

ABSTRACT
Proteins that form the reovirus outer capsid play an active role in the entry of reovirus into host cells. Among these, the σ1 protein mediates attachment of reovirus particles to host cells via interaction with cell surface glycans or the proteinaceous receptor junctional adhesion molecule A (JAM-A). The μ1 protein functions to penetrate the host cell membrane to allow delivery of the genome-containing viral core particle into the cytoplasm to initiate viral replication. We demonstrate that a reassortant virus that expresses the M2 gene-encoded μ1 protein derived from prototype strain T3D in an otherwise prototype T1L background (T1L/T3DM2) infects cells more efficiently than parental T1L. Unexpectedly, the enhancement in infectivity of T1L/T3DM2 is due to its capacity to attach to cells more efficiently. We present genetic data implicating the central region of μ1 in altering the cell attachment property of reovirus. Our data indicate that the T3D μ1-mediated enhancement in infectivity of T1L is dependent on the function of σ1 and requires the expression of JAM-A. We also demonstrate that T1L/T3DM2 utilizes JAM-A more efficiently than T1L. These studies revealed a previously unknown relationship between two nonadjacent reovirus outer capsid proteins, σ1 and μ1.
IMPORTANCE How reovirus attaches to host cells has been extensively characterized. Attachment of reovirus to host cells is mediated by the σ1 protein, and properties of σ1 influence the capacity of reovirus to target specific host tissues and produce disease. Here, we present new evidence indicating that the cell attachment properties of σ1 are influenced by the nature of μ1, a capsid protein that does not physically interact with σ1. These studies could explain the previously described role for μ1 in influencing reovirus pathogenesis. These studies are also of broader significance because they highlight an example of how genetic reassortment between virus strains could produce phenotypes that are distinct from those of either parent.
INTRODUCTION
Attachment of virus is the first step in the infection of host cells. Cell attachment occurs via interactions of viral attachment factors with host cell receptors. For enveloped viruses, viral glycoproteins embedded in the lipid membrane serve as attachment factors (1). For nonenveloped viruses, specific structural features on the capsid or sequences within the exposed portion of the viral structural proteins bind host receptors (1). Mutations within the receptor-binding site can alter the efficiency with which virus attaches to host cells and consequently modulate the capacity of the virus to establish infection. In viral systems where capsids are formed from multiple structural proteins, these proteins fit together in a precise geometric arrangement. Thus, changes to the properties of one capsid protein can influence the function of other capsid proteins. In this report, we highlight one such example by demonstrating a previously unknown functional relationship between two nonadjacent viral capsid proteins of mammalian orthoreovirus (reovirus).
Reovirus forms virions comprised of two concentric capsid shells (2). The inner capsid or core encapsidates the 10 segments of genomic double-stranded RNA (dsRNA) and contains enzymes needed to launch virus replication upon entry into cells (2). The viral outer capsid contains 3 capsid proteins, σ1, σ3, and μ1, that play important roles in cell entry (3). The σ3 and μ1 proteins form heterohexamers, 200 of which decorate the outer capsid (4, 5). Among them, the σ3 protein masks the cell penetration function of the μ1 protein until the virion is proteolytically disassembled (3). Attachment of the virion to the host cell occurs via trimers of the σ1 protein (6, 7), which are held onto virus particles at the icosahedral vertices of the particle via interaction with the turret-forming λ2 protein (4, 5, 8).
The σ1 protein interacts with host cells by associating with at least two types of receptors. σ1 proteins from all serotypes of reovirus engage proteinaceous receptor junctional adhesion molecule A (JAM-A) (9, 10). In addition, σ1 engages a serotype-specific glycan receptor. Whereas serotype 1 (T1) σ1 engages GM2, T3 σ1 engages glycans that terminate in sialic acid (11–14). Two other cell surface-localized host molecules, β1 integrin(s) and Ngr1, have also been implicated in facilitating reovirus entry and infection (15, 16). Whether β1 integrin interacts with viral components is not known. Though Ngr1 has been demonstrated to interact directly with virus particles (16), viral structures or proteins that participate in the interaction with Ngr1 remain to be identified.
We have previously characterized reovirus M2 gene reassortants in vitro to evaluate the conformational flexibility and membrane penetration properties of the M2-encoded μ1 protein (17, 18). Here we sought to examine the infectious properties of these viruses. We found that a reassortant type 1 reovirus with a type 3 M2 gene (T1L/T3DM2) establishes infection with greater efficiency than the parental T1L strain. Surprisingly, the enhanced infectivity of T1L/T3DM2 was related to an increase in its efficiency of binding to host cells in comparison to that of T1L. Our data suggest that the central region of the T3D-derived μ1 protein affects the attachment efficiency of the virus. The increased infectivity of T1L/T3DM2 requires the function of the σ1 attachment protein and expression of its cellular binding partner, JAM-A. Our studies revealed for the first time that the properties of the reovirus μ1 protein affect viral infectivity by impacting the receptor-binding function of the nonadjacent σ1 attachment protein.
MATERIALS AND METHODS
Cells.
Spinner-adapted murine L929 cells were maintained in Joklik's minimum essential medium (MEM) (Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM l-glutamine (Invitrogen), 100 U/ml penicillin (Invitrogen), 100 μg/ml streptomycin (Invitrogen), and 25 ng/ml amphotericin B (Sigma-Aldrich). Spinner-adapted L929 cells were used for cultivating, purifying, and measuring the titers of viruses. L929 cells obtained from ATCC were maintained in Eagle's MEM (Lonza) supplemented to contain 5% fetal bovine serum (FBS) (Sigma-Aldrich) and 2 mM l-glutamine (Invitrogen). Experiments to measure attachment, infectivity, and cell death were done using L929 cells from ATCC. CHO cells transfected with empty vector (pCDNA3.1) or with pCDNA3.1 encoding human JAM-A were maintained in Ham's F12 medium supplemented to contain 5% fetal bovine serum (FBS) (Sigma-Aldrich), 2 mM l-glutamine (Invitrogen), and 1 mg/ml G418 (Sigma-Aldrich). HeLa cells were maintained in Dulbecco's MEM supplemented to contain 5% fetal bovine serum (FBS) (Sigma-Aldrich) and 2 mM l-glutamine (Invitrogen). In experiments to determine the effect of GM2 binding, cells were pretreated at 37°C for 1 h with 100 mU/ml neuraminidase (MP Biomedicals or Roche) in serum-free media.
Generation of recombinant viruses.
Recombinant strains T1L/T3DM2, T1L/(DLD)M2, T1L/(LDL)M2, T1L/T3DM2 A305V, T1L/T3DM2 R327K, T1L/T3DM2 S340T, T1L/T3DM2 N342G, T1L/T3DM2 D517E, T1L/T3DS1, and T1L/T3DS1M2, which contain a wild-type T3D gene, a chimeric or mutated M2 gene, or a T3D S1 gene in an otherwise T1L background, were generated by using a plasmid-based reverse-genetics strategy (19). To confirm sequences of mutant viruses, viral RNA was extracted from infected cells and subjected to reverse transcription-PCR using three sets of M2-specific or S1-specific primer. PCR products were resolved on Tris-acetate-EDTA agarose gels, purified, and confirmed by sequence analysis.
Purification of viruses.
Purified reovirus virions were generated using second- or third-passage L-cell lysate stocks of reovirus. Viral particles were extracted from infected cell lysates by the use of Vertrel-XF (Dupont), layered onto 1.2- to 1.4-g/cm3 CsCl gradients, and centrifuged at 187,183 × g for 4 h. Bands corresponding to virions (1.36 g/cm3) were collected and dialyzed in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) (20). The concentration of reovirus virions in purified preparations was determined from an equivalence of 1 optical density (OD) unit at 260 nm equaling 2.1 × 1012 virions/ml (21). Virus titers were determined using a standard plaque assay method and spinner-adapted L929 cells. Four different preparations of T1L and T1L/T3DM2 were used for the experiments described in this paper. These are designated A, B, C, and D. The matched sets (particle-to-PFU ratios) were as follows: T1L-A (47) and T1L/T3DM2-A (42.5); T1L-B (262) and T1L/T3DM2-B (630); T1L-C (656) and T1L/T3DM2-C (367); and T1L-D (438) and T1L/T3DM2-D (750). Three preparations of T1L/T3DS1 and T1L/T3DS1M2 were used. The matched sets (particle-to-PFU ratios) were as follows: T1L/T3DS1-A (3,330) and T1L/T3DS1M2-A (4,090); T1L/T3DS1-B (715) and T1L/T3DS1M2 (757); and T1L/T3DS1-C (542) and T1L/T3DS1M2-C (453). A single preparation of DLDM2 and a single preparation of LDLM2 were used, and their particle-to-PFU ratios were 1,640 and 889, respectively. The single amino substitutions (particle-to-PFU ratios) were as follows: A305V (20), R327K (34), S340T (42.7), N342G (44.6), and D517E (46.1).
Dynamic light scattering (DLS).
The indicated virus stocks (2 × 1012 particles/ml) were analyzed using a Zetasizer Nano S dynamic light scattering system (Malvern Instruments). All measurements were made in a quartz Suprasil cuvette with a 3.00-mm-path length (Hellma Analytics). For each sample, the hydrodynamic diameter (DH) was determined by averaging readings across 15 iterations.
Generation of ISVPs in vitro.
Intermediate subviral particles (ISVPs) were generated in vitro by incubation of 2 × 1012 virions with 200 μg/ml of TLCK (Nα-p-tosyl-l-lysine chloromethyl ketone)-treated chymotrypsin (CHT) in a total volume of 0.1 ml at 32°C in virion storage buffer (150 mM NaCl, 15 mM MgCl2, 10 mM Tris-HCl [pH 7.4]) for 60 min. Proteolysis was terminated by addition of 2 mM phenylmethylsulfonyl fluoride (PMSF) and incubation of reaction mixtures on ice. Generation of ISVPs was confirmed by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie brilliant blue staining.
Assessment of infectivity by indirect immunofluorescence.
Monolayers of L929 cells, CHO cells, or HeLa cells (4 × 104) in 96-well plates were washed with phosphate-buffered saline (PBS) and infected with virions or ISVPs of the indicated reovirus strain at 4°C for 1 h. In experiments to examine the role of glycans in infection, the cells were pretreated with serum-free Eagle's MEM containing 100 mU/ml neuraminidase (Roche) at 37°C for 1 h prior to virus attachment. In experiments to examine the effect of 5C6 monoclonal Ab (MAb) or JAM-A glutathione S-transferase (GST–JAM-A) on virus infectivity, 1 × 1011 virus particles/ml were incubated at 4°C with various amounts of 5C6 MAb or GST–JAM-A overnight (22, 23). The mixture was used to initiate infection as described above. Following removal of the inoculum, cells were washed with PBS and incubated in media appropriate to the cell type at 37°C for 18 h. Monolayers were fixed with 100 μl of methanol at −20°C for a minimum of 30 min, washed twice with PBS, blocked with 2.5% immunoglobulin-free bovine serum albumin (BSA; Invitrogen)–PBS, and incubated with polyclonal rabbit anti-reovirus serum at a 1:5,000 dilution in PBS–0.25% Triton X-100 at room temperature for 30 min (24). Monolayers were washed twice with PBS and incubated with a 1:5,000 dilution of Alexa Fluor 488-labeled anti-rabbit immunoglobulin G. Cells were stained with 0.5 μg/ml 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen) at a concentration of 1:10,000. Monolayers were washed with PBS, and infected cells were visualized by indirect immunofluorescence using a fluorescein isothiocyanate (FITC)/DAPI filter set on an Olympus 1X71 microscope. Infected cells were identified by the presence of intense cytoplasmic fluorescence that was excluded from the cells. No background staining of uninfected control monolayers was noted. Reovirus antigen-positive cells were quantified by counting fluorescent cells in random fields in triplicate wells at ×20 magnification. Percentages of reovirus-positive cells were calculated by counting infected cells per total (DAPI-positive) cells.
Quantitation of cell death by acridine orange and ethidium bromide (AOEB) staining.
L929 cells grown in 96-well plates were adsorbed with virus at room temperature for 1 h. The percentage of dead cells was determined after incubation at 37°C for 48 or 72 h postinfection using AOEB staining. For each sample, a total of 600 cells were counted, and the percentage of isolated cells exhibiting orange staining (EB positivity) was determined by fluorescence microscopy using an FITC/tetramethyl rhodamine isocyanate (TRITC) filter set on Olympus IX71 microscope. As a control, cell death in mock-infected cells was also determined. Since cell death under these conditions was minimal (<3%), these data are not included.
Assessment of reovirus attachment to cells by flow cytometry.
Confluent 150-mm-diameter dishes of L929 cells (2 × 107) were washed with chilled PBS. The cells were detached using PBS supplemented with 20 mM EDTA (PBS-EDTA) and centrifuged at 1,000 × g at 4°C for 5 min. The cells were resuspended to obtain 2 × 106 cells per ml. Cell were adsorbed with virus particles at 4°C for 1 h with continuous rotation on a tube rotator. After removal of the inoculum, the cells were harvested and resuspended in PBS supplemented with 5% BSA (PBS-BSA). Reovirus-specific rabbit polyclonal antiserum at 1:2,500 was added, and the reaction mixture was incubated at 4°C for 30 min with continuous rotation. The cells were washed twice with PBS-BSA followed by incubation at 4°C for 30 min with Alexa Fluor 488-labeled goat anti-rabbit antibody at 1:1,000 in PBS-BSA with continuous rotation. The cells were washed twice with PBS-BSA prior to fixing with cold 1% paraformaldehyde–PBS. Viral attachment to cells was quantified using a BD FACSCalibur cell analyzer and the CellQuestPro software.
Assessment of reovirus attachment to cells by on-cell Western blotting.
L929 cells grown in 96-well plates were chilled at 4°C for 15 min and then adsorbed with virions at 4°C for 1 h. Cells were washed with chilled PBS and blocked with PBS-BSA at 4°C for 10 min. Cells were then incubated with polyclonal rabbit anti-reovirus serum at a 1:2,500 dilution in PBS-BSA at 4°C for 30 min. The cells were washed twice with PBS-BSA followed by incubation with a 1:1,000 dilution of Alexa Fluor 750-labeled goat anti-rabbit antibody at 4°C for 30 min. After two washes with PBS-BSA, cells were stained with a 1:1,000 dilution of a DNA stain, DRAQ5 (Cell Signaling Technology), at 4°C for 5 min. Cells were washed and then fixed with 4% formaldehyde at room temperature for 20 min. The plate was scanned using an Odyssey infrared imager (Li-COR). Binding index was quantified by quantifying the ratio of green (reovirus) and red fluorescence (DRAQ5) using Image Studio Lite software (Li-COR).
Comparing reactivity of anti-reovirus polyclonal sera.
High-affinity-binding polystyrene plates (Pierce) were coated at 4°C overnight with the indicated amount of T1L or T1L/T3DM2 resuspended in 0.1 M carbonate-bicarbonate buffer at pH 9.5. Plates were blocked at 4°C for 1 h with 2.5% BSA in virion storage buffer, followed by two washes with wash buffer (0.1% BSA, 0.05% Tween, virion storage buffer). The plates were stained at room temperature for 1 h with reovirus-specific rabbit polyclonal antiserum (1:5,000) followed by Alexa Fluor 750-labeled anti-rabbit antibody (1:1,000). The plate was scanned using an Odyssey infrared imager (Li-COR).
Assessment of virus attachment by plaque assay.
L929 cells grown in 24-well plates were chilled at 4°C for 15 min and then adsorbed with 5 × 104 virions/cell at 4°C for 1 h. Cells were washed with chilled PBS, overlaid with 0.5 ml PBS, and frozen at −80°C. Following two cycles of freeze-thaw, the cell-associated virus was quantified by plaque assay.
σ1/μ1 ratio by Western blot analysis.
Purified T1L or T1L/T3DM2 virions (2 × 1010 particles) from 4 independent viral preparations were resolved on 10% SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% milk–Tris-buffered saline (TBS) at room temperature for 1 h followed by incubation with 4A3 mouse anti-μ1 MAb (1:500) and rabbit anti-T1L σ1 head antibody (1:750) at room temperature for 1 h in blocking buffer (22, 25). The membranes were washed with TBS supplemented with 0.1% Tween 20 (TBS-T) twice for 15 min and then incubated with Alexa Fluor-conjugated anti-mouse IgG (for 4A3) or with Alexa Fluor-conjugated anti-rabbit IgG (for σ1 head) in blocking buffer. Following three washes, membranes were scanned using an Odyssey infrared imager (LI-COR). σ1/μ1 ratios were calculated from the intensities of the σ1 and μ1 bands using Image Studio Lite software (Li-COR).
Hemagglutination (HA) assay.
Purified T1L or T1L/T3DM2 virions (1 × 1011 particles) were serially diluted in 50 μl of PBS in 96-well round-bottom microtiter plates (Corning-Costar). Human erythrocytes (Innovative Research Company) were washed twice with chilled PBS and were resuspended at a concentration of 1% (vol/vol) in PBS. Washed erythrocytes (50 μl) were added to wells containing virus and incubated at 4°C overnight. HA titers were determined by calculating HA units. One HA unit is equal to the number of particles of virus in the well with the lowest dilution of virus that shows a “shield.”
Expression and purification of GST–JAM-A.
The plasmid for expressing the extracellular region of human JAM-A fused to the C terminus of glutathione S-transferase (GST) (GST–JAM-A) (23) was obtained from Terence Dermody (University of Pittsburgh School of Medicine). The vector encodes a thrombin cleavage site between GST and JAM-A. The GST–JAM-A expression plasmid was transformed into Rosetta-gami 2 chemically competent cells (Novagen). For large-scale protein expression and purification, cells were grown in Terrific Broth medium supplemented with 100 μg/ml ampicillin and 37 μg/ml chloramphenicol at 37°C. When the optical density at 600 nm reached 0.4 to 0.6, JAM-A GST expression was induced with 1 mM isopropyl-β-d-thiogalactopyranoside and the cells were grown at 16°C for an additional 18 h. Following protein expression, the cells were pelleted at 5,000 × g at 4°C for 15 min. The cell pellets were resuspended in a reaction mixture containing 50 mM Tris (pH 8), 150 mM NaCl, 100 μg/ml lysozyme, 1 mM phenylmethylsulfonyl fluoride, and 0.1% Triton X-100. The cells were then lysed by sonication. Unlysed cells and cell debris were removed by ultracentrifugation at 125,000 × g at 4°C for 30 min, and the clarified lysates were filtered through a 0.2-μm-pore-size syringe filter. GST–JAM-A was purified by affinity chromatography using Pierce glutathione agarose (Thermo Fisher Scientific) following the manufacturer's instructions. GST–JAM-A was eluted from the glutathione agarose with 50 mM Tris (pH 8), 150 mM NaCl, and 25 mM reduced glutathione. Fractions containing JAM-A GST were exchanged into 50 mM Tris (pH 8) and 150 mM NaCl by dialysis. The concentration of purified GST–JAM-A was determined by DC protein assay (Bio-Rad). JAM-A GST purity was assessed by SDS-PAGE and Coomassie brilliant blue staining. For experiments utilizing JAM-A, JAM-A GST that had been affinity purified using glutathione agarose was treated with 15 units of thrombin (GE Healthcare) at 16°C overnight with continuous rotation. The released material was separated from the beads, and thrombin was inactivated using 0.3 mM PMSF.
In vitro binding assay.
High-affinity-binding polystyrene plates (Pierce) were coated at 4°C overnight with 20 μg/ml of purified JAM-A protein diluted in 0.1 M carbonate-bicarbonate buffer at pH 9.5. Plates were blocked at 4°C for 1 h with 2.5% BSA in virion storage buffer, followed by two washes with wash buffer (0.1% BSA, 0.05% Tween, virion storage buffer). T1L or T1L/T3DM2 virions were allowed to bind at 4°C for 2 h. After two washes with wash buffer, the plates were stained at room temperature for 1 h with reovirus-specific rabbit polyclonal antiserum (1:5,000) followed by Alexa Fluor 750-labeled anti-rabbit antibody (1:1,000). The plate was scanned using Odyssey infrared imager (LI-COR).
Clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 deletion of JAM-A.
HeLa S3 cells were transfected with pSpCas9(BB)-2A-Puro (Addgene 48131) or pSpCas9(BB)-2A-Puro-JAM-A using Fugene 6 (Promega). To generate pSpCas9(BB)-2A-Puro-JAM-A, the guide sequence targeting human JAM-A was inserted into pSpCas9(BB)-2A-Puro by annealing forward (5′-CACCGGGACAAAGGCGCAAGTCGAG-3′) and reverse (5′-AAACCTCGACTTGCGCCTTTGTCC-3′) primers and insertion into the Bbs1 site. At 48 h posttransfection, cells were incubated for 2 passages with media containing 1 μg/ml puromycin (Sigma-Aldrich) and media without puromycin thereafter. Cell surface JAM-A expression was confirmed by flow cytometry using JAM-A-specific antibody (J10.4).
Statistical analysis.
The statistical significance of results of comparisons between experimental groups was determined by one-way analysis of variance (ANOVA) with Bonferroni's multiple-comparison test using Graphpad Prism software. When only two groups of samples were analyzed, the unpaired t test function was used.
RESULTS
The infectivity of T1L is enhanced by the presence of the T3D μ1 protein.
T1L/T3DM2 (a recombinantly generated monoreassortant virus that contains a M2 gene segment from strain T3D in an otherwise T1L background) displays a particle-to-PFU-ratio comparable to the parental T1L strain ratio (specific particle-to-PFU ratios are included in Materials and Methods). During characterization of its properties in L929 cells, we observed that T1L/T3DM2 produces significantly more infected cells at 18 h postinfection than T1L (Fig. 1A). This phenomenon was also observed in HeLa cells (Fig. 1B), indicating that the differences in infectivity are not restricted to a single cell type. Infection of a variety of host cells with reovirus results in cell death (26–29). In parallel with the greater capacity of T1L/T3DM2 to establish infection, we found that T1L/T3DM2 induced a higher level of cell death in L929 cells than T1L/T3DM2 (Fig. 1C). To rule out the possibility that the aggregation of viruses contributed to the observed increase in the infectivity and cytotoxicity of T1L/T3DM2, we compared virions of T1L and T1L/T3DM2 using dynamic light scattering. We detected only one peak with the expected hydrodynamic diameter for both virus preparations, with no evidence of aggregation in either sample (Fig. 1D). Thus, our data indicate that the presence of the T3DM2 gene segment in an otherwise T1L-specific genetic background enhances the capacity of T1L to establish infection and kill host cells.
FIG 1.
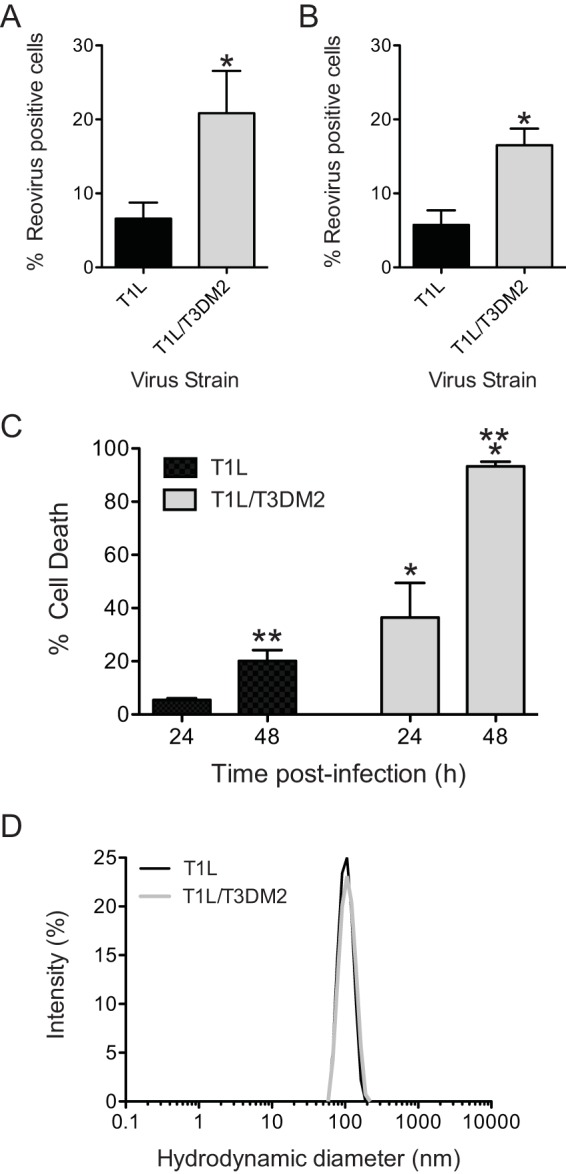
T1L/T3DM2 shows enhanced infectivity and cytotoxicity compared to T1L. (A and B) L929 cells (A) and HeLa cells (B) were adsorbed with 2 PFU per cell of the indicated virus strains. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antisera and a secondary antibody. Results are expressed as mean percent reovirus-positive cells (right panel) for three independent samples. Error bars indicate standard deviations (SD). *, P < 0.05 (as determined by Student's t test in comparison to T1L). (C) L929 cells were adsorbed with 10 PFU per cell of the indicated virus strains. The percentage of dead cells was determined after incubation for 48 and 72 h postinfection using AOEB staining. Results are expressed as mean percent cell death for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by one-way ANOVA with Bonferroni multiple-comparison test in comparison to T1L at the equivalent time point). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the same virus at 24 h). (D) Histograms of data from dynamic light scattering of a representative purified preparation of the indicated viruses are shown. The average hydrodynamic diameters for T1L and T1L/T3DM2 were 104 (±22) and 110 (± 25) nm, respectively.
T1L/T3DM2 attaches to host cells more efficiently.
Based on the superior capacity of T3D μ1-containing viruses to complete events required for cell penetration (17, 30, 31), we expected that the enhanced infectivity of T1L/T3DM2 would be due to its capacity to more efficiently cross cell membranes and initiate infection. However, to rule out the possibility that events prior to membrane penetration were impacted by the presence of T3DM2, we first assessed the capacity of T1L and T1L/T3DM2 to attach to host cells using flow cytometry. We observed that the mean fluorescence intensity of binding of T1L/T3DM2 was, surprisingly, greater than that of T1L (Fig. 2A and B). Using a fluorescence-based quantitative binding assay on adherent cells, we also observed that at lower multiplicities of infection (MOIs), T1L/T3DM2 bound to cells more efficiently than T1L in L929 cells (Fig. 2C and D). These data indicate that the presence of T3D μ1 in a T1L virus enhances attachment to cells. When the MOI was increased to 1 × 106 particles per cell or more, the two viruses bound to cells equivalently. These data indicate that the difference in binding efficiency can be overcome at higher concentrations. In the experiments described above, bound virus was detected by anti-reovirus polyclonal serum. To ensure that the differences in binding efficiency were not a function of the capacity of the polyclonal anti-reovirus serum to detect T1L and T1L/T3DM2, we tested the reactivity of these antisera to plate-coated T1L and T1L/T3DM2. We found that the antisera bound T1L ∼1.2 times better than T1L/T3DM2 (Fig. 2E). Thus, the increased attachment efficiency of T1L/T3DM2 was not an artifact of the method of detection. As an independent measure of virus attachment efficiency, we measured the amount of infectious virus attached to cells using plaque assay. Consistent with our measurement with other assays, we found that the cell-associated titer of T1L/T3DM2 was significantly higher than that of T1L. Together, these results indicate that T1L/T3DM2 attaches to cells more efficiently than T1L.
FIG 2.

T1L/T3DM2 displays enhanced attachment in comparison to T1L. (A and B) L929 cells were adsorbed with 5 × 104 particles per cell. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and a secondary antibody. (A) A representative histogram of attachment efficiency of T1L and T1L/T3DM2 obtained by flow cytometry is shown. MFI, mean fluorescence intensity. (B) Viral attachment efficiency was expressed as mean fluorescence intensity quantified by flow cytometry for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by Student's t test in comparison to T1L). (C and D) L929 cells were adsorbed with increasing multiplicities of infection of the indicated viruses. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. (C) A representative infrared scan of three wells of a plate adsorbed with 5 × 104 particles/cell of each virus strain is shown. (D) Results are expressed as mean binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by Student's t test in comparison to T1L). (E) Increasing numbers of particles of the indicated virus strain were coated onto plates. Plate-bound virus was detected using reovirus-specific rabbit polyclonal antisera and a secondary antibody. Mean intensities of staining as determined on an infrared scanner from three independent samples were plotted. Error bars indicate SD. *, P < 0.05 (as determined by Student's t test in comparison to T1L). (F) L929 cells were adsorbed with 5 × 104 particles per cell. Attached virus was detected using plaque assay. Mean titers for attached virus from three independent samples are shown. Error bars indicate SD. *, P < 0.05 (as determined by Student's t test in comparison to T1L).
The central portion of the μ1 protein impacts virus attachment.
With the goal of identifying determinants within T3D μ1 that affect the binding and infectivity of T1L viruses, we used viruses expressing chimeric T1L-T3D μ1 proteins (Fig. 3A) (17). The μ1 protein generates three cleavage fragments, μ1N, δ, and ϕ, during the course of infection (3). The μ1N portions of T1L and T3D μ1 are identical (17). We observed that a strain encoding DLD μ1 (T1L/DLDM2), where the N-terminal portion of δ (δN) is from T3D, the C-terminal portion of δ (δC) is from T1L, and ϕ is from T3D (17), bound to cells as efficiently as T1L. In contrast, a strain encoding LDL μ1 (T1L/LDLM2) displayed the binding efficiency characteristic of T1L/T3DM2 (Fig. 3B). These data indicate that δC is responsible for the difference in the levels of attachment efficiency of the virus strains. The δC portion of μ1 forms a jelly-roll beta barrel domain (32). The sequences of δC in T1L and T3D differ by five amino acids (17, 18). To test the contribution of each of the T1L-T3D polymorphic residues, we compared the cell attachment capacities of T1L viruses expressing T3D μ1 substituted with one of the five corresponding T1L residues. We found that each of the five substitution mutants displayed enhanced attachment similar to that seen with T1L/T3DM2 (Fig. 3C). Though we were unable to identify a single residue in δC that is responsible for the increase in attachment of T1L/T3DM2, these studies indicate that the presence of the T3D-derived δC domain enhanced the capacity of T1L to attach cells.
FIG 3.

The C-terminal portion of μ1 δ controls viral attachment efficiency. (A) Schematic of the reovirus μ1 proteins indicating cleavage fragments μ1N, δ, and ϕ. Designs of the chimeric μ1-expressing viruses are shown. (B and C) L929 cells were adsorbed with 5 × 104 particles per cell of the indicated viruses. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L).
The μ1-mediated increase in attachment efficiency is a function of T1L σ1.
Thus far, we have demonstrated that the presence of T3D μ1 in a T1L virus (T1L/T3DM2) enhances the attachment of virions to the host cell. The σ1 protein mediates attachment of reovirus to host cells (6, 7). Though the T1L and T3D σ1 polypeptide chains have low sequence homology, they fold into nearly identical trimeric fibers (10, 33). The σ1 proteins of both the T1L and T3D prototype reovirus strains engage JAM-A via their head domains (13, 33, 34). However, the T1L and T3D σ1 proteins engage distinct glycan receptors via disparate portions of the σ1 trimer (13, 14). We therefore asked if the presence of T3D μ1 influences the attachment of viruses containing T3D σ1 to host cells. For these experiments, we utilized recombinantly generated T1L/T3DS1 and T1L/T3DS1M2 and tested their capacity to infect cells. We observed that T1L/T3DS1 and T1L/T3DS1M2 were equally infectious (Fig. 4A). Moreover, these two viruses attached to cells with equivalent levels of efficiency (Fig. 4B). These studies suggested that the μ1-dependent enhancement in attachment efficiency is manifested only in viruses expressing T1L σ1.
FIG 4.
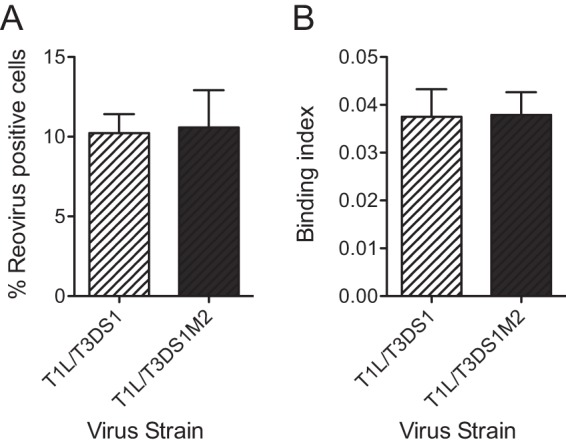
μ1-mediated increase in attachment efficiency is a function of T1L σ1. (A) L929 cells were adsorbed with 2 PFU per cell of the indicated virus strains. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus-positive cells (right panel) for three independent samples. Error bars indicate SD. (B) L929 cells were adsorbed with 5 × 104 particles per cell of the indicated viruses. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as mean binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD.
To directly determine if attachment of T1L/T3DM2 was dependent on T1L σ1, we assessed the capacity of a T1L σ1 head-specific monoclonal antibody, 5C6, to block infection of T1L and T1L/T3DM2 (22). For these experiments, virions incubated with 5C6 were allowed to attach to and infect L929 cells. We observed a decrease in the infectivity and attachment of both viruses (Fig. 5). These data suggest that attachment and infection by both T1L and T1L/T3DM2 are σ1 dependent and that the difference in the levels of infectivity of these two viruses cannot be attributed to the use of an alternative, as-yet-unidentified viral attachment factor.
FIG 5.

MAb 5C6 blocks reovirus attachment and infection. (A) L929 cells were adsorbed with 2 PFU per cell of the indicated virus strains preincubated with 5C6 MAb. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus-positive cells for independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the same virus incubated with 0 ng/ml of 5C6). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L treated with a similar concentration of 5C6). (B) L929 cells were adsorbed with 5 × 104 virus particles per cell of the indicated viral strains incubated with the indicated concentration of 5C6 MAb. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as mean binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the same virus incubated with 0 ng/ml of 5C6). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L treated with a similar concentration of 5C6).
The enhanced capacity of T1L/T3DM2 to infect and bind cells is lost upon ISVP formation.
Though a complete virus particle is expected to have 12 σ1 trimers, virus strains with fewer σ1 molecules remain infectious and may even display greater infectivity under some conditions (35, 36). Thus, one possible reason for the difference in the affinities of T1L and T1L/T3DM2 for host cells could be that these virus strains encapsidate different amounts of σ1 proteins. To test this idea, we measured the σ1/μ1 ratio in three independent freshly purified virus preparations using a quantitative immunoblot method. We found that the σ1/μ1 ratios of T1L and T1L/T3DM2 are equivalent (Fig. 6A and B). These data suggest that T1L and T1L/T3DM2 contain equivalent levels of σ1. Analysis of the glycan binding property of reovirus can be used as an alternative method to quantify σ1 levels (37). For these experiments, the capacity of T1L and T1L/T3DM2 to agglutinate human erythrocytes was measured. Our data indicate that the hemagglutination titers of T1L and T1L/T3DM2 are equivalent (Fig. 6C). Together, our data indicate that the difference in the levels of attachment efficiency of T1L and T1L/T3DM2 is not related to differences in the levels of the viral attachment protein.
FIG 6.

The enhanced capacity of T1L/T3DM2 to infect and bind cells is lost upon ISVP formation. (A and B) Virions (2 × 1010 particles) of the indicated virus strains were resolved on SDS-PAGE and immunoblotted using μ1- and σ1-specific antibodies and appropriate secondary antibodies. (A) A representative immunoblot is shown. (B) The results are plotted as ratios of σ1 and μ1 band intensities from three independent virus preparations. Error bars indicate SD. (C) T1L or T1L/T3DM2 virions from a representative virus preparation serially diluted in PBS and mixed with human erythrocytes were incubated overnight. Results are expressed as HA titer, the lowest dilution of virus that was capable of showing a shield of hemagglutination. (D) L929 cells were adsorbed with 0.02 PFU per cell of ISVPs of the indicated viruses. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus-positive cells for three independent samples. Error bars indicate SD. (E) L929 cells were adsorbed with 5 × 104 ISVPs per cell of the indicated virus strains. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD.
The σ1 proteins of T1L and T1L/T3DM2 are identical. Moreover, these virus strains contain the same amount of the σ1 protein (Fig. 6A to C). One possible reason for the differences in the capacities of these two viruses to interact with cells could be that their σ1 proteins are in different conformations. When virions are converted to ISVPs, σ1 assumes a more extended conformation (5, 8). We therefore tested whether the differences in infectivity and cell attachment efficiency were affected by ISVP formation. In contrast with what we observed with virions, the infectivities of T1L and T1L/T3DM2 ISVPs were equivalent (Fig. 6D). Correspondingly, the cell binding efficiencies of ISVPs of the two virus strains were also similar (Fig. 6E). These data suggest that the difference in the conformational states of σ1 on virions of T1L and T1L/T3DM2 may contribute to the differences in their efficiencies in binding host cell receptors and consequently in starting infections.
Enhanced infection by T1L/T3DM2 requires JAM-A.
The sensitivity of T1L/T3DM2 to 5C6 suggests that T1L/T3DM2 binds to cells via σ1. 5C6 blocks attachment of σ1 to both the T1 glycan receptor GM2 and the proteinaceous receptor JAM-A (9, 14). One possible explanation for the increased attachment of T1L/T3DM2 could be that T1L/T3DM2 binds to one or both of these receptors more efficiently than T1L. To determine if this is the case, we first assessed whether T1L and T1L/T3DM2 are differentially sensitive to enzymatic removal of GM2. For these experiments, cells were treated with Arthrobacter ureafaciens neuraminidase. While this treatment significantly reduced infection by T3D, we observed that the infectivity of both T1L and T1L/T3DM2 was unaffected by neuraminidase treatment (Fig. 7A), consistent with previous reports (14). We next tested the capacity of GST–JAM-A to block infection by T1L and T1L/T3DM2. GST–JAM-A is composed of an N-terminal fusion of GST with the extracellular portion of JAM-A and has previously been used to measure the binding affinity of reovirus to JAM-A (23). For these experiments, GST–JAM-A was incubated with each virus strain overnight and the capacity of these viruses to establish infection in L929 cells was measured by indirect immunofluorescence. We observed equivalent (∼ 5-fold to 7-fold) decreases in infection of T1L and T1L/T3DM2 with GST–JAM-A (Fig. 7B). The decrease in infectivity of T1L and T1L/T3DM2 matched the diminishment in the attachment efficiency of both of these viruses (Fig. 7C). These data indicate that both T1L and T1L/T3DM2 utilize JAM-A as the receptor to start infection.
FIG 7.

Enhanced infection by T1L/T3DM2 requires JAM-A. (A) L929 cells pretreated with 0 or 100 mU/ml neuraminidase were adsorbed with 2 PFU per cell of the indicated viruses. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent infected cells for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to 0 mU/ml neuraminidase-treated cells infected with the same virus). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to similarly treated cells infected with T1L). (B and C) L929 cells were adsorbed with 5 × 104 virus particles per cell incubated with the indicated concentration of GST–JAM-A. (B) After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent infected cells for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the same virus incubated with 0 μg/ml of GST–JAM-A). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L incubated with similar a concentration of GST–JAM-A. (C) Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the same virus incubated with 0 μg/ml of GST–JAM-A). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L incubated with similar a concentration of GST–JAM-A. (D) Increasing numbers of particles of the indicated virus strain were added to plates coated with purified JAM-A. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and a secondary antibody. Mean intensity of staining as determined on an infrared scanner from three independent samples was plotted. Error bars indicate SD.
Based on the results presented above, we hypothesized that T1L/T3DM2 might engage JAM-A more efficiently than T1L. To test this idea, we performed a plate-based in vitro binding assay. For these experiments, the capacity of increasing amounts of reovirus particles to bind plates coated with the extracellular portion of JAM-A (lacking GST) was assessed. At each virion concentration used, we found that T1L and T1L/T3DM2 bound to JAM-A with equivalent levels of efficiency (Fig. 7D). These data indicate that virions of T1L and T1L/T3DM2 do not display differences in the capacity to bind JAM-A in vitro.
The results presented above indicate that σ1 and JAM-A contribute to the differences in the attachment and infectivity of T1L and T1L/T3DM2 (Fig. 5 and 7B and C). However, in vitro data suggest that the binding efficiencies of T1L and T1L/T3DM2 for the extracellular portion of JAM-A are comparable (Fig. 7D). One possible explanation for these incongruent results may be that the amount of JAM-A needed for in vitro experiments far exceeds the amount of JAM-A present on cells. Indeed, we have observed no detectable binding of virions to a plate coated with lower concentrations of JAM-A (data not shown). It is also conceivable that the bacterially expressed extracellular portion of JAM-A differs from that of the native full-length JAM-A present on cells based on its posttranslational modification, the presence of the transmembrane and cytoplasmic portion of JAM-A, or its capacity to interact with other cellular molecules.
To evaluate the latter possibility, we utilized CHO cells that are poorly permissive with respect to infection by type 1 reovirus strains (38). In CHO cells transfected with vector alone, the infectivity of both T1L and T1L/T3DM2 was low, with T1L/T3DM2 displaying slightly greater infectivity at higher MOIs (Fig. 8A). Expression of human JAM-A in CHO cells increased their susceptibility to infection by 100 PFU/cell of both T1L and T1L/T3DM2 (Fig. 8A, left panel). Interestingly, JAM-A-expressing CHO cells supported infection by T1L/T3DM2 to a greater extent than infection by T1L. When the MOI was reduced to 20 PFU/cell, JAM-A expression appeared to increase infection only by T1L/T3DM2, further indicating that JAM-A expression affects infection of T1L and T1L/T3DM2 to different magnitudes. In some cell types, T1L can use GM2 as an attachment receptor (14). To rule out the possibility that the infectivity differences between T1L and T1L/T3DM2 are a function of GM2 engagement, we also compared the infectivities of these two viruses in JAM-A-expressing CHO cells treated with neuraminidase. Neuraminidase treatment diminished the infectivity of both T1L and T1L/T3DM2 in CHO cells to equivalent extents (Fig. 8B). Consistent with this, the attachment efficiency of T1L/T3DM2 exceeded that of T1L in JAM-A-expressing cells that were pretreated with neuraminidase (Fig. 8C). These data suggest that expression of JAM-A is sufficient to produce the observed difference in the levels of attachment and infectivity of T1L and T1L/T3DM2.
FIG 8.

Enhanced infectivity of T1L/T3DM2 is dependent on JAM-A expression (A) CHO cells stably transfected with an empty vector or a JAM-A expression vector were adsorbed with 100 (left panel) or 20 (right panel) PFU per cell of the indicated virus strains. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus-positive cells for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to empty vector transfected cells infected with the same virus). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L in the same cell type). (B) JAM-A-expressing CHO cells pretreated with 0 or 100 mU/ml neuraminidase were adsorbed with 20 PFU per cell of the indicated virus strains. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus positive for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the 0 mU/ml neuraminidase-treated cells infected with the same virus). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to similarly treated cells infected with T1L). (C) JAM-A-expressing CHO cells pretreated with 0 or 100 mU/ml neuraminidase were adsorbed with 5 × 104 particles per cell of the indicated virus strains. Attached virus was detected using reovirus-specific rabbit polyclonal antisera and secondary antibody. Cells were counterstained with a DNA stain. Viral attachment was quantified using an infrared scanner. Results are expressed as mean binding index (ratio of fluorescence of attached virus to that of cellular DNA) for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to the 0 mU/ml neuraminidase-treated cells infected with the same virus). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to similarly treated cells infected with T1L). (D) CRISPR-Cas9 control and JAM-A-deleted HeLa cells were adsorbed with 2 PFU per cell of the indicated viruses. After incubation at 37°C for 18 h, reovirus-positive cells were identified using reovirus-specific rabbit polyclonal antiserum and a secondary antibody. Results are expressed as mean percent reovirus-positive cells for three independent samples. Error bars indicate SD. *, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to control cells infected by the same virus). **, P < 0.05 (as determined by ANOVA with Bonferroni multiple-comparison test in comparison to T1L in the equivalent cell type).
To corroborate these results, we utilized HeLa cells from which endogenous JAM-A was deleted using CRISPR-Cas9 editing. Flow cytometric analysis of JAM-A-deleted cells indicated that surface expression of JAM-A was undetectable in >99% of cells (data not shown). In comparison to the native cells, which displayed differences in the infectivity of T1L and T1L/T3DM2 (analogous to Fig. 1B), cells lacking JAM-A were minimally infected by both T1L and T1L/T3DM2 (Fig. 8D). Together, these data indicate that the μ1-dependent difference in the levels of infectivity of T1L viruses is mediated by the JAM-A reovirus proteinaceous receptor.
DISCUSSION
Using a monogenic reassortant of prototype reovirus strains T1L and T3D, we present new evidence indicating that the reovirus μ1 protein influences the infectivity of reovirus. Our work demonstrates that the presence of the reovirus T3D-derived μ1 protein in a T1L background promotes infection by increasing the cell attachment efficiency of the virus. The infection-enhancing property of μ1 was attributable to its central jelly-roll β-barrel domain and required the presence of T1L σ1. Viruses expressing this part of μ1 utilized the cellular receptor JAM-A (engaged by σ1) more efficiently and were therefore capable of establishing infection more readily. This work highlights that the μ1 reovirus outer-capsid protein affects the function of another outer-capsid protein, reovirus attachment factor σ1.
The μ1 and σ1 proteins are not adjacent on the reovirus particle and do not make physical contact with each other (4, 5). Therefore, how μ1 can affect the function of σ1 is not clear. Our data indicate a role for the central portion of μ1 in controlling the attachment efficiency of the reovirus particle. The central portion of μ1 also controls the strain-specific differences in the capacity of reovirus to penetrate cell membranes during cell entry (17, 18). Penetration of cell membranes by reovirus is mediated by the μ1 N-terminal fragment, μ1N (39–41). μ1N is buried in the native particle, and its release for membrane interaction requires the proteolytic removal of the σ3 protector protein and conformational transitions in the trimer formed by μ1 (31, 42). The central region of μ1 that we identified as controlling the cell attachment efficiency of T1L σ1 also affects μ1 intratrimer interactions. Changes in this region alter the efficiency with which μ1 undergoes conformational transitions (17, 18, 43–46). We previously demonstrated that a single polymorphic difference between T1L and T3D μ1 (at amino acid 305) within this region controls strain-specific differences in the cell penetration function of T1L and T3D μ1 (18). However, our results presented here indicate that T1L-T3D polymorphic changes at this residue are not sufficient to influence the cell attachment efficiency of an otherwise T1L virus (Fig. 3C). Thus, the effect of μ1 on cell attachment properties and membrane penetration efficiency are genetically separable. However, the possibility that differences in the interactions between μ1 monomers within a μ1 trimer influence the structure of the viral particle cannot be ruled out. Indeed, this region of μ1 can also influence interactions between μ1 and the σ3 protector protein (4, 5). Though not via this central region, μ1 also makes contact with the λ2 protein and the reovirus core (4, 5). Whether any of these interactions are altered in T1L particles expressing T3D μ1 and, if so, how such modified interactions can affect the function of σ1 remain to be determined.
The σ1- and μ1-encoding gene segments (S1 and M2) have been previously implicated in controlling the same phenomena. The tropism of reovirus to the intestine and the central nervous system is linked to S1 and M2 (47–50). The viral S1 and M2 genes also control the capacity of reovirus to infect the liver and produce oily fur syndrome (51, 58). Similarly, the induction of immune tolerance following oral administration of reovirus is also determined by the S1 and M2 gene segments (52). While reovirus replication efficiency in the intestine is correlated with the sensitivity of the μ1 protein to digestion by intestinal proteases (50), the molecular basis of how μ1 affects these predominantly σ1-dependent properties remains unknown. In addition to these in vivo phenomena, strain-specific differences in the capacity of reovirus to elicit apoptosis and block host DNA synthesis in cell culture are also linked to S1 and M2 gene segments (26–28). Whether these cellular responses to infection are independently controlled by S1 and M2 gene products or whether S1 and M2 gene products cooperate to control the same event in viral replication and thereby produce these effects on the cells has not been elucidated. Previous work suggests that appropriate subcellular localization of μ1 fragments released from incoming virus particles or newly synthesized μ1 may directly influence proapoptotic signaling (53–55). Our work presented here indicates that μ1 can also affect cell death by potentiating the attachment function of σ1. We speculate that a few of the other phenotypes that are controlled by both S1 and M2 may also be consequences of the effect of μ1 on σ1 function.
Reassortment between related strains of segmented viruses occurs both in cell culture and in vivo (56). In a previous study of reovirus reassortment in vivo, mice were perorally inoculated with a mixture of T1L and T3D and the virus clones produced from such a mixed infection were analyzed (57). It was determined that ∼10% of the ∼1,200 virus clones analyzed were reassortants of T1L and T3D. The frequencies of reassortment between pairs of genes appear to be nonrandom, and only 5 types of reassortants were isolated. Among these, a reassortant containing a T1L S1 gene and a T3D M2 gene (similar to what we analyzed and characterized in this study) was recovered with high frequency. Whether the unusually high representation of this reassortant in the progeny is related to the replication advantage that we have described remains to be determined.
Analysis of reassortant viruses has been invaluable in assignment of function to many viral proteins. Reassortment can produce progeny viruses with new properties that differ from those of either parent. In some cases, these phenotypes are additive such that a reassortant virus that receives different virulence-enhancing genes from two parents is more pathogenic than each of the parental strains. In other cases, a newly generated reassortant strain can display properties that are distinct from those of either parent. The mechanistic basis of how reassortment produces a completely new phenotype is not known. One possible explanation for these observations is the generation of additional accommodating mutations within the viral genome that contribute to the new phenotype. Another possibility is that the new phenotypes are produced as a consequence of the presence of a particular combination of gene products from different parental strains. Data presented here highlight the latter, often-underappreciated effect of reassortment.
ACKNOWLEDGMENTS
We thank members of our laboratory for helpful suggestions and reviews of the manuscript. Flow cytometry was performed with assistance from Christiane Hassel in the Indiana University Flow Cytometry Core Facility. Dynamic light scattering was performed in the Indiana University Bloomington Physical Biochemistry Instrumentation Facility.
This work was supported by funds from Public Health Service award 1R01AI110637 and Indiana University. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Flint SJ, Enquist LW, Racaniello VR, Skalka AM. 2009. Principles of virology, 3rd ed ASM Press, Washington, DC. [Google Scholar]
- 2.Dermody TS, Parker JC, Sherry B. 2013. Orthoreoviruses. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed (electronic) Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Danthi P, Guglielmi KM, Kirchner E, Mainou B, Stehle T, Dermody TS. 2010. From touchdown to transcription: the reovirus cell entry pathway. Curr Top Microbiol Immunol 343:91–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. 2005. Features of reovirus outer capsid protein m1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure 13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. 1993. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol 122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner HL, Ault KA, Fields BN. 1980. Interaction of reovirus with cell surface receptors. I. Murine and human lymphocytes have a receptor for the hemagglutinin of reovirus type 3. J Immunol 124:2143–2148. [PubMed] [Google Scholar]
- 7.Lee PWK, Hayes EC, Joklik WK. 1981. Protein s1 is the reovirus cell attachment protein. Virology 108:156–163. doi: 10.1016/0042-6822(81)90535-3. [DOI] [PubMed] [Google Scholar]
- 8.Furlong DB, Nibert ML, Fields BN. 1988. Sigma 1 protein of mammalian reoviruses extends from the surfaces of viral particles. J Virol 62:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. 2001. Junction adhesion molecule is a receptor for reovirus. Cell 104:441–451. doi: 10.1016/S0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 10.Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. 2005. Junctional adhesion molecule A serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol 79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gentsch JR, Pacitti AF. 1985. Effect of neuraminidase treatment of cells and effect of soluble glycoproteins on type 3 reovirus attachment to murine L cells. J Virol 56:356–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul RW, Choi AH, Lee PWK. 1989. The a-anomeric form of sialic acid is the minimal receptor determinant recognized by reovirus. Virology 172:382–385. doi: 10.1016/0042-6822(89)90146-3. [DOI] [PubMed] [Google Scholar]
- 13.Reiter DM, Frierson JM, Halvorson EE, Kobayashi T, Dermody TS, Stehle T. 2011. Crystal structure of reovirus attachment protein sigma1 in complex with sialylated oligosaccharides. PLoS Pathog 7:e1002166. doi: 10.1371/journal.ppat.1002166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reiss K, Stencel JE, Liu Y, Blaum BS, Reiter DM, Feizi T, Dermody TS, Stehle T. 2012. The GM2 glycan serves as a functional coreceptor for serotype 1 reovirus. PLoS Pathog 8:e1003078. doi: 10.1371/journal.ppat.1003078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maginnis MS, Forrest JC, Kopecky-Bromberg SA, Dickeson SK, Santoro SA, Zutter MM, Nemerow GR, Bergelson JM, Dermody TS. 2006. Beta1 integrin mediates internalization of mammalian reovirus. J Virol 80:2760–2770. doi: 10.1128/JVI.80.6.2760-2770.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Konopka-Anstadt JL, Mainou BA, Sutherland DM, Sekine Y, Strittmatter SM, Dermody TS. 2014. The Nogo receptor NgR1 mediates infection by mammalian reovirus. Cell Host Microbe 15:681–691. doi: 10.1016/j.chom.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarkar P, Danthi P. 2010. Determinants of strain-specific differences in efficiency of reovirus entry. J Virol 84:12723–12732. doi: 10.1128/JVI.01385-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Madren JA, Sarkar P, Danthi P. 2012. Cell entry-associated conformational changes in reovirus particles are controlled by host protease activity. J Virol 86:3466–3473. doi: 10.1128/JVI.06659-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kobayashi T, Ooms LS, Ikizler M, Chappell JD, Dermody TS. 2010. An improved reverse genetics system for mammalian orthoreoviruses. Virology 398:194–200. doi: 10.1016/j.virol.2009.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berard A, Coombs KM. 2009. Mammalian reoviruses: propagation, quantification, and storage. Curr Protoc Microbiol 14:C:15C.1:15C.1.1–15C.1.18. doi: 10.1002/9780471729259.mc15c01s14. [DOI] [PubMed] [Google Scholar]
- 21.Smith RE, Zweerink HJ, Joklik WK. 1969. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 39:791–810. doi: 10.1016/0042-6822(69)90017-8. [DOI] [PubMed] [Google Scholar]
- 22.Virgin HW IV, Mann MA, Fields BN, Tyler KL. 1991. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J Virol 65:6772–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guglielmi KM, Kirchner E, Holm GH, Stehle T, Dermody TS. 2007. Reovirus binding determinants in junctional adhesion molecule-A. J Biol Chem 282:17930–17940. doi: 10.1074/jbc.M702180200. [DOI] [PubMed] [Google Scholar]
- 24.Wetzel JD, Chappell JD, Fogo AB, Dermody TS. 1997. Efficiency of viral entry determines the capacity of murine erythroleukemia cells to support persistent infections by mammalian reoviruses. J Virol 71:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boehme KW, Guglielmi KM, Dermody TS. 2009. Reovirus nonstructural protein sigma1s is required for establishment of viremia and systemic dissemination. Proc Natl Acad Sci U S A 106:19986–19991. doi: 10.1073/pnas.0907412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tyler KL, Squier MK, Rodgers SE, Schneider BE, Oberhaus SM, Grdina TA, Cohen JJ, Dermody TS. 1995. Differences in the capacity of reovirus strains to induce apoptosis are determined by the viral attachment protein sigma 1. J Virol 69:6972–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyler KL, Squier MK, Brown AL, Pike B, Willis D, Oberhaus SM, Dermody TS, Cohen JJ. 1996. Linkage between reovirus-induced apoptosis and inhibition of cellular DNA synthesis: role of the S1 and M2 genes. J Virol 70:7984–7991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rodgers SE, Barton ES, Oberhaus SM, Pike B, Gibson CA, Tyler KL, Dermody TS. 1997. Reovirus-induced apoptosis of MDCK cells is not linked to viral yield and is blocked by Bcl-2. J Virol 71:2540–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Connolly JL, Rodgers SE, Clarke P, Ballard DW, Kerr LD, Tyler KL, Dermody TS. 2000. Reovirus-induced apoptosis requires activation of transcription factor NF-kappaB. J Virol 74:2981–2989. doi: 10.1128/JVI.74.7.2981-2989.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lucia-Jandris P, Hooper JW, Fields BN. 1993. Reovirus M2 gene is associated with chromium release from mouse L cells. J Virol 67:5339–5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandran K, Farsetta DL, Nibert ML. 2002. Strategy for nonenveloped virus entry: a hydrophobic conformer of the reovirus membrane penetration protein m1 mediates membrane disruption. J Virol 76:9920–9933. doi: 10.1128/JVI.76.19.9920-9933.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liemann S, Chandran K, Baker TS, Nibert ML, Harrison SC. 2002. Structure of the reovirus membrane-penetration protein, m1, in a complex with its protector protein, s3. Cell 108:283–295. doi: 10.1016/S0092-8674(02)00612-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stettner E, Dietrich MH, Reiss K, Dermody TS, Stehle T. 2015. Structure of serotype 1 reovirus attachment protein sigma1 in complex with junctional adhesion molecule A reveals a conserved serotype-independent binding epitope. J Virol 89:6136–6140. doi: 10.1128/JVI.00433-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirchner E, Guglielmi KM, Strauss HM, Dermody TS, Stehle T. 2008. Structure of reovirus sigma1 in complex with its receptor junctional adhesion molecule-A. PLoS Pathog 4:e1000235. doi: 10.1371/journal.ppat.1000235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larson SM, Antczak JB, Joklik WK. 1994. Reovirus exists in the form of 13 particle species that differ in their content of protein sigma 1. Virology 201:303–311. doi: 10.1006/viro.1994.1295. [DOI] [PubMed] [Google Scholar]
- 36.Mohamed A, Teicher C, Haefliger S, Shmulevitz M. 2015. Reduction of virion-associated sigma1 fibers on oncolytic reovirus variants promotes adaptation toward tumorigenic cells. J Virol 89:4319–4334. doi: 10.1128/JVI.03651-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lerner AM, Cherry JD, Finland M. 1963. Haemagglutination with reoviruses. Virology 19:58–65. doi: 10.1016/0042-6822(63)90024-2. [DOI] [PubMed] [Google Scholar]
- 38.Danthi P, Hansberger MW, Campbell JA, Forrest JC, Dermody TS. 2006. JAM-A-independent, antibody-mediated uptake of reovirus into cells leads to apoptosis. J Virol 80:1261–1270. doi: 10.1128/JVI.80.3.1261-1270.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Agosto MA, Ivanovic T, Nibert ML. 2006. Mammalian reovirus, a nonfusogenic nonenveloped virus, forms size-selective pores in a model membrane. Proc Natl Acad Sci U S A 103:16496–16501. doi: 10.1073/pnas.0605835103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ivanovic T, Agosto MA, Zhang L, Chandran K, Harrison SC, Nibert ML. 2008. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. EMBO J 27:1289–1298. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang L, Agosto MA, Ivanovic T, King DS, Nibert ML, Harrison SC. 2009. Requirements for the formation of membrane pores by the reovirus myristoylated micro1N peptide. J Virol 83:7004–7014. doi: 10.1128/JVI.00377-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chandran K, Parker JS, Ehrlich M, Kirchhausen T, Nibert ML. 2003. The delta region of outer-capsid protein m1 undergoes conformational change and release from reovirus particles during cell entry. J Virol 77:13361–13375. doi: 10.1128/JVI.77.24.13361-13375.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Middleton JK, Agosto MA, Severson TF, Yin J, Nibert ML. 2007. Thermostabilizing mutations in reovirus outer-capsid protein m1 selected by heat inactivation of infectious subvirion particles. Virology 361:412–425. doi: 10.1016/j.virol.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Middleton JK, Severson TF, Chandran K, Gillian AL, Yin J, Nibert ML. 2002. Thermostability of reovirus disassembly intermediates (ISVPs) correlates with genetic, biochemical, and thermodynamic properties of major surface protein mu1. J Virol 76:1051–1061. doi: 10.1128/JVI.76.3.1051-1061.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Danthi P, Kobayashi T, Holm GH, Hansberger MW, Abel TW, Dermody TS. 2008. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol 82:161–172. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarkar P, Danthi P. 2013. The mu1 72-96 loop controls conformational transitions during reovirus cell entry. J Virol 87:13532–13542. doi: 10.1128/JVI.01899-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Weiner HL, Drayna D, Averill DR Jr, Fields BN. 1977. Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci U S A 74:5744–5748. doi: 10.1073/pnas.74.12.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiner HL, Powers ML, Fields BN. 1980. Absolute linkage of virulence and central nervous system tropism of reoviruses to viral hemagglutinin. J Infect Dis 141:609–616. doi: 10.1093/infdis/141.5.609. [DOI] [PubMed] [Google Scholar]
- 49.Hrdy DB, Rubin DH, Fields BN. 1982. Molecular basis of reovirus neurovirulence: role of the M2 gene in avirulence. Proc Natl Acad Sci U S A 79:1298–1302. doi: 10.1073/pnas.79.4.1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubin DH, Fields BN. 1980. Molecular basis of reovirus virulence: role of the M2 gene. J Exp Med 152:853–868. doi: 10.1084/jem.152.4.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Derrien M, Hooper JW, Fields BN. 2003. The M2 gene segment is involved in the capacity of reovirus type 3Abney to induce the oily fur syndrome in neonatal mice, a S1 gene segment-associated phenotype. Virology 305:25–30. doi: 10.1006/viro.2002.1723. [DOI] [PubMed] [Google Scholar]
- 52.Rubin DH, Weiner HL, Fields BN, Greene MI. 1981. Immunologic tolerance after oral administration of reovirus: requirement for two viral gene products for tolerance induction. J Immunol 127:1697–1701. [PubMed] [Google Scholar]
- 53.Coffey CM, Sheh A, Kim IS, Chandran K, Nibert ML, Parker JS. 2006. Reovirus outer capsid protein m1 induces apoptosis and associates with lipid droplets, endoplasmic reticulum, and mitochondria. J Virol 80:8422–8438. doi: 10.1128/JVI.02601-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Danthi P, Coffey CM, Parker JS, Abel TW, Dermody TS. 2008. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog 4:e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wisniewski ML, Werner BG, Hom LG, Anguish LJ, Coffey CM, Parker JS. 2011. Reovirus infection or ectopic expression of outer capsid protein micro1 induces apoptosis independently of the cellular proapoptotic proteins Bax and Bak. J Virol 85:296–304. doi: 10.1128/JVI.01982-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McDonald SM, Nelson MI, Turner PE, Patton JT. 2016. Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat Rev Microbiol 14:448–460. doi: 10.1038/nrmicro.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wenske EA, Chanock SJ, Krata L, Fields BN. 1985. Genetic reassortment of mammalian reoviruses in mice. J Virol 56:613–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barton ES, Connolly JL, Forrest JC, Chappell JD, Dermody TS. 2001. Utilization of sialic acid as a coreceptor enhances reovirus attachment by multistep adhesion strengthening. J Biol Chem 276:2200–2211. doi: 10.1074/jbc.M004680200. [DOI] [PubMed] [Google Scholar]