

1. Disease Characteristics
1.1 Name of the disease (synonyms)
Wolfram syndrome (WFS). Clinically, WFS presents with two clinical subtypes, namely WFS1 (diabetes insipidus and mellitus with optic atrophy and deafness, DIDMOAD), and WFS2.
1.2 OMIM# of the disease
222300 – Wolfram syndrome 1; 604928 – Wolfram syndrome 2 and 598500 – Mitochondrial form.
1.3 Name of the analysed genes or DNA/chromosome segments
Genes implicated in Wolfram syndrome type 1: WFS1; genes implicated in Wolfram syndrome type 2: CISD2.
1.4 OMIM# of the gene(s)
WFS1 MIM# 606201; CISD2 MIM# 611507.
1.5 Mutational spectrum
Wolfram syndrome 1 (WFS1) is an autosomal recessive progressive neurodegenerative disease characterised by early-onset type 1 diabetes mellitus (DM) and bilateral optic neuropathy (OA) with a wide spectrum of associated clinical conditions described below. Over 90% of variants are found in the WFS1 gene, which spans 33.4 kb on chromosome 4p16.1, and consists of 8 exons encoding the 890-amino acid Wolframin protein (NCBI reference sequence NM_006005.3, NM_001145853), that localises to the endoplasmic reticulum (ER). Current evidence suggests that Wolframin is a component of mitochondria-associated membranes and may play an important role in regulating ER–mitochondria homoeostasis.1 There have been over 250 variants in WFS1 described in patients with Wolfram syndrome, WFS1 (https://lovd.euro-wabb.org). Reported variants are mainly point mutations (missense, nonsense, frameshift mutations), but also small deletions, insertions and duplications.
Wolfram syndrome 2 (WFS2) is also recessively inherited with considerable overlap of clinical features with WFS1, it is classically associated with peptic ulcer disease and bleeding tendencies without diabetes insipidus (DI). It is caused by variants in the CISD2 (CDGSH Iron Sulfur Domain 2) gene on chromosome 4q24, which consists of 3 exons encoding the ER intermembrane small protein. Reported variants include a missense mutation in Jordanian families suggestive of a founder event and a deletion in one non-consanguineous Italian family.2, 3, 4
There has been a suggested link between mitochondrial DNA mutations and WFS.5 A 7.6-kb heteroplasmic deletion (spanning nucleotides 6465–14135) has been reported,6 in addition to multiple deletions of mitochondrial DNA and a point mutation (m.3337G>A) in the mitochondrial gene encoding subunit ND1 in a Tunisian patient.7 In some patients with WFS1 variants, secondary mitochondrial DNA instability can be found particularly in post-mitotic tissues such as skeletal muscle, and this may contribute to the more severe clinical manifestations.8, 9
It is important to note that Wolfram-like syndrome (OMIM 614296) also exists with overlapping features. This is an autosomal dominant disorder caused by heterozygous variants in WFS1, resulting in sensorineural hearing loss, DM, psychiatric illness and variable optic atrophy within the first decade of life.10, 11
1.6 Analytical methods
Bi-directional fluorescent Sanger sequencing of coding and intron–exon boundaries of WFS1 is the mainstay analytical method as an initial analysis. CISD2 screening can be performed if WFS2 is suspected, as this is rare. However, WFS1 and CISD2 screening is being included on next-generation sequencing panels in some laboratories.
1.7 Analytical validation
Parallel bi-directional fluorescent Sanger sequencing of known controls is required to validate procedures. Diagnostic testing must be carried out within a laboratory environment working to standards compliant with the ISO 15189. The majority of variants reported until now in the WFS1 gene causing autosomal recessive Wolfram syndrome result in loss-of-function.12
1.8 Estimated frequency of the disease
(incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report)
Estimated prevalence of 1 in 770 000 in the UK,13 1 in 100 000 in North America,14 1 in 500 000 in children15 and 1 in 68 000 in the Lebanese population (possibly attributable to high rates of consanguinity).16 Carrier frequency is 1 in 354 patients.13
1.9 Diagnostic setting

Comment: If a family has an affected child and wishes to have more children, prenatal diagnosis should be discussed in detail during genetic counselling.17
2. Test characteristics
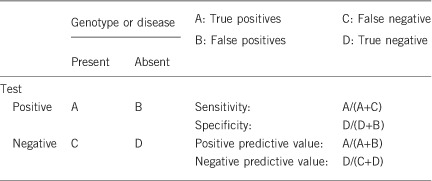
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
We estimate that the analytical sensitivity and specificity of the test used (bi-directional Sanger sequencing) will be >98%. A small loss of sensitivity may be due to intronic or other variants missed through exonic analysis. The proportion of such cases is not known.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
See above. We estimate analytical specificity of >98% given current testing methodologies, based on the false positives that can rarely occur in Sanger sequencing.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
If a patient has both DM and OA before 16 years of age, in the presence of a positive genetic test, the clinical sensitivity and specificity are both high as WFS type 1 and 2 are not genetically heterogeneous, with WFS1 accounting for >90% of WFS1 and CISD2 causing WFS2.18 However, due to the variable order and age of onset of different clinical features, care has to be taken with the interpretation of heterozygous variants in WFS1, which cause Wolfram-like syndrome disorders, including missense mutations associated with autosomal dominant OA and sensorineural hearing loss,10, 19 autosomal dominant nonsyndromic adult-onset diabetes,20 psychiatric symptoms and autosomal dominant low-frequency nonsyndromic sensorineural hearing loss.21
In a systematic review, analysing the published clinical data in 392 patients with WFS, 98.2% had DM and 82.14% developed OA.12 By age 18, the probability of having developed the DM is 93.60%, OA 79.06%, sensorineural hearing loss 40.56%, DI 35.20%, urinary defects 11.42% and neurological, psychiatric or developmental problems 7.57%.12
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
An individual without signs of DM and OA is unlikely to have a positive test as both clinical manifestations can be seen in the majority (90%) by the second decade (14–15 years of age for DM and at 25–26 years for OA), this increases to 95% probability at 23–24 years for DM and at 40–41 years for OA,12 and so the clinical specificity will be high. However, in some cases the onset of clinical features is variable and this can lower the clinical specificity.
2.5 Positive clinical predictive value
(life time risk to develop the disease if the test is positive)
Estimated >99% for two pathogenic alleles in WFS1 and CISD2. A genotype–phenotype correlation has been suggested for WFS1 variants in determining the age at onset of DM and DI in type 1 WFS.12
2.6 Negative clinical predictive value
(probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
For known pathogenic changes, or novel null mutations, the negative predictive value will be approaching 100%.
Index case in that family had not been tested:
If the index case is asymptomatic by 16 years of age and has a negative test result, it is highly predictive of unaffected status, but will fall short of 100% due to the analytical specificity noted above.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
WFS1 is a progressive neurodegenerative disorder characterised by the onset of DM around the age of 6 (range: 3 weeks—16 years), with optic atrophy developing typically by age 11 (range: 6 weeks—19 years).22 It is commonly associated with high-frequency sensorineural hearing loss (62%), which presents around age 16 (range: 5–19 years).13 Progressive neurologic abnormalities (60%, including cerebellar ataxia, peripheral neuropathy, dementia and psychiatric illness), urinary tract defects (60–90%, including ureteric obstruction, bladder atony and sphincter dyssynergia, and incontinence) and other endocrine abnormalities associated with pituitary dysfunction, such as hypogonadism and DI (51–87%) presenting around age 14 (range: 3 months–40 years).22 The median age of death is 27±11.4 years.12
Patients with WFS2 have overlapping features with WFS1, plus defective platelet aggregation resulting in peptic ulcer bleeding, but importantly an absence of DI.2, 23
Children who are suspected of having WFS will undergo a number of investigations including MRI of the brain and orbit to look for generalised brain atrophy (cerebellum, medulla and pons), absence of signal from the posterior pituitary and reduced signal from the optic nerve.24 Ancillary testing can be useful to confirm primary retinal ganglion cell dysfunction. Electrophysiology tests such as visual evoked potentials and the pattern electroretinogram provide objective measures of optic nerve funcion, and optical coherence tomography is a non-invasive ocular imaging modality that is frequently used to quantify and monitor progressive thinning of the retinal nerve fibre layer. Hearing tests such as pure tone audiometry document affected frequencies and progression of hearing loss. Tests for DI include urine analysis, the water deprivation test, blood levels of antidiuretic hormone and the antidiuretic hormone test to differentiate cranial versus nephrogenic DI.
Genetic testing can assist the clinical surveillance as pathogenic variants in WFS1 or CISD2 would justify pre-symptomatic regular follow-up by ophthalmologists, audiologists, endocrinologists and neurologists in order to provide the appropriate support to the patient and their family.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Although WFS is a rare disorder, it is associated with significant multisystem co-morbidity and a short life expectancy. Making the correct diagnosis early is therefore important to optimise the management of neurological and endocrine complications, which can be life-threatening and ensure appropriate support and rehabilitation. Clinical recognition can be challenging due to the varying onset of signs and symptoms, especially for physicians in non-specialist centres who do not manage the disease regularly. Patients will often require tertiary referral for accurate diagnosis. Genetic testing is important for accurate diagnosis, however, multidisciplinary team input will still be required for regular monitoring of clinical manifestations.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?

3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Patients with WFS have a poor visual prognosis, usually >6/60 (or 20/200) secondary to optic atrophy, therefore professions requiring perfect vision are impossible. Hence, a clinically confirmed diagnosis can already help in providing guidance regarding career choice.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes, a molecular diagnosis in an affected individual can resolve the genetic situation in that family, determine recessive segregation unambiguously and is a pre-requisite for genetic counselling for family members. For Wolfram-like syndrome, where de novo heterozygous variants in WFS1 are found, the recurrence risk is low but there is a high offspring risk of 50%.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
If molecular testing has identified a WFS1 mutation in the index patient, depending on age, examination can identify and exclude disease in at-risk relatives. However, further genetic tests are required to determine the carrier status. It is important to consider that heterozygous variants in WFS1 can cause Wolfram-like syndrome (section 2.3), and autosomal dominant cataracts,26 so patients must be examined to exclude any manifestations. This must be undertaken following genetic counselling and arguably when the patient can make their own decision.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Genetic testing for WFS1 variants will provide a molecular diagnosis. This yields information regarding onset of symptoms, recurrence risk, carrier status and hence will provide choices that would not otherwise be available to facilitate decision making for the patient and their family. Gene testing is essential in defining inheritance patterns and enabling effective genetic counselling. A positive gene test will preclude the need for further genetic testing.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. MM gratefully acknowledges the support of the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology.
The authors declare no conflict of interest.
References
- Poston CN, Krishnan SC, Bazemore-Walker CR: In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J Proteomics 2013; 79: 219–230. [DOI] [PubMed] [Google Scholar]
- Amr S, Heisey C, Zhang M et al: A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am J Hum Genet 2007; 81: 673–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzillo E, Delvecchio M, Carella M et al: A novel CISD2 intragenic deletion, optic neuropathy and platelet aggregation defect in Wolfram syndrome type 2. BMC Med Genet 2014; 15: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli M, Novara F, Calcaterra V, Zuffardi O, Genovese S: Wolfram syndrome 2: a novel CISD2 mutation identified in Italian siblings. Acta Diabetol 2014; 52: 175–178. [DOI] [PubMed] [Google Scholar]
- Barrientos A, Volpini V, Casademont J et al: A nuclear defect in the 4p16 region predisposes to multiple mitochondrial DNA deletions in families with Wolfram syndrome. J Clin Invest 1996; 97: 1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A, Cormier V, Chatelain P et al: Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (Wolfram syndrome, MIM 222300). J Clin Invest 1993; 91: 1095–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezghani N, Mnif M, Mkaouar-Rebai E et al: The mitochondrial ND1 m.3337G>A mutation associated to multiple mitochondrial DNA deletions in a patient with Wolfram syndrome and cardiomyopathy. Biochem Biophys Res Commun 2011; 411: 247–252. [DOI] [PubMed] [Google Scholar]
- Barrett TG, Scott-Brown M, Seller A, Bednarz A, Poulton K, Poulton J: The mitochondrial genome in Wolfram syndrome. J Med Genet 2000; 37: 463–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber DS, Vafai SB, Horton LC et al: Atypical case of Wolfram syndrome revealed through targeted exome sequencing in a patient with suspected mitochondrial disease. BMC Med Genet 2012; 13: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valero R, Bannwarth S, Roman S, Paquis-Flucklinger V, Vialettes B: Autosomal dominant transmission of diabetes and congenital hearing impairment secondary to a missense mutation in the WFS1 gene. Diabet Med 2008; 25: 657–661. [DOI] [PubMed] [Google Scholar]
- Chaussenot A, Bannwarth S, Rouzier C et al: Neurologic features and genotype-phenotype correlation in Wolfram syndrome. Ann Neurol 2011; 69: 501–508. [DOI] [PubMed] [Google Scholar]
- de Heredia ML, Cleries R, Nunes V: Genotypic classification of patients with Wolfram syndrome: insights into the natural history of the disease and correlation with phenotype. Genet Med 2013; 15: 497–506. [DOI] [PubMed] [Google Scholar]
- Barrett TG, Bundey SE, Macleod AF: Neurodegeneration and diabetes: UK nationwide study of Wolfram (DIDMOAD) syndrome. Lancet 1995; 346: 1458–1463. [DOI] [PubMed] [Google Scholar]
- Fraser FC, Gunn T: Diabetes mellitus, diabetes insipidus, and optic atrophy. An autosomal recessive syndrome? J Med Genet 1977; 14: 190–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S: Wolfram syndrome: important implications for pediatricians and pediatric endocrinologists. Pediatr Diabetes 2010; 11: 28–37. [DOI] [PubMed] [Google Scholar]
- Medlej R, Wasson J, Baz P et al: Diabetes mellitus and optic atrophy: a study of Wolfram syndrome in the Lebanese population. J Clin Endocrinol Metab 2004; 89: 1656–1661. [DOI] [PubMed] [Google Scholar]
- Domenech E, Kruyer H, Gomez C, Calvo MT, Nunes V: First prenatal diagnosis for Wolfram syndrome by molecular analysis of the WFS1 gene. Prenat Diagn 2004; 24: 787–789. [DOI] [PubMed] [Google Scholar]
- (US) NLoM: Genetics Home Reference [Internet]. Bethesda (MD): The Library; 2015 Sep. Wolfram Syndrome. Available from: http://ghr.nlm.nih.gov/condition/wolfrum-syndrome.
- Rendtorff ND, Lodahl M, Boulahbel H et al: Identification of p.A684V missense mutation in the WFS1 gene as a frequent cause of autosomal dominant optic atrophy and hearing impairment. Am J Med Genet A 2011; 155A: 1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnycastle LL, Chines PS, Hara T et al: Autosomal dominant diabetes arising from a Wolfram syndrome 1 mutation. Diabetes 2013; 62: 3943–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Lv H, Zhang F et al: Identification of a novel missense mutation in the WFS1 gene as a cause of autosomal dominant nonsyndromic sensorineural hearing loss in all-frequencies. Am J Med Genet A 2014; 164A: 3052–3060. [DOI] [PubMed] [Google Scholar]
- Rigoli L, Di Bella C: Wolfram syndrome 1 and Wolfram syndrome 2. Curr Opin Pediatr 2012; 24: 512–517. [DOI] [PubMed] [Google Scholar]
- al-Sheyyab M, Jarrah N, Younis E et al: Bleeding tendency in Wolfram syndrome: a newly identified feature with phenotype genotype correlation. Eur J Pediatr 2001; 160: 243–246. [DOI] [PubMed] [Google Scholar]
- Ito S, Sakakibara R, Hattori T: Wolfram syndrome presenting marked brain MR imaging abnormalities with few neurologic abnormalities. AJNR Am J Neuroradiol 2007; 28: 305–306. [PMC free article] [PubMed] [Google Scholar]
- Lu S, Kanekura K, Hara T et al: A calcium-dependent protease as a potential therapeutic target for Wolfram syndrome. Proc Natl Acad Sci USA 2014; 111: E5292–E5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry V, Gregory-Evans C, Emmett W et al: Wolfram gene (WFS1) mutation causes autosomal dominant congenital nuclear cataract in humans. Eur J Hum Genet 2013; 21: 1356–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]