

Abstract
Asbestos in combination with tobacco smoke exposure reportedly leads to more severe physiological consequences than asbestos alone; limited data also show an increased disease risk due to environmental tobacco smoke (ETS) exposure. Environmental influences during gestation and early lung development can result in physiological changes that alter risk for disease development throughout an individual’s lifetime. Therefore, maternal lifestyle may impact the ability of offspring to subsequently respond to environmental insults and alter overall disease susceptibility. In this study, we examined the effects of exposure to ETS in utero and during early postnatal development on asbestos-related inflammation and disease in adulthood. ETS exposure in utero appeared to shift inflammation towards a Th2 phenotype, via suppression of Th1 inflammatory cytokine production. This effect was further pronounced in mice exposed to ETS in utero and during early postnatal development. In utero ETS exposure led to increased collagen deposition, a marker of fibrotic disease, when the offspring was later exposed to asbestos, which was further increased with additional ETS exposure during early postnatal development. These data suggest that ETS exposure in utero alters the immune responses and leads to greater disease development after asbestos exposure, which is further exacerbated when exposure to ETS continues during early postnatal development.
Keywords: Asbestos, environmental tobacco smoke, inflammation, fibrosis
Introduction
Asbestos is a naturally occurring silicate mineral fiber that, once inhaled, leads to disease. Due to asbestos’ insulating and fireproof properties, it has been used extensively for building and manufacturing globally. One major source of asbestos exposure is from a unique amphibole fiber associated with vermiculite, that is used predominately for insulation; there are an estimated 35 million homes with vermiculite containing asbestos (Gunter et al., 2005). Additionally, many developing countries have a large demand for affordable, mass produced building materials, which keeps global use of asbestos high (Ewell, 2011). Therefore, the incidence of asbestos-related disease is expected to rise substantially in these countries in years to come (McCulloch and Tweedale, 2008). Given the current health risk for asbestos-exposed individuals, it is imperative that we improve our understanding of the mechanisms behind asbestos-related disease. This will, in turn, provide information needed to reduce the risk for developing and improve the treatment of asbestos-related disease in exposed individuals.
Smoking has been identified as a risk factor that dramatically increases the likelihood of asbestos-related disease development. Several studies have reported increased risk of cancer as well as an increase in overall mortality for smokers exposed to asbestos (Savastano et al., 2004, Veglia et al., 2007, Hammond et al., 1979). Less is known about how tobacco smoke affects risk for the development of pleural disorders after asbestos exposure, but one study reported increased pleural thickening in smokers exposed to asbestos (Christensen and Kopylev, 2012). Another study examined the effect of secondhand smoke exposure in adults exposed to asbestos and reported increased lung opacity, an indicator of inflammation and/or fibrotic interstitial thickening in the lung, in dually exposed individuals (Vierikko et al., 2008). However, the mechanisms by which tobacco smoke increases disease risk in asbestos-exposed individuals are not clear.
In order to better understand how tobacco smoke affects asbestos-induced disease it is important to consider the type of inflammatory response induced by each exposure. Two types of inflammatory responses, Th1 and Th2 are in balance with one another and may shift more towards one direction or another depending on what is causing an inflammatory response (Berger, 2000). Th1 immune responses are upregulated when fighting off pathogens and are associated with a strong inflammatory response. Th2 immune responses can resolve inflammation, but also have the potential to lead to autoimmunity if the balance shifts too far towards Th2 inflammation and can increase risk for tissue fibrosis (Charlton and Lafferty, 1995; Barron and Wynn, 2011). A number of studies have shown that smoking leads to a reduction in Th1 type immune response and an increase in Th2 type immune response (de Heens et al., 2009, Cozen et al., 2004, Slavinsky et al., 2002), including polarization of macrophages from M1 towards M2 (Shaykhiev et al., 2009). These alterations in inflammatory response increase risk for tissue fibrosis (Barron and Wynn), which has been observed in studies showing an increased risk of idiopathic pulmonary fibrosis in smokers (Baumgartner et al., 1997). It is likely that these alterations in smokers would increase risk for the development of tissue fibrosis associated with asbestos exposure as well.
Exposure to tobacco smoke in early life can be especially detrimental as it affects the lung before it is fully mature, and has the potential to cause non-reversible alterations during development that impact an individual’s disease risk throughout their lifetime (Lantz et al., 2009). For example, exposure in utero to environmental tobacco smoke (ETS) has been reported to have deleterious consequences on lung development and function (Stocks and Dezateux, 2003), and these alterations affect an individual’s risk for disease development later (Stein et al., 1999). To our knowledge no studies have assessed the effect of ETS exposure in utero and/or during early postnatal development on altering risk for disease development due to asbestos exposure later in life.
Our study objective was to determine whether in utero and/or early life ETS exposure alters asbestos-induced inflammation and lung disease development. Most asbestos exposures occur during adulthood, so it is important to examine how adult populations respond to asbestos exposure, after early life ETS exposures, to better understand the role of ETS on asbestos-induced disease. Therefore, pregnant C57BL/6 mice were exposed to either filtered air (FA), ETS during gestation (prenatal ETS), or ETS during gestation and the first 3 weeks of life (pre/postnatal ETS). When offspring from all three groups reached adulthood (twelve weeks of age), they were exposed to asbestos for either 24 hr to determine the effect of ETS on acute asbestos-induced inflammation or they were exposed to asbestos using a chronic model to determine the effect of ETS on asbestos-induced disease development.
Methods
Animals
C57BL/6 mice were housed in an SPF facility with controlled environmental conditions (22 ± 2°C; 30–40% humidity, 12 hr light: 12 hr dark cycle) and provided food and water ad libitum. All procedures were performed under protocols approved by the IACUC of the University of Montana.
Breeding and ETS Exposure
Pregnant C57BL/6 mice (8–9 week-old) were either exposed to FA, prenatal ETS, or pre/postnatal ETS; offspring from all three groups were exposed to asbestos at 12 weeks of age as shown in Figure 1.
Figure 1. A model showing ETS and asbestos exposures.

Group 1: FA, Group 2: prenatal ETS, Group 3: pre/postnatal ETS exposed to asbestos at 12 weeks of age. WLL fluid was collected 24 hr after exposure for the acute study. Mice in the chronic study were exposed to asbestos once per week for an additional three weeks. One month later lung tissue was collected for histological analysis.
For the purposes of breeding, two female mice were paired with one male mouse to create a timed-pregnant exposure scenario. Breeding and ETS exposure was carried out in the Center for Health and the Environment’s animal facilities at University of California-Davis. Following verification of a vaginal plug, at approximately day 1 of gestation, four and eight female mice were exposed to either FA or ETS throughout gestation, respectively. For the control group, timed-pregnant mice were exposed to only FA for 24 hr 7d/week for the duration of the study. For the ETS-exposed group, timed-pregnant dams were exposed daily to approximately1 mg/m3 of tobacco smoke for 6 hr/day for 7days/week. Since an active smoker can attain a personal cloud of particulate levels as high as 2.0 mg/m3 (Jinot and Bayard, 1994) the total concentration of suspended particulates was maintained at 1.0 ± 0.17 mg/m3 for this study to mimic environmental tobacco smoking concentrations. Research cigarettes (3R4F, University of Kentucky) were burned at a rate of two cigarettes every 10 min with a puff volume of 35 mL over 2 sec, once per minute. Both side stream and mainstream cigarette smoke were collected and passed to a dilution and aging chamber to achieve the target concentration of ETS. The carbon monoxide level was 4.8 ± 0.8 ppm, and the average temperature was 73°F. Once the dams gave birth, four dams and their offspring were moved out of the ETS chamber and subsequently exposed only to FA. Another four dams and offspring were further exposed to ETS under identical conditions for an additional 3 weeks. Following this additional period of ETS or FA exposure, the dams and pups were exposed to only FA until weaning and then shipped to the University of Montana via air. Upon arriving at the University of Montana, the dams and the offspring were quarantined for 3–4 weeks.
The mean litter size per FA- and ETS-exposed dams was 6.9 ± 0.3. The litter size and sex-ratio of the pups were not significantly different among groups (data not shown). Among total offspring, 24 pups from each group (group 1, 2, and 3) were randomly selected with the same sex ratio. These 24 pups were again split into 4 groups (asbestos vs. dispersion media (DM), and acute vs. chronic exposure), and 6 pups per each subgroup were used for further analyses.
Particle Collection and Characterization
Libby 6-mix was obtained from the U.S. Geological Survey. These amphibole fibers have been previously chemically and physically characterized in detail (Gunter ME, 2003, Meeker GP, 2003). The respirable fraction of amphibole fibers was obtained via an elutriation process, and the size of the fibers was verified using transmission electron microscopy, as previously described (Webber et al., 2008).
Asbestos Instillations
As shown in Figure 1, at twelve weeks old, offspring were anesthetized using isoflurane, and exposed to 50 ug of the Libby respirable fraction of asbestos by oropharyngeal aspiration that was suspended at a concentration of 2 mg/ml in DM (60% Phosphate Buffered Saline, 39 % Mouse Serum Albumin, 1 % distearoylphosphatidylcholine) or to DM alone. Asbestos suspensions were sonicated for 30 seconds at half maximum power in a Masonix cup-horn sonicator (XL2020, Farmingdale, NY) attached to a Forma circulating water bath at 550 watts and 20 Hz (8000 Joules). A volume of 25 μl of particle suspension (50 μg) or DM was delivered into the back of the throat. Holding the tongue to the side results in aspiration of the solution into the lungs.
Acute exposures to asbestos consisted of a single dose (50 μg of asbestos). Assessment of acute inflammatory response was determined 24 hr after asbestos exposure. The chronic exposure model used in this study was first developed for silica in our laboratory and modified for the asbestos experiments (Beamer et al., 2012). Specifically, the mice were exposed once/week for 4 weeks using the same asbestos concentration for each instillation as was used in the acute study; 28 days after the last exposure the mice were examined for chronic effects on the lung. Dose and durations of asbestos exposure were selected based on preliminary studies determining maximal acute inflammatory reaction at 24 hr, and chronic studies were based on a model of chronic-silica induced inflammation developed in our laboratory (Beamer et al., 2012).
Tissue Collection
Asbestos-exposed mice were euthanized with sodium pentobarbital (Euthasol™) 24 hr after exposure for the acute study and 56 days after exposure for the chronic study. For the acute study, whole lung lavage (WLL) fluid was collected from all mice using 1 ml of ice-cold PBS (pH 7.4) in order to be used for cytokine analysis as well as cell counts and differentials. One ml of lavage fluid was inserted and removed from the lungs 3 times in order to increase cytokine concentration. For the chronic study, lung tissues from mice were collected and fixed in 1 ml of 3% paraformaldehyde-PBS and submerged in the same fixative overnight at 4°C. Further histological preparation is described in more detail below.
Cytokine Release
A meso-scale discovery V-PLEX Proinflammatory Panel 1 (mouse) kit (MSD Rockville, Maryland) was used to determine cytokine concentrations according to manufacturer’s instructions.
Inflammatory Cell Counts and Differentials
Cell differentials were determined by centrifuging a small sample (35 × 103 cells) onto positively charged glass slides in a cytocentrifuge at 1500 rpm for 5 min (Shandon Cytospin 3, Thermo Fisher, Houston, TX). The slides were then stained in a Hematek slide stainer (Bayer Diagnostics, Dublin, Ireland) using a modified Wright-Giemsa stain (Protocol, Fisher, Houston, TX). Differentials were conducted using a Zeiss microscope at 400× and 200 cells counted per slide.
Histology and Tri-Chrome Staining
The lungs from each mouse were fixed, sectioned, and stained as previously described (Hamilton et al., 2012, Beamer et al., 2010). In brief, the lungs from each mouse were inflation-fixed overnight, then rinsed the following day with PBS 3 times and placed in 70% ethanol. The trachea and heart were removed and the lung tissues were placed in labeled cassettes. Processing was completed in a Leica ASP 300 tissue processor (Buffalo Grove, IL) on a 7.25 hour program: 30 minutes in 70% and 95% ethanol, two 1 hour changes in 100% ethanol, three changes of xylene for 30 minutes each and three paraffin changes at 45 minutes for the first bath and 1 hour in the second and third changes under vacuum. Tissues were sectioned at a thickness of 5 microns using a Leica RM2235 microtome (Buffalo Grove, IL). Both the H&E and trichrome staining were done in a Shandon 24-4 autostainer (GMI, Ramsey, MN). Mayer’s hematoxylin (Richard-Allan Scientific, Kalamazoo, MI) and alcoholic eosin (Thermo Shandon Limited, Runcorn, UK) were used for the H & E program. Weigert’s hematoxylin (Electron Microscopy Sciences, Hatfield, PA) and Gomori Trichrome (Harleco, EMD, VWR Randor, PA) were used for the trichrome staining. Histological analysis was done using stained tissues by a certified veterinary pathologist. Additionally, assessments of collagen deposition were obtained from the stained lung tissue.
Collagen Deposition
“Phantom” contours were used to divide up the tissue into very small (8 μm diameter) circles. If a contour showed blue staining, indicating collagen deposition via the Trichrome stain, it was counted as a collagen-positive event. A percent of positive contours over total tissue contours was derived to compare one tissue to the next. Given that airways and blood vessels naturally have collagen in them for structural purposes, all airways and blood vessels were isolated and removed from the calculation of total collagen deposition. Two sections using one lobe of lung tissue, between 21–28 microns apart, were analyzed and averaged.
Statistical analysis
A 2-way ANOVA was used in cases where there was more than 1 variable using the Tukey test. Post-hoc analysis was done comparing multiple variables using a 2-sided test, except in certain cases where a 1-sided test was used. There was unequal variance in at least one variable for both the neutrophil data and the cell count data. Therefore, data were log transformed when there was unequal variance in at least one level of a variable. In cases where one level of a variable had a zero, data were transformed using (log +1) as recommended by Freeman (David Freedman, 2007). Statistical power was greater than 0.8. Statistical significance was defined as a probability of type I error occurring at less than 5% (P < 0.05). The minimum number of experimental replications was 3 or greater. Graphics and analyses were performed on PRISM 5.0.
Results
Cytokine Production
Cytokine production was used as an indicator of the degree of inflammation induced by asbestos and to determine how ETS exposure in utero and during early postnatal development altered that response (Figure 2). For the non-asbestos exposed mice, relatively low levels of cytokines were observed for all groups (FA, prenatal ETS, and pre/postnatal ETS). However, for several cytokines, including IL-1β, TNF-α, and IL-6, the mice exposed to ETS pre/postnatal trended towards a higher overall average of cytokine production when compared to the FA mice (Figure 2A, B &C), although it was not significant. This may suggest that some degree of background inflammation occurred in these mice 9 weeks post-ETS exposure.
Figure 2. Inflammatory cytokine release for FA, prenatal, and pre/postnatal ETS-exposed mice in WLL after 24 hr asbestos exposure.

Alteration in level of (A) IL-1β, (B) TNF-α, (C) IL-6, (D) IL-5, (E) KC/GRO, and (F) IL-10. Data are expressed as mean ± SEM. Asterisks indicate significance ** at P < 0.01, * at P < 0.05 compared to FA, no particle. n = 6 mice per condition.
Asbestos exposure significantly increased IL-1β, TNF-α, IL-6, and IL-5 in the FA mice (Figure 2A, B, C, & D). Both ETS groups led to a significant increase in IL-6 and IL-5 after asbestos exposure when compared to the FA, no particle group (Fig. 2C & D), but not IL-1 β, or TNF- α as was seen in the FA mice exposed to asbestos. These data show that there were lower levels of Th1-associated inflammatory cytokine production after asbestos exposure in the prenatal and pre/postnatal ETS exposed groups.
Inflammatory Cell Infiltration
Cell counts and differentials were determined 24 hr after asbestos exposure. The number of cells pulled from the WLL was the same for all groups, with the exception of the prenatal ETS exposure and no particle groups having fewer cells (Figure 3A). Asbestos exposure decreased the number of macrophages in the lung, presumably due to cell death after phagocytizing asbestos fibers (Figure 3B). The number of macrophages in the lung after asbestos exposure was not further affected by either ETS exposure. Asbestos-exposure also led to a significant increase in neutrophil influx; however, similar to the macrophage data, the ETS exposures had no additional affect on neutrophil influx (Figure 3C).
Figure 3. Inflammatory cell infiltration for FA, prenatal, and pre/postnatal ETS-exposed mice in WLL after 24 hr asbestos exposure.

(A) Total cell numbers, (B) macrophages, and (C) neutrophils. Data expressed as mean ± SEM. Asterisks indicate significance, ** at P < 0.01, * at P < 0.05 compared to FA, no particle. n = 6 mice per condition.
Histopathology and Collagen Deposition
Collagen deposition in the lungs was used as an indicator of the degree of tissue fibrosis. Lung tissue sections in the chronic asbestos-exposed groups were stained with trichrome and hemotoxylin and eosin (H & E). Figure 4 shows representative photomicrographs for each group. In the non-asbestos-exposed groups, both the FA and prenatal ETS exposed groups appear to have normal lung morphology (Figure 4A & B). However, the non-asbestos-exposed pre/postnatal ETS group showed areas of abnormal collagen deposition and lung morphology (Figure 4C), suggesting that prenatal plus postnatal ETS exposure has the potential to detrimentally impact lung pathology in the long-term.
Figure 4. Collagen deposition in ETS- and asbestos-exposed mice.

Representative pictures of interstitial histology for each group.
Asbestos exposure led to visible increases in abnormal lung pathology and inflammatory cell infiltration for all three FA and ETS groups (Figure 4D, E, & F). The ETS groups had higher levels of abnormal lung pathology than the FA group, with the pre/postnatal ETS group being the most affected. This suggests that exposure to ETS during development and early life can detrimentally affect the response to secondary exposures later in life.
The degree of visible disease pathology is reflected in the percent collagen deposition for the asbestos-exposed groups (Figure 5). All three groups showed an increase in collagen deposition, although only the prenatal ETS and the pre/postnatal ETS groups reached significance. The increase in collagen deposition for the prenatal ETS group suggests that alterations are occurring in utero that affect disease risk due to exposures much later in life. The pre/postnatal ETS group had the highest level of collagen deposition, suggesting that the early life exposures further contribute to alterations in disease risk.
Figure 5. Collagen deposition for FA, prenatal and pre/postnatal ETS-exposed mice after chronic asbestos exposure.
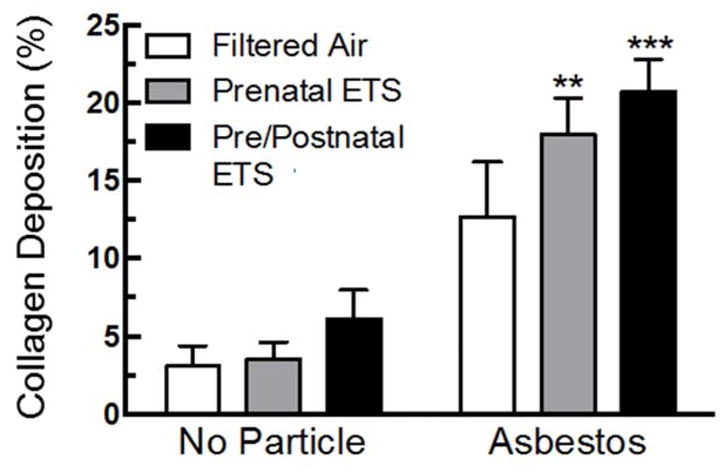
Asbestos exposure leads to a significant increase in collagen deposition for ETS groups, and the pre/postnatal ETS group appeared to have the highest level of abnormal lung pathology. Data expressed as mean ± SEM. Asterisks indicate significance *** at P < 0.001, ** at P <0.01 compared to FA, no particle. n = 3 mice per condition.
Discussion
The effect of in utero and/or early life ETS exposure on asbestos-induced inflammation and disease later in life was examined. The prenatal ETS group did not have as large of a response to asbestos exposure in terms of Th1 cytokine production when compared to the asbestos-exposed, FA group (Figure 2). The pre/postnatal ETS group had the lowest level of Th1 cytokine response to asbestos overall (Figure 2). The suppression of Th1-associated cytokine release indicates a likely shift in the lung inflammatory balance towards Th2 immunity due to ETS exposure, similar to what has been reported in other studies (Hagiwara et al., 2001, Ouyang et al., 2000). The shift in inflammatory phenotype has a number of implications including alterations in macrophage activation state, phagocytic function, fiber clearance, and degree of tissue fibrosis after exposure.
The inflammatory phenotype (Th1 vs Th2) is typically reflected in macrophage activation state (Martinez et al., 2008). Macrophages in the M1 activation state are associated with release of IFN-γ, IL-1β, TNF-α, and IL-6 (Mantovani et al., 2004). While IFN-γ did not appear above detectable limits after asbestos exposure, the other three cytokines were produced in response to asbestos exposure and to a lesser degree in the ETS exposed mice. Consequently, the shift in immune response could be explained by alterations in macrophage phenotype. A reduction in the number of M1 macrophage markers has previously been reported in smokers with chronic obstructive pulmonary disease (COPD) compared to smokers without COPD (Hodge et al., 2011). This supports the potential for smoking to alter the macrophage activation state, particularly in those with disease. While much of the cytokine data shows a similar trend, with less cytokines being released in the ETS-exposed groups after asbestos exposure compared to the non-ETS exposed group after asbestos exposure, the level of cytokines detected in the lavage fluid was fairly low. It is possible that the asbestos fibers are binding cytokines in the lung lining creating lower levels of detectable cytokine concentrations. Future studies should confirm the cytokine release data by determining gene expression of the cytokines in the lung.
Recent evidence has emerged that a type of innate lymphoid cell, ILC2, may play a role in type 2 immune pathologies and could be affecting macrophage activation states (Scanlon and McKenzie, 2012, Artis and Spits, 2015). ILC2’s express the ST2 receptor, which when bound by IL-33, leads to release of type 2 cytokines such as IL-5 (Scanlon and McKenzie, 2012). Cigarette smoke exposure leads to increases in IL-33 release (Kearley et al., 2015). Reports have claimed that increases in IL-33 can polarize macrophages more toward a type 2 phenotype (Kurowska-Stolarska et al., 2009). These studies suggest that ETS exposure may increase IL-33 release which would activate ILC2 cells leading to greater release of type 2 cytokines such as IL-5 as well and polarize macrophages toward type 2. This is supported by our data which showed higher levels of IL-5 production in ETS-exposed mice compared to FA-exposed mice in response to asbestos. Future studies should be done to determine the primary cause of alterations in inflammatory cytokine production including whether or not a shift in macrophage phenotype occurs due to ETS exposure and what role ILC2 cells play in this process. This could include histological assessment of lymphocytes in the tissue of chronically-exposed mice to determine if the alterations in inflammatory phenotype (Th1 vs Th2) persist.
Macrophages are the primary phagocytic cell responsible for removing inhaled asbestos from the lungs. Cigarette smokers are reported to have reduced macrophage phagocytic function (Kirkham et al., 2004, Ortega et al., 1994). The ability of phagosomes and lysosomes to fuse is defective in macrophages from smokers compared to non-smokers; the reduced phagocytosis ability impairs the macrophage’s capacity to kill microbes and clear bacteria (Harris and Gonzalez-Rothi, 1984, Phipps et al., 2010). Given that asbestos triggers similar pathways for phagocytizing particles that are used for microbes, it is possible that this is also the case for these mice, reducing their ability to clear the fibers. Decreases in phagocytic function of neutrophils have also been reported in association with cigarette smoke (Mehta et al., 2008), and may also be playing a role in reduced particle clearance in this model. We tried to estimate the number of asbestos particles remaining in the lung tissue after the chronic asbestos exposure using CytoViva hyperspectral imaging; however, there was no significant effect of particle clearance in this study. This might be because of our small sample size (data not shown). In addition, there is often a marked difference in body weight and overall size of the lungs in males and female mice. Therefore, it could be that female mice actually received a slightly higher dose of asbestos than males (relative to the amount of lung tissue present) as they were treated with the same dose of asbestos. We found female mice were more susceptible to asbestos exposure, but it was not statistically significant due to small sample size. Therefore, caution is required when extrapolating this data, and additional study with larger samples would be needed.
The reduction in Th1-associated cytokine production in the group exposed to ETS in utero is an especially important finding. These data imply that maternal exposure to ETS during pregnancy can lead to similar alterations in immune response in offspring as was seen with direct exposure to ETS during early development. This evidence provides a potential mechanistic explanation for reported alterations in lung function and disease risk observed in offspring from maternal smokers (Hofhuis et al., 2003, Gilliland et al., 2002, Gilliland et al., 2001). Offspring from maternal smokers have an increased risk for asthma development and wheezing that is associated with increased Th2-type inflammatory responses (Gilliland et al., 2001, Truyen et al., 2006, Abrahamsson et al., 2011). Furthermore, given that the mothers in this study were only exposed to ETS, this implies that offspring of non-smokers developing in utero in an environment where mothers are exposed to second-hand smoke may have similar alterations in lung function and disease risk as offspring of mothers who are smokers.
While the acute inflammatory response to asbestos exposure was generally reduced in relation to cytokine production due to ETS exposure, the ETS exposure did not appear to affect inflammatory cell infiltration (Figure 3). It is likely that the reduction in cytokine release was not great enough to lead to a significant alteration in inflammatory cell infiltration, but as discussed below, it appears that the shift in inflammatory cytokine production was sufficient to alter disease development later on.
The photomicrographs and collagen deposition data of the lung tissue show an increase in disease development after chronic asbestos exposure, with the pre/postnatal ETS-exposed group affected most significantly (Figures 4 & 5). It is possible therefore, that the initial strong Th1 type immune response, including increased cytokine production, was needed in order to better resolve overall disease development. Given that ETS in this study appeared to shift the immune response from Th1 towards Th2 type disease, and that tissue fibrosis is associated with Th2 type inflammation (Barron and Wynn, 2011), it makes sense that greater levels of tissue fibrosis would be observed in ETS-exposed mice. The shift towards Th2 type immune response may be due to alternate activation of the ILC2s which has also been shown to increase risk for tissue fibrosis (Hams et al., 2015). Additional factors, such as altered macrophage activation state and fiber clearance could also be affecting the degree of tissue fibrosis in the mice. Similar to our findings, a previous study evaluating the effects of long term combined amosite asbestos and cigarette smoke found an additive effect of the two on renal fibrosis (Boor et al., 2009). Taken together, results from our study show that ETS exposure in utero and during early development alters the inflammatory response to asbestos exposure and increases the risk for disease development.
Conclusions
In this study, ETS exposure in utero appeared to shift inflammation towards a Th2 phenotype via suppression of Th1 inflammatory cytokine production. This effect was further pronounced in mice exposed to ETS both in utero and during early development. In utero and/or early life ETS exposure also led to increased collagen deposition, a marker of fibrotic disease, after asbestos exposure. Collectively, these data suggest that ETS exposure in utero alters immune response and leads to greater disease development after asbestos exposure, which is further exacerbated when exposure to ETS continues during early development.
Acknowledgments
The National Institute of General Medical Sciences (P30 GM10333) and the National Institute of Environmental Health Sciences (R01 ES023209) supported this work. The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH, NIGMS, or NIEHS. The authors thank Pam Shaw, Lou Herritt, Mary Buford, Britten Postma, Virginia Kay, Melisa Schelvan (Center for Environmental Health Sciences at University of Montana), Dale Uyeminami and Shanie McCarty (Center for Health and the Environment at University of California, Davis) for their expert technical assistance with various aspects of this manuscript. We also thank the work of Dr. Jack Harkema at Michigan State University who reviewed and scored lung tissue sections of the ETS and asbestos-exposed mice as well as provided a summary of the observed histopathology.
Footnotes
Declaration of interest
No potential conflicts of interest were disclosed.
References
- ABRAHAMSSON TR, SANDBERG ABELIUS M, FORSBERG A, BJORKSTEN B, JENMALM MC. A Th1/Th2-associated chemokine imbalance during infancy in children developing eczema, wheeze and sensitization. Clin Exp Allergy. 2011;41:1729–39. doi: 10.1111/j.1365-2222.2011.03827.x. [DOI] [PubMed] [Google Scholar]
- ARTIS D, SPITS H. The biology of innate lymphoid cells. Nature. 2015;517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- BARRON L, WYNN TA. Fibrosis is regulated by Th2 and Th17 responses and by dynamic interactions between fibroblasts and macrophages. Am J Physiol Gastrointest Liver Physiol. 2011;300:G723–8. doi: 10.1152/ajpgi.00414.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAUMGARTNER KB, SAMET JM, STIDLEY CA, COLBY TV, WALDRON JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–8. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- BEAMER CA, MIGLIACCIO CT, JESSOP F, TRAPKUS M, YUAN D, HOLIAN A. Innate immune processes are sufficient for driving silicosis in mice. J Leukoc Biol. 2010;88:547–57. doi: 10.1189/jlb.0210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAMER CA, SEAVER BP, SHEPHERD DM. Aryl hydrocarbon receptor (AhR) regulates silica-induced inflammation but not fibrosis. Toxicol Sci. 2012;126:554–68. doi: 10.1093/toxsci/kfs024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERGER A. Th1 and Th2 responses: what are they? BMJ. 2000;321:424. doi: 10.1136/bmj.321.7258.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOOR P, CASPER S, CELEC P, HURBANKOVA M, BENO M, HEIDLAND A, AMANN K, SEBEKOVA K. Renal, vascular and cardiac fibrosis in rats exposed to passive smoking and industrial dust fibre amosite. J Cell Mol Med. 2009;13:4484–91. doi: 10.1111/j.1582-4934.2008.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHARLTON B, LAFFERTY KJ. The Th1/Th2 balance in autoimmunity. Curr Opin Immunol. 1995;7:793–8. doi: 10.1016/0952-7915(95)80050-6. [DOI] [PubMed] [Google Scholar]
- CHRISTENSEN KY, KOPYLEV L. Localized pleural thickening: smoking and exposure to Libby vermiculite. J Expo Sci Environ Epidemiol. 2012;22:320–3. doi: 10.1038/jes.2012.18. [DOI] [PubMed] [Google Scholar]
- COZEN W, DIAZ-SANCHEZ D, JAMES GAUDERMAN W, ZADNICK J, COCKBURN MG, GILL PS, MASOOD R, HAMILTON AS, JYRALA M, MACK TM. Th1 and Th2 cytokines and IgE levels in identical twins with varying levels of cigarette consumption. J Clin Immunol. 2004;24:617–22. doi: 10.1007/s10875-004-6247-0. [DOI] [PubMed] [Google Scholar]
- DAVID FREEDMAN RP. Statistics. W.W. Norton & Company; 2007. ROGER PURVES. [Google Scholar]
- DE HEENS GL, VAN DER VELDEN U, LOOS BG. Cigarette smoking enhances T cell activation and a Th2 immune response; an aspect of the pathophysiology in periodontal disease. Cytokine. 2009;47:157–61. doi: 10.1016/j.cyto.2009.05.006. [DOI] [PubMed] [Google Scholar]
- EWELL ME. Minerals Yearbook, 2008, V. 1, Metals and Minerals. 2011. MINING AND QUARRYING TRENDS—2008 [ADVANCE RELEASE] 3.1. [Google Scholar]
- GILLILAND FD, LI YF, DUBEAU L, BERHANE K, AVOL E, MCCONNELL R, GAUDERMAN WJ, PETERS JM. Effects of glutathione S-transferase M1, maternal smoking during pregnancy, and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2002;166:457–63. doi: 10.1164/rccm.2112064. [DOI] [PubMed] [Google Scholar]
- GILLILAND FD, LI YF, PETERS JM. Effects of maternal smoking during pregnancy and environmental tobacco smoke on asthma and wheezing in children. Am J Respir Crit Care Med. 2001;163:429–36. doi: 10.1164/ajrccm.163.2.2006009. [DOI] [PubMed] [Google Scholar]
- GUNTER ME, DD, TWAMLEY B, FOIT FF, JR, CORNELIUS S. Composition, Fe+3/Fe and crystal structure of non-asbestiform and asestiform amphiboles from Libby, Montana, USA. AM Mineralogist. 2003;89:1579. [Google Scholar]
- GUNTER ME, SINGLETON E, BANDLI BR, LOWERS HA, MEEKER GP. Differentiation of commercial vermiculite based on statistical analysis of bulk chemical data: Fingerprinting vermiculite from Libby, Montana USA. American Mineralogist. 2005;90:749–754. [Google Scholar]
- HAGIWARA E, TAKAHASHI KI, OKUBO T, OHNO S, UEDA A, AOKI A, ODAGIRI S, ISHIGATSUBO Y. Cigarette smoking depletes cells spontaneously secreting Th(1) cytokines in the human airway. Cytokine. 2001;14:121–6. doi: 10.1006/cyto.2001.0860. [DOI] [PubMed] [Google Scholar]
- HAMILTON RF, JR, BUFORD M, XIANG C, WU N, HOLIAN A. NLRP3 inflammasome activation in murine alveolar macrophages and related lung pathology is associated with MWCNT nickel contamination. Inhal Toxicol. 2012;24:995–1008. doi: 10.3109/08958378.2012.745633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMMOND EC, SELIKOFF IJ, SEIDMAN H. Asbestos exposure, cigarette smoking and death rates. Ann N Y Acad Sci. 1979;330:473–90. doi: 10.1111/j.1749-6632.1979.tb18749.x. [DOI] [PubMed] [Google Scholar]
- HAMS E, BERMINGHAM R, FALLON PG. Macrophage and Innate Lymphoid Cell Interplay in the Genesis of Fibrosis. Front Immunol. 2015;6:597. doi: 10.3389/fimmu.2015.00597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRIS JO, GONZALEZ-ROTHI RJ. Abnormal phagolysosome fusion in pulmonary alveolar macrophages of rats exposed chronically to cigarette smoke. Am Rev Respir Dis. 1984;130:467–71. doi: 10.1164/arrd.1984.130.3.467. [DOI] [PubMed] [Google Scholar]
- HODGE S, MATTHEWS G, MUKARO V, AHERN J, SHIVAM A, HODGE G, HOLMES M, JERSMANN H, REYNOLDS PN. Cigarette smoke-induced changes to alveolar macrophage phenotype and function are improved by treatment with procysteine. Am J Respir Cell Mol Biol. 2011;44:673–81. doi: 10.1165/rcmb.2009-0459OC. [DOI] [PubMed] [Google Scholar]
- HOFHUIS W, DE JONGSTE JC, MERKUS PJ. Adverse health effects of prenatal and postnatal tobacco smoke exposure on children. Arch Dis Child. 2003;88:1086–90. doi: 10.1136/adc.88.12.1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JINOT J, BAYARD S. Respiratory health effects of passive smoking: EPA’s weight-of-evidence analysis. J Clin Epidemiol. 1994;47:339–49. doi: 10.1016/0895-4356(94)90154-6. discussion 351–3. [DOI] [PubMed] [Google Scholar]
- KEARLEY J, SILVER JS, SANDEN C, LIU Z, BERLIN AA, WHITE N, MORI M, PHAM TH, WARD CK, CRINER GJ, MARCHETTI N, MUSTELIN T, ERJEFALT JS, KOLBECK R, HUMBLES AA. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. 2015;42:566–79. doi: 10.1016/j.immuni.2015.02.011. [DOI] [PubMed] [Google Scholar]
- KIRKHAM PA, SPOONER G, RAHMAN I, ROSSI AG. Macrophage phagocytosis of apoptotic neutrophils is compromised by matrix proteins modified by cigarette smoke and lipid peroxidation products. Biochem Biophys Res Commun. 2004;318:32–7. doi: 10.1016/j.bbrc.2004.04.003. [DOI] [PubMed] [Google Scholar]
- KUROWSKA-STOLARSKA M, STOLARSKI B, KEWIN P, MURPHY G, CORRIGAN CJ, YING S, PITMAN N, MIRCHANDANI A, RANA B, VAN ROOIJEN N, SHEPHERD M, MCSHARRY C, MCINNES IB, XU D, LIEW FY. IL-33 amplifies the polarization of alternatively activated macrophages that contribute to airway inflammation. J Immunol. 2009;183:6469–77. doi: 10.4049/jimmunol.0901575. [DOI] [PubMed] [Google Scholar]
- LANTZ RC, CHAU B, SARIHAN P, WITTEN ML, PIVNIOUK VI, CHEN GJ. In utero and postnatal exposure to arsenic alters pulmonary structure and function. Toxicol Appl Pharmacol. 2009;235:105–13. doi: 10.1016/j.taap.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MANTOVANI A, SICA A, SOZZANI S, ALLAVENA P, VECCHI A, LOCATI M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- MARTINEZ FO, SICA A, MANTOVANI A, LOCATI M. Macrophage activation and polarization. Front Biosci. 2008;13:453–61. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- MCCULLOCH J, TWEEDALE G. Defending the indefensible: the global asbestos industry and its fight for survival. Oxford; New York: Oxford University Press; 2008. [Google Scholar]
- MEEKER GP, BA, BROWNFIELD IK, LOWERS HA, SUTLEY SJ, HOEFEN TM, VANCE JS. The composition and morphology of amphiboles from the Rainy Creek Complex, Near Libby, Montana. AM. Mineralogist. 2003;88:1955–1969. [Google Scholar]
- MEHTA H, NAZZAL K, SADIKOT RT. Cigarette smoking and innate immunity. Inflamm Res. 2008;57:497–503. doi: 10.1007/s00011-008-8078-6. [DOI] [PubMed] [Google Scholar]
- ORTEGA E, BARRIGA C, RODRIGUEZ AB. Decline in the phagocytic function of alveolar macrophages from mice exposed to cigarette smoke. Comp Immunol Microbiol Infect Dis. 1994;17:77–84. doi: 10.1016/0147-9571(94)90009-4. [DOI] [PubMed] [Google Scholar]
- OUYANG Y, VIRASCH N, HAO P, AUBREY MT, MUKERJEE N, BIERER BE, FREED BM. Suppression of human IL-1beta, IL-2, IFN-gamma, and TNF-alpha production by cigarette smoke extracts. J Allergy Clin Immunol. 2000;106:280–7. doi: 10.1067/mai.2000.107751. [DOI] [PubMed] [Google Scholar]
- PHIPPS JC, ARONOFF DM, CURTIS JL, GOEL D, O’BRIEN E, MANCUSO P. Cigarette smoke exposure impairs pulmonary bacterial clearance and alveolar macrophage complement-mediated phagocytosis of Streptococcus pneumoniae. Infect Immun. 2010;78:1214–20. doi: 10.1128/IAI.00963-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAVASTANO L, BONACCI S, SARACINO V, LONGO M. The association of lung cancer with asbestos and tobacco smoking. Clin Ter. 2004;155:69–74. [PubMed] [Google Scholar]
- SCANLON ST, MCKENZIE AN. Type 2 innate lymphoid cells: new players in asthma and allergy. Curr Opin Immunol. 2012;24:707–12. doi: 10.1016/j.coi.2012.08.009. [DOI] [PubMed] [Google Scholar]
- SHAYKHIEV R, KRAUSE A, SALIT J, STRULOVICI-BAREL Y, HARVEY BG, O’CONNOR TP, CRYSTAL RG. Smoking-dependent reprogramming of alveolar macrophage polarization: implication for pathogenesis of chronic obstructive pulmonary disease. J Immunol. 2009;183:2867–83. doi: 10.4049/jimmunol.0900473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLAVINSKY J, 3RD, MYERS T, SWOBODA RK, LEIGH JE, HAGER S, FIDEL PL., JR Th1/Th2 cytokine profiles in saliva of HIV-positive smokers with oropharyngeal candidiasis. Oral Microbiol Immunol. 2002;17:38–43. doi: 10.1046/j.0902-0055.2001.00080.x. [DOI] [PubMed] [Google Scholar]
- STEIN RT, HOLBERG CJ, SHERRILL D, WRIGHT AL, MORGAN WJ, TAUSSIG L, MARTINEZ FD. Influence of parental smoking on respiratory symptoms during the first decade of life: the Tucson Children’s Respiratory Study. Am J Epidemiol. 1999;149:1030–7. doi: 10.1093/oxfordjournals.aje.a009748. [DOI] [PubMed] [Google Scholar]
- STOCKS J, DEZATEUX C. The effect of parental smoking on lung function and development during infancy. Respirology. 2003;8:266–85. doi: 10.1046/j.1440-1843.2003.00478.x. [DOI] [PubMed] [Google Scholar]
- TRUYEN E, COTEUR L, DILISSEN E, OVERBERGH L, DUPONT LJ, CEUPPENS JL, BULLENS DM. Evaluation of airway inflammation by quantitative Th1/Th2 cytokine mRNA measurement in sputum of asthma patients. Thorax. 2006;61:202–8. doi: 10.1136/thx.2005.052399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VEGLIA F, VINEIS P, OVERVAD K, BOEING H, BERGMANN M, TRICHOPOULOU A, TRICHOPOULOS D, PALLI D, KROGH V, TUMINO R, LINSEISEN J, STEINDORF K, RAASCHOU-NIELSEN O, TJONNELAND A, GONZALEZ CA, MARTINEZ C, DORRONSORO M, BARRICARTE A, CIRERA L, QUIROS JR, DAY NE, SARACCI R, RIBOLI E. Occupational exposures, environmental tobacco smoke, and lung cancer. Epidemiology. 2007;18:769–75. doi: 10.1097/ede.0b013e318142c8a1. [DOI] [PubMed] [Google Scholar]
- VIERIKKO T, JARVENPAA R, UITTI J, VIRTEMA P, OKSA P, JAAKKOLA MS, AUTTI T, VEHMAS T. The effects of secondhand smoke exposure on HRCT findings among asbestos-exposed workers. Respir Med. 2008;102:658–64. doi: 10.1016/j.rmed.2007.12.021. [DOI] [PubMed] [Google Scholar]
- WEBBER JS, BLAKE DJ, WARD TJ, PFAU JC. Separation and characterization of respirable amphibole fibers from Libby, Montana. Inhal Toxicol. 2008;20:733–40. doi: 10.1080/08958370801932544. [DOI] [PMC free article] [PubMed] [Google Scholar]