Lazier CB, Gupta R, Norman RW, Troyer DA, et al. Differential alterations in 5 alpha-reductase type 1 and type 2 levels during development and progression of prostate cancer. Prostate. 2005;63:231–9. doi: 10.1002/pros.20188. [DOI] [PubMed] [Google Scholar]
- 19.Fleshner NE, Lucia MS, Egerdie B, Aaron L, Eure G, et al. Dutasteride in localised prostate cancer management: the REDEEM randomised, double-blind, placebo-controlled trial. Lancet. 2012;379:1103–11. doi: 10.1016/S0140-6736(11)61619-X. [DOI] [PubMed] [Google Scholar]
- 20.Schroder FH, Bangma CH, Wolff JM, Alcaraz A, Montorsi F, et al. Can dutasteride delay or prevent the progression of prostate cancer in patients with biochemical failure after radical therapy. Rationale and design of the Avodart after Radical Therapy for Prostate Cancer Study (ARTS)? BJU Int. 2009;103:590–6. doi: 10.1111/j.1464-410X.2009.08373.x. [DOI] [PubMed] [Google Scholar]
- 21.Schroder F, Bangma C, Angulo JC, Alcaraz A, Colombel M, et al. Dutasteride treatment over 2 years delays prostate-specific antigen progression in patients with biochemical failure after radical therapy for prostate cancer: results from the randomised, placebo-controlled Avodart after Radical Therapy for Prostate Cancer Study (ARTS) Eur Urol. 2013;63:779–87. doi: 10.1016/j.eururo.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Niu YN, Ge RB, Hu LB, Diaz C, Wang ZW, et al. Reduced levels of 5-alpha reductase 2 in adult prostate tissue and implications for BPH therapy. Prostate. 2011;71:1317–24. doi: 10.1002/pros.21348. [DOI] [PubMed] [Google Scholar]
- 23.Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, et al. Tissue distribution and ontogeny of steroid 5 alpha-reductase isozyme expression. J Clin Invest. 1993;92:903–10. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayne CW, Ross M, Inglis NF. Induction of 5 alpha-reductase type II mRNA transcription in primary cultured prostate epithelial cells by a soluble factor produced by primary cultured prostate fibroblast cells. Eur J Cancer. 2003;39:1004–11. doi: 10.1016/s0959-8049(03)00071-6. [DOI] [PubMed] [Google Scholar]
- 25.Koivisto PA, Schleutker J, Helin H, Ehren-van Eekelen C, Kallioniemi OP, et al. Androgen receptor gene alterations and chromosomal gains and losses in prostate carcinomas appearing during finasteride treatment for benign prostatic hyperplasia. Clin Cancer Res. 1999;5:3578–82. [PubMed] [Google Scholar]
- 26.Thakur N, Gudey SK, Marcusson A, Fu JY, Bergh A, et al. TGFbeta-induced invasion of prostate cancer cells is promoted by c-Jun-dependent transcriptional activation of Snail1. Cell Cycle. 2014;13:2400–14. doi: 10.4161/cc.29339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen SY, Cai C, Fisher CJ, Zheng Z, Omwancha J, et al. c-Jun enhancement of androgen receptor transactivation is associated with prostate cancer cell proliferation. Oncogene. 2006;25:7212–23. doi: 10.1038/sj.onc.1209705. [DOI] [PubMed] [Google Scholar]
- 28.Elahy M, Dass CR. Dz13: c-Jun downregulation and tumour cell death. Chem Biol Drug Des. 2011;78:909–12. doi: 10.1111/j.1747-0285.2011.01166.x. [DOI] [PubMed] [Google Scholar]
- 29.Tan ML, Choong PF, Dass CR. Direct anti-metastatic efficacy by the DNA enzyme Dz13 and downregulated MMP-2, MMP-9 and MT1-MMP in tumours. Cancer Cell Int. 2010;10:9. doi: 10.1186/1475-2867-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia S, Gao X, Lee SH, Maira SM, Wu X, et al. Opposing effects of androgen deprivation and targeted therapy on prostate cancer prevention. Cancer Discov. 2013;3:44–51. doi: 10.1158/2159-8290.CD-12-0262. [DOI] [PMC free article] [PubMed] [Google Scholar]