

Abstract
Cupredoxins are electron transfer proteins that have active sites containing a monomeric Cu center with an unusual trigonal monopyramidal structure (Type 1 Cu). A single Cu-Scys bond is present within the trigonal plane that is responsible for its unique physical properties. We demonstrate that a cysteine-containing variant of streptavidin (Sav) can serve as a protein host to model the structure and properties of Type 1 Cu sites. A series of artificial Cu proteins are described that rely on Sav and a series of biotinylated synthetic Cu complexes. Optical and EPR measurements highlight the presence of a Cu–Scys bond and XRD analysis provide structural evidence. We further provide evidence that changes in the linker between the biotin and Cu complex within the synthetic constructs allows for small changes in the placement of Cu centers within Sav that have dramatic effects on the structural and physical properties of the resulting artificial metalloproteins. These findings highlight the utility of the biotin-Sav technology as an approach for simulating active sites of metalloproteins.
Metalloproteins perform a wide range of chemical transformations whose functions have yet to be achieved by artificial systems. The discrepancy in reactivity can be directly linked to the inability of most unnatural systems to duplicate the precise structural control of the primary and secondary coordination spheres that are present within the active sites of proteins. Numerous synthetic systems and artificial metallo-protein (ArMs) have been developed to regulate both coordination spheres and notable successes have been achieved.1-9 Biotin-streptavidin (Sav) technology has been found to be an effective method for placing metal complexes in specific locations within a protein thanks to Sav's unusually high affinity for biotin (Kb > 1014 M−1).10 For instance, ArMs comprised of biotinylated organometallic complexes and Sav catalyze a variety of organic transformations, many of which having impressive selectivities.11,12 We have extended this approach toward developing artificial Cu-proteins whose primary and secondary coordination sphere can be systematically modulated. We show herein that biotinylated Cu complexes can be confined within Sav to prepare ArMs that have properties similar to those of cupredoxins.
Cupredoxins have a central role in biological electron transfer processes and contain active sites with a single copper center (referred to as Type I Cu centers, Figure 1A) with unusual structural and spectroscopic properties, and relatively high redox potentials (180-800 mV vs NHE).3,13 Detailed structure-function studies have found that “classic” Type 1 centers, such as that in Alcaligenes denitrificans azurin, have Cu sites with unique coordination geometries that are best characterized as distorted trigonal monopyramidal. The trigonal plane contains two N-atom donors from histidine residues and one S-atom donor from a cysteine thiolate; the axial ligand can vary but is often an S-atom from a methionine thioether that only weakly interacts with the Cu center (Cu–Smet > 2.8 Å). The highly covalent Cu(II)–S(thiolate) bond is responsible for the distinct color that arises from an intense Sπ–Cu ligand-to-metal charge transfer (LMCT) band at λmax ~ 600 nm (εM = 3000-6000), as well as the small Cu hyperfine coupling of Az ~ 180 MHz. Variations within cupredoxins exist that are manifested in different structural and spectroscopic properties. These “perturbed” sites (e.g., Rhus vernicifera stellacyanin) have an absorbance band at λmax ~ 600 nm but exhibit an additional feature at λmax ~ 450 nm.14,15 Solomon has defined the parameter Rε = ε450/ε600 to distinguish between the various Type 1 Cu sites: an Rε of less than 0.15 is considered a classical site whereas those with larger values are perturbed sites.13 In addition to their unique primary coordination spheres, the secondary coordination spheres about the Cu centers in cupredoxins impact function. These effects are elegantly illustrated in the work of Lu who used mutagenesis methods to re-engineer the active site in azurin to vary the reduction potential by nearly 2 V.9
Figure 1.

Primary coordination sphere of Type 1 Cu center (A) and biotinylated ligands used in this study (B).
It has been a challenge to reproduce the structural, spectroscopic, and redox properties of Type 1 Cu sites within artificial systems. Attempts to model these active sites include metallopeptides with sequences that contain the loop of cupredoxins16 and three-helix bundles17 that assemble to form a Type 1 Cu binding site. In addition, synthetic Cu complexes have been prepared with sterically constrained ligands that have been useful in investigating the details of the CuII–S(thiolate) interaction.18-20 However, there are few synthetic examples that can simulate the properties of Type 1 Cu sites, but none that incorporates a biologically relevant S-donor from cysteine.
We selected the biotinylated di[2-(2-pyridyl)ethyl]amine (dpea), which is a tridentate ligand that Karlin has shown to tightly bind to CuII ions.21 Three constructs were prepared that differed by the linker group between the biotin and dpea (biot-R-dpea, where R = et, pr, bu) via a 3-step route from dpea (Scheme S1). Formation of the CuII complexes was achieved by treating biot-R-dpea with CuCl2 in CH3CN to afford [CuII(biot-R-dpea)(Cl)(H2O)]Cl. The ArMs [CuII(biot-R-dpea)(H2O)2]2+ ⊂ Sav WT (R = et (1a); pr (1b); bu (1c)) were prepared by incubating Sav WT at pH 6 in MES buffer (50 mM) with a DMF solution of [CuII(biot-R-dpea)(Cl)(H2O)]Cl for 5 min. A 4:1 ratio of each [CuII(biot-R-dpea)(Cl)(H2O)]Cl complex to Sav WT was determined using the (2-(4′-hydroxyazo-benzene)benzoic acid assay22 (Figure S1), indicating that each subunit contains one Cu complex. The spectroscopic properties support the formation of the artificial Cu proteins; protein solutions have weak bands at λmax ~ 650 (εM ~ 50) that are assigned to d-d transitions for the immobilized Cu(II) centers (Figures 2 & S2A, Table S1). The EPR spectra for 1a-c are nearly identical and were simulated for species with S = 1/2 spin ground states. In addition, each spectrum contained two sets of signals of equal intensity (Figure S3) that we suggest are caused by the immobilized Cu(II) complex adopting two orientations (Table S1).
Figure 2.
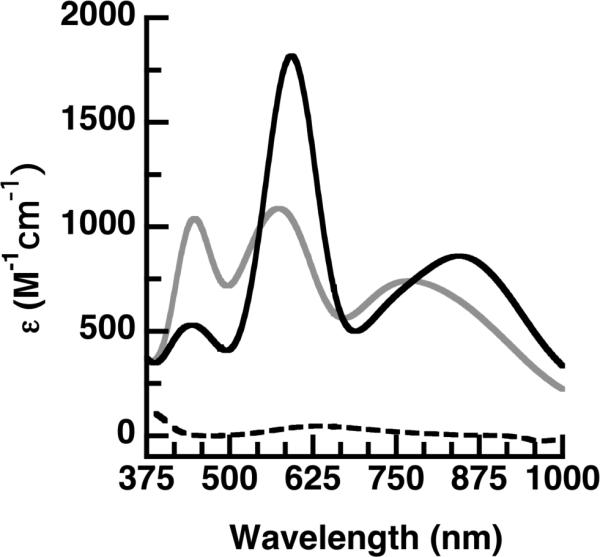
Absorbance spectra for 1a (---), 2a (gray), and 2b (black).
Single crystals of 1a were prepared by soaking crystals of apo-Sav WT with [CuII(biot-et-dpea)(H2O)2]2+. Its structure was solved to 1.72 Å resolution23 to reveal a single Cu(II) complex housed within each subunit of Sav WT arranged in a trigonal bipyramidal geometry, in which the primary coordination sphere is composed of 3 nitrogen atoms from the dpea ligand and 2 oxygen donors from aquo ligands (Figure S4A, Table S2). The dpea ligand binds in a meridional fashion to the copper center with an average Cu–N bond length of 1.98 (1) Å.24 Note that O1 is also part of an H-bonding network that includes S112, the carbonyl of A86, and a structural water molecule.
The structural results for 1a indicate that targeting position 112 may offer a suitable site to install an endogenous donor that could bind to the immobilized Cu complex. Previous work has shown that the imidazole residue in the S112H variant can coordinate to and immobilize a biotinylated Rh complex.25 Rather than using S112H as the host, we prepared new artificial Cu proteins using S112C variant with the goal to design ArMs with Cu sites that are similar to those found in cupredoxins. The confinement of complexes within a subunit should prevent intermolecular interactions that often hinder the formation of discrete Cu–Scys complexes in synthetic systems. For instance, reactions with free [CuII(biot-pr-dpea)(H2O)Cl]Cl complex and various thiols including the cysteine derivative N-(tert-butoxycarbonyl)-L-cysteine methyl ester in DMF, led to a loss of the CuII complex presumably through reduction and formation of disulfide species (Figure S5). To form discrete and stable complexes with Cu–Scys bonds, the S112C variant was incubated with [CuII(biot-R-dpea)(Cl)(H2O)]Cl using the conditions described above for Sav WT to produce [CuII(biot-R-dpea)Scys112+ ⊂ Sav S112C (R = et, (2a), pr, (2b), bu, (2c)).
ArM 2a exhibited optical features that were significantly different from those of 1 with an electronic absorption spectrum containing intense bands at λmax (εM) = 445 (1020), 570 (1010), and 770 (700) (Figure 2, Table 2). These data produced an Rε = 0.95, which is similar to the value of 0.82 reported for the purple cupredoxin Nmar1307.29 Structural information obtained from XRD studies on 2a support thiolate coordination. Analysis of single crystals that diffracted to 1.70 Å resolution revealed formation of a mononuclear Cu complex within each subunit in which the Cu center has an N3S primary coordination sphere (Figure S4B). The dpea ligand adopts a facial orientation around the immobilized Cu center in 2a producing an average Cu–N bond distance of 2.10(2) Å and a Cu–S1 bond length of 2.18(2) Å (Table 2). The CuII complex has N1-Cu-N2, S1-Cu-N1, and S1-Cu-N2 bond angles of 92(3)°, 129(2)°, and 113(2)° that forms a distorted trigonal plane with the Cu positioned 0.62 Å from the plane. In addition, a relatively long N2-Cu-N3 bond angle of 127(2)° was observed.
Table 2.
Selected Bond Lengths (Å) and Angles (°) for 2a-c, Stellacyanin, and M121H Azurin.
| Host | S112C |
Stellacyaninb | Azurin,c,d | ||
|---|---|---|---|---|---|
| n | et (2a) | pr (2b) | bu (2c) | M121H | |
| Cu-N1 | 2.19(2) | 2.24(2) | 2.28(2) | 2.04(2) | 2.02(1) |
| Cu-N2 | 2.03(2) | 2.05(2) | 2.10(2) | 1.96(2) | 2.06(1) |
| Cu-N3 | 2.09(2) | 2.20(2) | 2.12(2) | – | 2.24(1) |
| Cu-S1 | 2.18(2) | 2.11(2) | 2.09(2) | 2.18(2) | 2.25(1) |
| d[Cu-(N/S)t]a | 0.62 | 0.41 | 0.66 | 0.32 | 0.58 |
| N1-Cu-N2 | 92(3) | 92(3) | 89(3) | 101(3) | 98(3) |
| N1-Cu-S1 | 129(3) | 124(3) | 124(3) | 118(3) | 109(3) |
| N2-Cu-S1 | 113(3) | 132(3) | 118(3) | 134(3) | 130(3) |
| N1-Cu-N3 | 97(3) | 94(3) | 87(3) | – | 126(3) |
| N2-Cu-N3 | 127(3) | 96(3) | 105(3) | – | 89(3) |
| N3-Cu-S1 | 100(3) | 110(3) | 124(3) | – | 106(3) |
More intense bands at λmax ~ 600 nm were obtained for 2b and 2c and the higher energy bands at λmax ~ 445 nm were weaker in intensity compared to 2a (Figure 2, Table 1).
Table 1.
Properties for 2a-c and selected cupredoxins.
| Host | S112C |
Stellacyanina | Azurin | ||
|---|---|---|---|---|---|
| n | et (2a) | pr (2b) | bu (2c) | M121Hb,c | |
| λmax,nm (εM) | 448 (1040) | 443 (530) | 437 (780) | 440 (1090) | 439 (NR) |
| 574 (1090) | 593 (1820) | 611 (1790) | 595 (4970) | 593 (NR) | |
| 771 (740) | 846 (860) | 891 (840) | 781(690) | ||
| 893(580) | |||||
| Rε | 0.95 | 0.29 | 0.44 | 0.22 | 1.8 |
| g | 2.02 | 2.05 | 2.06 | 2.02, 2.08 | 2.05 |
| 2.07 | 2.06 | 2.08 | 2.05 | ||
| 2.19 | 2.18 | 2.19 | 2.29 | 2.25 | |
| A, MHz | nd, nd, 353 | 17 | 4 | 171 | 27, 27, 305 |
| 264 | 225 | 87 | |||
| 71 | 79 | 105 | |||
| E1/2 mVd | 140 | 110 | 95 | 184 | <200 |
These new optical data produce Rε values of 0.29 for 2b and 0.44 for 2c both of which are significantly lower than that in 2a and are approaching values that normally are associated with classical blue copper sites. Structural studies on 2b support that the Cu centers in these ArMs are distinct from that in 2a and more comparable to classical Type I Cu sites. The molecular structure of 2b from XRD data collected to 1.70 Å resolution, again, revealed a mononuclear, 4-coordinate Cu complex with an N3S primary coordination sphere (Figure 3A). However, noticeable differences were observed in the structure of the Cu complex in 2b compared to those in 2a. The Cu center in 2b is positioned 1.3 Å further from the biotin binding site, an adjustment that places the pyridine ring containing N2 nearer to residue L124 and that places N3 nearer to residue T114 (Figure 3C). To avoid steric clashing, a significant contraction of the N2-Cu-N3 angle results from 127° in 2a to 96° in 2b (Table 2). These changes in structure for 2b result in an immobilized trigonal monopyramidal Cu complex that has similar properties to Type I Cu centers. For instance, the N1-Cu-N2, S1-Cu-N1, and S1-Cu-N2 bond angles of 92(3)°, 124(2)°, and 132(2)° are nearly equivalent to the related angles found in the azurin M121H32 and stellacyanin (Table 2).30 In addition, the Cu–S1 bond length in 2b has contracted to 2.11(2) Å and the Cu center is displaced only 0.41 Å from the trigonal plane, which is closer to those found in Type 1 Cu sites (Table 2). We have also solved the molecular structure of 2c to 1.40 Å resolution and the structure of the Cu complex is similar, but not identical to 2b, (Table 2, Figure S4D) with the Cu center moving 0.82 Å further from the biotin binding site.
Figure 3.

Structure of 2b (A), structural overlay of 2a (white) and 2b (gray) (B), and space-filling representation of 2b highlighting interactions with residues (yellow) of the protein host (C).
The properties of 2a-c determined from S- and X-band EPR spectroscopy exhibited considerable differences in comparison to ArMs with Sav WT as the host (1a-c).33,34 New methods we introduce here use the spectra from two microwave frequencies, paired with simulation, to determine the A-tensors in the absence of resolved hyperfine splittings. The EPR spectra for 2a showed a 4-line hyperfine pattern at gz = 2.19 with a reduced Az value of 353 MHz compared to the average Az value of 523 MHz obtained for 1a. The lowering of the Az-value in 2a is consistent with Cu-Scys interaction in the ArM. The EPR spectra for 2b and 2c are dramatically different from that of 2a. Normally for Cu(II) complexes, the large hyperfine splitting for Az = A∥ is associated with gz = g∥. This is not the case for 2b and 2c, in which the gz-values of 2.18 and 2.19 have associated Az-values that are low (71 or 79 MHz, respectively). The largest hyperfine splitting values were found in the perpendicular direction: Ay–values of 264 (2b) and 225 MHz (2c) were determined from our simulations with Ax-values closer to zero (Table 1). These values indicate a rhombic A-tensor that deviates significantly from axial symmetry, in contrast to the g-tensor that is nearly axial. The combination of low Az-values and rhombic A-tensors for 2b and 2c is rare and is directly linked to their protein-induced coordination spheres. The small Az-value for 2b and 2c is indicative of relatively large covalency that is consistent with the short Cu–Scys bond lengths observed in the XRD studies.20 The rhombic A-tensor indicates a significant admixture of the dz2 orbital into the singly occupied dx2-y2. Our findings for 2b and 2c are similar to those reported for stellacyanin and the M121H variant of azurin (Table 1).28
The redox properties of the Cu ArMs were measured using cyclic voltammetry (Figure S6): 1a contains a reversible one-electron process for the Cu(II)/(I) couple at E1/2 = 70 mV versus NHE that shifts to 140 mV in 2a. This positive shift for the S112C variant is expected for a thiolate-ligated Cu center, and the potential of 140 mV approaches those reported for Type I proteins. The CVs of 1b-c and 2b-c are less reversible than 1a and 2a but exhibit a similar trend with an increase in Ea of ~70 mV for 2b versus 1b and ~35 mV for 2c versus 1c (Figure S7). This shift to positive potentials most likely reflects a more covalent Cu–Scys bond in 2b2c.
The results for our artificial Cu proteins revealed important design features for using Sav to confine coordination complexes. ArMs with Sav WT produced Cu complexes having nearly identical properties that are similar to those found in synthetic Cu species. However, the S112C variant of Sav produced artificial Cu proteins 2a-c that have markedly different properties caused by the presence of an endogenous thiolate ligand. Moreover, we were able to tune the position of the Cu centers within Sav S112C by varying the linker between the biotin and the metal complex that produced commensurate changes in the properties of the Cu complexes. This method was ineffective with Sav WT in which the the linker length had little effect on the confined species, suggesting that one-point binding through the biotin-Sav interaction may not be enough to regulate the placement of a metal complex within Sav. Coupling the ability to vary the linker with additional anchoring via endogenous ligand coordination offers a versatile means to fine-tune the properties of the metal cofactors.
The properties of 2b and 2c also illustrate the potential of this approach to model active sites of metalloproteins. The ability to modify the Sav host to engineer endogenous ligands, combined with synthetic ligands, produces new types of coordination spheres around the immobilized Cu centers that cannot be readily accessed in a pure synthetic construct. The formation of stable complexes with discrete Cu-Scys bonds in 2a-c was achieved by taking advantage of a Sav variant as the protein host. These results further highlight the benefits of this approach in modulating the structure and properties of an immobilized metal complex to obtain new structures.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the NIH (GM50781-21S1 to ASB and TRW and GM77387 to MPH) for financial support. We thank T. Poulos, H. Li and J. Klehr for assistance.
Footnotes
ASSOCIATED CONTENT
Experimental details for all reactions, physical and XRD measurements, Figures S1-S8, and electron density overlays for all protein structures. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Cook SA, Borovik AS. Acc. Chem. Res. 2015;48:2407. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shook RL, Borovik AS. Inorg. Chem. 2010;49:3646. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu J, Chakraborty S, Hosseinzadeh P, Yu Y, Tian S, Petrik I, Bhagi A, Lu Y. Chem. Rev. 2014;114:4366. doi: 10.1021/cr400479b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DeGrado WF, Summa CM, Pavone V, Nastri F, Lombardi A. Annu. Rev. Biochem. 1999;68:779. doi: 10.1146/annurev.biochem.68.1.779. [DOI] [PubMed] [Google Scholar]
- 5.Bos J, Browne WR, Driessen A, Roelfes G. J. Am. Chem. Soc. 2015;137:9796. doi: 10.1021/jacs.5b05790. [DOI] [PubMed] [Google Scholar]
- 6.Delapierre CM, Rondot L, Cavazza C, Ménage S. Israel J. Chem. 2015;55:61. [Google Scholar]
- 7.Song WJ, Tezcan FA. Science. 2014;346:1525. doi: 10.1126/science.1259680. [DOI] [PubMed] [Google Scholar]
- 8.Berggren G, Adamska A, Lambertz C, Simmons TR, Esselborn J, Atta M, Gambarelli S, Mouesca JM, Reijerse E, Lubitz W, Happe T, Artero V, Fontecave M. Nature. 2013;499:66. doi: 10.1038/nature12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hosseinzadeh P, Marshall NM, Chacón KN, Yu Y, Nilges MJ, New SY, Tashkov SA, Blackburn NJ, Lu Y. Proc. Natl. Acad. Sci. USA. 2016;113:262. doi: 10.1073/pnas.1515897112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber PC, Ohlendorf DH, Wendoloski JJ, Salemme FR. Science. 1989;243:85. doi: 10.1126/science.2911722. [DOI] [PubMed] [Google Scholar]
- 11.Wilson ME, Whitesides GM. J. Am. Chem. Soc. 1978;100:306. [Google Scholar]
- 12.Dürrenberger M, Ward TR. Curr. Opin. Chem. Biol. 2014;19:99. doi: 10.1016/j.cbpa.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 13.Solomon EI, Hadt RG. Coord. Chem. Rev. 2011;255:774. [Google Scholar]
- 14.LaCroix LB, Randall DW, Nersissian AM, Hoitink CWG, Canters GW, Valentine JS, Solomon EI. J. Am. Chem. Soc. 1998;120:9621. [Google Scholar]
- 15.Olesen K, Veselov A, Zhao Y, Wang Y, Danner B, Scholes CP, Shapleigh JP. Biochemistry. 1998;37:6086. doi: 10.1021/bi971603z. [DOI] [PubMed] [Google Scholar]
- 16.Daugherty RG, Wasowicz T, Gibney BR, DeRose VJ. Inorg. Chem. 2002;41:2623. doi: 10.1021/ic010555a. [DOI] [PubMed] [Google Scholar]
- 17.Plegaria JS, Duca M, Tard C, Friedlander TJ, Deb A, Penner-Hahn JE, Pecoraro VL. Inorg. Chem. 2015;54:9470. doi: 10.1021/acs.inorgchem.5b01330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kitajima N, Fujisawa K, Tanaka M, Moro-oka Y. J. Am. Chem. Soc. 1992;114:9232. [Google Scholar]
- 19.Holland PL, Tolman WB. J. Am. Chem. Soc. 1999;121:7270. [Google Scholar]
- 20.Randall DW, George S, Hedman B, Hodgson KO, Fujisawa K, Solomon EI. J. Am. Chem. Soc. 2000;122:11620. [Google Scholar]
- 21.Karlin KD, Kaderli S, Zuberbühler AD. Acc. Chem. Res. 1997;30:139–147. [Google Scholar]
- 22.Skander M, Humbert N, Collot J, Gradinaru J, Klein G, Loosli A, Sauser J, Zocchi A, Gilardoni F, Ward TR. J. Am. Chem. Soc. 2004;126:14411. doi: 10.1021/ja0476718. [DOI] [PubMed] [Google Scholar]
- 23.Crystals of 1b and 1c did not provide suitable data.
- 24.Thyagarajan S, Murthy NN, Sarjeant AAN, Karlin KD, Rokita SE. J. Am. Chem. Soc. 2006;128:7003. doi: 10.1021/ja061014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zimbron JM, Heinisch T, Schmid M, Hamels D, Nogueira ES, Schirmer T, Ward TR. J. Am. Chem. Soc. 2013;135:5384. doi: 10.1021/ja309974s. [DOI] [PubMed] [Google Scholar]
- 26.Malmström BG, Reinhammar B, Vänngård T. Biochim. Biophys. Acta. 1970;205:48. doi: 10.1016/0005-2728(70)90060-5. [DOI] [PubMed] [Google Scholar]
- 27.Reinhammar BR. Biochim. Biophys. Acta. 1972;275:245. doi: 10.1016/0005-2728(72)90045-x. [DOI] [PubMed] [Google Scholar]
- 28.Kroes SJ, Hoitink CW, Andrew CR, Ai J, Sanders-Loehr J, Messerschmidt A, Hagen WR, Canters GW. Eur. J. Biochem. 1996;240:342. doi: 10.1111/j.1432-1033.1996.0342h.x. [DOI] [PubMed] [Google Scholar]
- 29.Hosseinzadeh P, Tian S, Marshall NM, Hemp J, Mullen T, Nilges MJ, Gao Y-G, Robinson H, Stahl DA, Gennis RB, Lu Y. J. Am. Chem. Soc. 2016;138:6324. doi: 10.1021/jacs.5b13128. [DOI] [PubMed] [Google Scholar]
- 30.Hart PJ, Nersissian AM, Herrmann RG, Nalbandyan RM, Valentine JS, Eisenberg D. Protein Sci. 1996;5:2175. doi: 10.1002/pro.5560051104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Messerschmidt A, Prade L, Kroes SJ, Sanders-Loehr J, Huber R, Canters GW. Proc. Natl. Acad. Sci. USA. 1998;95:3443. doi: 10.1073/pnas.95.7.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker EN. J. Mol. Biol. 1988;203:1071. doi: 10.1016/0022-2836(88)90129-5. [DOI] [PubMed] [Google Scholar]
- 33.Lancaster KM, Zaballa M-E, Sproules S, Sundararajan M, DeBeer S, Richards JH, Vila AJ, Neese F, Gray HB. J. Am. Chem. Soc. 2012;134:8241. doi: 10.1021/ja302190r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.All EPR spectra of the Cu ArMs with S112C contained varying amounts (20 to 50%) species with features the same as those found in 1a-1c that were subtracted from the spectra in Figure 4. Complete details are found in the supporting information.
Figure 4.

X- (A) and S-band (B) difference EPR spectra for 2a (blue) and 2b (black). Simulations are dashed lined. Minor amounts of WT ArMs were subtracted from each spectrum.28 The g-values and (A-values in MHz) as indicated.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.