

ABSTRACT
Cytomegalovirus (CMV) is a ubiquitous human pathogen that increases the morbidity and mortality of immunocompromised individuals. The current FDA-approved treatments for CMV infection are intended to be virus specific, yet they have significant adverse side effects, including nephrotoxicity and hematological toxicity. Thus, there is a medical need for safer and more effective CMV therapeutics. Using a high-content screen, we identified the cardiac glycoside convallatoxin as an effective compound that inhibits CMV infection. Using a panel of cardiac glycoside variants, we assessed the structural elements critical for anti-CMV activity by both experimental and in silico methods. Analysis of the antiviral effects, toxicities, and pharmacodynamics of different variants of cardiac glycosides identified the mechanism of inhibition as reduction of methionine import, leading to decreased immediate-early gene translation without significant toxicity. Also, convallatoxin was found to dramatically reduce the proliferation of clinical CMV strains, implying that its mechanism of action is an effective strategy to block CMV dissemination. Our study has uncovered the mechanism and structural elements of convallatoxin, which are important for effectively inhibiting CMV infection by targeting the expression of immediate-early genes.
IMPORTANCE Cytomegalovirus is a highly prevalent virus capable of causing severe disease in certain populations. The current FDA-approved therapeutics all target the same stage of the viral life cycle and induce toxicity and viral resistance. We identified convallatoxin, a novel cell-targeting antiviral that inhibits CMV infection by decreasing the synthesis of viral proteins. At doses low enough for cells to tolerate, convallatoxin was able to inhibit primary isolates of CMV, including those resistant to the anti-CMV drug ganciclovir. In addition to identifying convallatoxin as a novel antiviral, limiting mRNA translation has a dramatic impact on CMV infection and proliferation.
INTRODUCTION
Cytomegalovirus (CMV) is an extremely large betaherpesvirus with over 230,000 bp and over 200 open reading frames. While the majority of CMV infections in immunocompetent hosts are asymptomatic (1), the phenomenon of latency allows the virus to establish a permanent and persistent infection. CMV is highly seroprevalent in the human population (>60%), with broad cell tropism, and can be transmitted through bodily fluids and organ transplantation and transplacentally from mother to fetus (2). Reactivation of latent CMV can cause disease in any population but is a particular source of morbidity and mortality for immunocompromised patients (3). It is estimated that treatment of CMV costs the United States $4.4 billion every year (4), and congenital CMV infections are the leading cause of permanent congenital neurological disabilities in the United States (5). The current FDA-approved therapies all target the viral DNA polymerase, and none are approved for use in pregnant women or newborns. These compounds can cause severe side effects, including neurological and vascular defects, multifaceted frailty syndrome, hepatitis, colitis, and pneumonia, as well as give rise to antiviral-resistant strains (6–11). To identify novel drug targets for CMV infection, we performed a high-content screen of bioactive compounds that included FDA-approved drugs for various disorders. This approach of repurposing existing drugs has multiple benefits, including lower development costs (12) and providing new tools for elucidating the mechanisms by which viruses manipulate the host cell. We utilized a reporter virus expressing the chimera consisting of the immediate-early 2 gene product fused with the yellow fluorescent protein (IE2-YFP) (13), a protein that is expressed during the IE phase of infection and prior to DNA replication, to identify compounds that target the early steps of infection. The cardiac glycoside convallatoxin was thus identified as a potent and selective inhibitor of CMV infection and subsequent proliferation (14).
Cardiac glycosides, currently being utilized to treat congestive heart failure and angina pectoris (15), are effective inhibitors of CMV infection, but the mechanism of action has yet to be fully characterized. Proposed targets include downstream manipulation of Src kinase upon altering Na+/K+-ATPase activity (14) or by manipulating hERG expression (16). Modulating the sugar residues of cardiac glycosides can alter their inhibitory activity, and the presence of l-sugar residues and multiple-sugar residues enhances their anti-CMV activity (17). In the current study, different cardiac glycoside variants were analyzed to identify the key side chain moieties of convallatoxin that are important for anti-CMV activity. Furthermore, we demonstrated that the inhibition of CMV infection by convallatoxin is due to decreased translation of viral proteins secondary to a reduction in the cellular import of methionine.
MATERIALS AND METHODS
Cell lines, antibodies, viruses, and chemicals.
Human lung fibroblasts (MRC5; ATCC, Manassas, VA) were cultured in complete Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin-streptomycin/ml, and 20 mM HEPES/ml at 37°C in a humidified incubator with 5% CO2. CMV AD169IE2-YFP was propagated as previously described (13). The infectious-virus yield was assayed on human fibroblasts by median tissue culture infective dose (TCID50). The TCID50 results were used to estimate the number of infectious particles per milliliter needed to infect cells at the desired multiplicity of infection (MOI). Cycloheximide was purchased from MP Biomedicals, Solon, OH (catalog no. 100183). All the cardiac glycosides were purchased from Sigma-Aldrich (St. Louis, MO), except for ouabain, which was purchased from Acros Organics (New Jersey).
Human foreskin fibroblasts (HFFs) were cultured in Eagle's minimal essential medium (MEM) supplemented with 10% FBS, 20 mM HEPES, 2 mM l-glutamine, 2.5 μg/ml amphotericin B, and 50 μg/ml gentamicin. HFF monolayers were inoculated with cells infected with each of four low-passage-number clinical isolates that are phenotypically cell associated. Two of the isolates, BI-4 and BI-6, are susceptible to the approved CMV antiviral agents: ganciclovir, foscarnet, and cidofovir. Isolate CH13 has a deletion of codons 597 to 603 in the CMV UL97 kinase gene, and CH19 has an A594V point mutation in the same gene. Both mutations confer resistance to ganciclovir.
Determining the EC50.
MRC5 cells (10,000 in 100 μl) were plated in each well of a 96-well plate (Greiner, Monroe, NC). The following day, the medium was replaced with DMEM with various concentrations (0 to 5 μM) of the compounds (final volume, 100 μl) in sextuplicate (1 h at 37°C), and then the cells were infected with AD169IE2-YFP (MOI, 3). At 18 h postinfection (hpi), the plates were analyzed with an Acumen eX3 cytometer for the number of IE2-YFP-positive cells per well based on the nuclear IE2-YFP fluorescence intensity per well (13, 14). The Acumen settings were previously utilized to measure the exclusive nuclear fluorescence intensity of infected cells (13). Fluorescence emission >2 standard deviations above background was registered as a positive signal. Any fluorescent signal larger than 5 μm, smaller than 300 μm, and separated from any other emission by at least 0.5 μm in both the x and y axes was classified as an event. Using cells treated with dimethyl sulfoxide (DMSO) equivalent to the highest dose of drug and infected with AD169IE2-YFP as 100% infection, the percent infection of cells pretreated with increasing amounts of compound was determined. The half-maximal effective concentrations (EC50s) were calculated using Prism5's nonlinear fit log (inhibitor)-versus-response (three parameters) analysis of these averages and extrapolation to the concentration that would produce 50% infection relative to DMSO treatment.
Determining the CC50.
MRC5 cells (10,000 in 100 μl) were plated in each well of a 96-well plate. The following day, the medium was replaced with DMEM with various concentrations (0 to 5 μM) of the compounds (final volume, 100 μl) in sextuplicate. After 19 h, NucGreenDead 488 (referred to here as NucGreen) (Life Technologies, Grand Island, NY) was directly added to five out of six wells per condition (10 min at room temperature). NucGreen is a dye to which living cells, but not dead cells, are impermeable and emits a fluorescent signal when bound to DNA (14, 18, 19). The number of NucGreen-positive cells was determined by measuring the fluorescence intensity per well using a fluorescent-plate reader. To determine the total number of cells/well, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for up to 2 h, the medium was replaced with PBS (100 μl), and NucGreen was added to the 5 wells for 10 min. The numbers of NucGreen-positive cells were determined by measuring the fluorescence intensity detected by the plate reader. The cell toxicity of the compound-treated cells was calculated as percent cell viability using the total number of NucGreen-positive cells treated with DMSO equivalent to the highest dose of drug as 100% compared to the number of NucGreen-positive cells from compound-treated cells. The half-maximal toxicity concentration (CC50) values were calculated using Prism5's nonlinear fit log (inhibitor)-versus-response (three parameters) analysis of these averages and extrapolation to the concentration that would produce cell toxicity of 50% above that with DMSO alone.
Time-of-addition experiments.
MRC5 cells (10,000 in 100 μl) were plated in a 96-well plate. The following day, the medium was replaced with 95 μl of DMEM. Compound (5 μl of 20× stock) was added to the wells at the designated time points relative to virus infection (range, −1 hpi to 8 hpi) in sextuplicate. The cells were infected at 0 hpi with AD169IE2-YFP (MOI, 3), and at 18 hpi, the plates were analyzed with an Acumen eX3 cytometer for the number of IE2-YFP-positive cells per well based on IE2-YFP fluorescence intensity per well (13). Using convallatoxin-treated cells at 0 hpi as 100% relative maximum inhibition, the percent relative maximum inhibition was determined from cells treated with drug at different time points relative to infection.
In silico structure analysis.
The Na+/K+-ATPase–ouabain complex structure (Protein Data Bank [PDB] code 4hyt) was prepared by optimizing H-bond assignments and energy minimization (to a root mean square deviation [RMSD] of 0.3 Å) using the Protein Preparation Wizard in Maestro (Schrödinger). Ligand files were prepared using MarvinSketch (ChemAxon) and LigPrep (Schrödinger) with standard parameters. Individual ligands were docked into the minimized pump structure using XP precision in Glide (Schrödinger).
Quantifying viral mRNA accumulation.
MRC5 cells (250,000 in 2 ml) were plated in a 6-well plate. The following day, the cells were treated with convallatoxin (5 nM or 50 nM) or the known mRNA synthesis inhibitor flavopiridol (20) (5 nM or 5 μM) and infected with wild-type (WT) AD169 (AD169WT) (MOI, 5) for 24 h. The cells were washed with PBS and then removed by trypsinization, and total RNA was harvested from the cells using the Promega (Madison, WI) mini RNA isolation kit. Reverse transcription (RT) was performed using the Roche First Strand cDNA synthesis kit and the provided oligo(dT) primers to ensure that only mRNA was reverse transcribed to cDNA. Quantitative PCR (qPCR) was performed in a LightCycler 480 using SYBR green and previously published primers targeting human GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and CMV UL122 (21). The cycle threshold (CT) value for each sample was calculated by LightCycler 480's Abs-Quant 2nd-derivative function, and using those values, the amount of UL122 cDNA in each sample relative to GAPDH cDNA was calculated and viral mRNA was expressed relative to DMSO treatment.
Measuring the impact of convallatoxin on [35S]methionine protein incorporation.
MRC5 cells (500,000 in 2 ml) were plated in a 6-well plate. The following day, the cells were treated with 0 to 50 nM convallatoxin for 24 h. The cells were trypsinized, pelleted, and resuspended in 250 μl of starvation medium (free of methionine and cysteine) with 5% [35S]methionine for 1.5 h. The cells were lysed in NP-40, and 20 μl of each sample's cell lysate was run on SDS-PAGE under reducing conditions overnight. The polyacrylamide gel was dried for 90 min at 72°C, and proteins were resolved using autoradiography. Additionally, 10 μl of each sample's cell lysate was applied to Whatman paper in triplicate, and protein complexes were precipitated by washing the Whatman paper in 10% trichloroacetic acid (TCA) for 10 min. The Whatman paper was air dried overnight before being placed in 2 ml of scintillation fluid, and scintillation counts per million were determined with a liquid scintillation counter (Beckman Coulter, Brea, CA). The average counts per million with the standard error of the triplicates were plotted, with DMSO-treated cells' average counts per million set as 100%.
In vitro translation inhibition experiment.
In vitro translation inhibition was determined using the TnT T7 Coupled Reticulocyte Lysate system (Promega; catalog no. L4611) according to the manufacturer's protocol for luciferase control experiments, with the following modifications. Reticulocyte lysates were pretreated for 30 min on ice with DMSO or 5 μM convallatoxin or cycloheximide (2.5 μl of DMEM containing 30 μM compound plus 12.5 μl of reticulocyte lysate), and to quench any translation prior to measuring luciferase activity, 5 μl of 30 μM cycloheximide was added to the 25-μl reaction mixture after a 90-min incubation at 30°C. Luciferase activity was then measured on a Cytation3 imaging reader in triplicate using luciferase assay reagent II (Promega, catalog no. E1960). The average luminescence with the standard error of each triplicate was plotted as relative luminescence, with DMSO-treated lysate as 100.
Differentiating protein synthesis inhibition and [35S]methionine uptake inhibition.
MRC5 cells (1,000,000 in 2 ml) were treated for 2 h in DMEM containing 50 nM convallatoxin or DMSO. The cells were pelleted, and each sample was split in half so that there were 4 samples of 500,000 cells each. Each sample was then incubated for 20 min in starvation medium (free of methionine and cysteine) with 5% [35S]Met and either DMSO alone, 50 μg/ml cycloheximide alone, 50 nM convallatoxin alone, or 50 nM convallatoxin and 50 μg/ml cycloheximide. The cells were lysed in 0.5% NP-40 (500 μl), and 10 μl of each sample's cell lysate was analyzed for scintillation counts as described above or without TCA precipitation to examine total cell lysate.
Clinical-strain plaque assay and qPCR experiments.
HFF monolayers were cultured in 24-well plates. The medium was removed and replaced with medium containing a range of convallatoxin nanomolar concentrations in triplicate wells for 7 to 10 days without refreshing the compound. For the control, DMSO was added in a volume equal to the largest volume of drug. Cell-associated stocks of each clinical strain were added to the wells in a volume calculated to contain 40 to 100 infected cells per well. The plaques in each well were counted under phase-contrast microscopy at 7 to 10 days postinoculation. On the same day that the plaques were read, the medium was removed, and the cells in the monolayers were lysed using the lysis buffer in the Qiagen DNA minikit. The monolayers were also scraped with a micropipette tip to ensure that all infected material was harvested. DNA was extracted from the lysate and cell scrapings using the Qiagen DNA minikit protocol. DNA from each well was quantitated by a validated in-house CMV real-time PCR assay, which targets the viral DNA polymerase gene. The EC50 was calculated using the average plaque count or CMV DNA copy number in the triplicate DMSO control wells as 100%.
Long-term, low-dose [35S]methionine scintillation experiments.
MRC5 cells (50,000 in 2 ml) were plated on a 6-well plate. The following day, the medium was replaced, and the cells were treated for 7 days in DMEM containing 0 to 15 nM convallatoxin without removing or replenishing the drug. Cells were harvested via trypsinization and resuspended in 250 μl of starvation medium (free of methionine and cysteine) with 0 to 15 nM drug and 5% [35S]Met for 1.5 h. The cells were lysed in 0.5% NP-40, and 10 μl of each sample's total cell lysate was analyzed for scintillation counts as described above.
RESULTS
Specific convallatoxin residues dictate potency against CMV.
We performed a high-content screen to identify novel inhibitors of CMV that target the virus at a step prior to immediate-early protein expression and identified convallatoxin as a potent selective inhibitor of CMV infection (14). Based on convallatoxin's efficacy to limit virus infection, we examined its mode of action and its potential role as an antiviral compound.
We initially defined the chemical moieties of convallatoxin that are important for inhibition of a CMV infection using 12 structurally diverse cardiac glycosides, including convallatoxin, that have various sugar residues and side chains of the steroidal core (Fig. 1). We utilized the expression of the fluorescent IE2-YFP chimera as a readout (reporter virus, CMV AD169IE2-YFP) for a CMV infection (13). IE2-YFP expression is localized entirely in the nucleus, allowing us to quantify virus infection with the Acumen eX3 high-throughput cytometer. MRC5 lung fibroblasts pretreated for 1 h with various doses of each compound (0 to 5 μM) were infected with AD169IE2-YFP (MOI, 3) (14) and analyzed 18 hpi for the number of fluorescent cells, which represents the number of infected cells (Fig. 2, black lines). The concentration of drug required to reduce the number of infected cells by 50% (EC50) was determined using nonlinear regression. The EC50s could be determined for only 10 of the 12 compounds, with a range of 0.01 μM to 3.4 μM. The three compounds with the lowest EC50s (convallatoxin, 0.01 μM; ouabain, 0.03 μM; and β-antiarin, 0.04 μM) (Fig. 2A to C) all contained a 6-deoxy-α-l-mannopyranoside sugar functionality on the C-3 hydroxy group of the steroid A ring and the C-17 butenolide in the β-conformation, indicating that this combination of moieties is critical for inhibition of a viral infection.
FIG 1.
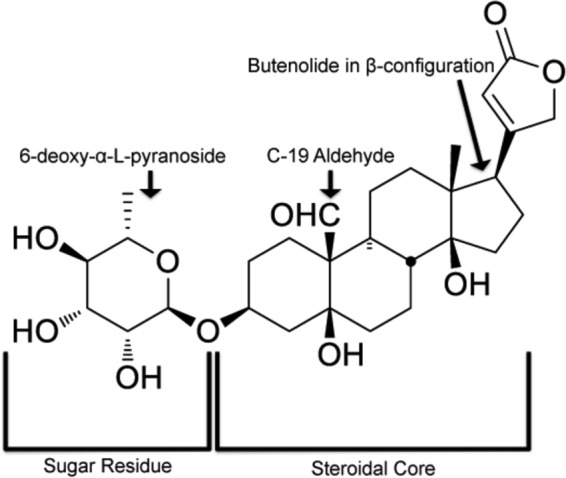
Chemical structure of convallatoxin. Convallatoxin consists of two functional domains: the steroidal core and the glycan chain. The arrows indicate the important structures identified for anti-CMV efficacy (oxygenated alkyl at C-19) and for maintaining efficacy during the infectious life cycle (6-deoxy-α-l-mannopyranoside).
FIG 2.

Determination of EC50 and CC50 values of diverse cardiac glycoside compounds. MRC5 cells treated with DMSO and increasing amounts of glycoside variants (0 to 5 μM) were infected with AD169IE2-YFP (MOI, 3) and subjected to analysis using an Acumen eX3 cytometer for YFP+ cells. The percent infection (left axis, black lines and circles) was determined based on the number of YFP+ cells from compound-treated cells, setting cells treated with DMSO equivalent to the highest dose of drug as 100% infection. Intranuclear cell staining using NucGreen fluorescent cell dye was utilized to determine cell viability (right axis, gray lines and squares), with cells treated with DMSO equivalent to the highest dose of drug as 100% cell viability. The error bars represent standard errors of the mean.
In order to determine that the inhibition of virus infection was not due to toxicity, drug-treated cells were stained with the cell-impermeable dye NucGreenDead 488 (Life Technologies) (Fig. 2, gray lines). The dye binds to double-stranded DNA and is able to enter the cell only upon the breakdown of the plasma membrane (14, 18, 19), a condition consistent with cell death. The fluorescence signal thus localizes to the nuclei of dead cells, which can be analyzed using the Acumen eX3 cytometer. The concentration of drug required to induce cell death in 50% of the cells above background (CC50) was determined using nonlinear regression. Strikingly, all of the compounds had minimal toxicity up to the maximum dose tested (5 μM), and thus, an exact CC50 value could not be accurately calculated (Fig. 2). This striking lack of toxicity speaks to the selective nature of cardiac glycosides' inhibition of CMV infection.
The compounds were classified into four general classes based on antiviral potency (Fig. 2 and 3). For one group of compounds, including convallatoxin, ouabain, and β-antiarin, the EC50s below 50 nM (Fig. 2A to C and 3) demonstrated high potency. These compounds all contain an aldehyde or hydroxide functionality at the C-19 position of the steroid, butenolide functionality in the β-configuration of C-17, and a 6-deoxy-α-l-mannopyranoside sugar. The next group of compounds have moderate antiviral selectivity, with EC50s in the 50 nM to 1 μM range (Fig. 2D to G and 3). Compounds of this type, including cymarin, K-strophanthin, mansonin, and strophanthidin, all contain an aldehyde functionality at the C-19 position of the steroid and butenolide functionality in the β-configuration of C-17 but either have methylated sugars attached to the steroid core or lack the sugar entirely. The third group shows minimal antiviral selectivity, with EC50s greater than 2 μM (Fig. 2H to J and 3). Compounds of this type (strophanthidol, strogoside, and glucostrebloside) all have a hydroxy substituent or lactone functionality at the C-19 position of the steroid and have a butenolide functionality in the β-configuration of C-17 but lack the 6-deoxy-α-l-mannopyranoside sugar. In fact the only difference between strophanthidin and strophanthidol is at C-19, and it reduces its antiviral potency by over 5-fold. The last group shows no measurable antiviral activity (Fig. 2K to L) and either lacks the C-19 oxidized alkyl group or has the butenolide functionality in the α-configuration of C-17 (allo-emicymarin and 17-epi-strophanthidin, respectively). These data support the model in which specific side chains dictate the efficacy of CMV inhibition.
FIG 3.

Structures of the cardiac glycoside variants and their potencies to inhibit a CMV infection. The variants were classified as high-potency, medium-potency, low-potency, and no-potency compounds.
Analysis of the anti-CMV pharmacodynamics properties of cardiac glycosides.
To further evaluate the 10 effective compounds, we performed a time-of-addition assay, in which a single dose of each compound (EC75 values) was added to infected cells at various time points relative to the time of infection (starting 1 h prior to infection [−1] up to 8 h postinfection) and evaluated for infection at 18 hpi to identify virus-infected cells (Fig. 4). We tested the 10 compounds in this assay and found that 4 of them (cymarin, K-strophanthin, mansonin, and strophanthidin) were quite effective at inhibiting a virus infection when added up to 1 hpi, with a statistically significant increase in infection beginning at 2 h postinfection (Fig. 4A to D). Ouabain, glucostrebloside, strogoside, and strophanthidol were quite effective at inhibiting a virus infection when added up to 2 h postinfection, with a statistically significant increase in infection beginning at 3 h postinfection (Fig. 4E to H), while β-antiarin and convallatoxin remained effective when added up to 4 h postinfection, with statistically significant increases in virus infection beginning at 6 h postinfection (Fig. 4I to J). The differences in the pharmacodynamics among the variants may be related to a combination of a compound's efficacy to modulate the activity of Na+/K+-ATPase, the Kd (dissociation constant) value, cell permeability properties, and half-life in cells and medium. Interestingly, this difference may be related to the combination of the configuration of the sugar moiety at C-3 and the presence of an aldehyde at C-19; the two compounds that maintained efficacy up to 4 h postinfection (i.e., convallatoxin and β-antiarin) both have glycosidic bonds in the α-configuration, while the other compounds, all of which no longer inhibited viral replication after 3 h, either have β-configured glycosidic bonds, lack the attached sugar moiety altogether, or do not have an aldehyde at C-19. The kinetics of cardiac glycosides' inhibition of CMV infection were altered by a combination of both the sugar residues and the steroidal core. Despite falling into 3 distinct groups, the fact that no cardiac glycoside lost efficacy prior to being added 2 h postinfection indicates that all 10 of the cardiac glycosides clearly inhibit CMV at a postentry step.
FIG 4.

Antiviral time-of-addition assay. MRC5 cells were infected with AD169IE2-YFP (MOI, 3) at time zero and treated with a single dose of each compound (cymarin, 500 nM; K-strophanthin, 500 nM; mansonin, 5,000 nM; strophanthidin, 5,000 nM; glucostrebloside, 5,000 nM; strogoside, 5,000 nM; strophanthidol, 5,000 nM; ouabain, 50 nM; β-antiarin, 50 nM; or convallatoxin, 50 nM) at various times relative to infection (−1 hpi to 8 hpi). At 18 hpi, the number of YFP+ cells was determined using an Acumen eX3 cytometer. The relative maximal inhibition was determined based on the 0-hpi sample as 100% relative maximal inhibition. The error bars represent standard errors of the mean. Statistical significance was determined by a two-tailed t test of means; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
In silico interaction studies of glycosides reveal differences in anti-CMV activity.
Crystal structures show that cardiac glycosides interact at the same site on the Na+/K+-ATPase, and minute structural differences in the compounds lead to interactions with different side chains on the transporter (22). To identify the details of ligand-receptor interactions, the ligands were docked into the binding pocket of the published crystal structure of the high-affinity Na+/K+-ATPase–ouabain complex (23). The high-efficacy ligands (ouabain, convallatoxin, and β-antiarin) were predicted to dock in the same pose, forming hydrogen bonds with Glu116, Glu117, and Thr797 of the protein (Fig. 5A). Ouabain is unique among the three in that it may hydrogen bond to Gln111 by virtue of having a hydroxy substituent on C-1. This structural difference probably explains the different time-of-addition profile for ouabain compared to convallatoxin and β-antiarin. Indeed, strogoside and gluostrebloside have the same C-1 hydroxy group and the same time-of-addition profile as ouabain. The moderate-efficacy ligands (strophanthidin, mansonin, and cymarin) have a pose similar to that of the high-efficacy ligands yet are unable to hydrogen bond with Glu116 (Fig. 5B), likely due to the variation within the sugar ring. The low-efficacy compounds (strophanthidol and strogoside) and the no-efficacy compounds (allo-emicymarin and 17-epi-strophanthidol) were unable to bind in the same manner and were incapable of forming the same hydrogen bonds as the high- and moderate-efficacy ligands. K-Strophanthin and glucostrebloside did not result in reliable docking models, probably due to their higher complexity (number of rotatable bonds) and unclear stereochemistry.
FIG 5.

In silico molecular modeling of convallatoxin with the Na+/K+-ATPase. (A) High-affinity ligands. (B) Medium-affinity ligands. The blue outlines represent the cardiac glycoside binding pockets of the Na+/K+-ATPase, and the arrows indicate noncovalent bonds with the indicated residues of the Na+/K+-ATPase.
Since all predictions are done using the high-affinity conformation of the protein, it is possible that there is a lower-affinity conformation that supports the binding of the lower-efficacy ligands when there is a high concentration of drug present. Future studies involving mutation of the Na+/K+-ATPase could evaluate these predictions. At this point, given that convallatoxin was the most potent inhibitor with a high affinity for the Na+/K+-ATPase and the latest time-of-addition profile, we decided to focus our efforts on determining the mechanism of action of convallatoxin's inhibition of CMV infection.
Convallatoxin does not inhibit mRNA accumulation.
To determine if the antiviral mechanism of convallatoxin targets a step upstream of mRNA expression, MRC5 cells were treated with DMSO, convallatoxin (5 nM or 50 nM), or flavopiridol (5 nM or 5 μM) and infected with AD169WT for 24 h prior to subjecting the total RNA to qRT-PCR for IE2 and GAPDH mRNAs. The relative IE2 mRNA levels were normalized to the GAPDH levels (Fig. 6). While flavopiridol, the known transcription inhibitor (20), was able to reduce the amount of IE2 transcripts by 76% (P < 0.01) relative to DMSO treatment, it was clear that convallatoxin does not inhibit IE2 mRNA accumulation, and it appeared to actually increase the amount of IE2 mRNA in a dose-dependent manner (at 5 nM, a 2.35-fold increase [P < 0.01]; at 50 nM, a 5.01-fold increase [P < 0.001]) (Fig. 6). This increase may be due to the lack of IE2 protein levels, which acts as a negative regulator of IE2 gene expression (24, 25), and confirms that convallatoxin inhibits CMV infection after virus entry. Therefore, these results indicate that convallatoxin inhibits CMV infection at a step after IE gene mRNA transcription.
FIG 6.
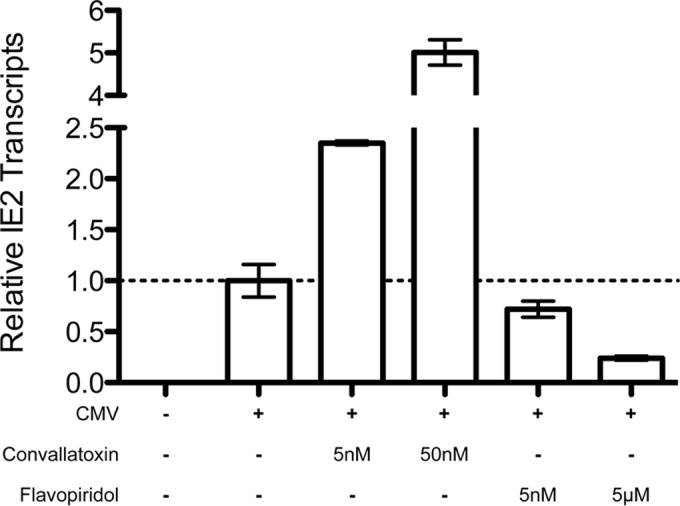
Convallatoxin does not inhibit IE2 gene transcription of virus-infected cells. MRC5 cells treated with DMSO, convallatoxin (5 nM or 50 nM), or flavopiridol (5 nM or 50 μM) were infected with AD169IE2-YFP and analyzed for mRNA transcripts of IE2 using qRT-PCR. Mock-infected cells were used as a control. Using the calculated CT values, the amount of IE2 transcripts relative to GAPDH transcripts was determined for each sample, and the average of the three replicates was plotted relative to DMSO. DMSO-treated cells were considered to have a relative IE2 transcript amount of 1, using standard errors (error bars).
Convallatoxin inhibits protein synthesis by altering methionine transport.
Convallatoxin does not impair IE2 mRNA levels (Fig. 6), yet it causes a dramatic decrease in IE2-YFP fluorescence and CMV US11 expression during infection (14). Collectively, the results suggest that convallatoxin treatment targets protein synthesis. To address this possibility at a global level, MRC5 cells were treated with 0 to 50 nM convallatoxin under conditions similar to those of the infection studies and were metabolically labeled with [35S]methionine ([35S]Met) to measure the synthesis of newly synthesized proteins (Fig. 7). Proteins with [35S]Met incorporated were analyzed by measuring TCA-precipitated counts, and the cell lysates were resolved by SDS-PAGE (Fig. 7A and B). Convallatoxin reduced the amount of [35S]Met incorporation into proteins as measured by a scintillation counter (Fig. 7A) and autoradiography (Fig. 7B). Low concentrations (3.125 nM or 6.25 nM) of convallatoxin had little to no impact on [35S]Met incorporation (17% reduction relative to DMSO [P < 0.01] and 10% increase relative to DMSO [P = 0.16], respectively), while convallatoxin at 12.5 nM and above had a significant reduction in [35S]Met incorporation (69% reduction relative to DMSO [P < 0.001]). These data were consistent with the level of [35S]Met-labeled polypeptides in infected cells analyzed under similar conditions (data not shown). The results demonstrate that convallatoxin treatment likely inhibits CMV infection by limiting the global expression of newly synthesized proteins and thus preventing the production of viral proteins during a virus infection. In addition, the depletion of methionine may trigger an amino acid response that would further inhibit the early steps of a virus infection.
FIG 7.

Convallatoxin reduces methionine uptake. (A and B) The cell lysates from [35S]Met-labeled MRC5 cells (1.5 h) treated with convallatoxin (0, 3.125 nM, 6.25 nM, 12.5 nM, 25 nM, or 50 nM) for 24 h were subjected to liquid scintillation (A) and resolved by SDS-PAGE (B) (lanes 1 to 5 correspond to lysates from cells treated with increasing concentrations of convallatoxin). (C) Luciferase activity was measured with the ProMega TnT in vitro transcription and translation kit pretreated with 5 μM cycloheximide or 5 μM convallatoxin (30 min) upon addition of luciferase-expressing vector. (D and E) The cell lysates from [35S]Met-labeled MRC5 cells (20 min), untreated or treated with convallatoxin (50 nM; 2 h), cycloheximide (50 μg/ml; 2 h), or both, were subjected to liquid scintillation directly (white bars) or as TCA-precipitated proteins (gray bars). The normalized values represent the averages of 3 replicates normalized to DMSO-treated cells as 100%. The error bars are standard errors.
In order to examine the possibility that convallatoxin directly targets the translational machinery, luciferase translation in vitro was analyzed based on luciferase activity using Promega's TnT T7 Coupled Reticulocyte Lysate system (Fig. 7C). The rabbit reticulocyte lysate was treated with DMSO, convallatoxin, or the translation inhibitor cycloheximide, followed by the addition of a plasmid that expressed luciferase under the control of the T7 promoter. The luciferase activity was measured after 2 h, and DMSO-treated samples were determined to be 100% activity. As expected, cycloheximide treatment caused a dramatic decrease in luciferase activity, likely due to the inhibition of mRNA translation (99.7% reduction [P < 0.001]) (Fig. 7C). On the other hand, convallatoxin did not reduce luciferase activity, demonstrating that the compound does not directly target the translational machinery (Fig. 7C). These results imply that convallatoxin's reduction of protein synthesis likely occurs via indirect means by possibly altering the pools of available methionine.
Methionine is an essential amino acid and is required to be actively transported across the membrane bilayer by neutral amino acid transport systems known as the sodium-dependent system A and the sodium-independent systems L and ASC (26). To examine whether convallatoxin inhibits methionine transport into cells, MRC5 cells were pretreated with DMSO or convallatoxin, followed by the addition of [35S]Met in the presence of DMSO, cycloheximide, or convallatoxin under conditions that effectively block virus infection (Fig. 7D and E). The amount of [35S]Met in the cell lysate corresponds to free [35S]Met and [35S]Met-labeled protein, while TCA-precipitated counts measure only the proteins with [35S]Met incorporated (Fig. 7D). Compared to DMSO, cycloheximide treatment alone reduced the free [35S]Met in cell lysate by only 12% (P < 0.01) while reducing the [35S]Met in proteins by 69% (P < 0.001) (Fig. 7E), indicating, as expected, a significant reduction of methionine incorporation into proteins. In contrast, convallatoxin treatment caused a 60% (P < 0.001) reduction of free [35S]Met in the cell lysate and 71% (P < 0.001) reduction of methionine in proteins (Fig. 7E). These results indicate that convallatoxin's reduction in mRNA translation limits protein synthesis by inhibiting the transport of methionine into the cell.
Convallatoxin effectively inhibits clinical strains of CMV.
We next examined whether convallatoxin was effective at limiting the dissemination of clinical cell-associated virus (Fig. 8). In a clinical setting, drug treatment is more likely to be a relatively constant low-dose treatment over the course of disease conditions. To better replicate this treatment scenario, HFFs infected with 4 clinical CMV strains (BI-4, BI-6, CH13, and CH19) that propagate exclusively by cell-to-cell spread were treated with convallatoxin (0 to 64 nM) for 7 days and then quantified by plaque assay and qPCR of viral DNA (Fig. 8). Among the 4 clinical strains tested, the EC50s ranged from 4.8 to 7.7 nM (Table 1 and Fig. 8A to D). Strikingly, the low EC50s demonstrated the effectiveness of convallatoxin at limiting CMV replication. The slightly lower EC50 values in the replication studies than in the infection studies may be due to the prolonged treatment time of convallatoxin. Remarkably, replication studies that added convallatoxin at 24 and 48 h following cell plating yielded ∼5 nM EC50 values for clones Bl-6 and CH3 (Table 1). These results support the model in which convallatoxin can effectively block viral propagation at different times during the virus life cycle. Of greater significance is the fact that two ganciclovir-resistant strains, CH13 and CH19, are sensitive to convallatoxin at EC50s in the nanomolar range.
FIG 8.

Convallatoxin is effective at limiting dissemination of clinical strains. (A to D) HFFs infected with the clinical strains BI-4 (A), BI-6 (B), CH13 (C), and CH19 (D) were treated with convallatoxin at various concentrations for up to 7 days. The number of plaques and viral DNA copies were analyzed and determined, setting DMSO-treated cells as 100%. (E) MRC5 cells treated with various doses of convallatoxin (7 days) were radiolabeled with [35S]Met. The cell lysates were directly subjected to liquid scintillation, and the normalized values represent averages of 3 replicates, with DMSO-treated cells set as 100%. The error bars are standard errors.
TABLE 1.
EC50s of ganciclovir and convallatoxin for CMV clinical strains
| Virus strain | EC50 (nM) |
||
|---|---|---|---|
| Ganciclovir plaque assay | Convallatoxin |
||
| Plaque assay | qPCR | ||
| BI-4 | <3,000 | 6.8 | 9.9 |
| BI-6 | <3,000 | 6.8 | 6.3 |
| BI-6 | NDc | 4.8a | ND |
| BI-6 | ND | 4.6b | ND |
| CH13 | 11,500 | 7.7 | 7.0 |
| CH19 | 15,700 | 4.8 | 5.0 |
| CH19 | ND | 4.9a | ND |
| CH19 | ND | 4.5b | ND |
Convallatoxin was added at 24 h postplating.
Convallatoxin was added at 48 h postplating.
ND, not determined.
Does convallatoxin alter methionine levels under the prolonged treatment conditions used in virus replication studies? To address this point, fibroblasts treated with 0 to 15 nM convallatoxin for 7 days were radiolabeled with [35S]Met for 1 h (Fig. 8D). The cell lysates were directly analyzed with a scintillation counter to determine the level of free [35S]Met and [35S]Met-incorporating proteins. While the EC50s for the plaque assays ranged from 4.8 nM to 7.7 nM (Table 1), 3.75 nM convallatoxin reduced methionine transport by ∼20% (Fig. 8E) but caused a 30 to 40% reduction in the number of plaques, further supporting the model in which a decrease in expression of viral protein may have a dramatic impact on virus replication. Further, higher-concentration convallatoxin treatment (7.5 nM and 15 nM) significantly decreased plaque numbers and viral loads (Fig. 8A to D), consistent with the effectiveness of convallatoxin at blocking virus dissemination. The data indicate that CMV replication was dramatically impacted by decreased methionine levels by limiting the expression of the immediate-early proteins, which are required for expression of early and late gene products. Low levels of IE1 and IE2 proteins may not be sufficient to induce the expression of the early and late proteins. Additionally, a decrease in methionine levels may trigger metabolic changes that would further inhibit virus replication.
DISCUSSION
All of the currently FDA-approved therapies for CMV target the DNA polymerase, and there is an urgent need to expand the repertoire of CMV therapeutics. The present study proposes a novel model of how the cardiac glycoside convallatoxin effectively inhibits CMV infection by limiting the pool of methionine for the expression of the IE genes. The efficacy of convallatoxin appears to be related to its chemical moieties. The convallatoxin structure is comprised of a steroid ring structure containing an aldehyde at C-19, a butenolide functionality in the β-configuration on C-17, and a 6-deoxy-α-l-mannopyranoside sugar functionality at C-3 (Fig. 1). The butenolide's orientation permits hydrogen bonding between hydroxyl residues on the steroid backbone of the compound with amino acids Thr797 and Glu117 of the Na+/K+-ATPase, and the 6-deoxy-α-l-mannopyranoside sugar forms hydrogen bonds with Glu116 (Fig. 5B). These interactions modulate the activity of the Na+/K+ pump to reduce the amount of sodium export from the cell. We propose that this in turn reduces the sodium gradient across the cell membrane and thus leads to a decrease in sodium-dependent methionine cotransport into the cell (Fig. 7E) (27). Remarkably, convallatoxin is effective at inhibiting CMV cell-free infections, as well as cell-associated infections, at nanomolar concentrations in both the short and long term compared to other cardiac glycosides (Fig. 2 and 8A to D). Additionally, in contrast to most of the other cardiac glycosides tested, convallatoxin maintains its efficacy when added several hours postinfection (Fig. 4). This is most likely due to its affinity with the Na+/K+ pump rather than representing a different mechanism of action or a greater potency at actually inhibiting the movement of ions, especially since convallatoxin was abandoned as a therapeutic cardiac glycoside in favor of ouabain.
Previous work has shown that ouabain inhibited CMV from upregulating hERG synthesis and decreased IE protein expression (16). It is now clear that this may be due to a reduction in the import of methionine (Fig. 7E) and that while the cell can tolerate a slight reduction in protein synthesis, the virus is unable to compensate for the reduction in its own protein synthesis (Fig. 8A to D). Because of this, IE2 mRNA production is actually increased in the presence of convallatoxin (Fig. 6) as the virus tries to increase expression of IE2. This mechanism is further supported by the fact that convallatoxin is effective when added up to 4 hpi (Fig. 4); infection is not rescued by the addition of wortmannin and PP2 (data not shown), known inhibitors of the proteins SRC and phosphatidylinositol 3-kinase (PI3K), which act downstream of Na+/K+-ATPase (28); and convallatoxin is unable to inhibit in vitro transcription and translation (Fig. 7C). Methionine, an essential amino acid that the cell cannot synthesize, is actively transported into cells in both sodium-dependent and sodium-independent manners (27). Since CMV replication is entirely dependent on IE gene expression, this reduction in methionine import (Fig. 7E) and protein synthesis (Fig. 7A and B) causes the drastic reduction in virus replication of primary clinical isolates (Table 1 and Fig. 8A to D). Ouabain has been shown to reduce protein synthesis in herpes simplex virus infection (29) and was suspected to inhibit the canine parasite Babesia gibsoni in an amino acid-dependent manner, as well (30). In contrast, HIV is inhibited by digoxin via disruption of RNA processing (31), most likely in a calcium-dependent manner (32), and ouabain inhibits the entry of several coronaviruses (28), as well as tick-borne encephalitis virus (33). There are viruses that are most likely resistant to glycosides, as well. Sindbis virus and coxsackie B virus downregulate Na+/K+-ATPase and remove the sodium gradient (34, 35) and are thus unlikely to be inhibited by a cardiac glycoside. Collectively, convallatoxin may be an effective broad-spectrum antiviral due to the high demand for newly synthesized viral and cellular proteins during virus infection.
This mechanism might also explain why convallatoxin has been a successful cancer treatment in vitro, preferentially killing cancer cells versus healthy cells, and appears to have a mechanism distinct from those of all other classical cancer drugs (36). In fact, the gene expression changes in the cell are similar in CMV infection and cancer (37). Convallatoxin can be safely consumed orally by rats at a daily dosage of roughly 0.76 mg/kg of body weight (38), which would translate to a little over 50 mg/day orally in a 70-kg person. While comparing plasma levels and in vitro dosing is not entirely accurate due to the effects of the volume of distribution (36), the toxic serum convallatoxin concentrations in mice are 18 to 180 μM (39, 40), well above 1,000 times what would be necessary to prevent CMV replication.
Using a panel of a dozen cardiac glycosides, we were able to discern some of the important structural elements for inhibition of CMV infection by cardiac glycosides. The presence of an oxidized methyl group at C-19, the chirality of the butenolide at C-17, and a sugar moiety with a glycosidic bond in the α-configuration were shown to be significant for the efficacy and pharmacodynamics of cardiac glycosides as antiviral agents, and the mechanism of action includes the inhibition of methionine transport. Convallatoxin, ouabain, and β-antiarin have almost identical structures (differing from convallatoxin only by the addition of a hydroxy group at the C-12 position in β-antiarin and the replacement of the aldehyde with a hydroxymethyl group at C-19 and additional hydroxy groups at C-1 and C-11 in ouabain) and are by far the most potent of the compounds at inhibiting a virus infection. It appears that the combination of both the C-19 oxygen species and the presence of a sugar in the α-configuration are key to a highly potent cardiac glycoside-based CMV inhibitor. Our work also begins to identify the structures associated with toxicity in cardiac glycosides. For example, strophanthidol was the most toxic of the compounds, but not the most effective (Fig. 2), indicating that the toxicity associated with cardiac glycosides is not directly linked to its efficacy as an anti-CMV agent. This means that more effective antiviral cardiac glycosides could possibly be designed without necessarily increasing their toxicity.
The time-of-addition data for all of the anti-CMV glycosides (Fig. 4) indicated that they were all working after CMV entry into the cell, since they maintained efficacy when added up to 1 hpi (K-strophanthin, strophanthidin, mansonin, and cymarin), 2 hpi (ouabain, glucostrebloside, strogoside, and strophanthidol), or 4 hpi (convallatoxin and β-antiarin), and we have previously shown that antivirals that maintain efficacy when added up to 1 hpi do not inhibit virus entry (T. Cohen, F. Vigant, B. Lee, and D. Tortorella, submitted for publication). Despite IE2-YFP's half-life of ∼2.5 h (41), the addition of the compound at the later time points did not inhibit the detection of IE2-YFP fluorescent species at 18 hpi. This may be due to the inhibition kinetics of the compounds and the utilization of the intracellular methionine prior to the effective inhibition of methionine transport. The differences in times of addition are most likely not differences in modes of action, since they are generally linked with antiviral potency. The 4 compounds that work when added until 1 hpi all have medium antiviral potency, 3/4 compounds that work when added until 2 hpi have low potency, and 2/3 compounds that work when added until 4 hpi have high potency. The one compound whose antiviral potency and timing appear to be incongruent is ouabain. Ouabain is unique among the 3 high-potency ligands in that it has a hydroxyl group at C-1 and is thus able to hydrogen bond with Gln111 of the Na+/K+-ATPase (Fig. 5A). It is possible that this interaction with the protein is responsible for this difference, and the low-potency ligands may have a similar interaction; however, we are unable to verify this due to the undefined stereocenters of glucostrebloside and because we have only the high-affinity protein binding crystal for the Na+/K+-ATPase. Future mutational studies and/or more crystallography data would allow greater confidence in the explanation of the relationship among antiviral potency, time-of-addition data, and molecular protein-ligand interactions.
Our work lays the foundation for the directed design of future cardiac glycoside analogues with improved anti-CMV activity, as well as for elucidating the role of the Na+/K+-ATPase in infection. The cardiac glycosides could be used in combined-therapy regimens with polymerase-targeting agents, such as ganciclovir (42). In vitro experiments have demonstrated that the combination of convallatoxin and ganciclovir has at least additive, if not synergistic, effects in blocking CMV proliferation (T. Cohen and D. Tortorella, unpublished data), as others have shown with ouabain and ganciclovir (42). Currently, combinatorial therapy for CMV is not effective, since all the currently FDA-approved therapies target the same step in the viral life cycle. In a clinical setting, this combination of a DNA polymerase inhibitor and a protein expression inhibitor could help to limit the emergence of resistant strains or to lower the dosage of ganciclovir required for treatment and thus minimize adverse events in patients.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants AI101820, AI112318, and AI113971. T.C. is a predoctoral trainee supported by USPHS Institutional Research Training Award T32-AI07647.
REFERENCES
- 1.Cohen JI, Corey GR. 1985. Cytomegalovirus infection in the normal host. Medicine 64:100–114. doi: 10.1097/00005792-198503000-00003. [DOI] [PubMed] [Google Scholar]
- 2.Mocarski ES, Shenk T, Pass RF. 2006. Cytomegaloviruses, p 2702–2772. In Knipe DM, Howley PM (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 3.Ho M. 1990. Epidemiology of cytomegalovirus infections. Rev Infect Dis 12(Suppl 7):S701–S710. doi: 10.1093/clinids/12.Supplement_7.S701. [DOI] [PubMed] [Google Scholar]
- 4.Dove A. 2006. A long shot on cytomegalovirus, vol 20, p 40–45. LabX Media Group, Midland, Ontario, Canada. [Google Scholar]
- 5.Cannon MJ, Davis KF. 2005. Washing our hands of the congenital cytomegalovirus disease epidemic. BMC Public Health 5:70. doi: 10.1186/1471-2458-5-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinberg A, Jabs DA, Chou S, Martin BK, Lurain NS, Forman MS, Crumpacker C. 2003. Mutations conferring foscarnet resistance in a cohort of patients with acquired immunodeficiency syndrome and cytomegalovirus retinitis. J Infect Dis 187:777–784. doi: 10.1086/368385. [DOI] [PubMed] [Google Scholar]
- 7.Jabs DA, Enger C, Dunn JP, Forman M. 1998. Cytomegalovirus retinitis and viral resistance: ganciclovir resistance. CMV Retinitis and Viral Resistance Study Group. J Infect Dis 177:770–773. [DOI] [PubMed] [Google Scholar]
- 8.Chou S, Marousek G, Guentzel S, Follansbee SE, Poscher ME, Lalezari JP, Miner RC, Drew WL. 1997. Evolution of mutations conferring multidrug resistance during prophylaxis and therapy for cytomegalovirus disease. J Infect Dis 176:786–789. doi: 10.1086/517302. [DOI] [PubMed] [Google Scholar]
- 9.Markham A, Faulds D. 1994. Ganciclovir. An update of its therapeutic use in cytomegalovirus infection. Drugs 48:455–484. [DOI] [PubMed] [Google Scholar]
- 10.Vittecoq D, Dumitrescu L, Beaufils H, Deray G. 1997. Fanconi syndrome associated with cidofovir therapy. Antimicrob Agents Chemother 41:1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagstaff AJ, Bryson HM. 1994. Foscarnet. A reappraisal of its antiviral activity, pharmacokinetic properties and therapeutic use in immunocompromised patients with viral infections. Drugs 48:199–226. [DOI] [PubMed] [Google Scholar]
- 12.Hahn KL. 2011. Old drugs are new again, vol 77. Intellisphere, LLC., Plainsboro, NJ. [Google Scholar]
- 13.Gardner TJ, Bolovan-Fritts C, Teng MW, Redmann V, Kraus TA, Sperling R, Moran T, Britt W, Weinberger LS, Tortorella D. 2013. Development of a high-throughput assay to measure the neutralization capability of anti-cytomegalovirus antibodies. Clin Vaccine Immunol 20:540–550. doi: 10.1128/CVI.00644-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardner TJ, Cohen T, Redmann V, Lau Z, Felsenfeld D, Tortorella D. 2015. Development of a high-content screen for the identification of inhibitors directed against the early steps of the cytomegalovirus infectious cycle. Antiviral Res 113:49–61. doi: 10.1016/j.antiviral.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Furstenwerth H. 2010. Ouabain—the insulin of the heart. Int J Clin Pract 64:1591–1594. doi: 10.1111/j.1742-1241.2010.02395.x. [DOI] [PubMed] [Google Scholar]
- 16.Kapoor A, Cai H, Forman M, He R, Shamay M, Arav-Boger R. 2012. Human cytomegalovirus inhibition by cardiac glycosides: evidence for involvement of the HERG gene. Antimicrob Agents Chemother 56:4891–4899. doi: 10.1128/AAC.00898-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai H, Wang HY, Venkatadri R, Fu DX, Forman M, Bajaj SO, Li H, O'Doherty GA, Arav-Boger R. 2014. Digitoxin analogues with improved anticytomegalovirus activity. ACS Med Chem Lett 5:395–399. doi: 10.1021/ml400529q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts KL, Manicassamy B, Lamb RA. 2015. Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol 89:1537–1549. doi: 10.1128/JVI.03306-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Formentin P, Alba M, Catalan U, Fernandez-Castillejo S, Pallares J, Sola R, Marsal LF. 2014. Effects of macro- versus nanoporous silicon substrates on human aortic endothelial cell behavior. Nanoscale Res Lett 9:421. doi: 10.1186/1556-276X-9-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao SH, Fujinaga K, Marion JE, Taube R, Sausville EA, Senderowicz AM, Peterlin BM, Price DH. 2000. Flavopiridol inhibits P-TEFb and blocks HIV-1 replication. J Biol Chem 275:28345–28348. doi: 10.1074/jbc.C000446200. [DOI] [PubMed] [Google Scholar]
- 21.Li G, Rak M, Nguyen CC, Umashankar M, Goodrum FD, Kamil JP. 2014. An epistatic relationship between the viral protein kinase UL97 and the UL133-UL138 latency locus during the human cytomegalovirus lytic cycle. J Virol 88:6047–6060. doi: 10.1128/JVI.00447-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laursen M, Gregersen JL, Yatime L, Nissen P, Fedosova NU. 2015. Structures and characterization of digoxin- and bufalin-bound Na+,K+-ATPase compared with the ouabain-bound complex. Proc Natl Acad Sci U S A 112:1755–1760. doi: 10.1073/pnas.1422997112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laursen M, Yatime L, Nissen P, Fedosova NU. 2013. Crystal structure of the high-affinity Na+K+-ATPase-ouabain complex with Mg2+ bound in the cation binding site. Proc Natl Acad Sci U S A 110:10958–10963. doi: 10.1073/pnas.1222308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulus C, Nevels M. 2009. The human cytomegalovirus major immediate-early proteins as antagonists of intrinsic and innate antiviral host responses. Viruses 1:760–779. doi: 10.3390/v1030760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pizzorno MC, Hayward GS. 1990. The IE2 gene products of human cytomegalovirus specifically down-regulate expression from the major immediate-early promoter through a target sequence located near the cap site. J Virol 64:6154–6165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krupsky M, Fine A, Berk JL, Goldstein RH. 1993. The effect of retinoic acid on amino acid uptake and protein synthesis by lung fibroblasts. J Biol Chem 268:23283–23288. [PubMed] [Google Scholar]
- 27.Sullivan JL, Debusk A. 1978. Transport of l-methionine in human diploid fibroblast strain WI38. Biochim Biophys Acta 508:389–400. doi: 10.1016/0005-2736(78)90341-3. [DOI] [PubMed] [Google Scholar]
- 28.Burkard C, Verheije MH, Haagmans BL, van Kuppeveld FJ, Rottier PJ, Bosch BJ, de Haan CA. 2015. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J Virol 89:4434–4448. doi: 10.1128/JVI.03274-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dodson AW, Taylor TJ, Knipe DM, Coen DM. 2007. Inhibitors of the sodium potassium ATPase that impair herpes simplex virus replication identified via a chemical screening approach. Virology 366:340–348. doi: 10.1016/j.virol.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamasaki M, Takada A, Yamato O, Maede Y. 2005. Inhibition of Na,K-ATPase activity reduces Babesia gibsoni infection of canine erythrocytes with inherited high K, low Na concentrations. J Parasitol 91:1287–1292. doi: 10.1645/GE-509R.1. [DOI] [PubMed] [Google Scholar]
- 31.Wong RW, Balachandran A, Ostrowski MA, Cochrane A. 2013. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog 9:e1003241. doi: 10.1371/journal.ppat.1003241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laird GM, Eisele EE, Rabi SA, Nikolaeva D, Siliciano RF. 2014. A novel cell-based high-throughput screen for inhibitors of HIV-1 gene expression and budding identifies the cardiac glycosides. J Antimicrob Chemother 69:988–994. doi: 10.1093/jac/dkt471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mal'dov DG, Gmyl LV, Karganova GG. 1997. Change in Na+,K+-ATPase activity during reproduction of the tick-borne encephalitis virus in SPEV cell culture. Vopr Virusol 42:23–26. (In Russian.) [PubMed] [Google Scholar]
- 34.Ulug ET, Garry RF, Bose HR Jr. 1996. Inhibition of Na+K+ATPase activity in membranes of Sindbis virus-infected chick cells. Virology 216:299–308. doi: 10.1006/viro.1996.0065. [DOI] [PubMed] [Google Scholar]
- 35.Modalsli K, Bukholm G, Mikalsen SO, Degre M. 1992. Coxsackie B1 virus-induced changes in cell membrane-associated functions are not responsible for altered sensitivity to bacterial invasiveness. Arch Virol 124:321–332. doi: 10.1007/BF01309812. [DOI] [PubMed] [Google Scholar]
- 36.Felth J, Rickardson L, Rosen J, Wickstrom M, Fryknas M, Lindskog M, Bohlin L, Gullbo J. 2009. Cytotoxic effects of cardiac glycosides in colon cancer cells, alone and in combination with standard chemotherapeutic drugs. J Nat Prod 72:1969–1974. doi: 10.1021/np900210m. [DOI] [PubMed] [Google Scholar]
- 37.McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. 2014. Global reprogramming of the cellular translational landscape facilitates cytomegalovirus replication. Cell Rep 6:9–17. doi: 10.1016/j.celrep.2013.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura M, Utsunomiya H, Nakamura M, Landon EJ. 2000. Effect of dietary cardiac glycosides on blood pressure regulation in rats. Can J Physiol Pharmacol 78:548–556. doi: 10.1139/y00-023. [DOI] [PubMed] [Google Scholar]
- 39.Forster W, Sziegoleit W, Guhlke I. 1965. Comparative study of some extracardiac effects of cardiac glycosides. Arch Int Pharmacodyn Ther 155:165–182. (In German.) [PubMed] [Google Scholar]
- 40.Fink SL, Robey TE, Tarabar AF, Hodsdon ME. 2014. Rapid detection of convallatoxin using five digoxin immunoassays. Clin Toxicol 52:659–663. doi: 10.3109/15563650.2014.932366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Teng MW, Bolovan-Fritts C, Dar RD, Womack A, Simpson ML, Shenk T, Weinberger LS. 2012. An endogenous accelerator for viral gene expression confers a fitness advantage. Cell 151:1569–1580. doi: 10.1016/j.cell.2012.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cai H, Kapoor A, He R, Venkatadri R, Forman M, Posner GH, Arav-Boger R. 2014. In vitro combination of anti-cytomegalovirus compounds acting through different targets: role of the slope parameter and insights into mechanisms of action. Antimicrob Agents Chemother 58:986–994. doi: 10.1128/AAC.01972-13. [DOI] [PMC free article] [PubMed] [Google Scholar]