

Abstract
High valent Fe–OH species are often invoked as key intermediates but have only been observed in Compound II of cytochrome P450s. To further address the properties of non-heme FeIV–OH complexes we demonstrate the reversible protonation of a synthetic FeIV–oxo species containing a tris-urea tripodal ligand. The same protonated FeIV–oxo species can be prepared via oxidation, suggesting a putative FeV–oxo species was initially generated. Computational, Mössbauer, XAS, and NRVS studies indicate that protonation of the FeIV–oxo complex most likely occur on the tripodal ligand, which undergoes a structural change that results in the formation of a new intramolecular hydrogen bond with the oxido ligand that aids in stabilizing the protonated adduct. We suggest that similar species for protonated high valent Fe–oxo species may occur in the active sites of proteins. This finding further argues for caution when assigning unverified high valent Fe–OH species to mechanisms.
Graphical Abstract
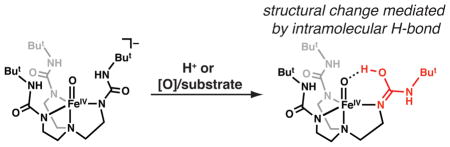
Metal–oxo/hydroxo species are important classes of intermediates that are involved in a variety of oxidative transformations in biology.1–3 The most well studied systems are produced from the activation of dioxygen or peroxide in heme enzymes. The paradigm systems are the cytochrome P450s (P450s), whose hydroxylase components have active sites that are composed of a single heme center and a hydrogen-bonding (H-bonding) network near the di-oxygen binding site.3–5 A common mechanistic proposal among P450s is that a high valent Fe–oxo intermediate is formed from the binding and activation of dioxygen, which then serves as the kinetically competent species in the oxidation of substrates.5,6 The successful trapping of this transient intermediate in P450s (denoted Compound I) has provided sufficient spectroscopic and kinetic evidence to definitively assign it as a high valent FeIV=O(ligand radical) species, supporting the premise that high valent iron–oxo species are formed in monooxygenases.7 The mechanism proposed for the reactivity of Compound I involves the generation of an FeIV–OH species and a carbon radical from an initial C–H bond cleavage step (Scheme 1).3 The FeIV–OH species (Compound II) in P450s has taken on considerable importance with the findings of Green who suggested that its axial coordination to a thiolate ligand both lowers the reduction potential and increases the FeIV–OH pKa value to ~12 in Compound II. The attenuation of the reduction potential of Compound I and pKa of Compound II prevents oxidation of neighboring tyrosine and tryptophan residues that would lead to protein destruction and facilitates C–H bond cleavage (Scheme 1).8
Scheme 1.
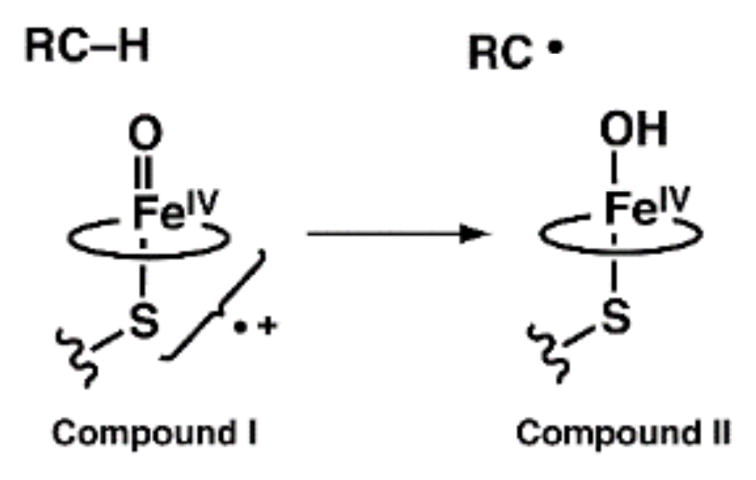
Proposed route for C–H bond cleavage by cytochrome P450. Circles represent protoporphyrin I.
The interplay between high valent Fe–oxo and Fe–OH species also has importance in the function of heme-containing peroxidases.9,10 Recent experimental findings have sparked discussions on whether protonation occurs at the FeIV–oxo species or at a proximal site that then forms a H-bond to the oxido ligand.11,12 The importance of protonated FeIV–oxo species extends beyond heme proteins to non-heme systems that include the Rieske-type monooxygenases and synthetic systems, such as the one suggested by Fukusumi and Nam.13,14 Unlike non-heme FeIV–oxo species in which considerable progress has been made in understanding their properties,15–20 there is a dearth of experimental evidence for protonated non-heme, FeIV–oxo complexes. Nevertheless, it is assumed that protonation of synthetic FeIV–oxo complex occurs at the oxido ligand even though most pKa values for the FeIV—OH units are not known or thought to be less than 5.10,21–23 Furthermore, most synthetic systems lack intramolecular H-bonding networks that are vital in regulating the site of protonation in proteins with FeIV–oxo units.11,12
This report describes methods for the preparation of a protonated FeIV–oxo complex (1). We have previously generated a series of synthetic high valent metal–oxo species supported by the [H3buea]3− ligand (Scheme 2).20,24–28 Included in this series is the high-spin, mononuclear FeIV–oxo complex, [FeIVH3buea(O)]− (2), which has local C3 symmetry that is enforced by the strong nitrogen donors of the deprotonated urea groups.20,29 In addition, the [H3buea]3− ligand promotes the formation of intramolecular H-bonds and thus aids in regulating the secondary sphere around the iron center in a manner reminiscent of active sites in proteins. We have now used this approach to prepare 1, the protonated congener of 2, and characterize its physical properties. Moreover, we show that the same species is obtained via oxidation of 2, implicating the formation of an FeV–oxo species. Our findings suggest that protonation of 1 does not occur at the oxido ligand, but rather at [H3buea]3−, which undergoes a major structural change that is stabilized by a strong intramolecular H-bond to the FeIV–oxo unit.
Scheme 2.

Reactivity of 2 at −80 °C in THF.
The starting point of our study was 2, whose conjugate acid has an experimentally estimated pKa value of ~11.20,30 Based on the work on heme proteins described above,8 we reasoned that this value was sufficient to produce a protonated form that was stable enough to be detected. Consistent with this prediction, treatment of 2 with one equiv [H3NPh]BF4 (pKa = 5.2 in THF, rt)31 at −80°C in THF produced a new species that we have assigned as the protonated analog 1 (Scheme 2). Spectrophotometric monitoring of the reaction showed replacement of the characteristic bands of 2 at λmax = 350, 430, 530, and 810 nm with new peaks for 1 at λmax = 380 and 540 nm; this conversion exhibited isosbestic behavior upon incremental addition of the acid to a solution of 2 with isosbestic points observed at λ = 320, 490 and 710 nm (Figure 1). The EPR signal from 2 vanished and a commensurate change was observed in the Mössbauer parameters from δ = 0.04 mm/s and ΔEQ = 0.50 mm/s for 2 to δ = 0.04 mm/s and ΔEQ = 0.87 mm/s for 1 (Figure 1). Variable field and temperature Mössbauer studies to be published later indicate an S = 2 spin ground state. We also explored this reaction with other acids at −80°C in THF: complete formation of 1 was observed with 1.5 equiv of [NH(CH2CH3)3]BF4 (pKa = 12.5 in THF, rt) but no reaction was found when 2 was treated with one equivalent of pyrrolidinium tetrafluoroborate (pKa = 13.5 in THF, rt).31 These observations suggest an approximate bracketing of the pKa value for 1 between 11–13, which is consistent with our previous thermodynamic predictions for the pKa value for 1 in DMSO (see above).
Figure 1.

Electronic absorbance spectra (left) for the protonation of 2 (dash) to 1 (solid) via sequential addition of 0.25, 0.50, 0.75, and 1.0 equiv of [H3NPh]+ at −80°C in THF. Inset: Oxidation of 2 to 1 with N(p-tol)3+• at −80°C in THF. Mössbauer spectra (right) of 2 (A) and 1 (B) recorded at 4 K in THF. Red lines are the least-square fits of the experimental data with linewidths: (A) 0.32 mm/s, (B) 0.37 mm/s.
The deprotonation of 1 was studied at −80°C in THF using the non-nucleophilic base, diazabicycloundecene (DBU, pKa 16.8 in THF, rt):31 both UV-vis and Mössbauer spectroscopies showed that 1 can be cleanly converted to 2 (Scheme 2, Figure S1) in a yield of 90%. These results demonstrate the reversibility of the protonation/deprotonation process. Moreover, if our assignment of 1 is correct, a comproportionation reaction should occur between 1 and [FeIIH3buea(OH)]2− (3, eq 1) to form [FeIIIH3buea(OH)]− (4), two complexes that we have previously prepared and characterized.24,36 This premise was successfully affirmed by mixing equimolar amounts of 1 and 3 to afford 4 in a concentration that was twice that of the starting complexes. This reaction was followed using UV-vis and EPR spectroscopies, and the final product had identical spectra to those published for 4 (Figures S2 and S3).36 We also found that 1 can be reduced to 4 in nearly quantitative yield using [CoCp2] as the reductant (eq 2, Figure S4). These results indicate that the molecular structure of 2 is not irreversibly changed upon protonation to form 1.
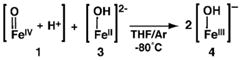 |
(1) |
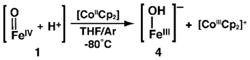 |
(2) |
The vibrational properties of 1 were evaluated using nuclear resonance vibrational spectroscopy (NRVS). Using a sample that contained 80% of 1(57Fe-16O)32 we obtained a NRVS spectrum with a peak at 800 cm−1 that is assigned to the Fe–O vibration (Figure 2). This band shifts to 764 cm−1 in the 1(57Fe-18O) isotopomer which is expected based on a harmonic Fe–O oscillator model that predicts a difference of 36 cm−1. This vibrational feature does not originate from 2 because the sample had only 10% of 2 remaining.30 The NRVS spectrum of 2(57Fe-16O) measured independently, and under identical conditions, showed a peak at 794 cm−1 that we had previously assigned to an FeIV–oxo vibration using FTIR spectroscopy. Furthermore, the lower energy features (450–200 cm−1) between 1(57Fe-16O) and 2(57Fe-16O) that arise from Fe–N vibrations are significantly different, indicative of a structural change upon protonation. We have also probed the structure of 1 using X-ray absorption spectroscopy. The XANES spectrum for 1 has an energy edge at 7122.9 eV that is similar to the 7123.3 eV value found for 2 (Figure S5); both edge energies are consistent with an FeIV center. EXAFS analysis found that the Fe–Ooxo bond lengths in 1 (1.65 Å) and 2 (1.67 Å) are the same within experimental error (Tables S1–S2, Figures S6–S7) with the distance in 2 matching that found by XRD on a crystalline sample.12 A Badger’s rule analysis predicted that this Fe–O bond distance measured for 1 would have an ν(Fe–O) of 799 cm−1 which is in excellent agreement with our NRVS data. Note also that the two complexes have statistically significant differences in the remaining components of their primary coordination spheres. In particular, the EXAFS data for 1 were best fit to three Fe–N bonds at a distance of 1.95 Å while for 2 the fit gave four FeN bonds at a distance of 2.00. The details of these metrical differences are not yet known but it clear that structural changes occur upon conversion of 2 to 1.
Figure 2.

NRVS spectra for 1(57Fe-16O) (A), 1(57Fe-18O) (B), and 2(57Fe-16O) (C). Samples were ~10 mM in THF.
We have used DFT methods to further understand the structural properties of 1.33 The DFT analysis suggests the added proton is located between the oxido group and one of the [H3buea]3− arms. Three low-energy optimized structures for 1 were found that differ by approximately 5 kcal/mol with 1a predicted to be the lowest energy structure. The two lowest energy tautomers have the proton localized on [H3buea]3− (Figure 3). The structure of 1a has one urea group rotated such that one carbonyl group is positioned within the cavity with the proton on O2 (Figure 3). The rotated urea in 1a adopts an imidic acid tautomer with a O2-C10 bond distance of 1.319 Å and a relatively short N2–C10 bond length of 1.329 Å (Table S3).34 The second tautomer 1b has the proton centered on one of the urea arms to form a quaternarized nitrogen center. The third structure 1c has the proton on the oxido ligand to form a hydroxo group. One urea arm in 1c is twisted, which is presumably to avoid steric clashing with the O–H bond.
Figure 3.

Geometry optimizations, relative free energies at RT, and computed Mössbauer parameters for 1a – 1c determined from DFT. The red segments represent key structural aspects of [H3buea]3−.
The Fe–O1 bond distances of 1.696 and 1.703 Å in 1a and 1b are not significantly changed from the analogous bond distance of 1.680(1) Å in 2, which was determined by XRD and supported by XAS and DFT analyses.20,29,35 The long O1⋯H1 distances of 1.450 Å in 1a and 1.549 Å in 1b suggest that the apical ligand is better described as a H-bonded oxido than a hydroxido. However, the O1⋯O2 distance in 1a (2.495 Å) and O1⋯N3 distance in 1b (2.614 Å) are shorter than in other characterized monomeric M–OH or M–oxo structures with [H3buea]3−, indicating that stronger H-bonds are formed because of the increased acidity of these new H-bond donors.36 For 1c, the Fe–O1 bond length is increased to 1.812 Å that reflects the formation of the hydroxido ligand. Moreover, DFT finds that in each tautomer an asymmetry exists within bond length being the equatorial plane that is caused by one Fe–Neq substantially larger (greater than 2 Å) than the others (Table S3); these bond length differences could explain the changes in the FeN vibrations observed between 1 and 2 by NRVS. Finally, DFT predicted that each tautomer has an S = 2 spin state with computed Mössbauer parameters comparable to our experimental findings (Figure 3).
Based on the results from spectroscopy and computations, we argue that the oxido ligand in 1 is the less likely site of protonation. This claim is based on the similarities of the Fe–O vibrations for 1 and 2 as determined by NRVS measurements and the small changes in the Fe–O/N bond lengths between the two FeIV complexes obtained from EXAFS analysis. We thus suggest that the protonation occurs on one of the urea arms of the [H3buea]3−, as illustrated in the structures of 1a and 1b. Based on relative free energy considerations, 1a and 1b are nearly equivalent and their Mössbauer parameters match well with values obtained from experiments. Tautomer 1c contains an FeIV–OH unit but with structural parameters that do not match those found by EXAFS. In addition, Badger’s rule suggests an Fe–O bond length of 1.81 Å as computed for 1c would have a ν(Fe–O) = 573 cm−1, which does not agree with experiment (Figure 2).37
Complex 1 also can be obtained via the oxidation of 2. We previously reported that the FeIV–oxo complex 2 exhibits an irreversible oxidation at 0.34 V vs. [FeCp2]+/0 in DMSO at room temperature that we now report shifts to 0.14 V in THF (Figure S8). To probe this oxidative event, the reaction between 2 and the radical amminium cation, N(p-tol)3+• (0.4 V vs. [FeCp2]0/+)38 was monitored spectrophotometrically at −80°C in THF. Rather than a new FeV species that would be expected for single electron transfer, this reaction gave 1 as the major iron species in 60% yield. The formation of 1 was supported by spectrophotometric (inset, Figure 1) and Mössbauer experiments (Figure S9). The same results were also obtained when the reaction was performed at −105°C in THF and −120°C in 2-MeTHF. A mechanistic route that is consistent with these observations is the initial formation of an FeV–oxo intermediate (6) that, in the absence of external substrates, rapidly reacts with solvent to form 1 (Scheme 2). Because we have been unable to detect any oxidized products from THF, we examined this reaction with external substrates. The oxidized form of 2 did not react with external compounds such as 9,10-dihydroanthracene, but detectable products were observed with phenols. For instance, when 100 equiv of 2,4,6-tri-tert-butylphenol (7) were present in the oxidation reaction, the formation of 1 and the corresponding phenoxy radical (9) was observed, as evidenced by the appearance of its optical bands at λmax = 382 and 402 nm (eq 3, Figures S10–S11). Moreover, 2,2′,6,6′-tetra-tert-butyl-4,4′-biphenol (10) was produced in a 20(5)% yield when 2,6-di-tert-butylphenol (8) was used instead of 7, as determined by gas chromatography (GC) and GC-MS (Figures S12–S13). These results provide evidence that an FeV–oxo species could initially be formed during the oxidative conversion of 2 to 1.
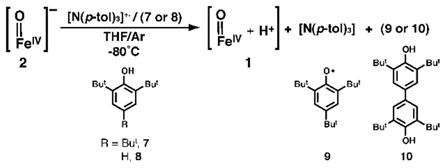 |
(3) |
Protonated FeIV–oxo complexes are difficult to prepare, yet such species are proposed to readily form within protein active sites in which intramolecular H-bonds within the secondary coordination sphere are prevalent. Using the synthetic ligand [H3buea]3− that also promotes intramolecular H-bonds, we have demonstrated that the well-characterized FeIV–oxo complex 2 reacts with acids to produce a new species that is assigned to the protonated FeIV–oxo species 1. The properties and reactivity of 1 are all consistent with this assignment, including its reversion back to 2 with the addition of the base DBU, its reaction with the FeII–OH complex 3 to produce the FeIII–OH species 4, and its conversion to 4 by reduction with [CoCp2]. Insights into the structure of 1 came from spectroscopy and DFT calculations, which suggested that the likely site of protonation is the [H3buea]3− ligand. The results from NRVS spectroscopy indicated that the FeIV–oxo unit remains intact. In the lowest energy complexes from DFT, one tripodal arm is protonated, resulting in a new intramolecular H-bonding interaction with the FeIV–oxo unit. The computational studies implicate the FeIV–O⋯H–X (X = O, N) intramolecular H-bond as a key contributor to the stability of this structure.
Treatment of 2 with oxidant did not yield a detectable FeV species that would be comparable to Compound I; instead, 1 was obtained in good yield. The detection of 1 provided the opportunity to examine its formation from routes similar to those used to produce Compound II in P450s. In the presence of phenolic substrates, the expected oxidation products were found, which indicated that a putative FeV–oxo species may have been produced. Taken together, our findings offer experimental and computational evidence that a synthetic protonated FeIV–oxo complex has been prepared via a route that is similar to that proposed in P450s.8
Our experimental results showed that 1 and 2 differ from one another by a single proton, but the combination of spectroscopy and theory suggests protonation of 2 most likely occurs on [H3buea]3− rather than forming an FeIV–OH unit as might be expected. The structure of 1a illustrates how an FeIV–O unit may assist in protonating a nearby functional group through formation of an intramolecular H-bond. We argue these types of interactions are not limited to just our system and may occur within the active site of a protein or a synthetic complex, especially those with presumed high valent Fe–OH units.11,12 For instance, mechanisms for non-heme iron enzymes and their synthetic models often invoke FeIV–OH and FeV–OH intermediates,14,39–41 but evidence for the protonation of an FeIV–oxo unit has only been observed in heme-containing proteins such as Compound II of P450s and the chloroperoxidases.8,42,43 Such reactivity has yet to be definitively observed in other metalloproteins that invoke high valent Fe–OH species such as the Rieske non-heme oxygenases.1,39 Our results question whether these high valent Fe–OH species are the only intermediates in mechanisms other than P450s, particularly when protonation at other sites within the complex (or an active site) cannot be ruled out.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health USA (GM050781 to ASB, GM49970 to MPH, and GM101390 to MTG), the Ministerio de Economia y Competitividad (CTQ2014-59212-P && 2015FIB00165), and the European Fund for Regional Development (UNGI10-4E-801) for financial support, and E. E. Alp, J. Zhao, and M. Hu from the APS in the Argonne National Laboratory for the support in collecting NRVS spectra. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource supported by DOE, OBES and NIH; we thank M Latimer and E. Nelson for assistance.
Footnotes
Experimental details for all reactions and physical measurements, and Figures S1–S12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- 2.Que L, Tolman WB. Nature. 2008;455:333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]
- 3.Mas-Ballesté R, Que L. Science. 2006;312:1885–1886. doi: 10.1126/science.1129814. [DOI] [PubMed] [Google Scholar]
- 4.Poulos TL. Chem Rev. 2014;114:3919–3962. doi: 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krest CM, Onderko EL, Yosca TH, Calixto JC, Karp RF, Livada J, Rittle J, Green MT. J Biol Chem. 2013;288:17074–17081. doi: 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 7.Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 8.Yosca TH, Rittle J, Krest CM, Onderko EL, Silakov A, Calixto JC, Behan RK, Green MT. Science. 2013;342:825–829. doi: 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gumiero A, Metcalfe CL, Pearson AR, Raven EL, Moody PCE. J Biol Chem. 2011;286:1260–1268. doi: 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behan RK, Green MT. J Inorg Biochem. 2006;100:448–459. doi: 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 11.Chreifi G, Baxter EL, Doukov T, Cohen AE, McPhillips SE, Song J, Meharenna YT, Soltis SM, Poulos TL. Proc Natl Acad Sci U S A. 2016;113:1226–1231. doi: 10.1073/pnas.1521664113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casadei CM, Gumiero A, Metcalfe CL, Murphy EJ, Basran J, Concilio MG, Teixeira SCM, Schrader TE, Fielding AJ, Ostermann A, Blakeley MP, Raven EL, Moody PCE. Science. 2014;345:193–197. doi: 10.1126/science.1254398. [DOI] [PubMed] [Google Scholar]
- 13.Wackett LP. Enzyme Microb Technol. 2002;31:577–587. [Google Scholar]
- 14.Park J, Lee YM, Nam W, Fukuzumi S. J Am Chem Soc. 2013;135:5052–5061. doi: 10.1021/ja311662w. [DOI] [PubMed] [Google Scholar]
- 15.Hohenberger J, Ray K, Meyer K. Nat Commun. 2012;3:1–13. doi: 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
- 16.McDonald AR, Que L. Coord Chem Rev. 2013;257:414–428. [Google Scholar]
- 17.Puri M, Que L. Acc Chem Res. 2015;48:2443–2452. doi: 10.1021/acs.accounts.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang D, Ray K, Collins MJ, Farquhar ER, Frisch JR, Gómez L, Jackson TA, Kerscher M, Waleska A, Comba P, Costas M, Que L. Chem Sci. 2013;4:282–291. doi: 10.1039/C2SC21318D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nam W, Lee YM, Fukuzumi S. Acc Chem Res. 2014;47:1146–1154. doi: 10.1021/ar400258p. [DOI] [PubMed] [Google Scholar]
- 20.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yosca TH, Behan RK, Krest CM, Onderko EL, Langston MC, Green MT. J Am Chem Soc. 2014;136:9124–9131. doi: 10.1021/ja503588n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jensen MP, Costas M, Ho RYN, Kaizer J, Mairata i Payeras A, Münck E, Que L, Rohde J-U, Stubna A. J Am Chem Soc. 2005;127:10512–10525. doi: 10.1021/ja0438765. [DOI] [PubMed] [Google Scholar]
- 23.Stoian SA, Xue G, Bominaar EL, Que L, Münck E. J Am Chem Soc. 2014;136:1545–1558. doi: 10.1021/ja411376u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.MacBeth CE, Golombek AP, Jr, VGY, Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 25.Borovik AS. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 26.Gupta R, Taguchi T, Lassalle-Kaiser B, Bominaar EL, Yano J, Hendrich MP, Borovik AS. Proc Natl Acad Sci U S A. 2015;112:5319–5324. doi: 10.1073/pnas.1422800112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cook SA, Borovik AS. Acc Chem Res. 2015;48:2407–2414. doi: 10.1021/acs.accounts.5b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shook RL, Borovik AS. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta R, Lacy DC, Bominaar EL, Borovik AS, Hendrich MP. J Am Chem Soc. 2012;134:9775–9784. doi: 10.1021/ja303224p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usharani D, Lacy DC, Borovik AS, Shaik S. J Am Chem Soc. 2013;135:17090–17104. doi: 10.1021/ja408073m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaljurand I, Kütt A, Sooväli L, Rodima T, Mäemets V, Leito I, Koppel IA. J Org Chem. 2005;70:1019–1028. doi: 10.1021/jo048252w. [DOI] [PubMed] [Google Scholar]
- 32.Percentages were determined using Mössbauer spectroscopy. The samples used in these studies had less than a 10% impurity of 2.
- 33.Swart M, Solà M, Bickelhaupt FM. J Chem Phys. 2009;131:094103. doi: 10.1063/1.3213193. [DOI] [PubMed] [Google Scholar]
- 34.Fairlie DP, Taube H. Inorg Chem. 1985;24:3199–3206. [Google Scholar]
- 35.Swart M. Chem Commun. 2013;49:6650. doi: 10.1039/c3cc42200c. [DOI] [PubMed] [Google Scholar]
- 36.MacBeth CE, Gupta R, Mitchell-Koch KR, Young VG, Lushington GH, Thompson WH, Hendrich MP, Borovik AS. J Am Chem Soc. 2004;126:2556–2567. doi: 10.1021/ja0305151. [DOI] [PubMed] [Google Scholar]
- 37.Green MT. J Am Chem Soc. 2006;128:1902–1906. doi: 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- 38.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 39.Prat I, Mathieson JS, Güell M, Ribas X, Luis JM, Cronin L, Costas M. Nat Chem. 2011;3:788–793. doi: 10.1038/nchem.1132. [DOI] [PubMed] [Google Scholar]
- 40.Tiago de Oliveira F, Chanda A, Banerjee D, Shan X, Mondal S, Que L, Bominaar EL, Münck E, Collins TJ. Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]
- 41.Prat I, Company A, Postils V, Ribas X, Que L, Luis JM, Costas M. Chemistry. 2013;19:6724–6738. doi: 10.1002/chem.201300110. [DOI] [PubMed] [Google Scholar]
- 42.Green MT, Dawson JH, Gray HB. Science. 2004;304:1653–1656. doi: 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- 43.Stone KL, Behan RK, Green MT. Proc Natl Acad Sci U S A. 2006;103:12307–12310. doi: 10.1073/pnas.0603159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.