

Abstract
Developing new strategies to rapidly incorporate the fac-[MI(CO)3]+ (M = Re, 99mTc) core into biological targeting vectors in radiopharmaceuticals continues to expand as molecules become more complex and as efforts to minimize nonspecific binding increase. This work examines a novel isothiocyanate-functionalized bifunctional chelate based on 2,2′-dipicolylamine (DPA) specifically designed for complexing the fac-[MI(CO)3]+ core. Two strategies (postlabeling and prelabeling) were explored using the isothiocyanate-functionalized DPA to determine the effectiveness of assembly on the overall yield and purity of the complex with amine containing biomolecules. A model amino acid (lysine) examined (1) amine conjugation of isothiocyanate-functionalized DPA followed by complexation with fac-[MI(CO)3]+ (postlabeling) and (2) complexation of fac-[MI(CO)3]+ with isothiocyanate-functionalized DPA followed by amine conjugation (prelabeling). Conducted with stable Re and radioactive 99mTc analogs, both strategies formed the product in good to excellent yields under macroscopic and radiotracer concentrations. A synthetic peptide (AE105) which targets an emerging biomarker in CaP prognosis, urokinase-type plasminogen activator receptor (uPAR), was also explored using the isothiocyanate-functionalized DPA strategy. In vitro PC-3 (uPAR+) cell uptake assays with the 99mTc-labeled peptide (8a) showed 4.2 ± 0.5% uptake at 4 h. In a murine model bearing PC-3 tumor xenografts, in vivo biodistribution of 8a led to favorable tumor uptake (3.7 ± 0.7% ID/g) at 4 h p.i. with relatively low accumulation (<2% ID/g) in normal organs not associated with normal peptide excretion. These results illustrate the promise of the isothiocyanate-functionalized approach for labeling amine containing biological targeting vectors with fac-[MI(CO)3]+.
Graphical abstract
INTRODUCTION
Molecular imaging techniques utilizing radioisotopes for single photon emission computed tomography (SPECT) and positron emission tomography (PET) are vital contributors to numerous diagnostic medical procedures. 99mTc [t1/2 = 6.02 h, 140 keV γ (89%)] remains the most widely used radionuclide in diagnostic radiopharmaceuticals due to its nearly ideal nuclear properties for SPECT and convenient on-site availability via 99Mo-99mTc generators. For targeted molecular imaging applications with 99mTc, the fac-[99mTcI(CO)3]+ core is particularly attractive because of its ease of preparation via an aqueous-based kit, versatile coordination chemistry, and formation of inert complexes with a broad spectrum of ligands which can be conjugated to targeting biomolecules.1–4 Despite these advantages, labeling with the fac-[99mTcI(CO)3]+ core often requires high temperatures (>90 °C) for quantitative complexation in acceptable time frames (30–60 min) for radiopharmaceutical preparation. The chelate, then click method, wherein metal complexation with a chelator occurs prior to conjugating the resulting complex to a biomolecule targeting vector via a click reaction, allows radiolabeling with temperature-sensitive biomolecules (e.g., antibodies) which would normally be denatured under the complexation conditions.
Several reports have highlighted the potential benefits of the chelate, then click strategy for labeling targeting biomolecules with the fac-[MI(CO)3]+ (M = Re, 99mTc) core.5,6 While the copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC) reaction has been heralded as the iconic click reaction,7 a growing body of literature has revealed the drawbacks of the copper catalyst in biological applications. The negative effects associated with the metal catalyst including Cu-mediated cleavage of peptides, coordination of Cu to targeting biomolecules, and biological toxicity remain considerable barriers to the use of the CuAAC reaction. Therefore, alternative click reactions which do not require a metal catalyst are being investigated for generating targeted radiopharmaceuticals and other applications.8–13 Among these reactions, isoxazole formation,14 thioether formation,15,16 and hydrazone generation have been successfully utilized in proof-of-concept studies with the fac-[MI(CO)3]+ core.17
In addition to click reactions, amine-reactive functionalities have also been used in facile approaches for labeling biomolecules with Re and 99mTc complexes in prelabeling or preconjugate strategies, which are analogous to the chelate, then click strategy. Isothiocyanate moieties and several types of activated esters (e.g., N-hydroxy succinimide, tetrafluorophenyl) have been employed to conjugate an array of metal complexes in several oxidation states (MI–V) to solvent-accessible amine groups in macromolecules, peptides, and small molecule targeting vectors.18–35 Conjugation reactions employing amine-reactive functionalities do not require a catalyst, and they proceed efficiently in mild conditions at physiological temperatures. While these coupling reactions lack the chemical selectivity of “ideal” click reactions, they are widely employed for conjugating amine-reactive bifunctional chelates with peptides and antibody targeting vectors to generate targeted imaging and therapeutic radiopharmaceuticals.36–38
Widespread use of amine-conjugation strategies with targeting biomolecules prompted the present study’s development of a novel bifunctional chelate containing an isothiocyanate moiety for generating targeted radiopharmaceuticals with the fac-[MI(CO)3]+ core. Although midvalent Re and 99mTc complexes containing isothiocyanates have been employed in prelabeling reactions with targeting vectors,20,21 the use of isothiocyanate-functionalized fac-[MI(CO)3]+ complexes in this labeling strategy has not been demonstrated in the literature. 2,2′-Dipicolylamine (DPA) was chosen as the chelate scaffold due to the favorable complexation and kinetic inertness of this chelate with the fac-[MI(CO)3]+ core.6,39–41 One of the main goals of the studies was to evaluate the use of the labeled complexes in the prelabeling strategy with compounds containing free amine groups. N-α-Boc-L-lysine-COOH (BocLys) was chosen as a model compound for analysis and was coupled with the bifunctional chelate and metal complexes via prelabeling and postlabeling strategies (Scheme 1).
Scheme 1.
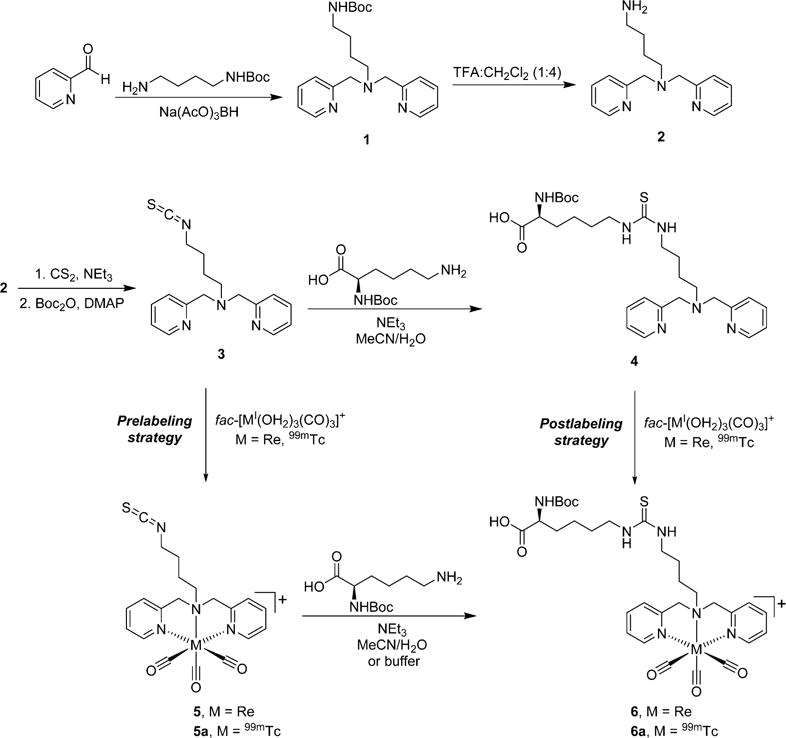
Reaction Scheme for Generating the Isothiocyanate-Functionalized Ligand, Metal Complexes, and BocLys Conjugates via prelabeling and postlabeling Strategies
The bifunctional chelate was also reacted with an established targeting peptide (AE105)42–46 which binds to the urokinase plasminogen activator receptor (uPAR) to generate a 99mTc-labeled analog for use in cancer applications. uPAR is an attractive biomarker due to its overexpression in a variety of malignancies including breast, pancreatic, colon, ovarian, and prostate cancer (CaP).47–49 Urokinase plasminogen activator (uPA) and uPAR are known components in numerous cell signaling pathways including cytoskeleton organization, vascularization, proteolysis of extracellular membrane components, cell growth, and motility.50–55 uPA/uPAR have also been implicated in cancer cell invasion, metastasis, and advanced disease.51,56–59 Various reports have targeted uPA/uPAR for preclinical imaging42–44,46,60,61 or therapeutic applications in cancer.48,62–66 Increased expression of uPAR has been associated with more aggressive and advanced CaP relative to indolent or localized disease.63,67–71 Developing uPAR-targeted analogs labeled with SPECT radionuclides would be desirable to aid CaP assessment in patients. In the present study, the 99mTc-labeled AE015 peptide was utilized in binding assays in vitro with PC-3 cells and in biodistribution analyses in vivo with mice bearing PC-3 xenografts.
RESULTS AND DISCUSSION
The ligands and metal complexes were prepared as shown in Scheme 1. tert-Butyl (4-aminobutyl)carbamate was chosen as the linker for generating the DPA bifunctional chelate precursor. This six-atom spacer was envisioned to separate the coordination center from the isothiocyanate functionality for biomolecule attachment and to prevent interactions of nonchelate donors (amine, isothiocyanate, thiourea) with the metal center in subsequent steps.
Reductive amination reactions between the linker and 2-pyridinecarboxaldehyde using NaBH(OAc)3 successfully generated the Boc-protected DPA precursor ligand 1 in 90% yield after purification by column chromatography. Removal of the Boc group using TFA/CH2Cl2 followed by basic extraction yielded 2 in its neutral form in high yield (90%).
The primary amine group in the deprotected ligand was converted to the isothiocyanate functionality by adopting a generalized literature procedure from Munch et al.72 Reaction of 2 in EtOH with NEt3 and excess CS2 formed a dithiocarbamate intermediate which upon addition of di-tert-butyl-dicarbonate and catalytic DMAP decomposed to the desired isothiocyanate, 3. The crude reaction was purified by column chromatography on basic alumina to give 3 in 70% isolated yield. All spectroscopic characterization results were consistent with the desired compound. Notably, strong bands at 2095 cm−1 in the IR spectra indicated the presence of the isothiocyanate group.
Ligand 3 was subsequently reacted with a protected amino acid to characterize the coupling reaction of this novel ligand with a small molecule model system prior to its use with more complicated targeting molecules. BocLys was chosen as the model amino acid since the unprotected ε-amine group would mimic the reactivity of solvent-exposed lysine side chains in macromolecules. Retention of the Boc protecting group was intended to prevent the α-amine group from reacting with a second ligand during conjugation. In a similar approach to the SAAC strategy,73 the lysine conjugate could also be directly utilized in solid phase peptide synthesis for future applications if desired.
The conjugation conditions avoided inorganic bases and minimized aqueous solvent to prevent hydrolysis of the isothiocyanate functional group. A mixture of MeCN and minimal H2O (~10%) with NEt3 as the base was found suitable to maintain solubility of all components and promote smooth conjugation to the intended thiourea product. RP-HPLC analysis during the reaction with 3 indicated formation of a single new product under these conditions. The BocLys conjugate, 4, was obtained in good yield (71%) following preparatory HPLC purification of the reaction mixture. All spectroscopic characterization data were as anticipated for the desired product.
Two separate routes were utilized to generate the metal complex of the BocLys conjugated chelate. In the prelabeling route, the isothiocyanate ligand 3 was complexed with fac-[ReI(OH2)3(CO)3]+ to examine the effect of metal complexation prior to coupling with BocLys. The main purpose of generating the rhenium complexes was to have characterized reference materials prior to performing tracer-scale reactions with 99mTc; therefore, macroscale reaction conditions were not optimized to improve the isolated yields of these species. Heating 3 with fac-[ReI(OH2)3(CO)3]+ at 40–50 °C in MeOH was found suitable for complexation without significant hydrolysis of the isothiocyanate to the amine. These conditions produced the desired product, 5, which was isolated as a pure species by preparatory RP-HPLC in a moderate yield of 34%. IR analysis showed the expected vCO stretching bands for the fac-[ReI(CO)3]+ core (2028 and 1903 cm−1) as well as the presence of the isothiocyanate group (2107 cm−1), and ESI-MS analysis was consistent with the desired complex (583.1 m/z). While the NMR spectra of 5 showed singlets for the methylene protons of the DPA chelate as opposed to AB quartets seen with several other fac-[MI(CO)3(DPA)] complexes,5,17 the downfield shifts of these protons in 5 (4.88 ppm) compared to the original ligand (3.79 ppm) indicated that metal complexation occurred at the DPA portion of the chelate as expected.
This rhenium complex was subsequently conjugated with BocLys using similar conditions as for the free ligand. The reaction proceeded smoothly to produce a single new species, 6, which was purified by preparatory HPLC with a favorable yield (64%). ESI-MS analysis confirmed the product was the desired complex (829.2 m/z). IR analysis indicated loss of the isothiocyanate group from the starting complex while the vCO stretching bands for the fac-[ReI(CO)3]+ core were retained in 6 as anticipated. Surprisingly, 1H NMR showed splitting for the methylene DPA protons as opposed to the singlets observed for these species in 5 prior to conjugation with BocLys. This suggests that these protons experienced slightly different environments upon conversion of the isothiocyanate group to the thiourea group despite the presence of the butyl linker between these functionalities and the DPA chelate area. Complex 6 was also generated via the postlabeling strategy by complexing the lysine-conjugated ligand, 4, with fac-[ReI(OH2)3(CO)3]+ in MeOH at 60–65 °C. These reaction conditions formed 6 as the predominant species with the same RP-HPLC tR as observed in the prelabeling pathway (11.0 min for both routes). The desired complex was isolated with a 32% yield following preparatory RP-HPLC purification of the postlabeling reaction. All spectroscopic data of 6 obtained by the prelabeling and postlabeling pathways were identical, confirming that both routes are viable for generating the desired metal complex on the macroscopic level with Re.
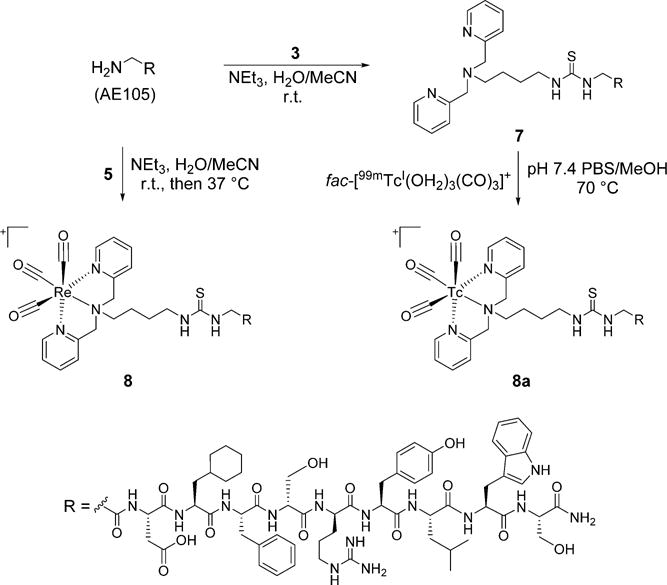
Analogous reactions as used with Re were subsequently explored on the tracer scale with 99mTc. In the prelabeling pathway, ligand 3 at 1 × 10−4 M was complexed with fac-[99mTcI(OH2)3(CO)3]+ in pH 7.4 sodium phosphate buffer at 70 °C for 1 h to produce the corresponding complex, 5a, in 92% radiochemical purity. As shown in Figure 1, the HPLC tR of this species (10.7 min) correlated with the rhenium complex, 5 (10.5 min), indicating the 99mTc complex was generated as anticipated. Under these conditions, ~5% hydrolysis of the isothiocyanate group was observed.
Figure 1.
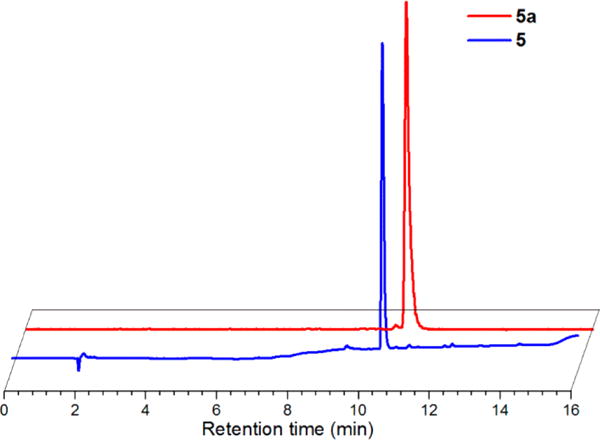
Normalized and offset RP-HPLC chromatograms of 5 (UV absorbance, 254 nm; earlier blue trace) at tR 10.5 min and 5a (radiodetector, counts per minute (cpm); later red trace) at tR 10.7 min.
Crude reaction mixtures of 5a were used in subsequent coupling reactions with BocLys. Several different reaction conditions were used to explore the efficiency of the isothiocyanate conjugations at the tracer level with 99mTc, although extensive investigations to probe the limits of product formation or maximize product yields were beyond the scope of these proof-of-concept studies. Performing the reactions at 37 °C in a mixed MeCN/aqueous solution with NEt3 as the base was intended to minimize hydrolysis of the isothiocyanate group and to mimic the conditions used with the rhenium complex at the macroscopic level. Since these conditions would denature or cause precipitation of proteins and other macromolecules used as targeting moieties, parallel reactions employed an aqueous NaHCO3 solution free of organic solvents to be compatible with these alternative targeting biomolecules. Both the aqueous and mixed organic/aqueous conditions were performed with BocLys at either 10 mM or 50 mM to probe the coupling efficiency at different nucleophilic amine concentrations.
As shown in Figure 2, the HPLC tR of the 99mTc complex generated by coupling 5a with BocLys correlated with the rhenium analog, 6 (11.0 min), thereby confirming the identity of the product as the desired complex, 6a (11.2 min). Table 1 shows the radiochemical yields of 6a from the various prelabeling reaction conditions explored. The yields of the final complex consistently increased by extending the reaction times from 30 to 60 min, although the rate of product production appeared to be slower during the 30–60 min period as would be expected from a bimolecular reaction. In general, utilizing the mixed organic/aqueous conditions with 50 mM BocLys led to the greatest yields, although the complex was obtained in higher yields at 10 mM BocLys than at 50 mM using the conditions stated above. This unexpected result was due to increased hydrolysis of 5a with the higher percentage of water from the aqueous BocLys stock solution in the 50 mM reaction compared to the 10 mM reaction. Further attempts were not made to minimize hydrolysis of the starting complex in the mixed organic/aqueous reactions. Greater amounts of the hydrolyzed products were observed in the aqueous conditions compared to the mixed organic/aqueous reactions, although increasing the concentration of BocLys in the aqueous reactions led to lower formation of the hydrolyzed species (data not shown). This is likely due to the greater efficiency of thiourea formation relative to isothiocyanate hydrolysis at higher BocLys concentrations. Time points beyond 60 min were not explored due to the diminishing changes in product yields and the increasing amounts of hydrolyzed byproduct observed with time. A more detailed kinetic analysis was not pursued in these studies as coupling reactions between isothiocyanate ligands and amines have been thoroughly investigated elsewhere.74
Figure 2.
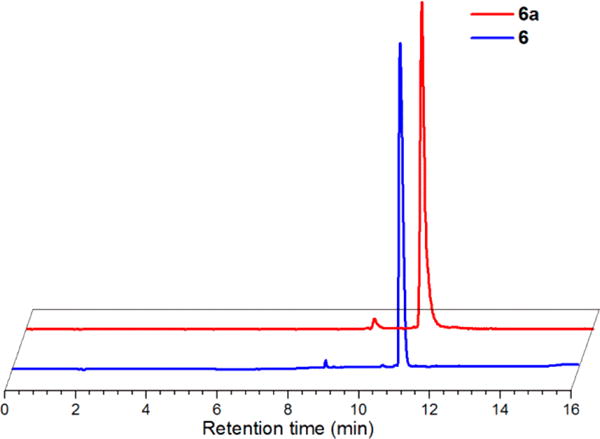
Normalized and offset RP-HPLC chromatograms of 6 (UV absorbance, 254 nm; earlier blue trace) at tR 11.0 min and 6a (radiodetector, counts per minute (cpm); later red trace) at tR 11.2 min.
Table 1.
Radiochemical Yields of 6a via the prelabeling Pathway from 5aa
| concentration of BocLys | 6ab
|
||
|---|---|---|---|
| 30 min | 60 min | ||
| Organic conditionsc | 10 mM | 90% | 94% |
| 50 mM | 79% | 85% | |
| Aqueous conditionsd | 10 mM | 31% | 46% |
| 50 mM | 65% | 68% | |
Reactions were heated at 37 °C for the times indicated and then allowed to cool briefly before aliquots were removed for analysis by radio-HPLC using HPLC method 1.
Organic conditions: mixed MeCN/phosphate buffer solutions with NEt3 as the base.
Aqueous conditions: aqueous pH 10.2 NaHCO3 solutions.
It is well-known that the pH of the reactions has a significant effect on the rate of both thiourea formation and isothiocyanate hydrolysis.20,21,74–76 The pH of the aqueous NaHCO3 conditions in the above studies was held above 10 due to the pKa of the ε-amine group of lysine. Preliminary studies in aqueous NaHCO3 at lower pH (~9) showed decreased thiourea product formation due to greater levels of protonation and diminished nucleophilicity of BocLys; greater levels of hydrolysis were also observed in these conditions (data not shown). The relationship between solution pH and the pKa of the reactive nucleophile should be considered in future applications of the prelabeling methodology with the complex explored here to optimize labeling yields of the intended amine-bearing moiety.
Complex 6a was also formed via the postlabeling strategy by heating 4 with fac-[99mTcI(OH2)3(CO)3]+ in pH 7.4 buffer at 70 °C for 30–60 min. As was observed for the rhenium analog, the HPLC tR of 6a produced via the postlabeling pathway (11.2 min) matched the product of the prelabeling pathway at the tracer level with 99mTc. Employing ligand concentrations of 1 × 10−4 M or 5 × 10−5 M led to quantitative conversion from the fac-[99mTcI(OH2)3(CO)3]+ precursor in 30–60 min with >85% radiochemical purities (Table 2) as anticipated from previous studies with analogous DPA chelates.5
Table 2.
Radiochemical Purities of 6a via the postlabeling Pathway from Ligand 4a
| ligand concentration | 6a
|
|
|---|---|---|
| 30 min | 60 min | |
| 1 × 10−4 M | 96% | – |
| 5 × 10−5 M | 87% | 91% |
Reactions were heated at 70 °C in 10 mM sodium phosphate pH 7.4 buffer for the times indicated and then allowed to cool briefly before aliquots were removed for analysis by radio-HPLC using HPLC method 1 or 2.
The above proof-of-concept studies show that while complex 5a can successfully be used to label molecules containing amine groups with the fac-[99mTcI(CO)3]+ core via the prelabeling route, overall radiochemical yields were superior with the postlabeling strategy in the model systems explored here. Hydrolysis and incomplete thiourea formation from the isothiocyanate precursor would not be optimal during biomolecule labeling as purification would be required and specific activities of the final radiopharmaceuticals would be lower compared to quantitative reactions with the metal complex. Utilizing high concentrations (>1 mM) of biomolecules in the labeling reaction would further decrease specific activities; if the unlabeled and labeled components are not separated prior to in vivo administration, the unlabeled biomolecules may block target receptors during molecular imaging applications. Despite these shortcomings, the prelabeling route with this complex may be advantageous for labeling temperature-sensitive biomolecules with the fac-[99mTcI(CO)3]+ core since the coupling reactions can be accomplished at 37 °C instead of the significantly higher temperatures required for quantitative complexation of the ligand. Furthermore, the prelabeling strategy prevents the possibility of mixed or undefined coordination of the fac-[99mTcI(CO)3]+ core with nonspecific sites in macromolecules employed as targeting agents, thereby minimizing in vivo transchelation of the radiolabel from the targeting molecule. The collective results obtained with the BocLys model studies indicated the novel isothiocyanate chelate (3) was worthy of further use with a medically relevant biomolecule.
While most applications with isothiocyanate-functionalized ligands use macromolecules (e.g., antibodies) as the targeting molecule, the in vivo circulation times of these targeting agents (days) are generally longer than desirable for the relatively short half-life of 99mTc (6 h). The radionuclide typically decays appreciably before optimal uptake at the target and clearance from the blood pool and nontarget organs occur, thus leading to poor signal-to-noise ratios in the region of interest. Peptides, on the other hand, exhibit relatively fast pharmacokinetics and are better suited for use with 99mTc for imaging applications. AE105, a peptide which binds to uPAR and contains a single amine group, was selected as the targeting vector for assessment with the bifunctional chelate above.
AE105 has been conjugated with several chelators and successfully radiolabeled with 64Cu,42,45,77 68Ga,43 18F,46 213Bi,64 and 177Lu.65,66 In vivo studies using xenograft tumor models have shown that uptake of the radiolabeled peptides within tumors correlates with uPAR expression.45,46 These constructs are thus potential candidates for noninvasive assessment of tumor status and disease progression over time. While Armstrong et al. previously generated a 99mTc-labeled peptide for assessment with uPAR, the analog was not used for in vivo analysis.78 The current work presents a first-generation AE105 analog labeled with 99mTc for in vivo assessment with CaP.
The bifunctional isothiocyanate ligand, 3, was used in slight excess compared to AE105 to ensure complete conjugation of the peptide (Scheme 2). Since the peptide contained a single nucleophilic amine group, excess ligand was not anticipated to lead to multiple products during conjugation. Similar to reactions with the BocLys model systems explored earlier, a single major peak was observed during the reaction when NEt3 and mixed H2O/MeCN conditions were employed overnight at room temperature. Purification of the reaction by RP-HPLC produced the desired peptide conjugate, 7, in 40% yield with high purity (>98%) as indicated by RP-HPLC analysis of the isolated material (Supporting Information Figure S1). MALDITOF MS analysis (1594.9 m/z) confirmed the identity of 7 as the desired product. The Re(CO)3-complexed peptide conjugate, 8, was prepared by reacting AE105 with complex 5 (Scheme 2). This reaction used the same conditions as employed for 7, although low conjugation efficiency prompted heating at 37 °C to help drive product formation. While partial hydrolysis of the isothiocyanate group of 5 was apparent in these conditions, further addition of the metal complex led to the peptide conjugate as the major species in the reaction. Purification of the mixture by RP-HPLC generated 8 in 43% yield with >97% purity as indicated by RP-HPLC analysis (Figure 3). MALDI-TOF MS analysis (1864.8 m/z) confirmed the identity of 8 as desired.
Scheme 2.
Reaction Schemes with uPAR Peptide AE105
Figure 3.
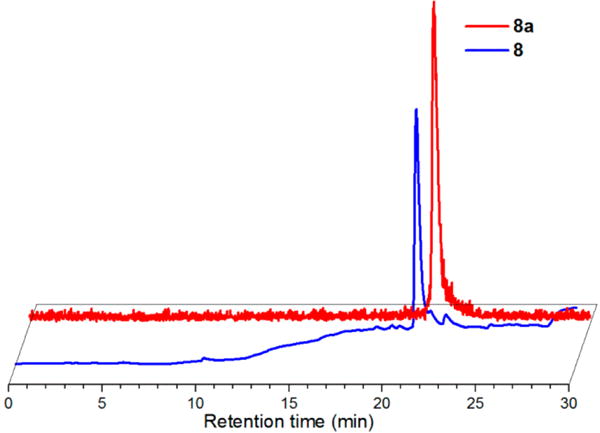
Normalized and offset RP-HPLC chromatograms of 8 (UV absorbance, 254 nm; earlier blue trace) at tR 21.5 min and 8a (radiodetector, counts per minute (cpm); later red trace) at tR 21.7 min.
AE105 conjugate 7 was subsequently radiolabeled with 99 mTc. Heating 7 at 5 × 10−5 M with fac-[99mTcI(OH2)3(CO)3]+ in PBS/MeOH at 70 °C for 60 min yielded a single species in >99% radiochemical purity by radio-HPLC analysis. Comparison of the tR of this species (21.7 min) with that of the Re analog, 8 (21.5 min), confirmed the radiolabeled peptide as the desired product, 8a (Figure 3). This species was isolated with decay-corrected yields of 59–60% following purification by HPLC method 3. 8a was observed to be >96% stable in mouse serum up to 6 h at 37 °C (Supporting Information Figure S3), indicating it would be a viable candidate for subsequent biological analyses.
In vitro cell uptake assays were performed to assess the uPAR-mediated uptake of 8a with PC-3 cells. 8a showed a time-dependent increase in cell uptake to yield 2.4 ± 0.3%, 3.3 ± 0.5%, and 4.2 ± 0.5% of the applied activity at 1, 2, and 4 h, respectively (Figure 4). The cell uptake of 8a decreased to 1.1 ± 0.9%, 1.3 ± 0.4%, and 1.5 ± 0.8% of the applied activity at 1, 2, and 4 h, respectively, in the presence of unlabeled AE105 to block uPAR-mediated binding. These results established the uptake of 8a with PC-3 cells was mediated by uPAR and indicated that the 99mTc labeled AE105 can specifically bind to its biological target.
Figure 4.
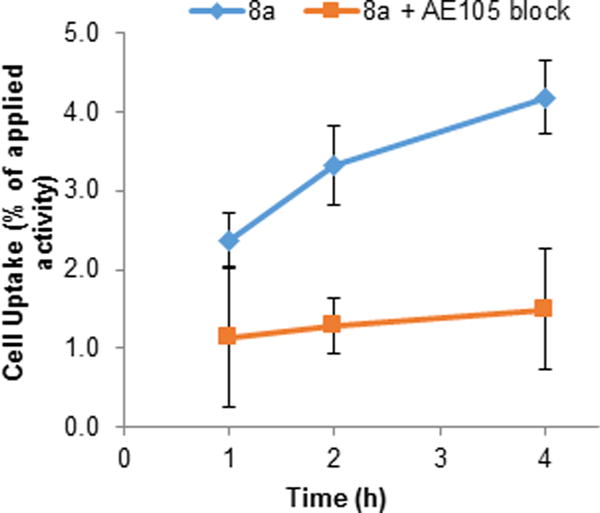
PC-3 in vitro cell uptake assay. PC-3 cells (1 × 105 per well in 24-well plates) were incubated at 37 °C for 1, 2, or 4 h with purified 8a (1 μCi) alone (blue diamonds) or with 2 μg AE105 (red squares) to block uPAR-mediated binding. Data represent the amount of activity associated with the cells as a percentage of the total activity applied ± standard deviation of quadruplicate wells.
The encouraging in vitro results with 8a prompted its further investigation in vivo with mouse biodistribution studies. Figure 5 shows the uptake of 8a in selected tissues at 1 and 4 h following tail vein injection of mice bearing PC-3 xenograft tumors. The radiolabeled peptide showed relatively low uptake (<2% ID/g) at 1 h p.i. in all normal tissues except those involved with excretion (liver, intestines, and kidneys with 12.0 ± 1.3%, 4.2 ± 0.8%, and 6.6 ± 0.7% ID/g, respectively) and the lungs (3.2 ± 0.4% ID/g). The high uptake of 8a in the liver follows the pattern observed with other small molecules and peptides labeled with the lipophilic fac-[99mTcI(CO)3(DPA)]+ moiety. Relative to the 1 h time point, the uptake values decreased by 4 h p.i. for all normal organs as would be expected from typical peptide pharmacokinetics. Favorable uptake and retention of 8a in the tumor was observed at both 1 and 4 h p.i., yielding 3.6 ± 0.6% and 3.7 ± 0.7% ID/g, respectively. Since the tumor uptake was higher than all other tissues except the liver, intestine, and kidneys at both time points, 8a may be an encouraging lead compound for future work as a SPECT imaging peptide for PC-3 tumors in vivo.
Figure 5.
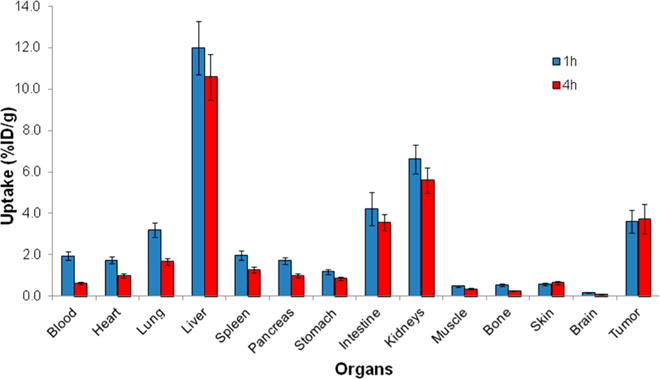
Biodistribution of 8a in male athymic nude mice bearing PC-3 tumors 1 and 4 h after injection of approximately 100 μCi of 8a via the tail vein. Data are expressed as % ID/g ± standard deviation (n = 4 per group).
While radiolabeled uPAR analogs have been assessed with several tumor types in vivo, few reports utilizing CaP models have been published. An 18F-AlF-NOTA conjugate of AE105 employed by Persson et al.46 demonstrated an uptake of 4.2 ± 0.1% ID/g in PC-3 xenograft tumors at 1 h p.i. by quantitative PET imaging in mice. Additional PET and biodistribution analysis indicated the uptake of the radiolabeled peptide had declined to ~1% ID/g after 2 h p.i. The comparable uptake of 8a within PC-3 tumors at both 1 and 4 h p.i. in the present study suggests the 99mTc analog may be more amenable for imaging uPAR-expressing tumors at later time points compared to the 18F analog. Future analysis with 8a is required to determine whether tumor uptake correlates with uPAR expression, as has been observed with related AE105 analogs labeled with other radionuclides.
In a subsequent study utilizing a transgenic PC-3M-LUC2 CaP model,66 Persson et al. demonstrated a DOTA-AE105 analog labeled with 177Lu (177Lu-DOTA-AE105) bound to cells in vitro to a significantly greater level than a nonspecific AE105 mutant analog. Furthermore, 177Lu-DOTA-AE105 inhibited metastatic spread of PC-3M-LUC2 in vivo, and PET imaging with 64Cu-DOTA-AE105 was able to detect metastatic lesions which were not detectable with bioluminescent imaging. Considering the widespread availability and clinical utilization of 99mTc in diagnostic imaging, these pioneering studies with radiolabeled AE105 analogs in CaP models supported the development of the 99mTc-labeled construct shown here. The collective results from the current study and previous publications suggest AE105 analogs labeled with a variety of radionuclides show promise for continued preclinical development and potential translation to clinical CaP assessment in the future.
CONCLUSIONS
This report presents the first example of an isothiocyanate-functionalized DPA ligand for complexation with the fac-[MI(CO)3]+ core. The bifunctional ligand was synthesized and readily complexed with fac-[MI(CO)3]+ at both macroscopic and tracer levels. The ligand and metal complexes were successfully reacted with BocLys via prelabeling and postlabeling strategies to generate the intended final products. Exploration of the prelabeling pathway showed the potential of the novel isothiocyanate complexes for reacting with solvent-accessible amine groups at ambient temperatures in relatively short (~30 min) time periods as would be desirable for developing a wide variety of targeted radiopharmaceuticals with the fac-[MI(CO)3]+ core. Application of the bifunctional ligand with AE105 successfully generated a novel fac-[MI(CO)3]+-labeled peptide for targeting uPAR. The radiolabeled analog (8a) showed uPAR-mediated uptake in PC-3 cells in vitro and favorable uptake and retention in PC-3 xenograft tumors in vivo. These results indicate that the novel chelate and uPAR-targeting construct shown here is worthy of future investigations for CaP assessment.
EXPERIMENTAL PROCEDURES
All reagents and solvents were of reagent grade or higher and obtained from commercial suppliers (Aldrich, Fluka, Acros, Fisher) and used as received unless noted otherwise. N-α-Boc-L-Lysine-COOH (BocLys) was from Chem-Impex International (Wood Dale, IL). AE105 (NH2-Gly-Asp-Cha-Phe-D-Ser-D-Arg-Tyr-Leu-Trp-Ser-NH2 was synthesized as previously described.46,79,80 fac-[ReI(OH2)3(CO)3](SO3CF3),81 and tert-butyl (4-aminobutyl)carbamate82 were prepared from literature procedures. [99mTcO4]− was obtained from Cardinal Health (Spokane, WA) or from Stanford Nuclear Medicine Clinic (Stanford, CA) and was used to prepare fac-[99mTcI(OH2)3(CO)3]+ via commercially available Isolink kits (Tyco, Inc.) as previously described.83 Nuclear magnetic resonance (NMR) spectra were recorded at 293 K on a 300 MHz Varian Mercury Vx spectrometer using 5 mm NMR tubes. 1H and 13C NMR spectra peak positions were referenced using residual solvent signals, and spectra were processed using Varian VNWR 6.1 software. Electrospray ionization mass spectrometry (ESI-MS) was performed by direct infusion of sample solutions on a Thermo-Finnigan LCQ Advantage instrument or an Agilent 1100 Ion Trap LC/MS/MS (scanned from 50 to 2000 m/z with the drying gas at 12 mL min−1 at 350 °C and nebulizer pressure set at 50 psig). Matrix assisted laser desorption/ionization time-of-flight (MALDI-TOF) MS was performed at the Washington State University Molecular Biology and Genomics Core (Pullman, WA). Elemental analyses were conducted by Atlantic Micro Laboratories in Norcross, GA. Infrared (IR) spectra were recorded on a Thermo Nicolett 6700 FTIR with an ATR cell and analyzed with OMNIC 7.1a software. UV/vis spectra were recorded on a Varian Cary 50 Bio spectrophotometer and analyzed with Cary WinUV 3.00 software. Solutions of the compounds were prepared in UV/vis quality methanol and measured in 1 cm path length quartz cuvettes. Analytical separation and identification of small molecules and peptides by reversed-phase high performance liquid chromatography (RP-HPLC) were conducted using an Agilent Zorbax SB-Aq column (3.5 μm, 4.6 × 150 mm) or a Varian Pursuit XRs column (C18, 5 μm, 4.6 × 250 mm) with a Phenomenex security guard cartridge (C18, 4.0 × 3.0 mm). A PerkinElmer Series 200 analytical chromatography system equipped with a PerkinElmer Radiomatic 610TR detector and a Hitachi D-7000 series analytical chromatography system (L-7100 pump, L-7400 UV detector) equipped with a Berthold FlowStar LB 513 radiodetector were used during analyses. Unless noted otherwise, elutions performed on these systems used the following gradient profiles (HPLC methods 1 and 2) with 0.05% TFA (trifluoroacetic acid) in H2O (solvent A) and methanol (MeOH, solvent B) as solvents at 1 mL/min and UV detection at 254 nm. HPLC method 1 (employing the Zorbax SB-Aq column): 0–1.5 min 100% A, 1.5–5 min linear gradient to 35% A/65% B, 5–10.5 min linear gradient to 25% A/75% B, 10.5–12 min isocratic 25% A/75% B, 12–15 min 100% B, return to 100% A at 15 min and equilibrate; UV was monitored at 254 nm; HPLC method 2 (employing the Pursuit XRs column): 0–3 min 100% A, 3–9 min isocratic 75% A/25% B, 9–20 min linear gradient to 100% B, 20–25 min 100% B, return to 100% A at 25 min and equilibrate. Chromatograms were plotted using OriginPro 8.5.1 (OriginLab Corporation, MA). Preparatory RP-HPLC separations and purifications of small molecules used an Agilent Zorbax SB-C18 PrepHT column (C18, 7 μm, 21.2 × 250 mm) fitted with a matching guard cartridge (5 μm, 21.2) on a Hitachi D-7000 series semipreparative chromatography system (L-7150 pump, L-7400 UV detector). Elutions were performed with solvents A and B; UV was monitored at 220 nm. The PC-3 prostate cancer cell line was obtained from American Type Culture Collection (Manassas, VA). Male athymic nude mice used for biodistribution analysis were purchased from Charles River Laboratory (Wilmington, MA).
Small Molecule Synthesis and Complexation Reactions
tert-Butyl (4-(bis(pyridin-2-ylmethyl)amino)butyl)-carbamate, 1
tert-Butyl (4-aminobutyl) carbamate (193 mg, 1.03 × 10−3 mol), dichloroethane (15 mL), and 2-pyridinecarboxyaldehyde (0.215 mL, 2.26 × 10−3 mol) were combined in a flask. The flask was purged with N2 and placed in an ice bath for 5 min. Sodium triacetoxyborohydride (NaBH-(OAc)3), 546 mg, 2.58 × 10−3 mol) was added and the reaction was stirred under N2 in the ice bath for an additional 5 min and then at room temperature for 2.5 h. The reaction was quenched with 8 mL H2O and stirred for 1 h. A 2.5 M NaOH solution was added to the reaction to reach pH ~ 10 and the reaction mixture was transferred to a separatory funnel. The organic layer was collected and the remaining aqueous layer was extracted with CH2Cl2 (3 × 15 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated to dryness under reduced pressure. The crude residue was purified by silica gel flash chromatography using 3–4% MeOH in CH2Cl2. Collected fractions containing the product (Rf = 0.06 in 3% MeOH/CH2Cl2 on silica gel) were combined and concentrated to dryness under vacuum to yield 1 as a dark yellow oil (343.5 mg, 90%).
Anal. Calcd for C21H30N4O2·0.5MeOH: C, 66.81; H, 8.35; N, 14.50. Found: C, 66.30; H, 8.11; N, 14.51. 1H NMR [δ (ppm), 300 MHz, CDCl3]: 8.49 (d, 2 H), 7.61 (dd, 2 H), 7.47 (d, 2 H), 7.10 (dd, 2 H), 4.83 (s, 1 H), 3.76 (s, 4 H), 3.02 (q, 2 H), 2.51 (t, 2 H), 1.53 (m, 2 H), 1.47–1.35 (m, 11 H). 13C NMR [δ (ppm), 75 MHz, CDCl3]: 159.1, 155.6, 148.2, 135.7, 122.3, 121.3, 77.8, 59.7, 53.3, 53.0, 27.9, 27.2, 23.7. MS (+ESI): 393.2 m/z; calculated for [M + Na]+: 393.2 m/z. IR (neat oil, cm−1): 2931, 1709, 1692, 1590, 1433, 1364, 1249, 1168, 758. UV/vis εmax (202 nm): 11,300 M−1 cm−1; ε261 nm: 5,600 M−1 cm−1.
N1,N1-Bis(pyridin-2-ylmethyl)butane-1,4-diamine, 2
Compound 1 (291.8 mg, 7.9 × 10−4 mol) was dissolved in a 1:4 TFA:CH2Cl2 (5 mL) mixture and the solution was stirred at room temperature for 90 min. The mixture was concentrated to dryness under vacuum to yield a red-brown oil. The residue was dissolved in CH2Cl2 (20 mL) and washed with H2O (20 mL) which had been adjusted to pH 10 with 1 M NaOH. The organic layer was then collected and the aqueous layer was extracted with CH2Cl2 (2 × 20 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated to dryness under vacuum to yield 2 as an amber-colored oil (192.2 mg, 90%).
Anal. Calcd for C16H22N4: C, 71.08; H, 8.20; N, 20.72. Found: C, 71.01; H, 8.14; N, 20.59. 1H NMR [δ (ppm), 300 MHz, CDCl3]: 8.45 (d, 2 H), 7.58 (dd, 2 H), 7.46 (d, 2 H), 7.07 (dd, 2 H), 3.74 (s, 4 H), 2.56 (t, 2 H), 2.48 (t, 2 H), 1.76 (br s, 2 H), 1.50 (m, 2 H), 1.35 (m, 2 H). 13C NMR [δ (ppm), 75 MHz, CDCl3]: 159.2, 148.3, 135.8, 122.3, 121.3, 59.8, 53.5, 41.2, 30.4, 23.8. MS (+ESI): 271.1 m/z; calculated for [M + H]+: 271.2 m/z. IR (neat oil, cm−1): 3051, 3010, 2929, 2815, 1691, 1589, 1432, 1200, 1125, 760. UV/vis εmax (204 nm): 12,500 M−1 cm−1; ε262 nm: 6,400 M−1 cm−1.
4-Isothiocyanato-N,N-bis(pyridin-2-ylmethyl)butan-1-amine, 3
Compound 2 (73 mg, 2.7 × 10−4 mol) was dissolved in absolute ethanol (EtOH, 1.5 mL), after which CS2 (162 μL, 2.7 × 10−3 mol) and triethylamine (NEt3, 37.6 μL, 2.7 × 10−4 mol) were added to the solution. The reaction was stirred at room temperature for 1.5 h and was then placed in an ice bath for 10 min. Di-tert-butyl-dicarbonate (57 mg, 2.6 × 10−4 mol) was added, followed immediately by 4-dimethylaminopyridine (DMAP, 1 mg, 8 × 10−6 mol) dissolved in absolute EtOH (0.25 mL). After 10 min, the reaction was allowed to come to room temperature and was stirred for 1 h. The mixture was then concentrated to dryness under vacuum. The crude residue was purified by flash chromatography on basic alumina using 1% MeOH/CH2Cl2. Collected fractions containing the product (Rf = 0.38 in 2% MeOH/CH2Cl2 on basic alumina) were combined and concentrated to dryness under vacuum to yield 3 as an amber-colored oil (59 mg, 70%).
Anal. Calcd for C17H20N4S: C, 65.35; H, 6.45; N, 17.93. Found: C, 64.45; H, 6.51; N, 17.13. 1H NMR [δ (ppm), 300 MHz, CDCl3]: 8.51 (d, 2 H), 7.65 (dd, 2 H), 7.48 (d, 2 H), 7.14 (dd, 2 H), 3.79 (s, 4 H), 3.40 (t, 2 H), 2.55 (t, 2 H), 1.63 (m, 4 H). 13C NMR [δ (ppm), 75 MHz, CDCl3]: 159.6, 149.0, 136.5, 123.0, 122.1, 60.5, 53.1, 44.8, 27.7, 24.2. MS (+ESI): 313.3 m/z; calculated for [M + H]+: 313.2 m/z. IR (neat oil, cm−1): 2932, 2814, 2176, 2095, 1588, 1432, 751. UV/vis εmax (203 nm): 19,200 M−1 cm−1; ε262 nm: 8,600 M−1 cm−1.
(S)-6-(3-(4-(Bis(pyridin-2-ylmethyl)amino)butyl)thioureido)-2-((tert-butoxycarbonyl)amino) hexanoic acid, 4
Compound 3 (40 mg, 1.28 × 10−4 mol), BocLys (47.3 mg, 1.92 × 10−4 mol), and NEt3 (52.3 μL, 3.75 × 10−4 mol) were dissolved in a 9:1 acetonitrile (MeCN):H2O mixture (5 mL), and the solution was stirred at room temperature overnight. The reaction was concentrated under reduced pressure and purified by preparatory HPLC using the following gradient method (prepHPLC method 1) with a flow rate of 12 mL/min: 0–3 min 100% A, 3–9 min isocratic 75% A/25% B, 9–25 min linear gradient to 100% B, 25–32 min 100% B, return to 100% A at 32 min and equilibrate. Fractions containing pure product were combined and concentrated to dryness under vacuum to yield 4 as an off-white solid (72 mg, 71%).
Anal. Calcd for C28H42N6O4S: C, 60.19; H, 7.58; N, 15.04. Found: C, 60.11; H, 7.51; N, 15.12. 1H NMR [δ (ppm), 300 MHz, CD3OD]: 8.66 (d, 2 H), 7.89 (dd, 2 H), 7.51 (d, 2 H), 7.45 (dd, 2 H), 4.62 (s, 4 H), 4.06 (dd, 1 H), 3.50 (m, 2 H), 3.45–3.32 (m, 4 H), 1.85 (m, 4 H), 1.74–1.53 (m, 6 H), 1.44 (s, 9 H). 13C NMR [δ (ppm), 75 MHz, CD3OD]: 176.2, 163.1, 162.6, 158.1, 151.8, 150.3, 139.4, 125.5, 80.4, 58.5, 56.0, 54.8, 44.0, 32.5, 29.7, 28.7, 27.4, 24.2, 22.4. MS (+ESI): 559.3 m/z; calculated for [M + H]+: 559.3 m/z. IR (solid, cm−1): 3292, 2933, 1671, 1552, 1166, 1129, 761, 719. UV/vis: εmax (203 nm): 35,000 M−1 cm−1; ε242 nm: 20,000 M−1 cm−1.
fac-[ReI(CO)3(3)](C2F3O2), 5
Compound 3 (33 mg, 1.05 × 10−4 mol) was dissolved in 4 mL of MeOH in a 50 mL flask. A 0.1 M solution of fac-[ReI(OH2)3(CO)3](SO3CF3) (1.1 mL, 1.1 × 10−4 mol) was added and the reaction was stirred at 50 °C under a condenser for 1.75 h. The reaction was then concentrated under reduced pressure and purified by preparatory HPLC using the following gradient method (prepHPLC method 2) with a flow rate of 12.5 mL/min: 0–4 min 100% A, 4–12 min linear gradient to 40% A/60% B, 12–33 min linear gradient to 15% A/85% B, 33–36 min linear gradient to 100% B, 36–43 min 100% B, return to 100% A at 43 min and equilibrate. Fractions containing pure product were combined and concentrated to dryness under vacuum to yield 5 as an off-white crystalline residue (24.9 mg, 34%).
Anal. Calcd for C22H20F3N4O5ReS: C, 37.98; H, 2.90; N, 8.05. Found: C, 37.86; H, 2.77; N, 7.87. 1H NMR [δ (ppm), 300 MHz, CD3OD]: 8.87 (d, 2 H), 7.94 (dd, 2 H), 7.55 (d, 2 H), 7.38 (dd, 2 H), 4.88 (s, 4 H), 3.88 (m, 2 H), 3.75 (t, 2 H), 2.08 (m, 2 H), 1.83 (m, 2 H). 13C NMR [δ (ppm), 75 MHz, CD3OD]: 197.1, 162.1, 153.2, 141.6, 126.9, 124.5, 71.0, 68.6, 45.8, 28.1, 23.5. MS (+ESI): 583.1 m/z; calculated for [M]+: 583.1 m/z. IR (solid, cm−1): 2951, 2107, 2028, 1903, 1734, 1683, 1135, 763, 705. UV/vis εmax (235 nm): 9,900 M−1 cm−1; ε261 nm: 7,800 M−1 cm−1.
fac-[ReI(CO)3(4)](C2F3O2), 6. Postlabeling Pathway from 4
Compound 4 (36.4 mg, 4.62 × 10−5 mol) was dissolved in 0.5 mL of MeOH and 3 mL of H2O in a 25 mL flask. A 0.1 M solution of fac-[ReI(OH2)3(CO)3](SO3CF3) (0.53 mL, 5.3 × 10−5 mol) was added and the reaction was stirred at 60 °C under a condenser for 3.5 h. The reaction was then diluted in MeOH to dissolve the creamy precipitate, filtered, and purified by preparatory HPLC using the following gradient method (prepHPLC method 3) with a flow rate of 12.5 mL/min: 0–3 min 100% A, 3–10 min isocratic 75% A/25% B, 10–32 min linear gradient to 100% B, 32–40 min 100% B, return to 100% A at 40 min and equilibrate. Fractions containing pure product were combined and concentrated to dryness under vacuum to yield 6 as a whitish semisolid residue (14.1 mg, 32%).
Prelabeling Pathway from 5
Complex 5 (22.5 mg, 3.2 × 10−5 mol), BocLys (15.9 mg, 6.4 × 10−5 mol), and NEt3 (18.5 μL, 1.3 × 10−4 mol) were combined in a flask containing 4 mL of MeCN and 15 drops H2O, and the reaction was stirred at room temperature overnight. The solution was then concentrated under reduced pressure and purified by preparatory HPLC using prepHPLC method 3 with a flow rate of 12.5 mL/min. Fractions containing pure material were combined and concentrated to dryness under vacuum to yield 6 as a white residue (19.5 mg, 64%).
Anal. Calcd for C33H42F3N6O9ReS: C, 42.08; H, 4.49; N, 8.92. Found: C, 41.98; H, 4.45; N, 8.85. 1H NMR [δ (ppm), 300 MHz, CD3OD]: 8.87 (d, 2 H), 7.93 (dd, 2 H), 7.54 (d, 2 H), 7.37 (dd, 2 H), 4.99–4.79 (m, 4 H), 4.07 (dd, 1 H), 3.95 (m, 2 H), 3.68 (br s, 2 H), 3.44 (br s, 2 H), 1.99 (m, 2 H), 1.92–1.51 (m, 8 H), 1.44 (s, 9 H). 13C NMR [δ (ppm), 75 MHz, CD3OD]: 197.2, 176.2, 162.2, 158.2, 153.1, 141.6, 126.9, 124.6, 80.5, 71.6, 68.7, 54.8, 44.1, 32.5, 29.7, 28.7, 27.8, 24.3, 22.7. MS (+ESI): 829.2 m/z; calculated for [M]+: 829.2 m/z. IR (solid, cm−1): 3284, 2936, 2029, 1909, 1689, 1163, 765. UV/vis εmax (203 nm): 37,000 M−1 cm−1; ε240 nm: 22,200 M−1 cm−1.
AE105 Peptide Conjugation and Complexation Reactions
AE105 Conjugate of Ligand 3, 7
0.8 mg AE105 (5.3 × 10−7 mol) was dissolved in 100 μL of H2O. A solution of 0.4 mg 3 (1.28 × 10−6 mol) dissolved in 40 μL of MeCN and 5 μL of NEt3 (3.59 × 10−5 mol) were added, and the reaction was stirred at room temperature overnight. The following day, 7.5 μL of 10% TFA in H2O was added and the reaction was purified by analytical RP-HPLC using the Varian Pursuit XRs column with solvents A and B according to the following gradient method with a flow rate of 1 mL/min (HPLC method 3): 0–3 min 95%A/5% B, 3–9.5 min linear gradient to 55% A/45% B, 9.5–24.5 min linear gradient to 10% A/90% B, 24.5–29 min 100% B, return to 95% A/5% B at 29 min and equilibrate; UV was monitored at 238 nm. Fractions containing pure product were combined, concentrated by rotary evaporation, frozen, and lyophilized to yield 0.4 mg (40%) of the desired product. MALDI-TOF MS: 1594.9 m/z; calcd for [M + H]+: 1594.8 m/z.
AE105 Conjugate of Complex 5, 8
0.5 mg AE105 (3.31 × 10−7 mol) was dissolved in 60 μL of H2O. A solution of 0.35 mg 5 (4.99 × 10−7 mol) dissolved in 26 μL of MeCN and 5 μL of NEt3 (3.59 × 10−5 mol) were added, and the reaction was stirred at room temperature overnight. The following day, additional 5 (1.04 × 10−7 mol) dissolved in 8 μL of MeCN and 2 μL of NEt3 (1.5 × 10−5 mol) was added, and the reaction was stirred at 37 °C for 4.5 h. 7 μL 10% TFA in H2O was then added and the reaction was purified by analytical RP-HPLC by HPLC method 3. Fractions containing pure product were combined, concentrated by rotary evaporation, frozen, and lyophilized to yield 0.3 mg (43%) of the desired product. MALDI-TOF MS: 1864.8 m/z; calcd for [M]+: 1864.7 m/z.
General Procedures for 99mTc Reactions
Complexation of Isothiocyanate Ligand 3 and Postlabeling Pathway with 4
Solutions of 3 or 4 in 10 mM pH 7.4 sodium phosphate buffer (250–450 μL) were sealed in 5 mL labeling vials and the vials were purged with N2 for 5–10 min. Solutions of fac-[99mTcI(OH2)3(CO)3]+ (50–250 μL, 0.13–1.6 mCi) were added to bring the final ligand concentrations to 1 × 10−4 or 5 × 10−5 M in final volumes of 500 μL, and the vials were heated at 70 °C for 30–60 min. The vials were cooled prior to analysis and purification by radio-HPLC using HPLC method 1 or 2.
Prelabeling Pathway with Complex 5a. Mixed Organic/Aqueous Conditions
Crude reaction mixtures of 5a (50 μL, 6–150 μCi) were added to 5 mL labeling vials containing MeCN (375 or 415 μL), NEt3 (25 μL), and 0.5 M BocLys in H2O (50 or 10 μL) to bring the final BocLys concentrations to 50 mM or 10 mM in final volumes of 500 μL. The vials were sealed, purged with N2 for 2–3 min, and heated at 37 °C for 30–60 min. The vials were analyzed by radio-HPLC using HPLC method 1 and purified by HPLC method 1 or 2.
Aqueous NaHCO3 Conditions
Crude reaction mixtures of 5a (50 μL, 6–10 μCi) were added to 5 mL labeling vials containing 0.1 M NaHCO3 pH 10.2 buffer (400 or 440 μL) and 0.5 M BocLys in H2O (50 or 10 μL) to bring the final BocLys concentrations to 50 mM or 10 mM in final volumes of 500 μL. The vials were sealed, purged with N2 for 2–3 min, and heated at 37 °C for 30–60 min. The vials were analyzed by radio-HPLC using HPLC method 1 and purified by HPLC method 1 or 2.
Peptide Radiolabeling
A solution of 48 μg 7 (2.5 × 10−8 mol) dissolved in 30 μL of H2O/MeOH, 220 μL of pH 7.4 PBS, and 50 μL of MeOH were combined in a 5 mL sealable labeling vial. The vial was sealed and sparged with N2 for 5 min, and 200 μL of fac-[99mTcI(OH2)3(CO)3]+ solution (79 μCi) was added to yield a final peptide concentration of 5 × 10−5 M in a final volume of 500 μL. The reaction was then heated at 70–80 °C with periodic analysis by radio-HPLC using HPLC method 3. For the in vitro and in vivo biological analyses, the above radiolabeling procedure was followed utilizing 10 μg 7 and 10 mCi fac-[99mTcI(OH2)3(CO)3]+ to yield the product peptide 8a. The reaction solution was purified by radio-HPLC with a C18 column at a flow rate of 1 mL/min using HPLC method 4: 0–3 min 95% A/5% C (MeCN with 0.1% TFA), 3–18 min linear gradient to 5% A/95% C, 18–20 min hold 5% A/95% C, 20–23 return to 95% A/5% C and equilibrate. The collected product fraction was diluted to ten times its volume, passed through a C18 cartridge, and eluted with EtOH (1 mL). The radioactive fractions (2 × 100 μL) were combined, adjusted to pH 7.4 with PBS (100 μL), diluted with saline (2 mL) to give a solution of 8a with <10% EtOH, and passed through a 0.22 μM filter for subsequent use. The purity of this solution was verified by radio-HPLC using HPLC method 4 (Supporting Information Figure S2).
Peptide Stability Analysis
The serum stability of 8a was determined as previously described.5 Briefly, HPLC-purified 8a (100 μCi) in 50 μL of PBS was incubated with mouse serum (500 μL) at 37 °C for a total of 6 h. At periodic time points, the solution was filtered through a centrifugal filter (10 K; Millipore Corp.) and the filtrate was analyzed by radio-HPLC using HPLC method 4.
In Vitro and In Vivo Biological Evaluations
Cell Culture and Animal Model
Cell growth and tumor implantation procedures were performed as previously described.84 Briefly, PC-3 cells were cultured in Ham’s F-12K (Kaighn’s) Medium (GIBCO, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified incubator containing 5% CO2 at 37 °C. A 70–80% confluent monolayer was detached with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) and dissociated into a single-cell suspension for further cell culture and assays. The animal procedures were performed according to a protocol approved by the Stanford University Institutional Animal Care and Use Committee. Approximately 2 × 106 cultured PC-3 cells were suspended in 100 μL of PBS and subcutaneously implanted in the left shoulders of male athymic nude mice. Tumors were grown to a size of 0.5–1 cm in diameter (5–6 weeks) prior to biodistribution studies.
In Vitro PC-3 Cell Uptake Assays
The uPAR-mediated binding of the radiolabeled peptide with PC-3 cells was performed as previously described.84 Briefly, PC-3 cells suspended in Ham’s F-12K (Kaighn’s) Medium containing 25 mM N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid), 0.2% bovine serum albumin, and 0.3 mM 1,10-phenanthroline were seeded at a density of 1 × 105 per well in 24-well plates and allowed to attach overnight. The cells were then incubated at 37 °C for 1, 2, or 4 h with purified 8a (1 μCi) alone or with 2 μg AE105 to block uPAR-mediated binding. The cells were then washed 3 times with ice-cold PBS, lysed with 1 mL of 0.1 M NaOH containing 0.1% sodium dodecyl sulfate, and transferred to γ-counter tubes. Radioactivity was measured by a γ-counter (PerkinElmer model 1470), and cell uptake was expressed as the percentage of added radioactivity. The experiment was performed in quadruplicate wells.
In Vivo PC-3 Xenograft Mouse Biodistribution Analysis
For biodistribution studies, male athymic nude mice bearing PC-3 xenografts (n = 4 per group) were injected via the tail vein with approximately 100 μCi of 8a and were euthanized at 1 or 4 h post injection (p.i.). Tumor and normal tissues of interest were removed and weighed, and their radioactivity was measured using a γ-counter. Radioactivity uptake was expressed as a percentage of the injected radioactive dose per gram of tissue (% ID/g).
Supplementary Material
Acknowledgments
The authors wish to thank Mary Dyszlewski of Covidien, Inc., for the Isolink kits and Dr. Gerhard Munske of the Washington State University Molecular Biology and Genomics Core for performing the MALDI-TOF analysis. This research was funded in part by the Office of Science (BER), U.S. Department of Energy (DE-SC0008397), the NIH/NIGMS Biotechnology Training Program at Washington State University (Institutional Award T32 GM008336), the Auvil Fellows Program, the College of Arts and Sciences, and the Chemistry Department at Washington State University.
ABBREVIATIONS
- BocLys
N-α-Boc-L-Lysine-COOH
- CaP
prostate cancer
- Cha
β-cyclohexyl-L-alanine
- cpm
counts per minute
- DMAP
4-dimethylaminopyridine
- DPA
2,2′-dipicolylamine
- ESI-MS
electrospray ionization mass spectrometry
- EtOH
ethanol
- HPLC
high performance liquid chromatography
- IR
infrared
- MALDI-TOF
matrix assisted laser desorption/ionization time-of-flight
- MeCN
acetonitrile
- MeOH
methanol
- MS
mass spectrometry
- NaBH(OAc)3
sodium triacetoxyborohydride
- NEt3
triethylamine
- NMR
nuclear magnetic resonance
- PBS
phosphate buffered saline
- RP-HPLC
reversed-phase HPLC
- SAAC
single amino acid chelate
- SPECT
single photon emission computed tomography
- TFA
trifluoroacetic acid
- tR
retention time
- uPAR
urokinase plasminogen activator receptor
- UV/vis
ultraviolet/visible spectroscopy
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bioconjchem.5b00531.
RP-HPLC chromatogram of 7. Radio-HPLC chromatograms of 8a after purification and following incubation in mouse serum for 6 h (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Waibel R, Alberto R, Willuda J, Finnern R, Schibli R, Stichelberger A, Egli A, Abram U, Mach JP, Plückthun A, et al. Stable one-step technetium-99m labeling of His-tagged recombinant proteins with a novel Tc(I)-carbonyl complex. Nat Biotechnol. 1999;17:897–901. doi: 10.1038/12890. [DOI] [PubMed] [Google Scholar]
- 2.Egli A, Alberto R, Tannahill L, Schibli R, Abram U, Schaffland A, Waibel R, Tourwe D, Jeannin L, Iterbeke K, et al. Organometallic 99mTc-aquaion labels peptide to an unprecedented high specific activity. J Nucl Med. 1999;40:1913–1917. [PubMed] [Google Scholar]
- 3.Alberto R, Schibli R, Egli A, Schubiger AP, Abram U, Kaden TA. A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc-(OH2)3(CO)3]+ from [99mTcO4]− in Aqueous Solution and Its Reaction with a Bifunctional Ligand. J Am Chem Soc. 1998;120:7987–7988. [Google Scholar]
- 4.Alberto R, Kyong Pak J, van Staveren D, Mundwiler S, Benny P. Mono-, bi-, or tridentate ligands? The labeling of peptides with 99mTc-carbonyls. Biopolymers. 2004;76:324–333. doi: 10.1002/bip.20129. [DOI] [PubMed] [Google Scholar]
- 5.Kasten BB, Ma X, Liu H, Hayes TR, Barnes CL, Qi S, Cheng K, Bottorff SC, Slocumb WS, Wang J, et al. Clickable, Hydrophilic Ligand for fac-[MI(CO)3]+ (M = Re/99mTc) Applied in an S-Functionalized α-MSH Peptide. Bioconjugate Chem. 2014;25:579–592. doi: 10.1021/bc5000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore AL, Bucar DK, MacGillivray LR, Benny PD. “Click” labeling strategy for M(CO)3 (M = Re, 99mTc) prostate cancer targeted Flutamide agents. Dalton Trans. 2010;39:1926–1928. doi: 10.1039/b921413e. [DOI] [PubMed] [Google Scholar]
- 7.Kolb HC, Finn MG, Sharpless KB. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Becer CR, Hoogenboom R, Schubert US. Click Chemistry beyond Metal-Catalyzed Cycloaddition. Angew Chem, Int Ed. 2009;48:4900–4908. doi: 10.1002/anie.200900755. [DOI] [PubMed] [Google Scholar]
- 9.Jewett JC, Bertozzi CR. Cu-free click cycloaddition reactions in chemical biology. Chem Soc Rev. 2010;39:1272–1279. doi: 10.1039/b901970g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wangler C, Schirrmacher R, Bartenstein P, Wangler B. Click-Chemistry Reactions in Radiopharmaceutical Chemistry: Fast & Easy Introduction of Radiolabels into Biomolecules for In Vivo Imaging. Curr Med Chem. 2010;17:1092–1116. doi: 10.2174/092986710790820615. [DOI] [PubMed] [Google Scholar]
- 11.Debets MF, van Berkel SS, Dommerholt J, Dirks AJ, Rutjes FPJT, van Delft FL. Bioconjugation with Strained Alkenes and Alkynes. Acc Chem Res. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- 12.Sletten EM, Bertozzi CR. From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Acc Chem Res. 2011;44:666–676. doi: 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeng D, Zeglis BM, Lewis JS, Anderson CJ. The Growing Impact of Bioorthogonal Click Chemistry on the Development of Radiopharmaceuticals. J Nucl Med. 2013;54:829–832. doi: 10.2967/jnumed.112.115550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bottorff SC, Kasten BB, Stojakovic J, Moore AL, MacGillivray LR, Benny PD. Cu-Free 1,3-Dipolar Cycloaddition Click Reactions To Form Isoxazole Linkers in Chelating Ligands for fac-[MI(CO)3]+ Centers (M = Re, 99mTc) Inorg Chem. 2014;53:1943–1945. doi: 10.1021/ic402825t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerjee SR, Babich JW, Zubieta J. Site directed maleimide bifunctional chelators for the M(CO)3+ core (M = 99mTc, Re) Chem Commun (Cambridge, U K) 2005:1784–1786. doi: 10.1039/b417588c. [DOI] [PubMed] [Google Scholar]
- 16.Hayes TR, Lyon PA, Silva-Lopez E, Twamley B, Benny PD. Photo-initiated Thiol-ene Click Reactions as a Potential Strategy for Incorporation of [MI(CO)3]+ (M = Re, 99mTc) Complexes. Inorg Chem. 2013;52:3259–3267. doi: 10.1021/ic302771f. [DOI] [PubMed] [Google Scholar]
- 17.Ganguly T, Kasten BB, Bučar DK, MacGillivray LR, Berkman CE, Benny PD. The hydrazide/hydrazone click reaction as a biomolecule labeling strategy for M(CO)3 (M = Re, 99mTc) radiopharmaceuticals. Chem Commun (Cambridge, U K) 2011;47:12846–12848. doi: 10.1039/c1cc15451f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritzberg AR, Abrams PG, Beaumier PL, Kasina S, Morgan AC, Rao TN, Reno JM, Sanderson JA, Srinivasan A, Wilbur DS. Specific and stable labeling of antibodies with technetium-99m with a diamide dithiolate chelating agent. Proc Natl Acad Sci U S A. 1988;85:4025–4029. doi: 10.1073/pnas.85.11.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eary JF, Schroff RW, Abrams PG, Fritzberg AR, Morgan AC, Kasina S, Reno JM, Srinivasan A, Woodhouse CS, Wilbur DS, et al. Successful Imaging of Malignant Melanoma with Technetium-99m-Labeled Monoclonal Antibodies. J Nucl Med. 1989;30:25–32. [PubMed] [Google Scholar]
- 20.Linder KE, Wen MD, Nowotnik DP, Ramalingam K, Sharkey RM, Yost F, Narra RK, Nunn AD, Eckelman WC. Technetium-labeling of monoclonal antibodies with functionalized BATOs: 2. TcCl(DMG)3CPITC labeling of B72.3 and NP-4 whole antibodies and NP-4 F(ab′)2. Bioconjugate Chem. 1991;2:407–414. doi: 10.1021/bc00012a005. [DOI] [PubMed] [Google Scholar]
- 21.Linder KE, Wen MD, Nowotnik DP, Malley MF, Gougoutas JZ, Nunn AD, Eckelman WC. Technetium labeling of monoclonal antibodies with functionalized BATOs. 1. TcCl(DMG)3PITC. Bioconjugate Chem. 1991;2:160–170. doi: 10.1021/bc00009a005. [DOI] [PubMed] [Google Scholar]
- 22.Kasina S, Rao TN, Srinivasan A, Sanderson JA, Fitzner JN, Reno JM, Beaumier PL, Fritzberg AR. Development and Biologic Evaluation of a Kit for Preformed Chelate Technetium-99m Radiolabeling of an Antibody Fab Fragment Using a Diamide Dimercaptide Chelating Agent. J Nucl Med. 1991;32:1445–1451. [PubMed] [Google Scholar]
- 23.Salmain M, Gunn M, Gorfti A, Top S, Jaouen G. Labeling of proteins by organometallic complexes of rhenium(I). Synthesis and biological activity of the conjugates. Bioconjugate Chem. 1993;4:425–433. doi: 10.1021/bc00024a003. [DOI] [PubMed] [Google Scholar]
- 24.Liu S, Edwards DS, Looby RJ, Poirier MJ, Rajopadhye M, Bourque JP, Carroll TR. Labeling Cyclic Glycoprotein IIb/IIIa Receptor Antagonists with 99mTc by the Preformed Chelate Approach: Effects of Chelators on Properties of [99mTc]Chelator–Peptide Conjugates. Bioconjugate Chem. 1996;7:196–202. doi: 10.1021/bc9500958. [DOI] [PubMed] [Google Scholar]
- 25.Guhlke S, Schaffland A, Zamora PO, Sartor J, Diekmann D, Bender H, Knapp FF, Biersack HJ. 188Re- and 99mTc-MAG3 as prosthetic groups for labeling amines and peptides: Approaches with pre- and postconjugate labeling. Nucl Med Biol. 1998;25:621–631. doi: 10.1016/s0969-8051(98)00025-0. [DOI] [PubMed] [Google Scholar]
- 26.Salmain M, Gorfti A, Jaouen G. Side-chain selective and covalent labeling of proteins by organometallic complexes of heavy transition metals: possible application in radio-crystallography of proteins. Eur J Biochem. 1998;258:192–199. doi: 10.1046/j.1432-1327.1998.2580192.x. [DOI] [PubMed] [Google Scholar]
- 27.Arterburn JB, Rao KV, Goreham DM, Valenzuela MV, Holguin MS, Hall KA, Ott KC, Bryan JC. Functionalized Rhenium(V) Organoimido Complexes as Potential Radiopharmaceuticals. 2. Synthesis, Structural Characterization, and Reactivity of N-Succinimidyl Ester Derivatives with Amines. Organometallics. 2000;19:1789–1795. [Google Scholar]
- 28.Verbeke K, Verbeke A, Vanbilloen H, Verbruggen A. Preparation and preliminary evaluation of 99mTc-EC-For-MLFK. Nucl Med Biol. 2002;29:585–592. doi: 10.1016/s0969-8051(02)00321-9. [DOI] [PubMed] [Google Scholar]
- 29.Calderon Sanchez O, Mohammed A, Mier W, Graham K, Schuhmacher J, Arndt SO, Haberkorn U, Mocelo R, Eisenhut M. 2,3,5,6-Tetrafluorophenyl N-(S-Benzoylthioacetyl)glycylglycyl-p-aminobenzoate, a Heterobifunctional 99mTc Ligand for Precomplexed Antibody Labeling. Bioconjugate Chem. 2003;14:1209–1213. doi: 10.1021/bc034091b. [DOI] [PubMed] [Google Scholar]
- 30.Smith CJ, Gali H, Sieckman GL, Higginbotham C, Volkert WA, Hoffman TJ. Radiochemical Investigations of 99mTc-N3S-X-BBN[7–14]NH2: An in Vitro/in Vivo Structure-Activity Relationship Study Where X = 0-, 3-, 5-, 8-, and 11-Carbon Tethering Moieties. Bioconjugate Chem. 2003;14:93–102. doi: 10.1021/bc020034r. [DOI] [PubMed] [Google Scholar]
- 31.Misra P, Humblet V, Pannier N, Maison W, Frangioni JV. Production of Multimeric Prostate-Specific Membrane Antigen Small-Molecule Radiotracers Using a Solid-Phase 99mTc Preloading Strategy. J Nucl Med. 2007;48:1379–1389. doi: 10.2967/jnumed.107.040303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Misra P, Lebeche D, Ly H, Schwarzkopf M, Diaz G, Hajjar RJ, Schecter AD, Frangioni JV. Quantitation of CXCR4 Expression in Myocardial Infarction Using 99mTc-Labeled SDF-1α. J Nucl Med. 2008;49:963–969. doi: 10.2967/jnumed.107.050054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giblin MF, Jurisson SS, Quinn TP. Synthesis and Characterization of Rhenium-Complexed α-Melanotropin Analogs. Bioconjugate Chem. 1997;8:347–353. doi: 10.1021/bc9700291. [DOI] [PubMed] [Google Scholar]
- 34.Spradau TW, Katzenellenbogen JA. Protein and Peptide Labeling with (Cyclopentadienyl)tricarbonyl Rhenium and Technetium. Bioconjugate Chem. 1998;9:765–772. doi: 10.1021/bc980043t. [DOI] [PubMed] [Google Scholar]
- 35.Spradau TW, Edwards WB, Anderson CJ, Welch MJ, Katzenellenbogen JA. Synthesis and biological evaluation of Tc-99m-cyclopentadienyltricarbonyltechnetium-labeled octreotide. Nucl Med Biol. 1999;26:1–7. doi: 10.1016/s0969-8051(98)00060-2. [DOI] [PubMed] [Google Scholar]
- 36.Volkert WA, Hoffman TJ. Therapeutic Radiopharmaceuticals. Chem Rev. 1999;99:2269–2292. doi: 10.1021/cr9804386. [DOI] [PubMed] [Google Scholar]
- 37.Cooper MS, Sabbah E, Mather SJ. Conjugation of chelating agents to proteins and radiolabeling with trivalent metallic isotopes. Nat Protoc. 2006;1:314–317. doi: 10.1038/nprot.2006.49. [DOI] [PubMed] [Google Scholar]
- 38.Brechbiel MW. Bifunctional Chelates for Metal Nuclides. Q J Nucl Med Mol Imaging. 2008;52:166–173. [PMC free article] [PubMed] [Google Scholar]
- 39.Liu G, Dou S, He J, Vanderheyden JL, Rusckowski M, Hnatowich DJ. Preparation and Properties of 99mTc-(CO)3+-Labeled N,N-Bis(2-pyridylmethyl)-4-aminobutyric Acid. Bioconjugate Chem. 2004;15:1441–1446. doi: 10.1021/bc049866a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerjee SR, Schaffer P, Babich JW, Valliant JF, Zubieta J. Design and synthesis of site directed maleimide bifunctional chelators for technetium and rhenium. Dalton Trans. 2005:3886–3897. doi: 10.1039/b507096a. [DOI] [PubMed] [Google Scholar]
- 41.Maresca KP, Hillier SM, Femia FJ, Zimmerman CN, Levadala MK, Banerjee SR, Hicks J, Sundararajan C, Valliant J, Zubieta J, et al. Comprehensive Radiolabeling, Stability, and Tissue Distribution Studies of Technetium-99m Single Amino Acid Chelates (SAAC) Bioconjugate Chem. 2009;20:1625–1633. doi: 10.1021/bc900192b. [DOI] [PubMed] [Google Scholar]
- 42.Li ZB, Niu G, Wang H, He L, Yang L, Ploug M, Chen X. Imaging of Urokinase-Type Plasminogen Activator Receptor Expression Using a 64Cu-Labeled Linear Peptide Antagonist by microPET. Clin Cancer Res. 2008;14:4758–4766. doi: 10.1158/1078-0432.CCR-07-4434. [DOI] [PubMed] [Google Scholar]
- 43.Persson M, Madsen J, Østergaard S, Ploug M, Kjaer A. 68Ga-labeling and in vivo evaluation of a uPAR binding DOTA- and NODAGA-conjugated peptide for PET imaging of invasive cancers. Nucl Med Biol. 2012;39:560–569. doi: 10.1016/j.nucmedbio.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 44.Kriegbaum MC, Persson M, Haldager L, Alpizar-Alpizar W, Jacobsen B, Gaardsvoll H, Kjaer A, Ploug M. Rational targeting of the urokinase receptor (uPAR): development of antagonists and non-invasive imaging probes. Curr Drug Targets. 2011;12:1711–1728. doi: 10.2174/138945011797635812. [DOI] [PubMed] [Google Scholar]
- 45.Persson M, Madsen J, Oestergaard S, Jensen MM, Joergensen JT, Juhl K, Lehmann C, Ploug M, Kjaer A. Quantitative PET of human urokinase-type plasminogen activator receptor with 64Cu-DOTA-AE105: implications for visualizing cancer invasion. J Nucl Med. 2012;53:138–145. doi: 10.2967/jnumed.110.083386. [DOI] [PubMed] [Google Scholar]
- 46.Persson M, Liu H, Madsen J, Cheng Z, Kjaer A. First 18F-labeled ligand for PET imaging of uPAR: In vivo studies in human prostate cancer xenografts. Nucl Med Biol. 2013;40:618–624. doi: 10.1016/j.nucmedbio.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shariat SF, Semjonow A, Lilja H, Savage C, Vickers AJ, Bjartell A. Tumor markers in prostate cancer I: blood-based markers. Acta Oncol. 2011;50(Suppl 1):61–75. doi: 10.3109/0284186X.2010.542174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.O’Halloran TV, Ahn R, Hankins P, Swindell E, Mazar AP. The many spaces of uPAR: delivery of theranostic agents and nanobins to multiple tumor compartments through a single target. Theranostics. 2013;3:496–506. doi: 10.7150/thno.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nogueira L, Corradi R, Eastham JA. Other biomarkers for detecting prostate cancer. BJU Int. 2010;105:166–169. doi: 10.1111/j.1464-410X.2009.09088.x. [DOI] [PubMed] [Google Scholar]
- 50.Behrendt N, Roenne E, Daneo K. The structure and function of the urokinase receptor, a membrane protein governing plasminogen activation on the cell surface. Bio Chem Hoppe-Seyler. 1995;376:269–279. [PubMed] [Google Scholar]
- 51.Margheri F, Luciani C, Taddei ML, Giannoni E, Laurenzana A, Biagioni A, Chilla A, Chiarugi P, Fibbi G, Del Rosso M. The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget. 2014;5:1538–1553. doi: 10.18632/oncotarget.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Preissner KT, Kanse SM, May AE. Urokinase receptor: a molecular organizer in cellular communication. Curr Opin Cell Biol. 2000;12:621–628. doi: 10.1016/s0955-0674(00)00141-1. [DOI] [PubMed] [Google Scholar]
- 53.Chapman HA, Wei Y. Protease crosstalk with integrins: the urokinase receptor paradigm. Thromb Haemostasis. 2001;86:124–129. [PubMed] [Google Scholar]
- 54.Ploug M. Structure-function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr Pharm Des. 2003;9:1499–1528. doi: 10.2174/1381612033454630. [DOI] [PubMed] [Google Scholar]
- 55.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. doi: 10.1038/nrm2821. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y. The role and regulation of urokinase-type plasminogen activator receptor gene expression in cancer invasion and metastasis. Med Res Rev. 2001;21:146–170. doi: 10.1002/1098-1128(200103)21:2<146::aid-med1004>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 57.Deryugina EI, Quigley JP. Cell surface remodeling by plasmin: a new function for an old enzyme. J Biomed Biotechnol. 2012;2012:564259. doi: 10.1155/2012/564259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noh H, Hong S, Huang S. Role of urokinase receptor in tumor progression and development. Theranostics. 2013;3:487–495. doi: 10.7150/thno.4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sidenius N, Blasi F. The urokinase plasminogen activator system in cancer: Recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003;22:205–222. doi: 10.1023/a:1023099415940. [DOI] [PubMed] [Google Scholar]
- 60.Liu D, Overbey D, Watkinson L, Giblin MF. Synthesis and Characterization of an 111In-Labeled Peptide for the in Vivo Localization of Human Cancers Expressing the Urokinase-Type Plasminogen Activator Receptor (uPAR) Bioconjugate Chem. 2009;20:888–894. doi: 10.1021/bc800433y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun Y, Ma X, Cheng K, Wu B, Duan J, Chen H, Bu L, Zhang R, Hu X, Deng Z, et al. Strained cyclooctyne as a molecular platform for construction of multimodal imaging probes. Angew Chem, Int Ed. 2015;54:5981–5984. doi: 10.1002/anie.201500941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang L, Cao Z, Sajja HK, Mao H, Wang L, Geng H, Xu H, Jiang T, Wood WC, Nie S, et al. Development of Receptor Targeted Magnetic Iron Oxide Nanoparticles for Efficient Drug Delivery and Tumor Imaging. J Biomed Nanotechnol. 2008;4:439–449. doi: 10.1166/jbn.2008.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y, Cozzi PJ. Targeting uPA/uPAR in prostate cancer. Cancer Treat Rev. 2007;33:521–527. doi: 10.1016/j.ctrv.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 64.Knör S, Sato S, Huber T, Morgenstern A, Bruchertseifer F, Schmitt M, Kessler H, Senekowitsch-Schmidtke R, Magdolen V, Seidl C. Development and evaluation of peptidic ligands targeting tumour-associated urokinase plasminogen activator receptor (uPAR) for use in α-emitter therapy for disseminated ovarian cancer. Eur J Nucl Med Mol Imaging. 2008;35:53–64. doi: 10.1007/s00259-007-0582-3. [DOI] [PubMed] [Google Scholar]
- 65.Persson M, Rasmussen P, Madsen J, Ploug M, Kjaer A. New peptide receptor radionuclide therapy of invasive cancer cells: in vivo studies using 177Lu-DOTA-AE105 targeting uPAR in human colorectal cancer xenografts. Nucl Med Biol. 2012;39:962–969. doi: 10.1016/j.nucmedbio.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 66.Persson M, Juhl K, Rasmussen P, Brandt-Larsen M, Madsen J, Ploug M, Kjaer A. uPAR Targeted Radionuclide Therapy with 177Lu-DOTA-AE105 Inhibits Dissemination of Metastatic Prostate Cancer. Mol Pharmaceutics. 2014;11:2796–2806. doi: 10.1021/mp500177c. [DOI] [PubMed] [Google Scholar]
- 67.Shariat SF, Roehrborn CG, McConnell JD, Park S, Alam N, Wheeler TM, Slawin KM. Association of the circulating levels of the urokinase system of plasminogen activation with the presence of prostate cancer and invasion, progression, and metastasis. J Clin Oncol. 2007;25:349–355. doi: 10.1200/JCO.2006.05.6853. [DOI] [PubMed] [Google Scholar]
- 68.Miyake H, Hara I, Yamanaka K, Arakawa S, Kamidono S. Elevation of urokinase-type plasminogen activator and its receptor densities as new predictors of disease progression and prognosis in men with prostate cancer. Int J Oncol. 1999;14:535–541. doi: 10.3892/ijo.14.3.535. [DOI] [PubMed] [Google Scholar]
- 69.Miyake H, Hara I, Yamanaka K, Gohji K, Arakawa S, Kamidono S. Elevation of serum levels of urokinase-type plasminogen activator and its receptor is associated with disease progression and prognosis in patients with prostate cancer. Prostate. 1999;39:123–129. doi: 10.1002/(sici)1097-0045(19990501)39:2<123::aid-pros7>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 70.Piironen T, Haese A, Huland H, Steuber T, Christensen IJ, Brunner N, Danoe K, Hoeyer-Hansen G, Lilja H. Enhanced discrimination of benign from malignant prostatic disease by selective measurements of cleaved forms of urokinase receptor in serum. Clin Chem (Washington, DC, U S) 2006;52:838–844. doi: 10.1373/clinchem.2005.064253. [DOI] [PubMed] [Google Scholar]
- 71.Al-Janabi O, Taubert H, Lohse-Fischer A, Froehner M, Wach S, Stoehr R, Keck B, Burger M, Wieland W, Erdmann K, et al. Association of tissue mRNA and serum antigen levels of members of the urokinase-type plasminogen activator system with clinical and prognostic parameters in prostate cancer. BioMed Res Int. 2014;2014:1. doi: 10.1155/2014/972587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Munch H, Hansen JS, Pittelkow M, Christensen JB, Boas U. A new efficient synthesis of isothiocyanates from amines using di-tert-butyl dicarbonate. Tetrahedron Lett. 2008;49:3117–3119. [Google Scholar]
- 73.Stephenson KA, Zubieta J, Banerjee SR, Levadala MK, Taggart L, Ryan L, McFarlane N, Boreham DR, Maresca KP, Babich JW, et al. A New Strategy for the Preparation of Peptide-Targeted Radiopharmaceuticals Based on an Fmoc-Lysine-Derived Single Amino Acid Chelate (SAAC). Automated Solid-Phase Synthesis, NMR Characterization, and in Vitro Screening of fMLF(SAAC)G and fMLF[(SAAC-Re(CO)3)+]G. Bioconjugate Chem. 2004;15:128–136. doi: 10.1021/bc034128s. [DOI] [PubMed] [Google Scholar]
- 74.Mirzadeh S, Brechbiel MW, Atcher RW, Gansow OA. Radiometal labeling of immunoproteins: covalent linkage of 2-(4-isothiocyanatobenzyl)diethylenetriaminepentaacetic acid ligands to immunoglobulin. Bioconjugate Chem. 1990;1:59–65. doi: 10.1021/bc00001a007. [DOI] [PubMed] [Google Scholar]
- 75.Hedberg E, Långström B. Synthesis of 4-([18F]Fluoromethyl)phenyl Isothiocyanate and its Use in Labelling Oligonucleotides. Acta Chem Scand. 1997;51:1236–1240. [Google Scholar]
- 76.Rosa-Neto P, Wängler B, Iovkova L, Boening G, Reader A, Jurkschat K, Schirrmacher E. [18F]SiFA-isothiocyanate: A New Highly Effective Radioactive Labeling Agent for Lysine-Containing Proteins. ChemBioChem. 2009;10:1321–1324. doi: 10.1002/cbic.200900132. [DOI] [PubMed] [Google Scholar]
- 77.Persson M, Hosseini M, Madsen J, Jørgensen TJD, Jensen KJ, Kjaer A, Ploug M. Improved PET Imaging of uPAR Expression Using new 64Cu-labeled Cross-Bridged Peptide Ligands: Comparative in vitro and in vivo Studies. Theranostics. 2013;3:618–632. doi: 10.7150/thno.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Armstrong AF, Lemon JA, Czorny SK, Singh G, Valliant JF. Evaluation of single amino acid chelate derivatives and regioselective radiolabelling of a cyclic peptide for the urokinase plasminogen activator receptor. Nucl Med Biol. 2009;36:907–917. doi: 10.1016/j.nucmedbio.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 79.Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K. Peptide-Derived Antagonists of the Urokinase Receptor. Affinity Maturation by Combinatorial Chemistry, Identification of Functional Epitopes, and Inhibitory Effect on Cancer Cell Intravasation. Biochemistry. 2001;40:12157–12168. doi: 10.1021/bi010662g. [DOI] [PubMed] [Google Scholar]
- 80.Lin L, Gårdsvoll H, Huai Q, Huang M, Ploug M. Structure-based Engineering of Species Selectivity in the Interaction between Urokinase and Its Receptor: Implication for preclinical cancer therapy. J Biol Chem. 2010;285:10982–10992. doi: 10.1074/jbc.M109.093492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.He H, Lipowska M, Xu X, Taylor AT, Carlone M, Marzilli LG. Re(CO)3 Complexes Synthesized via an Improved Preparation of Aqueous fac-[Re(CO)3(H2O)3]+ as an Aid in Assessing 99mTc Imaging Agents. Structural Characterization and Solution Behavior of Complexes with Thioether-Bearing Amino Acids as Tridentate Ligands. Inorg Chem. 2005;44:5437–5446. doi: 10.1021/ic0501869. [DOI] [PubMed] [Google Scholar]
- 82.Poole LB, Klomsiri C, Knaggs SA, Furdui CM, Nelson KJ, Thomas MJ, Fetrow JS, Daniel LW, King SB. Fluorescent and Affinity-Based Tools To Detect Cysteine Sulfenic Acid Formation in Proteins. Bioconjugate Chem. 2007;18:2004–2017. doi: 10.1021/bc700257a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang H, Kasten BB, Liu H, Qi S, Liu Y, Tian M, Barnes CL, Zhang H, Cheng Z, Benny PD. Novel, Cysteine-Modified Chelation Strategy for the Incorporation of [MI(CO)3]+ (M = Re, 99mTc) in an α-MSH Peptide. Bioconjugate Chem. 2012;23:2300–2312. doi: 10.1021/bc300509k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu Y, Hu X, Liu H, Bu L, Ma X, Cheng K, Li J, Tian M, Zhang H, Cheng Z. A Comparative Study of Radiolabeled Bombesin Analogs for the PET Imaging of Prostate Cancer. J Nucl Med. 2013;54:2132–2138. doi: 10.2967/jnumed.113.121533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.