

ABSTRACT
Infected cell protein 0 (ICP0) of herpes simplex virus 1 (HSV-1) is an α gene product required for viral replication at low multiplicities of infection. Upon entry, nuclear domain 10 (ND10) converges at the incoming DNA and represses viral gene expression. ICP0 contains a RING-type E3 ubiquitin ligase that degrades the ND10 organizer PML and disperses ND10 to alleviate the repression. In the present study, we focused on understanding the regulation of ICP0 E3 ligase activity in the degradation of different ICP0 substrates. We report the following. (i) A SUMO interaction motif located at ICP0 residues 362 to 364 is required for the degradation of PML isoforms II, IV, and VI but not isoform I. This differentiation mechanism exists in both HEp-2 and U2OS cells, regardless of the cell's permissiveness to the ICP0-null virus. (ii) Physical interaction between SIM362–364 and PML II is necessary but not sufficient for PML II degradation. Both proximal sequences surrounding SIM362–364 and distal sequences located at the ICP0 C terminus enhance the degradation of PML II. (iii) The ICP0 C terminus is dispensable for PML I degradation. Instead, bipartite PML I binding domains located in the N-terminal half of ICP0 coordinate to promote the degradation of PML I. (iv) The stability of ICP0, but not its ND10 fusion ability, affects the rate of PML I degradation. Taken together, our results show that ICP0 uses at least two regulatory mechanisms to differentiate its substrates. The disparate recognition of the ICP0 E3 substrates may be related to the different roles these substrates may play in HSV-1 infection.
IMPORTANCE Viruses have a limited genetic coding capacity but must encounter a multilayered comprehensive host defense. To establish a successful infection, viruses usually produce multifunctional proteins to coordinate the counteractions. Here we report that an HSV-1 protein, ICP0, can recognize individual host factors and target them differently for destruction. We identified elements that are important for the ICP0 E3 ubiquitin ligase to differentially recognize two of its substrates, PML I and PML II. This is the first study that has systematically investigated how ICP0 discriminates two similar molecules by very different mechanisms. This work lays the foundation for understanding the role of host defensive factors and the mechanisms viruses use to take advantage of some host proteins while destroying others.
INTRODUCTION
Herpes simplex virus (HSV) causes a wide range of mild to severe herpetic diseases, including cold sores, genital lesions, stromal keratitis, and encephalitis. Following infection, HSV establishes a lifelong latency in ganglion neurons. Its sporadic and sometimes asymptomatic reactivation nourishes a wide spread of the virus, making it one of the most prevalent opportunistic pathogens that cause severe problems in immunocompromised individuals (1).
Upon HSV-1 infection, the incoming viral DNA encounters various host restrictive factors, namely, the intrinsic and innate antiviral responses. To establish effective replication, HSV-1 expresses multiple viral proteins to antagonize the many layers of host defenses. One of them, infected cell protein 0 (ICP0), is a key viral countermeasure that dismantles the cellular defense. This 775-amino-acid α (immediate-early) gene product plays a critical role in both lytic and latent infections. It enhances viral transcription by attacking host restrictive factors that silence the viral genome. ICP0 uses at least two modes of counteraction: (i) degradation of host restrictive factors and (ii) interaction with cellular repressive pathways. One of the most important domains in ICP0 is a RING-type E3 ubiquitin ligase located at residues 116 to 156 (2, 3). It targets several cellular regulators, including PML, Sp100, IFI16, DNA-PK, and RNF8 (4–7). Knockdown of these ICP0 substrates increases viral expression and replication (7–9), suggesting that these proteins are part of the antiviral defense. ICP0 also interacts with a diverse array of cellular partners, such as USP7, CoREST, and cyclin D3 (10–12). Some of these interactions disrupt cellular repression and consequently promote viral expression (13).
Because of ICP0's complex biochemical properties, the mechanism by which ICP0 domains coordinate in gene activation is not clear. We seek to dissect ICP0 functions in vivo and delineate the cooperativity among ICP0 domains. In preceding reports, we have studied the dynamic interaction between ICP0 and a nuclear structure called ND10. ND10 bodies are involved in many cellular pathways, such as gene regulation, cell cycle regulation, DNA repair, cell senescence, and antiviral defense (for reviews, see references 14 and 15). Upon HSV-1 infection, ND10 bodies converge at the incoming DNA (16, 17) to repress viral gene expression. Over 150 cellular proteins have been identified as ND10 components (18). One of them, PML, a substrate targeted by the ICP0 E3 ligase, is the key organizer of ND10 (19). Degradation of PML by ICP0 leads to dispersal of ND10 components and subsequent derepression of the HSV genome. We have reported that the interaction between ICP0 and ND10 consists of three sequential steps, adhesion, fusion, and retention, with different ICP0 domains being required for each step (20). We have identified three proline-rich ND10 fusion segments (ND10-FS1, ND10-FS2, and ND10-FS3), which redundantly drive ICP0 to merge with ND10. Each of the three ND10-FSs by itself is sufficient to facilitate the ICP0-ND10 fusion, but only when all three ND10-FSs are deleted can the fusion be abolished (21). Given the general rule of genetic economy in viruses, this redundancy indicates the extreme importance of the ICP0-ND10 fusion process in the HSV-1 life cycle. Presumably, ICP0-ND10 fusion can facilitate quick access of the ICP0 E3 ligase to its substrates located within ND10. In the present study, we focused on identifying ICP0 elements that regulate E3 ligase activity. We report that ICP0 recognizes and degrades individual PML isoforms via at least two different mechanisms. The PML protein has seven isoforms, all of which result from alternative splicing of a single PML gene (22, 23). Using PML isoforms I and II as model substrates, we have investigated the relationship between ND10 fusion and PML degradation, characterized elements that regulate substrate differentiation by ICP0, and examined how the inability to degrade certain substrates affects viral replication.
MATERIALS AND METHODS
Cells and viruses.
The sources and cultural conditions for HEp-2, HEL, and U2OS cells were described previously (21). U2OS-TetOn cells were grown in McCoy's 5A medium (Sigma) supplemented with 10% fetal bovine serum (FBS) and 6 μg/ml blasticidin (Invitrogen). All of the recombinant viruses illustrated in Fig. 1A have been previously described (20, 21).
FIG 1.

ICP0 lacking segment C failed to degrade a subset of PML. (A) Schematic diagrams of the domain structures of ICP0 cDNA and its central region. Lines: 1, ICP0 cDNA; 2, closeup illustration of the central region of ICP0. The black oval with an R represents the RING domain; NLS represents the nuclear localization sequence; the gray ovals with FS1, -2, and -3 are the three ND10-FSs; and the letters A to E are the segments arbitrarily used to label the internal deletions of ICP0. Amino acid numbers marking the boundaries of these domains are illustrated above the gene. The ICP0s in all of the viruses used in this study are tagged with mCherry. (B and C) Effects of ICP0 internal deletions on PML degradation. HEL cells were infected with mutant viruses at 10 PFU/cell for 8 h (B) or pretreated with 10 μM MG132 and then infected with the viruses indicated at 10 PFU/cell in the presence of MG132 for 8 h (C). Infected cells were lysed, and total cell lysates were probed in Western blot assays with the antibodies indicated in each gel. (D) Different types of cells were infected with the mutant viruses at 10 PFU/cell for the times indicated and then subjected to Western blotting as described above. The values to the left of the gels are molecular sizes in kilodaltons.
Construction of TetOn cell lines stably expressing PML isoforms.
Plasmid pAcCMV-PML, which contains PML VI, was a generous gift from Bernard Roizman (University of Chicago) (24). The various C termini of PML isoforms I, II, and IV were generated by reverse transcription (RT)-PCR and ligated to the first five exons of PML VI to obtain the full-length cDNAs for PML I, II, and IV. All four isoforms were fused in frame with a myc tag and cloned into plasmid pcDNA4/To (Invitrogen).
The U2OS-TetOn cell line was a generous gift from Daniel R. Schoenberg (The Ohio State University) (25). The plasmids of pcDNA4/To/mycPML isoforms were transfected into U2OS-TetOn cells with Lipofectamine reagent (Invitrogen). Stably transfected colonies were selected in U2OS growth medium containing 6 μg/ml blasticidin and 100 μg/ml phleomycin D1 (Zeocin). Stable cell lines were then identified by Western blotting after a 12-h induction with 1 μg/ml doxycycline (Dox; Sigma-Aldrich).
The HEp-2-TetOn cell line was constructed by transfecting the pcDNA6/TR plasmid (Invitrogen) into HEp-2 cells. Stably transfected colonies were selected in DMEM (Invitrogen) supplemented with 10% FBS and 6 μg/ml blasticidin. Cells from individual colonies were transiently transfected with the pcDNA4/To/LacZ plasmid (Invitrogen) in the presence or absence of Dox and then stained for β-galactosidase. One line with the best induction rate was selected as the HEp-2-TetOn cells. The plasmids of pcDNA4/To/mycPML isoforms were transfected into the HEp-2-TetOn cells. Stably transfected colonies were selected in HEp-2 growth medium containing 6 μg/ml blasticidin and 100 μg/ml phleomycin D1. Stable cell lines were then identified by Western blotting after a 24-h Dox induction.
Confocal microscopy.
HEp-2-TetOn or U2OS-TetOn cells expressing PML isoforms grown on four-well glass slides (Thermo Fisher Scientific) were induced with 1 μg/ml Dox for 24 h. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with phosphate-buffered saline (PBS) containing 5% horse serum plus 1% bovine serum albumin. The cells were then reacted with rabbit anti-Sp100 and mouse anti-myc antibodies at 4°C overnight, rinsed, and reacted with fluorescein isothiocyanate-conjugated goat anti-rabbit (Sigma) and Texas Red-conjugated goat anti-mouse (Invitrogen) secondary antibodies. Images were taken with a Leica SP8 confocal microscope.
PML half-life assay.
HEp-2-TetOn or U2OS-TetOn cells expressing PML isoforms were induced with 1 μg/ml Dox overnight and then exposed to a test virus at 10 PFU/cell for 1 h. The inoculum was removed, and the cells were incubated in growth medium for 1 h before 100 μg/ml cycloheximide (CHX) was added. Cells were harvested at 2-h intervals and subjected to Western blotting. Bands of PML isoforms were scanned and quantitated with ImageJ software.
Coimmunoprecipitation.
HEp-2-TetOn cells expressing mycPML I and mycPML II were induced with 1 μg/ml Dox for 24 h before being exposed to a test virus at 2 PFU/cell for 18 h. The infected cells were harvested and washed with PBS, swelled in hypotonic buffer A (10 mM Tris-HCl [pH 7.4], 1.5 mM MgCl2,10 mM KCl, 0.5 mM dithiothreitol [DTT], 1× protease inhibitor cocktail [Sigma], 1× phosphatase inhibitor cocktail [Sigma]), and lysed by six strokes in a Dounce homogenizer. After removal of the cytoplasm, the nuclei were lysed in lysis buffer (10 mM Tris-HCl [pH 8], 140 mM NaCl, 1.5 mM MgCl2, 1 mM DTT, and 0.5% NP-40 supplemented with 1× protease inhibitor cocktail and phosphatase inhibitor cocktail) by brief sonication. Benzonase nuclease (Novagen) was added to the crude nuclear lysates at a final concentration of 0.1 U/μl to digest the nucleic acids at 16°C for 30 min. The nuclear lysates were then spun at 12,000 rpm for 15 min to remove the insoluble nuclear debris. The nuclear extracts were incubated with an anti-myc monoclonal antibody at 4°C overnight with gentle agitation. The immune complexes were captured by Pierce protein G agarose beads (Thermo Scientific). The immunoprecipitates were subjected to Western blotting.
Western blotting.
Total cell lysates extracted as described before (21) or immunoprecipitates prepared as described above were electrophoretically separated by SDS-PAGE, transferred onto a polyvinylidene difluoride membrane (Millipore), probed with antibodies, and visualized by ECL Western blotting detection reagent (GE Healthcare), as described elsewhere (21).
Antibodies.
An anti-PML polyclonal antibody, an anti-actin monoclonal antibody, and an anti-myc monoclonal antibody were purchased from Santa Cruz Biotechnology Inc. An anti-Sp100 polyclonal antibody was purchased from Abcam. Anti-mCherry polyclonal and monoclonal antibodies were purchased from Clontech.
qPCR.
HEp-2-TetOn cells expressing mycPML II grown on 35-mm plates were induced with 1 μg/ml Dox overnight before being infected with a test virus at 0.1 PFU/cell for 1 h. The inoculum was then removed, and cells were incubated in DMEM supplemented with 10% newborn calf serum. At 2 and 24 h postinfection (hpi), total DNA was extracted, quantitative PCR (qPCR) was performed, and the DNA fold increase was calculated as previously described (20).
qRT-PCR.
HEp-2-TetOn cells expressing mycPML I or mycPML II grown on 35-mm plates were induced with 1 μg/ml Dox overnight before being infected with a test virus at 10 PFU/cell for 1 h. The inoculum was then removed, and cells were incubated in DMEM supplemented with 10% newborn calf serum. At the times indicated, total RNA was extracted and cDNA was synthesized with the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific). Quantitative RT-PCR was performed with primers 5′-ATGGCATCAATGCAGAAGCTGATCT-3′ and 5′-CTGGAACTCCTCCTCCGAAG-3′ to detect mycPML isoforms. 18S rRNA primers (Ambion) were used for normalization.
RESULTS
PML is partially degraded by a mutant form of ICP0 lacking residues 343 to 391.
Previously, we identified three ND10-FSs located at ICP0 residues 242 to 291, 343 to 391, and 393 to 441 that redundantly facilitated the ND10 fusion of ICP0 (21). To assess whether ICP0-ND10 fusion is correlated with PML degradation by ICP0, we examined PML levels in cells infected with recombinant viruses containing deletions in the central region of ICP0. For simplicity, we arbitrarily divided the central region (residues 241 to 475) into five segments, A to E (Fig. 1A, line 2). Segment A, B, C, or D contains 50 amino acids, while segment E has 33 amino acids. The A, C, and D segments coincide with ND10-FS1, -2, and -3, respectively. The properties of the recombinant viruses used in this series of experiments, which have been previously reported (20, 21), are illustrated in Fig. 1A.
As shown before in HEL cells (20), wild-type ICP0 in recombinant virus RHG101 completely degraded PML at 8 hpi (Fig. 1B, lane 2), whereas ICP0 lacking the entire central region (RHG110) completely lost the ability to degrade PML, comparable to the mock-treated control (Fig. 1B, lanes 8 and 1). Consistent with our previous report (21), in cells infected with virus RHG113, which contains ICP0 lacking segment C, residual PML was observed at 8 hpi (Fig. 1B, lane 5), whereas ICP0 lacking segment A, B, D, or E (RHG111, -112, -114, or -115) degraded PML to the same extent as that of the wild type (RHG101). Interestingly, the PML level in RHG113-infected cells was considerably lower than that in mock- or RHG110-infected cells, suggesting partial PML degradation in cells infected with RHG113. By treating the cells with the proteasome inhibitor MG132, we confirmed that the decrease in PML in RHG113-infected cells was the result of proteasomal degradation (Fig. 1C). We then proceeded to investigate the cause of partial degradation. In a previous report, we showed that an increased multiplicity of infection did not improve the partial PML degradation in RHG113-infected cells (21). Here we tested whether prolonged infection can eventually complete PML degradation. As shown in Fig. 1D, in HEL cells, wild-type ICP0 (RHG101) partially degraded PML at 4 hpi (lane 2) and PML completely disappeared at 8 hpi (lane 3). As a negative control, ICP0 lacking the entire central region (RHG110) did not degrade PML at any time point (Fig. 1D, lanes 8 to 10). For RHG113, we found that partial PML degradation persisted at all three points tested (Fig. 1D, lanes 5 to 7), indicating that partial PML degradation is not a delayed effect and cannot be compensated for by prolonging the infection. Therefore, PML molecules are differentially recognized and a subset of PML was not degraded by RHG113.
We further investigated the cell type dependency of this partial degradation. In both U2OS cells, the permissive line for ICP0-null virus (26), and the nonpermissive HEp-2 cells tested (26), we found that PML was partially degraded by ICP0 lacking segment C in RHG113-infected cells (Fig. 1D, lanes 15 to 17 and 25 to 27), suggesting the presence of a similar mechanism that governs the differential PML degradation in all three cell types.
SIM362–364, located in segment C, is necessary for ICP0 to degrade PML isoforms II, IV, and VI but not PML isoform I.
PML has seven isoforms that have the same N terminus from exon 1 to exon 6 but assemble exons 7 to 9 differently to form the individual C termini (23). All of these isoforms can be recognized by the polyclonal anti-PML antibody used as described above. Two isoforms, PML I and PML II, have a molecular mass of ∼100 kDa, matching the size of the major band detected in Fig. 1, whereas all other isoforms are smaller and can only be detected in overexposed Western blots. We hypothesize that the partial degradation observed in Fig. 1 is the result of differential degradation of PML I and PML II by ICP0 in the absence of segment C. To test this, we individually expressed myc-tagged PML I and PML II to examine their half-lives in RHG113-infected cells. In order to obtain uniform expression that is suitable for quantitative assays, we established stable cell lines expressing mycPML I or mycPML II. Since PML is a cell cycle regulator (27), we adopted a TetOn system to avoid potential cell alteration induced by long-term PML overexpression in stable line selection. We received U2OS-TetOn cells from Daniel Schoenberg (25) and constructed a HEp-2-TetOn cell line by stably transfecting the pcDNA6/TR plasmid into HEp-2 cells. Using both HEp-2-TetOn and U2OS-TetOn as parental cell lines, we constructed mycPML I- and mycPML II-expressing cells.
As shown in Fig. 2A, the expression of exogenous PML I or II in both HEp-2-TetOn and U2OS-TetOn cells was tightly controlled, with no detection of mycPML in the absence of Dox (lanes 1, 5, 9, and 13). Upon 24 h of Dox induction, mycPML was highly expressed and became the major PML population in induced cells (lanes 4, 8, 12, and 16). Ectopic PML I or PML II was colocalized to ND10 bodies represented by another major ND10 component, Sp100, without dramatically changing the morphology of ND10 (Fig. 2B and C). One exception was U2OS-TetOn/mycPML II cells, in which overexpression of mycPML II apparently increased the number of ND10 bodies and deformed their structure into short twitchy lines (Fig. 2C, j to l). Therefore, in the next series of experiments we compared the differential PML degradation in both HEp-2-TetOn and U2OS-TetOn cells (Fig. 3) but analyzed the regulatory requirements of substrate differentiation mainly in HEp-2-TetOn cells.
FIG 2.
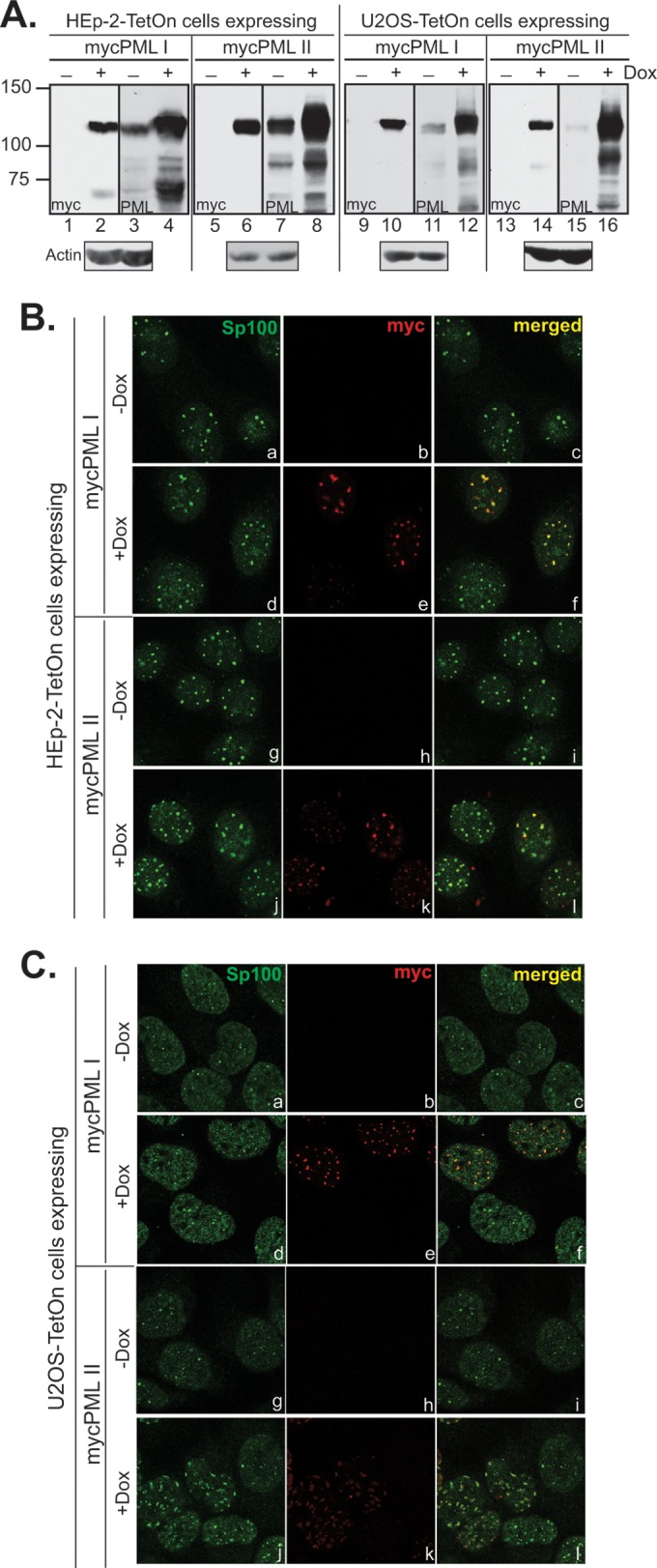
Expression and localization of ectopic PML I and PML II in HEp-2-TetOn and U2OS-TetOn cells. (A) Comparison of the endogenous PML level and the ectopic mycPML I or mycPML II level in stable cell lines. HEp-2-TetOn or U2OS-TetOn cells expressing mycPML I or mycPML II were either mock induced or induced with 1 μg/ml Dox overnight before the cells were harvested. Total cell lysates were subjected to Western blotting to examine ectopic expression with an anti-myc antibody and to compare the endogenous and exogenous PML levels with an anti-PML antibody. The loading control probed with an anti-actin antibody is shown below the induction gels. The values to the left are molecular sizes in kilodaltons. (B and C) Localization of mycPML I and mycPML II in HEp-2 (B) and U2OS (C) cells. HEp-2-TetOn or U2OS-TetOn cells expressing mycPML I or mycPML II were seeded onto four-well slides and mock induced or induced as described above. The primary antibodies used to stain cells are indicated at the top of the columns.
FIG 3.

Segment C is required for the degradation of PML II but not for that of PML I. (A) Schematic representation of the experimental scheme used to determine PML half-lives. TetOn cells carrying a PML isoform were seeded, induced with 1 μg/ml Dox, and then exposed to a test virus at 10 PFU/cell for 1 h. The inoculum was removed, and cells were incubated in growth medium for 1 h before CHX was added. Cells were harvested at 2-h intervals. (B) Samples taken at the 0-, 2-, 4-, and 6-h points were probed with an anti-myc antibody in Western blot assays. The cell lines, the PML isoforms, and the time points are indicated above the gel strips. Virus names (or mock infection) are listed to the left of the gels. Band densities were quantified by ImageJ, and PML half-lives are plotted below the corresponding gels. The subpanels are labeled with lowercase letters a to p.
To examine the degradation pattern of PML I and PML II early in HSV-1 infection, we established a half-life assay as illustrated in Fig. 3A. Briefly, stable TetOn cells were induced to express mycPML 24 h before infection. After viral penetration and removal of the inoculum, infected cells were incubated in growth medium for 1 h to allow the initial synthesis of ICP0. CHX was then added to block protein synthesis, and cells were harvested afterward at 2-h intervals. mycPML levels were quantitated by measuring the band densities in Western blot assays. The mycPML half-life in each infection was then calculated by normalizing the PML levels at different time points against that at the 0-h point, which was set as 100%. With this assay, we tested PML I and PML II degradation in RHG113-infected cells. In Fig. 3B, we found that PML I expressed in both HEp-2-TetOn and U2OS-TetOn cells was effectively degraded by RHG113 (Fig. 3B, c and k), comparable to degradation by RHG101 (Fig. 3B, b and j), whereas in mock-infected cells, PML I was not degraded (Fig. 3B, a and i). The PML I half-life in RHG101- or RHG113-infected cells was shorter than 2 h, whereas it was longer than 6 h in mock-infected cells (Fig. 3B, d and l). On the other hand, RHG113 did not degrade PML II in either HEp-2-TetOn or U2OS-TetOn cells (panels g and o) and the PML II half-life was longer than 6 h, similar to that in mock-infected cells (Fig. 3B, e and m), whereas wild-type ICP0 again quickly degraded PML II (Fig. 3B, f and n), with a PML II half-life shorter than 2 h (Fig. 3B, h and p).
In Fig. 3B, we noticed that in infected cells where ICP0 was capable of degrading PML, the mycPML I or mycPML II level at the 0-h point (lanes 1 in Fig. 3B, b, c, f, j, k, and n) was significantly lower than that in mock-infected or infected cells, where ICP0 was unable to degrade PML (lanes 1 in Fig. 3B, a, e, g, i, m, and o). Presumably, this was the result of rapid PML degradation upon ICP0 synthesis that occurred between the −2- and 0-h points. To prove this hypothesis, we first examined the mycPML I and mycPML II mRNA levels. We found minimal changes in mycPML I or mycPML II mRNA at 0 h in cells infected with different viruses (Fig. 4A), confirming that the variation in PML protein observed at the 0-h point was a posttranslational event. We then examined a −2-h sample to show that PML isoforms were induced to a comparable level before a virus was added (Fig. 4B, a to f, lanes 2, and g to l, lanes 1). When we added MG132 to one of the parallel infections at −2 h along with the virus and harvested those cells at 0 h, the proteasome inhibition between −2 and 0 h prevented the degradation of mycPML (Fig. 4B, a to f, lanes 1). We also included a −1-h point in an independent experiment and found that levels of mycPML I and mycPML II remained the same between −2 and −1 h in different infections (Fig. 4B, g to l, lanes 1 and 2). However, after the medium was supplemented with 10% serum, the mycPML level dropped significantly from −1 to 0 h when the viruses had an ICP0 capable of degrading that PML isoform (Fig. 4B, g, i, and j, lanes 2 and 3), whereas no degradation occurred if ICP0 was inactive for degradation (Fig. 4B, h, k, and l, lanes 2 and 3), suggesting that the supplement of 10% serum at −1 h triggered a robust expression of α genes, including that for ICP0, which led to rapid degradation of mycPML when the mutant ICP0 remained active for the isoform. Therefore, the decrease in the mycPML I or mycPML II level at the 0-h point in infections where ICP0 was capable of degrading the PML isoform was indeed the result of PML degradation by the newly synthesized ICP0. On the basis of these observations, we established two criteria to assess the ability of an ICP0 mutant to degrade PML isoforms: (i) whether degradation occurs between the −2- and 0-h points before the CHX treatment and (ii) whether the half-life of a PML isoform is more comparable to the nondegradation control or to the full-degradation control after CHX treatment.
FIG 4.

SIM362–364 is required for the degradation of PML II but not for that of PML I. (A) No substantial changes in mycPML I or mycPML II mRNA level at −2 or 0 h in infections. HEp-2-TetOn cells expressing mycPML I or mycPML II were induced and infected with the viruses indicated. At −2 and 0 h, total RNA was extracted, cDNA was synthesized, and quantitative RT-PCR was performed to detect mycPML isoforms. The cycle threshold (ΔCt) number of mycPML was normalized against the 18S rRNA. (B and C) HEp-2-TetOn cells expressing mycPML I or mycPML II were infected with the viruses indicated in the presence or absence of CHX or MG132. Samples were taken for Western blotting (WB) with an anti-myc antibody (Ab) (B), and the half-lives of PML isoforms were calculated as described above (C). (D) The same samples shown in panel B parts a to f were probed with an anti-mCherry antibody in Western blot assays.
Previously, Boutell et al. (28) identified seven SUMO interaction motif-like sequences (SLSs) scattering throughout ICP0. One of them, SLS4, located at residues 362 to 364, is responsible for the interaction with SUMO-2/3 and the ubiquitination of a poly-SUMO-2 substrate in vitro. Another report from the same group showed that a recombinant virus containing a mutant SLS4 degraded PML isoform I but not isoform II in the absence of all endogenous PMLs (29). SLS4 (we named it SIM362–364) resides in the center of segment C. To test whether SIM362–364 was responsible for the differential PML degradation observed above, we used the recombinant virus RHG130, in which ICP0 contains the I362G, V363A, and I364G substitutions, and examined the half-lives of mycPML I and mycPML II (Fig. 4B, a to f, lanes 3 to 6, and C). To further exclude potential variations caused by viral injection or virion tegument proteins, we used recombinant virus RHG120, which contains the C116G and C156A substitutions in the ICP0 RING finger and has completely lost E3 ligase activity (20), as the nondegradation control (Fig. 4B, b and e). Consistent with the RHG113 infection and the previous report (29), ICP0 containing mutated SIM362–364 (RHG130) was able to degrade mycPML I to the same extent as the wild type (Fig. 4B, a and c), showing a half-life shorter than 2 h (Fig. 4C, left). Moreover, mutations in SIM362–364 completely impaired ICP0's ability to degrade mycPML II (Fig. 4B, f). The half-life of mycPML II in RHG130-infected cells was longer than 6 h, similar to that of the E3 inactive mutant (RHG120) (Fig. 4C, right). In Fig. 4D, the control experiment shows that ICP0 in samples shown in Fig. 4B, a to f, was expressed to comparable levels at 0 h, suggesting that the differentiation in PML I and II degradation was solely the result of the I362G, V363A, and I364G substitutions. Consistently, mycPML I expressed in U2OS-TetOn cells was also rapidly degraded by RHG130 (Fig. 5A, c), whereas mycPML II was not at all degraded by RHG130 (Fig. 5A, f), again suggesting that the requirement of SIM362–364 in the differential degradation of PML I and II is cell type independent.
FIG 5.

Degradation of PML isoforms I, II, IV, and VI in U2OS-TetOn cells. U2OS-TetOn cells expressing individual PML isoforms were induced and infected with the viruses indicated. Samples were taken for Western blotting (WB) with an anti-myc (A) or an anti-mCherry (B) antibody (Ab) as described above.
To further examine whether the degradation of other PML isoforms by ICP0 relies on a similar mechanism, we constructed U2OS-TetOn cells that stably express mycPML IV or mycPML VI and examined their degradation patterns. Figure 5A shows that mutations in SIM362–364 also abolished the degradation of mycPML IV and mycPML VI (Fig. 5A, i and l), which led us to conclude that (i) ICP0 uses one mechanism that requires SUMO-SIM362–364 interaction to recognize and degrade PML II, IV, and VI while using a different regulation mechanism to degrade PML I and (ii) regardless of the permissiveness to the ICP0-null virus, HEp-2 and U2OS cells use the same mechanism to differentially recognize and degrade PML I and II.
Given these findings, we chose PML I and PML II as the model substrates to further investigate ICP0 elements that facilitate its substrate differentiation.
SIM362–364 is solely responsible for ICP0 interaction with PML II, but it is not sufficient for PML II degradation.
To fully understand PML II degradation by ICP0, we tested whether physical interaction between ICP0 and PML II is important for PML II degradation. Like endogenous PML, ectopically expressed mycPML was highly aggregated at ND10 (Fig. 2). Because of the association of ND10 with condensed nuclear chromatin and nuclear matrix (15), most of the mycPML expressed was insoluble and precipitated in the nuclear debris (data not shown). Therefore, we pretreated crude nuclear lysates with Benzonase before clearing the nuclear debris for coimmunoprecipitation assays. The results show that mycPML II interacted with full-length ICP0 containing active SIM362–364, with or without RING activity (Fig. 6A, lanes 3 and 8). With mutations in SIM362–364 or deletion of segment C, ICP0 lost the ability to interact with mycPML II (Fig. 6A, lanes 4 and 9), suggesting that SIM362–364 in segment C is necessary for ICP0 to interact with PML II.
FIG 6.

Segment C coordinates with the proximal sequences surrounding SIM362–364 and distal C-terminal sequences to regulate PML II degradation. (A) SIM362–364 in segment C is required for ICP0-PML II interaction. HEp-2-TetOn/PML II cells were induced and infected with the viruses indicated at 2 PFU/cell for 18 h. Coimmunoprecipitation was carried out with an anti-myc antibody (Ab), and the precipitates were probed in Western blot (WB) assays with the antibodies indicated to the left of the gels. (B and C) HEp-2-TetOn cells expressing mycPML II were induced, infected with the viruses indicated, and harvested for Western blotting with the antibodies indicated (B), and the PML II half-life was determined (C) as described above. (D) One-step growth curves of the viruses indicated on HEL cells. (E) HEp-2-TetOn cells expressing mycPML II were induced and infected with the viruses indicated at 0.1 PFU/cell. Total DNAs extracted at 2 and 24 hpi were subjected to qPCR with primers targeting the ICP27 or 18S rRNA gene to calculate the viral DNA fold increase within 24 h. (F) HEp-2-TetOn cells expressing mycPML II were induced, infected with the C-terminal truncation viruses, and harvested for Western blotting.
Previously, we showed that in cells infected with virus RHG136, in which ICP0 lacked all of the segments but C in the central region (Fig. 1A), the presence of a single ND10-FS2 (coinciding with segment C) was sufficient to drive the ICP0-ND10 fusion (21). Here we asked whether segment C alone is also sufficient for the interaction and degradation of PML II. We found that ICP0 containing C alone (RHG136) interacted with mycPML II (Fig. 6A, lane 10), but it failed to resuscitate PML II degradation (Fig. 6B, d). Interestingly, when we put the surrounding sequences of segment B or D back into the central region along with C (RHG121 and RHG111), PML II degradation was partially restored (Fig. 6B, e and f). Figure 6C shows that the half-life of mycPML II was close to 4 h in the presence of segments C to E (RHG121) and it was shortened to about 2 h when segments B to E were all restored (RHG111). Moreover, recombinant viruses RHG103 and RHG104, which contain ICP0 lacking the C-terminal 107 or 226 amino acids (20), also failed to degrade mycPML II (Fig. 6F, c and d), consistent with a previous report showing that ICP0 with the C-terminal 181 amino acids deleted did not degrade SUMOylated PML II (30). Therefore, we conclude that although the presence of segment C alone is sufficient for ICP0 to interact with PML II, the SUMO-SIM362–364 interaction alone is not sufficient for PML II to be degraded by ICP0. Sequences in the immediate vicinity of segment C or at the distal C-terminal end are both required for effective PML II degradation to occur.
To understand the biological significance of the inability to degrade a selective set of PML isoforms, we further examined the impact of segment C deletion on viral replication. Figure 6D shows that in HEL cells, RHG113 had a 38-fold decrease in viral yield at 24 hpi compared to that of wild-type ICP0 containing RHG101, whereas virus RHG112, in which ICP0 lacks only segment B, behaved very much like the wild type. Consistent with this result, qPCR analysis also shows that in HEp-2-TetOn cells expressing mycPML II, viral DNA had a 2,624-fold increase for RHG101 within 24 h but only a 780-fold increase for RHG113, just slightly higher than the 610-fold increase for RHG120 (Fig. 6E). Therefore, although RHG113 maintains the ability to degrade PML I (Fig. 3), the inability of RHG113 to degrade PML II, IV, and VI greatly impaired its viral DNA replication.
PML I degradation is not affected by ICP0-ND10 fusion.
To further identify elements important for PML I degradation, we tested two potential mechanisms that may regulate the degradation of PML I. (i) ND10 fusion may control the access of ICP0 to PML I, and (ii) direct interaction may occur between PML I and ICP0 domains other than SIM362–364.
To test whether a failure in ND10 fusion can affect PML I degradation, ND10 fusion-competent virus RHG136 and fusion-incompetent virus RHG135, in which all three ND10-FSs of ICP0 have been deleted and ICP0 cannot fuse with ND10 (21), were used for PML I half-life assays. Figure 7 shows that compared to the nondegradation control RHG120 (lanes 1 to 4) and the full-degradation control RHG101 (lanes 5 to 8), effective PML I degradation was detected in both RHG135- and RHG136-infected cells (lanes 9 to 16), suggesting that mycPML I was degraded in the absence or presence of functional ND10-FS. Therefore, proline-rich ND10-FSs play no role in PML I degradation, indicating that PML I can be degraded by ICP0 either inside or outside ND10 bodies. In addition, since RHG135 does not contain SIM362–364, PML I degradation is regulated by events other than the SUMO-SIM362–364 interaction.
FIG 7.

The ND10 fusion ability of ICP0 does not affect PML I degradation. HEp-2-TetOn cells expressing mycPML I were induced and infected with the viruses indicated. Samples taken at the 0-, 2-, 4-, and 6-h points were probed by Western blotting. The nature of cell line, the names of the viruses, and the time points are indicated above the gel strips. The antibodies used for Western blotting are indicated in the gel strips.
Bipartite PML I binding domains in the ICP0 N terminus coordinate the degradation of PML I, while the C terminus has trivial effects.
To identify additional elements regulating PML I recognition and degradation, we performed deletion mapping. In contrast to PML II, ICP0 lacking the C-terminal 107 or 226 amino acids maintained the ability to degrade mycPML I (Fig. 8A, lanes 12 to 15 and 17 to 20), with slight delays. The PML I half-lives in RHG103- and RHG104-infected cells were 2 to 4 h (Fig. 8B).
FIG 8.

Bipartite ICP0-PML I interaction is required for PML I degradation, while the ICP0 C terminus has trivial effects. (A and B) HEp-2-TetOn cells expressing mycPML I were induced, infected with the C-terminal truncation viruses, and harvested for Western blotting (A), and the PML I half-life was determined (B) as described above. (C) Domains important for ICP0-PML I interaction. HEp-2-TetOn cells expressing mycPML I were induced and infected with the viruses indicated at 2 PFU/cell for 18 h. Coimmunoprecipitation (IP) was carried out with the anti-myc antibody (Ab), and the precipitates were probed in Western blot assays with the antibodies indicated to the right of the gels. (D) HEp-2-TetOn cells expressing mycPML I were induced, infected with the N-terminal truncation viruses, and harvested for Western blotting.
We further examined the physical interaction between ICP0 and PML I by coimmunoprecipitation. Again, we found that full-length ICP0, regardless of whether it contained an active RING (RHG101) or a mutant RING (RHG120), had the ability to interact with mycPML I (Fig. 8C, lanes 4 and 12), suggesting that the RING finger itself was not the interaction domain. In this series of experiment, MG132 was added to infections where ICP0 had the ability to degrade PML I so that the bait protein (mycPML I) was preserved in coimmunoprecipitation. In line with the above observation that SIM362–364 in segment C was not necessary for PML I degradation, Fig. 8C shows that mycPML I was able to pull down ICP0 lacking segment C in the presence of MG132 (lane 5). In the absence of MG132, however, mycPML I was largely degraded and ICP0 was not pulled down without the bait (Fig. 8C, lane 13). These results suggest that (i) the coimmunoprecipitation assay has reliable specificity and (ii) segment C is not required for physical interaction between ICP0 and PML I.
We also tested the binding between C-terminally truncated ICP0 and mycPML I. In agreement with the finding that the ICP0 C terminus is dispensable in PML I degradation (Fig. 8A and B), ICP0 interacted with mycPML I in the absence of the entire C terminus (Fig. 8C, lane 6), suggesting that the PML I interaction domain is located in the N-terminal half of ICP0. Next, we conducted additional deletion mapping to identify the N-terminal domains that are important for the ICP0-PML I interaction. N-terminal residues 1 to 83 and internal residues 245 to 474 are the sequences flanking the RING domain but not participating in the formation of the C3HC4 RING (31). We used recombinant viruses RHG105 (single deletion of residues 1 to 83), RHG110 (single deletion of residues 245 to 474), and RHG118 (double deletion of both residues 1 to 83 and 245 to 474) to map the ICP0-PML I interaction. Figure 8C shows that ICP0 lacking either residues 1 to 83 or residues 245 to 474 was coimmunoprecipitated with mycPML I (lanes 14 and 15) but double deletion ICP0 lacking both regions did not interact with mycPML I (lane16).
Consistent with the bipartite ICP0-PML I interaction, in PML I half-life assays, the mycPML I level at 0 h in both RHG105- and RHG110-infected cells was significantly lower than in the −2-h sample, suggesting that PML I degradation occurred between the −2- and 0-h points (Fig. 8D, top, lanes 12 and 17). However, after CHX was added to each infection, progressive loss of mycPML I was not observed in either RHG105- or RHG110-infected cells (lanes 13 to 15 and 18 to 20). An examination of the mutant ICP0 level revealed that in both RHG105- and RHG110-infected cells, ICP0 disappeared quickly after CHX treatment (Fig. 8D, bottom, lanes 12 to 15 and 17 to 20). The dramatic loss of ICP0 in the absence of residues 1 to 83 or residues 245 to 474 resulted in the withdrawal of E3 ligase upon CHX treatment and prevented the continuous degradation of PML I in these infections. One interesting phenomenon is the accumulation of modified mycPML I with lower mobility at the 0-h point in both RHG105- and RHG110-infected cells (Fig. 8D, top, lanes 12 and 17), which was never observed in either the full-degradation control (RHG101) or the nondegradation control (RHG120) (Fig. 8D, top, lanes 2 and 7), suggesting that a single deletion of residues 1 to 83 or residues 245 to 474 likely did not affect the ubiquitination of PML I but hampered the proteasomal degradation of ubiquitinated PML I instead. In RHG118-infected cells, the deletion of both PML I binding domains completely abolished PML I degradation. Neither a decrease in mycPML between the −2- and 0-h points nor progressive disappearance of mycPML I was observed (Fig. 8D, top, lanes 21 to 25), as in RHG120-infected cells.
DISCUSSION
ICP0 is a key regulator in both the lytic and latent life cycles of HSV-1. By degrading host restrictive factors with its E3 ubiquitin ligase or by directly interacting with host regulatory complexes, ICP0 plays a vital role in disarming host antiviral responses (13, 32, 33). Because of ICP0's complex biochemical properties, the cooperativity of its functional domains is not clearly understood. We seek to dissect the involvement of ICP0 elements in individual ICP0 functions and delineate the coordination of different elements in the course of infection. In this study, we focused on understanding how different domains of ICP0 regulate its E3 ubiquitin ligase activity. We found that ICP0 uses different mechanisms to distinguish two of its substrates, PML I and PML II.
First, ICP0 relied solely on SIM362–364 to interact with PML II. However, this interaction alone was not sufficient for PML II to be degraded by ICP0. Proximal sequences surrounding SIM362–364 and distal sequences located at the ICP0 C terminus were both involved in supporting effective PML II degradation. This means that the overall structure of ICP0 is likely important for ICP0 to ubiquitinate and degrade PML II, even when SUMO-SIM362–364 is intact. The RING structure has been determined with the purified peptide of amino acids 105 to 177 (31). Preliminary in silico remodeling of ICP0 showed many unstructured regions in ICP0, which may be compatible with the complex interactions between ICP0 and its numerous partners but also makes it difficult to unveil a true mechanism. Structural studies of ICP0 are now necessary and urgent.
The SUMO-SIM362–364 interaction is not a determining factor for ICP0 to recognize and degrade PML I. Instead, two ICP0 domains located upstream and downstream of the RING finger participate in a bipartite interaction with PML I that coordinates effective PML I degradation. Either part of the ICP0-PML I interaction may be sufficient to bring PML I into the vicinity of ICP0 for ubiquitination to occur, but proteasomal degradation is greatly decelerated without cooperation between the two arms of the bipartite interaction. Everett's group has previously shown interaction between PML I and ICP0 residues 1 to 388 by yeast two-hybrid screening, but they did not detect any interaction between PML I and ICP0 residues 1 to 241 (30). Although it does not directly participate in the ICP0-PML I interaction, the C-terminal half of ICP0, may help to maintain the integrity of the individual PML I binding domains when only one of the two is left in ICP0. Again, structural studies are critical for understanding the coordination among these ICP0 domains.
Naturally, the next interesting question is why ICP0 treats individual substrates differently. Is it because of the differences in the biochemical properties of these substrates? For example, individual PML isoforms may occupy different premises of ND10, such as the ND10 surface or core area. We examined the effects of ND10 fusion on PML I degradation and found that PML I can be degraded regardless of whether ICP0 is able to fuse with ND10 or not. SIM362–364 is located in the center of segment C, surrounded by multiple prolines before and after. Therefore, to test whether the ND10 fusion process has any role in PML II degradation, we need to carefully dissect the segment C sequence so we can abolish the ND10 fusion process without changing the SUMO-SIM362–364 interaction or vice versa.
A second possible reason for ICP0 to recognize its substrates differently can be that the roles these substrates playing in HSV infection are quite different. Xu et al. showed that PML gene knockout via CRISPR/Cas causes reduced induction of Sp100 upon interferon treatment but also causes wild-type HSV-1 to replicate ineffectively, suggesting that the PML gene products have both positive and negative influences on HSV replication (34). That ICP0 uses different mechanisms to degrade PML isoforms may be a result of evolutionary adaptation of HSV to host responses.
In this study, we used tetracycline-inducible PML isoforms I and II as model E3 substrates to characterize the ICP0 elements regulating differential PML degradation. The advantage of using a TetOn system is the tight regulation for ectopic PML and the minimum impact on regular cell cycles when stable cell lines are being screened and passaged. We have noticed that overexpression of PML II, but not PML I, altered the number and shape of ND10 bodies in U2OS cells, but not in HEp-2 cells (Fig. 2), suggesting that short-term PML overexpression may still change ND10 composition and function in a cell type-dependent manner. Although we have shown that the differential regulation of PML I and II degradation by ICP0 is a mechanism common to both HEp-2 and U2OS cells, we still need to be cautious when interpreting the overall roles of PML or other ND10 components in future studies.
ACKNOWLEDGMENTS
We thank Bernard Roizman and Daniel Schoenberg for reagents. We thank Gilberto Bezerra and Binh Ha for their assistance in stable line screening and viral titration. We thank the Microscopy, Imaging, and Cytometry Resources (MICR) Core facility at Wayne State University for technical support.
These studies were supported by an NIH grant (RO1AI118992) and Wayne State University startup funds awarded to Haidong Gu.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology 6th ed. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Everett R, O'Hare P, O'Rourke D, Barlow P, Orr A. 1995. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interaction with PML-containing nuclear structures. J Virol 69:7339–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lium EK, Silverstein S. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J Virol 71:8602–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chelbi-Alix MK, de The H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941. doi: 10.1038/sj.onc.1202366. [DOI] [PubMed] [Google Scholar]
- 5.Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc Natl Acad Sci U S A 109:E3008–E3017. doi: 10.1073/pnas.1211302109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chaurushiya MS, Lilley CE, Aslanian A, Meisenhelder J, Scott DC, Landry S, Ticau S, Boutell C, Yates JR III, Schulman BA, Hunter T, Weitzman MD. 2012. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol Cell 46:79–90. doi: 10.1016/j.molcel.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Everett RD, Parada C, Gripon P, Sirma H, Orr A. 2008. Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J Virol 82:2661–2672. doi: 10.1128/JVI.02308-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Orzalli MH, Conwell SE, Berrios C, DeCaprio JA, Knipe DM. 2013. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc Natl Acad Sci U S A 110:E4492–4501. doi: 10.1073/pnas.1316194110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. 1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J 16:1519–1530. doi: 10.1093/emboj/16.7.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc Natl Acad Sci U S A 104:17134–17139. doi: 10.1073/pnas.0707266104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu H. 2016. Infected cell protein 0 functional domains and their coordination in herpes simplex virus replication. World J Virol 5:1–13. doi: 10.5501/wjv.v5.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 15.Gu H, Zheng Y. 2016. Role of ND10 nuclear bodies in the chromatin repression of HSV-1. Virol J 13:62. doi: 10.1186/s12985-016-0516-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75. doi: 10.1006/viro.1996.0094. [DOI] [PubMed] [Google Scholar]
- 17.Sourvinos G, Everett RD. 2002. Visualization of parental HSV-1 genomes and replication compartments in association with ND10 in live infected cells. EMBO J 21:4989–4997. doi: 10.1093/emboj/cdf458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Damme E, Laukens K, Dang TH, Van Ostade X. 2010. A manually curated network of the PML nuclear body interactome reveals an important role for PML-NBs in SUMOylation dynamics. Int J Biol Sci 6:51–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh ET, Strauss JF III, Maul GG. 1999. PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J Cell Biol 147:221–234. doi: 10.1083/jcb.147.2.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gu H, Zheng Y, Roizman B. 2013. The interaction of herpes simplex virus ICP0 with ND10 bodies: a sequential process of adhesion, fusion and retention. J Virol 87:10244–10254. doi: 10.1128/JVI.01487-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng Y, Gu H. 2015. Identification of three redundant segments responsible for herpes simplex virus 1 ICP0 to fuse with ND10 nuclear bodies. J Virol 89:4214–4226. doi: 10.1128/JVI.03658-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fagioli M, Alcalay M, Pandolfi PP, Venturini L, Mencarelli A, Simeone A, Acampora D, Grignani F, Pelicci PG. 1992. Alternative splicing of PML transcripts predicts coexpression of several carboxy-terminally different protein isoforms. Oncogene 7:1083–1091. [PubMed] [Google Scholar]
- 23.Jensen K, Shiels C, Freemont PS. 2001. PML protein isoforms and the RBCC/TRIM motif. Oncogene 20:7223–7233. doi: 10.1038/sj.onc.1204765. [DOI] [PubMed] [Google Scholar]
- 24.Lopez P, Jacob RJ, Roizman B. 2002. Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J Virol 76:9355–9367. doi: 10.1128/JVI.76.18.9355-9367.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otsuka Y, Kedersha NL, Schoenberg DR. 2009. Identification of a cytoplasmic complex that adds a cap onto 5′-monophosphate RNA. Mol Cell Biol 29:2155–2167. doi: 10.1128/MCB.01325-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J Virol 69:6249–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bernardi R, Pandolfi PP. 2003. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22:9048–9057. doi: 10.1038/sj.onc.1207106. [DOI] [PubMed] [Google Scholar]
- 28.Boutell C, Cuchet-Lourenço D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog 7:e1002245. doi: 10.1371/journal.ppat.1002245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Everett RD, Boutell C, Pheasant K, Cuchet-Lourenço D, Orr A. 2014. Sequences related to SUMO interaction motifs in herpes simplex virus 1 protein ICP0 act cooperatively to stimulate virus infection. J Virol 88:2763–2774. doi: 10.1128/JVI.03417-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuchet-Lourenço D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett RD, Barlow P, Milner A, Luisi B, Orr A, Hope G, Lyon D. 1993. A novel arrangement of zinc-binding residues and secondary structure in the C3HC4 motif of an alpha herpes virus protein family. J Mol Biol 234:1038–1047. doi: 10.1006/jmbi.1993.1657. [DOI] [PubMed] [Google Scholar]
- 32.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 33.Lanfranca MP, Mostafa HH, Davido DJ. 2014. HSV-1 ICP0: An E3 ubiquitin ligase that counteracts host intrinsic and innate immunity. Cells 3:438–454. doi: 10.3390/cells3020438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu P, Mallon S, Roizman B. 2016. PML plays both inimical and beneficial roles in HSV-1 replication. Proc Natl Acad Sci U S A 113:E3022–3028. doi: 10.1073/pnas.1605513113. [DOI] [PMC free article] [PubMed] [Google Scholar]