

ABSTRACT
Tat protein, the HIV transactivator, regulates transcription of the HIV genome by the host transcription machinery. Efficient inhibitors of HIV transcription that target Tat or the cellular cofactor NF-κB are well known. However, inhibition of HIV Tat-dependent transcription by targeting the general transcription and DNA repair factor II human (TFIIH) has not been reported. Here, we show that spironolactone (SP), an aldosterone antagonist approved for clinical use, inhibits HIV-1 and HIV-2 infection of permissive T cells by blocking viral Tat-dependent transcription from the long terminal repeat (LTR). We found that treatment of Jurkat and primary CD4+ T cells with SP induces degradation of the XPB cellular helicase, a component of the TFIIH complex, without affecting cellular mRNA levels, T cell viability, or T cell proliferation. We further demonstrate that the effect of SP on HIV infection is independent of its aldosterone antagonist function, since the structural analogue, eplerenone, does not induce XPB degradation and does not inhibit HIV infection. Rescue experiments showed that the SP-induced block of HIV infection relies, at least partially, on XPB degradation. In addition, we demonstrate that SP specifically inhibits Tat-dependent transcription, since basal transcription from the LTR is not affected. Our results demonstrate that SP is a specific inhibitor of HIV Tat-dependent transcription in T cells, which additionally suggests that XPB is a cofactor required for HIV infection. Targeting a cellular cofactor of HIV transcription constitutes an alternative strategy to inhibit HIV infection, together with the existing antiretroviral therapy.
IMPORTANCE Transcription from the HIV promoter is regulated by the combined activities of the host transcription machinery and the viral transactivator Tat protein. Here, we report that the drug spironolactone—an antagonist of aldosterone—blocks viral Tat-dependent transcription, thereby inhibiting both HIV-1 and HIV-2 infection of permissive T cells. This inhibition relies on the degradation of the cellular helicase XPB, a component of the TFIIH transcription factor complex. Consequently, XPB appears to be a novel HIV cofactor. Our discovery of the HIV-inhibitory activity of spironolactone opens the way for the development of novel anti-HIV strategies targeting a cellular cofactor without the limitations of antiretroviral therapy of drug resistance and high cost.
INTRODUCTION
Human immunodeficiency virus types 1 and 2 (HIV-1 and HIV-2) are members of the family Retroviridae and are the causative agents of AIDS. The viral RNA of retroviruses is reverse transcribed into double-stranded DNA and integrated into the cellular chromosome, generating a provirus. Transcription from the provirus promoter in the long terminal repeat (LTR) depends on the combined activities of the host transcription machinery and the HIV transcription activator Tat.
The general transcription and DNA repair factor II human (TFIIH) plays a key role in unwinding DNA for transcription, as well as for nucleotide excision repair (1). TFIIH is also involved in cell cycle regulation and chromosome segregation, as recently reviewed by Compe and Egly (2). During transcription of protein-coding genes by RNA polymerase (Pol) II, TFIIH is involved in DNA opening of the promoter and is required for the transition from initiation to early elongation of Pol II (3). TFIIH is a 10-subunit complex (4); its core is formed by the subunits xeroderma pigmentosum group B (XPB), p62, p52, p44, p34, and trichothiodystrophy A (TTDA/p8). Xeroderma pigmentosum group D (XPD) links the core with the cyclin-dependent kinase (CDK)-activating kinase (CAK) complex (composed of CDK7, ménage à trois 1 [MAT1], and cyclin H).
XPB is an ATP-dependent DNA helicase with 3′-5′ polarity (5). During transcription initiation, the ATPase activity of XPB is required for promoter opening and escape (6). TFIIH rotates and threads the double-stranded DNA (dsDNA) into the active-site cleft of Pol II, where upstream DNA at the promoter region is melted by the molecular-wrench action of XPB (7). XPB-mediated promoter opening is followed by serine 5 phosphorylation of the heptapeptide repeat of the carboxy-terminal domain (CTD) of Pol II by the CDK7 subunit of TFIIH (8). Pol II is paused within 20 to 40 nucleotides downstream from the transcription start site. Pol II release for productive transcription elongation starts after phosphorylation at serine 2 of the CTD by the human positive transcription elongation factor complex, called P-TEFb. This complex is composed of CDK9 and cyclin T1. It has been proposed that XPB ensures that the transition from initiation to elongation proceeds in an efficient, programmed manner by inhibiting CDK9 phosphorylation (9).
The HIV-1 transcription activator Tat is a small protein (101 amino acids) required for efficient transcription of viral genes (10, 11). Tat binds to the transactivation response element (TAR) present in the nascent viral RNA (12). Tat also transactivates transcription in a TAR-independent manner by stimulating nuclear translocation of NF-κB (13). Whether Tat stimulates initiation or elongation of transcription has long been debated, but its major role in regulation of elongation is well established. Tat interacts with several basal transcription factors at the promoter, and it is involved in transcriptional complex assembly and transcription initiation complex stability (14). Tat may play a role in the transition from initiation to elongation by binding directly to the CAK complex of TFIIH (15, 16). The interaction of Tat with the P-TEFb complex (17) and the role of Tat during transcription elongation are well documented (18–20). Tat binding to TAR enhances P-TEFb recruitment and release of paused Pol II at the HIV-1 promoter by activating Pol II CTD phosphorylation.
XPB has been reported both to suppress and to be required for HIV-1 infection. In XPB-deficient fibroblast lines, it was postulated that XPB is a defense against retroviral infection (21, 22). XPB was identified as a factor required for HIV-1 in two genome scale screens in HIV-1-susceptible indicator cells for HIV-1 IIIB and for HXB2 (23, 24). These contradictory data have led us to revisit the role of XPB in HIV infection in relevant target cells for the virus, namely, in T cells. To achieve this aim, we focused on the effect of XPB depletion on HIV infection by taking advantage of the property of spironolactone (SP) to induce rapid and reversible degradation of XPB by the 26S proteasome (25). SP blocks the main mineralocorticoid hormone aldosterone by binding to the mineralocorticoid receptor and acting as a receptor antagonist.
Here, we show that SP inhibits HIV-1 and HIV-2 infection of permissive T cells by blocking viral Tat-dependent transcription. This inhibition relies on the degradation of XPB. Thus, we provide evidence for revisiting the role of XPB as an HIV cofactor required for HIV infection.
MATERIALS AND METHODS
Cell lines.
Lymphocytic Jurkat T cells (CD4+/CXCR4+; clone 20) were a kind gift of O. Schwartz (Institut Pasteur, Paris, France). Jurkat LTR-GFP (JLTRG-R5) cells expressing the HIV LTR promoter and the green fluorescent protein (GFP) reporter gene were obtained through the NIH AIDS Reagent Program, Division of AIDS (from O. Kutsch; catalog number 11586). Jurkat T cells were maintained in complete RPMI medium (RPMI 1640 GlutaMax-I; Gibco) with 10% heat-inactivated fetal calf serum (FCS) (Eurobio) and 1% antibiotics (penicillin, streptomycin, and amphotericin B; Gibco). HeLa TZM-bl reporter cells were obtained through the NIH AIDS Reagent Program, Division of AIDS (from J. C. Kappes, X. Wu, and Tranzyme Inc.; catalog number 8129). 293T cells (American Type Culture Collection [ATCC]) and HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Gibco) supplemented with glutamine (Gibco), 10% heat-inactivated FCS (Eurobio), and 1% antibiotics (as described above). SP (Sigma) and eplerenone (EPL) (Sigma) were diluted in dimethyl sulfoxide (DMSO) (Sigma) to 5 mM and then added to the culture medium at 10 μM.
Purification and activation of primary CD4+ T lymphocytes.
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood from anonymous donors (obtained from the French Blood Agency [EFS], Rungis, France, in accordance with the ethical guidelines of the Institut Cochin, Paris, France) by Ficoll (Sigma) density gradient separation. Primary CD4+ T cells were isolated by positive selection with CD4 magnetic Microbeads (Miltenyi Biotec). The primary CD4+ T cells were activated with Dynabead Human T-activator CD3/CD28 T cell expander (Gibco) and cultured in complete RPMI medium containing interleukin-2 (IL-2) (30 IU/ml; PeproTech) for 3 days prior to infection.
Cell viability and proliferation.
For viability, MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] (Sigma) assays were performed with 30,000 Jurkat or primary CD4+ T cells plated in 96-well flat-bottom plates and cultured in complete RPMI medium containing the tested drugs (final volume, 250 μl per well). At different time points, 50 μl of 5-mg/ml MTT solution in phosphate-buffered saline (PBS) (Gibco) was added to each well for 4 h. After removal of the medium, 100 μl of DMSO was added to each well to dissolve the formazan crystals, and the absorbance at 540 nm was determined using an Infinite F200 Pro plate reader (Tecan).
To assess the proliferation of Jurkat T cells, the cells were labeled with a Cell Trace carboxyfluorescein succinimidyl ester (CFSE) cell proliferation kit (Invitrogen), incubated for 3 days with drugs in RPMI complete medium, and analyzed every day by flow cytometry (Accuri C6; BD Biosciences).
Viruses.
The NL4.3-Luc ΔenvΔnef plasmid, here designated HIV-1 Luc, was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS (from N. Landau [26]; catalog number 3418). The plasmid HIV-2-ROD9-ΔenvΔnef-GFP (HIV-2 ROD9-GFP) was kindly provided by N. Manel. The construct (pRRLsin) coding for the HIV-1-derived vector containing the human cytomegalovirus (CMV) immediate-early gene promoter driving the transcription of GFP (HIV-1-CMV-GFP) was obtained from A. Cimarelli. The replicative proviral clone HIV-1-NL4.3 was obtained through the NIH AIDS Reagent Program, Division of AIDS (from M. Martin; catalog number 114). The HIV-2 GL-AN proviral clone pGL-AN was kindly provided by A. Adachi.
Virus production.
To produce single-cycle pseudoviruses, plasmids coding for HIV-1 Luc and for the vesicular stomatitis virus glycoprotein (pVSV-G) were transfected by the calcium-phosphate method in 293T cells. Similarly, to produce HIV-2 ROD9, the cells were transfected with pVSV-G and the plasmid HIV-2-VSV-G-ROD9-ΔenvΔnef-GFP. To produce HIV-1-CMV-GFP, the cells were transfected with plasmids pVSV-G and psPAX2 (kindly provided by Nicolas Manel) and with HIV-1-CMV-GFP. To produce the replicative viruses HIV-1-NL4.3 and HIV-2 GL-AN, proviral plasmids were transfected in the 293T cells. The culture supernatants were collected 48 h and 72 h after transfection and centrifuged at 300 × g. The viral particles were then concentrated in 4% sucrose gradients (Euromedex) by ultracentrifugation at 100,000 × g for 2 h. The viruses were titrated using TZM-bl reporter cells.
Transduction and infection.
Cells were transduced with the single-cycle virus HIV-1-Luc (using 0.6 ng of p24 for 500 cells), HIV-2 ROD9 (using 0.05 ng of p24 for 500 cells), and HIV-1-CMV-GFP (using a multiplicity of infection [MOI] of 0.1) or infected with viruses (HIV-1 NL4.3 or HIV-2 GL-AN at an MOI of 0.1) for 3 h at 37°C in complete RPMI medium containing 5 mM HEPES and 2 μg/ml dextran (Sigma). The cells were then cultured in complete RPMI medium with or without the drugs (SP or EPL at the indicated concentrations) or the carrier (DMSO). The percentages of infected cells were determined by Gag labeling in cells infected with NL4.3 and GL-AN viruses by labeling with anti-HIV Gag antibody (Kc57-fluorescein isothiocyanate [FITC]; Beckman Coulter), followed by analysis by flow cytometry (Accuri C6; BD Biosciences). The level of infection was measured by the luciferase activity in cells transduced with HIV-1-Luc by the luciferase assay system (Promega), following the manufacturer's protocol, and luminescence was analyzed with the Infinite F200 Pro plate reader (Tecan). Normalization of luciferase units was performed using the BCA (bicinchoninic acid) kit assay (Thermo Fisher) on sample lysates. The HIV-1 yield was determined by quantifying p24 levels using an enzyme-linked immunosorbent assay (ELISA) (Innotest; Fujirebio).
Analysis of mRNA levels by reverse transcription and real-time PCR.
Total RNA was extracted with the NucleoSpin RNAII kit (Macherey-Nagel, France). cDNA was synthesized with the Maxima first-strand cDNA synthesis kit (Thermo Scientific, France) and quantified by real-time PCR with a LightCycler 2.0 (Roche, France). Oligonucleotide sequences used are listed in Table 1. The Sybr green technology was employed to amplify all the genes under the following conditions: a first step of denaturation at 95°C for 8 min, followed by 40 cycles of denaturation (95°C for 10 s), annealing (60°C for 10 s), and extension (72°C for 8 s). The second-derivative-maximum method provided by the Light Cycler 480 SW 1.5 quantification software (Roche Diagnostics) was used.
TABLE 1.
Oligonucleotide sequences
| Gene | Primera | Sequence (5′–3′) |
|---|---|---|
| GFP | F (453) | CTATATCATGGCCGACAAGC |
| R (561) | GGTGTTCTGCTGGTAGTGGT | |
| Tat | F (32) | GGAAGCATCCAGGAAGTCAG |
| R (175) | GAGCTCTTCGTCGCTGTCTC | |
| XPB | F (692) | CCACTTCCCGAGTGACAGAT |
| R (1,036) | GTCACACCAACCAGGGACTT | |
| XPD | F (100) | TACCCCGAGCAGTTCTCCTA |
| R (263) | CTGGCACAGTTCTTGAGCAG | |
| Rad52 | F (352) | GAGTACCAGGCCATCCAGAA |
| R (485) | CCCAGCCATTGTAACCAAAC | |
| EEF1G | F | AGATGGCCCAGTTTGATGCTAA |
| R | GCTTCTCTTCCCGTGAACCCT | |
| HPRT | F | TGACACTGGCAAAACAATGCA |
| R | GGTCCTTTTCACCAGCAAGCT |
F, forward; R, reverse. Numbers indicate the primer’s first nucleotide position in the corresponding gene.
Western blot analysis and antibodies.
Transduced, transfected, or infected cells were lysed in adequate volumes of radioimmunoprecipitation assay (RIPA) buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.5), 2 mM EDTA, 0.5% NP-40, 10% glycerol, and endonuclease (benzonase; Sigma). Protein extracts were separated by SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF) membranes, and revealed by chemiluminescence (CDPStarW; Applied Biosystems). Signals were acquired with a LAS 4000 apparatus (Fujifilm) for further analysis using Multigauge software (Fujifilm). The following antibodies were used: anti-XPB (TFIIH p89 S-19; Santa Cruz Biotechnology [SCB]), anti-XPD (TFIIH p80 ab54676; Abcam), anti-CDK7 (C-4; SCB), anti-CDK9 (C-20; SCB), anti-Pol II Ser5 (Activ Motif), anti-GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (6C5; SCB), and anti-Tat (02-001; SCB). All secondary antibodies were coupled with alkaline phosphatase (Life Technologies).
Transfection for promoter activity.
Jurkat T cells or JLTRG-R5 cells were transfected with DMRIE-C (Life Technologies) in Opti-MEM medium (Gibco). Jurkat cells (300,000) were transfected with 200 ng of plasmid expressing different promoters controlling the luciferase reporter gene. The pGL3 plasmid encodes CMV-Luc (a kind gift of Daniel Vaiman, Institut Cochin). The pLTR-Luc plasmid (a kind gift of Stephane Emiliani, Institut Cochin) encodes the complete HIV-1 LTR (nucleotides 1 to 791) driving the luciferase reporter gene. The pHTLV-Luc plasmid encodes the LTR promoter of human T-lymphotropic virus (HTLV) driving the luciferase reporter gene (kindly provided by C. Pique, Institut Cochin). These plasmids are transfected with different concentrations of the HIV Tat-encoding plasmid pEV280 (obtained through the NIH AIDS Reagent Program, Division of AIDS, from E. Verdin; catalog number 10453). The plasmid pTK-Renilla (kindly provided by C. Pique, Institut Cochin) was transfected in 1/10 concentration for normalization. After 6 h of transfection, complete RPMI medium was added, and the cells were incubated overnight at 37°C. The next day, complete RPMI medium was supplemented with SP or DMSO for an additional 24 h. The cells were lysed in 100 μl passive lysis buffer (Promega). The Dual Luciferase assay system (Promega) was used to determine the luminescence of firefly and Renilla luciferases. The data were analyzed with the Infinite F200 Pro plate reader (Tecan).
Similarly, 300,000 JLTRG-R5 cells were transfected with 100 ng of plasmid pEV280 encoding the HIV-1 Tat protein. The cells were cultured as described above, and after SP or DMSO incubation, were fixed in 4% paraformaldehyde (PFA) (Electron Microscopy Sciences) and analyzed by flow cytometry (Accuri C6; BD Biosciences).
RESULTS
Spironolactone inhibits HIV-1 and HIV-2 transduction in Jurkat T cells.
Rapid and reversible proteasome-dependent degradation of XPB induced by SP was reported in HeLa cells (25); therefore, we investigated this property of SP in T cells. XPB was strongly degraded in Jurkat cells after 10 μM SP treatment (Fig. 1A, lane 2). To confirm the observation of Yoder et al. (21, 22) that in lymphocytes XPB is a defense against retroviral infection, we performed SP treatment of Jurkat T cells transduced with the same single-cycle virus, HIV-1-CMV-GFP, used in that study. This virus leads to CMV-driven GFP expression following integration into the host genome. In agreement with their results, SP treatment led to a 30% increase of HIV-1-CMV-GFP transduction in Jurkat T cells (Fig. 1B). Given that XPB belongs to the TFIIH complex, which is involved in transcription, we investigated whether the degradation of XPB induced by SP could affect gene expression under the control of the HIV-1 LTR promoter. For this purpose, a single-cycle HIV-1 encoding luciferase directly under the control of the HIV-1 LTR promoter (HIV-1-Luc) was used. Strikingly, in Jurkat T cells, a 3-fold reduction of HIV-1 transduction was observed in the presence of SP (Fig. 1C). XPB protein levels were similar in nontransduced and HIV-1-transduced Jurkat T cells (Fig. 1D, lanes 1 and 2), and HIV-1 transduction did not affect SP-induced degradation of XPB (Fig. 1D, lane 3). Unlike SP, EPL, a compound derived from SP and also an antagonist of aldosterone, did not induce XPB degradation (Fig. 1A, lane 3) and had no effect on HIV-1 transduction (Fig. 1E). These observations correlated with complete XPB degradation during the 2 days of transduction in the presence of SP, but not in the presence EPL (Fig. 1A). Increasing doses of SP resulted in dose-dependent inhibition of HIV-1 transduction; the 50% inhibitory concentrations (IC50s) of SP for two different amounts of inoculum were 1.4 μM and 5.0 μM (Fig. 1F). Therefore, these results indicate that SP inhibition of HIV-1 transduction, as well as the SP-induced degradation of XPB, is independent of the mineralocorticoid receptor antagonist activity of SP and that this inhibition is associated with degradation of XPB.
FIG 1.

Spironolactone, but not eplerenone, inhibits HIV-1 transduction. (A) XPB protein levels in Jurkat T cells treated with DMSO, SP, or EPL. (B) Jurkat T cell transduction with the single-cycle HIV-1-CMV-GFP. Cells were cultured with or without SP. The number of GFP+ cells was analyzed by flow cytometry. P = 0.005. (C) Jurkat T cell transduction with the pseudotyped single-cycle HIV-1-Luc (HIV-1-VSV-G-NL4.3-luciferase-Δenv-Δnef). Cells were cultured or not with SP. P = 0.0004. (D) XPB protein level in Jurkat T cells after HIV-1-Luc transduction and cell culture with DMSO, SP, or EPL. X/G represents the ratio of XPB to GAPDH protein levels. (E) Jurkat T cell transduction with the single-cycle HIV-1 Luc. Cells were treated or not with EPL. P = 0.1977. ns, no statistically significant difference. (F) Jurkat T cell transduction with two different amounts of the single-cycle HIV-1-Luc. Cells were cultured with increasing concentrations of SP, and the IC50 was determined. (G) Jurkat cell transduction with the single-cycle HIV-2 ROD9 (HIV-2-Rod9-VSV-G-GFP-Δenv). Cells were treated or not with SP. The results are the means of three independent experiments performed in duplicate. P = 0.0004. (H) Jurkat cell transduction with the single-cycle HIV-2 ROD9, treated or not with EPL. The results are the means of three independent experiments performed in duplicate. P = 0.1977. The values were normalized, taking as 100% the value obtained for one replicate of control cells treated with DMSO. The error bars represent standard errors of the mean (SEM). Unless otherwise indicated, SP or EPL was added to the culture medium at a concentration of 10 μM for 48 h. **, P < 0.01; ***, P < 0.001.
To investigate if SP has an effect on HIV-2 transduction, Jurkat T cells were transduced with single-cycle HIV-2 ROD9-GFP, followed by treatment with SP. A 30% reduction of HIV-2 transduction was observed under SP treatment (Fig. 1G). As for HIV-1, no significant difference in HIV-2 transduction was observed between EPL-treated Jurkat and control cells (Fig. 1H). Together, these results show that SP inhibits both HIV-1 and HIV-2 transduction in Jurkat T cells simultaneously with XPB degradation.
Spironolactone inhibits HIV-1 transduction in primary CD4+ T cells.
Importantly, a 3-fold reduction of HIV-1 transduction was observed in SP-treated primary CD4+ T cells transduced with HIV-1-Luc (Fig. 2A). As for Jurkat T cells, XPB protein levels were similar in nontransduced and in HIV-1-trasduced primary CD4+ T cells (Fig. 2B, lanes 1 and 2). Therefore, in primary CD4+ T cells, as in Jurkat T cells, HIV-1 transduction is inhibited by SP, and this inhibition is associated with degradation of XPB.
FIG 2.
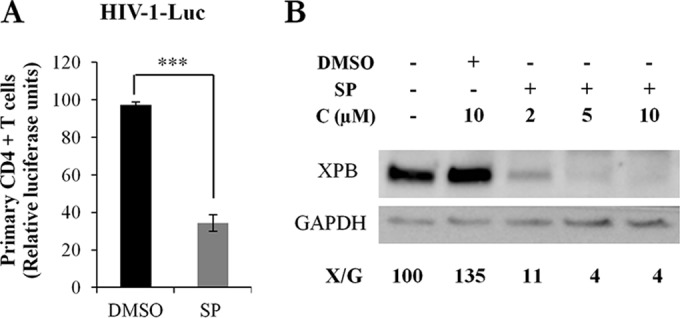
Spironolactone inhibits HIV-1 transduction in primary CD4+ T cells. (A) Primary CD4+ T cell transduction with the single-cycle HIV-1 Luc. Cells were treated or not with 10 μM SP for 48 h. The results are the means of experiments with 4 different donors performed in triplicate. P < 0.0001. (B) XPB levels in primary CD4+ T cells transduced and cultured in the presence of DMSO or increasing concentrations of SP. X/G represents the ratio of XPB to GAPDH protein levels. ***, P < 0.001.
Spironolactone does not affect general transcription components and cell survival.
While XPB was strongly degraded in Jurkat cells after 10 μM SP treatment, other components of the TFIIH complex, such as XPD and CDK7, or proteins involved in RNA transcription, such as serine 5-phosphorylated Pol II and CDK9, were not affected (Fig. 3A). EPL did not induce degradation of XPB or any of the proteins analyzed (Fig. 3A). XPB and XPD mRNA levels measured by RT-quantitative PCR (qPCR) were unchanged in the presence of SP or EPL (Fig. 3B). Similarly, no difference in mRNA levels was observed for other cellular genes, such as the GAPDH housekeeping gene or the gene coding for the DNA repair protein Rad52 (Fig. 3B). In addition, SP did not affect cell viability measured using the MTT assay in Jurkat cells (Fig. 3C) or primary CD4+ T cells (Fig. 3D) or cell proliferation measured by CFSE labeling (data not shown).
FIG 3.

Spironolactone induces degradation of XPB, but not of other general transcription components, without affecting cell viability or proliferation. (A) TFIIH protein levels in Jurkat T cells cultured in the presence of DMSO, SP, or EPL. (Left) Representative Western blot. (Right) Quantification of the results of four independent experiments. (B) XPB, XPD, GAPDH, and Rad52 mRNA levels in Jurkat T cells treated with DMSO, SP, or EPL. rq, relative quantity; NF, normalization factor. (C) Viability of Jurkat T cells using the MTT test. The cells were treated with DMSO or SP for up to 3 days. (D) Viability of primary CD4+ T cells using the MTT test. The cells were treated with DMSO or SP. Unless otherwise indicated, SP or EPL was added to the culture medium at a concentration of 10 μM for 48 h. The error bars represent SEM.
Altogether, the results show that SP treatment of CD4+ Jurkat and primary T cells results in degradation of XPB without affecting cellular mRNA levels, T cell viability, or T cell proliferation.
Inhibition of HIV-1 transduction by SP relies on XPB degradation.
To directly assess the role of XPB, HeLa cells, in which XPB DNA can be easily transfected for rescue experiments, were used. Higher doses of SP were necessary to reach optimal XPB degradation (Fig. 4A), and no change in cell viability measured using the MTT test was seen at these concentrations (Fig. 4B). In addition, higher doses are necessary to inhibit HIV-1-Luc transduction in HeLa cells (Fig. 4C) than in Jurkat T cells (Fig. 2B). Nonetheless, exogenous XPB expression partially restored HIV-1 transduction in SP-treated cells (Fig. 4D and E). Therefore, the inhibition of HIV-1 infection induced by SP relies at least partially on XPB degradation. Together, these results support the conclusion that XPB is a cofactor required for HIV infection.
FIG 4.

Spironolactone induces XPB degradation and inhibits HIV-1 transduction in HeLa cells. (A) XPB protein levels in HeLa cells treated with increasing concentrations of DMSO or SP. X/G represents the ratio of XPB to GAPDH protein levels. A vertical white space was added because a lane of noninfected sample treated with SP was deleted from the blot. (B) Viability of HeLa cells treated with increasing concentrations of DMSO or SP for up to 3 days; the MTT test was used. Abs, absorbance. (C) HeLa cell transduction with the single-cycle HIV-1-Luc. Cells were treated or not with increasing doses of SP. (D) SP inhibition of HIV transduction is partially restored by addition of exogenous XPB. HeLa cells were transfected with pcDNA3-XPB for 24 h, transduced with HIV-1-Luc, and cultured with DMSO or SP for an additional 24 h, and the luciferase activity was determined. The values were normalized, taking as 100% the value obtained for one replicate of control cells treated with DMSO. The results are the means of three independent experiments performed in triplicate. The error bars represent SEM. (E) XPB protein levels of the transduction experiment shown in panel D. Unless otherwise indicated, SP was added to the culture medium for 48 h.
Spironolactone inhibits infection of T cells with replicative HIV-1 and HIV-2.
We next analyzed whether SP could inhibit replicative HIV-1 and HIV-2 during multiple rounds of infection. Jurkat T cells infected with the replicative HIV-1 NL4.3 and treated with SP showed, at 3 days postinfection (dpi), a 4-fold reduction in Gag-positive cells (Fig. 5A) and a 7-fold reduction of the mean of Gag-FITC fluorescence (data not shown). Unlike SP, no inhibition, but rather a small increase, of HIV-1 replication was observed with EPL (Fig. 5B). SP led to a dose-dependent reduction of HIV-1 infection, with an IC50 of 4.2 μM for an MOI of 0.05 (Fig. 5C). Similarly to HIV-1, SP induced 3-fold inhibition of HIV-2 infection during multiple rounds of virus infection in Jurkat T cells infected with the replicative HIV-2 GL-AN (Fig. 5D). Together, the results show that SP-mediated inhibition of HIV-1 and HIV-2 infection is amplified after several rounds of viral multiplication in T cells.
FIG 5.

Spironolactone inhibits HIV-1 and HIV-2 infection of Jurkat T cells. (A) HIV-1 NL4.3 infection of Jurkat T cells treated or not with SP. The number of infected cells was measured by Gag labeling followed by flow cytometry analysis. P = 0.005. (B) HIV-1 NL4.3 infection of Jurkat T cells treated or not with EPL. P = 0.0027. (C) Jurkat T cell infection with HIV-1 NL4.3 at an MOI of 0.05. SP was added to the culture medium at the concentrations indicated. The number of infected cells was measured by Gag labeling followed by flow cytometry analysis, and the IC50 was determined. (D) HIV-2 GL-AN infection of Jurkat T cells treated or not with SP. P = 0.005. (E) Kinetics of HIV-1 infection in the presence or absence of SP for 12 days. Jurkat T cells were infected with HIV-1 NL4.3 and cultured with DMSO or SP; the medium was changed every 3 days. For some cells at 3 dpi, SP was replaced by RPMI medium without further addition of the drug. For some cells at 6 dpi, DMSO was replaced by SP. The results are the means of four independent experiments performed in duplicate (A and D) and of two independent experiments performed in triplicate (B, C, and E). (A, B, and C) The values were normalized, taking as 100% the value obtained for one replicate of control cells treated with DMSO. Unless otherwise indicated, SP or EPL was added to the culture medium at a concentration of 10 μM for 48 h. The error bars represent SEM. **, P < 0.01.
To determine if SP acts after HIV-1 infection is established, we examined the kinetics of HIV-1 infection in the presence or absence of SP for 12 days. Jurkat T cells were infected with HIV-1 NL4.3 and cultured with DMSO or SP, and the medium was changed every 3 days to maintain XPB depletion. Cells cultured with SP for 12 days showed complete inhibition of HIV-1 replication (Fig. 5E, solid gray line), while during this time, the percentage of living cells remained unaffected. For cells cultured without SP, 7% and 30% Gag-positive cells were obtained at 6 dpi and 12 dpi, respectively (Fig. 5E, black lines). When SP was added to the medium at 6 dpi, the virus stopped replicating (Fig. 5E, dashed gray line). These results show that SP can act after HIV-1 infection has been established.
Tat-dependent HIV-1 transactivation is inhibited by spironolactone.
Since XPB belongs to the TFIIH complex that is involved in transcription, we investigated whether SP could affect viral RNA transcription. To study viral gene expression, Jurkat T cells were transfected with three plasmids, one coding for the HIV-1 LTR promoter driving expression of luciferase, a second for the Renilla luciferase, and different concentrations of a third coding for HIV-1 Tat. Without Tat, luciferase expression was identical in cells treated or not with SP. Tat efficiently transactivated the LTR in the absence of SP, but not in its presence (Fig. 6A). Similar transfection experiments showed that SP had no effect on CMV (Fig. 6B) and on HTLV-1 LTR (Fig. 6C) promoters, indicating that SP specifically inhibits HIV-1 transcription from its own LTR. In addition, we confirmed an interaction between Tat and XPB (data not shown) that was previously revealed (16).
FIG 6.

HIV-1 Tat-dependent transcription is inhibited by spironolactone. (A) Effect of SP on the transfected HIV LTR promoter in the presence of Tat. Jurkat T cells were cotransfected with plasmids encoding HIV-LTR-Luc, Renilla luciferase (Ren), and Tat. After 24 h of plasmid expression, the cells were treated with DMSO or SP. P = 0.0121. (B) Effect of SP on transfected CMV promoter in the presence of Tat. Jurkat cells were cotransfected with a plasmid containing CMV-Luc, a second coding for Ren, and 100 ng of a plasmid coding for Tat. After 24 h of plasmid expression, the cells were treated with DMSO or SP. P = 0.1977. ns, no statistically significant difference. (C) Effect of SP on transfected LTR promoter in the presence of Tat. Jurkat cells were cotransfected with a plasmid containing HTLV-LTR-Luc, a second coding for Ren, and 100 ng of a plasmid coding for Tat. After 24 h of plasmid expression, the cells were treated with DMSO or SP. P = 0.1977. (D) Partial rescue of SP-mediated Tat transactivation inhibition by exogenous XPB. Jurkat T cells were transfected with 4 plasmids encoding HIV-LTR-Luc, Ren, XPB, and Tat. After 24 h of transfection, the cells were treated with DMSO or SP. In panels A, B, C, and D, Renilla luciferase was used for normalization. (E) Tat-mediated activation of transcription of a chromosomally integrated HIV-1 LTR promoter. JLTRG-R5 cells were transfected with different concentrations of a plasmid coding for Tat and cultured or not with SP. P = 0.0051. (F) TNF-α activation of transcription of a chromosomal HIV-1 LTR promoter. JLTRG-R5 cells were stimulated with TNF-α and cultured or not with SP before analysis by flow cytometry to determine the percentage of GFP+ cells. The results are the means of three experiments performed in triplicate. Unless otherwise indicated, SP was added to the culture medium at a concentration of 10 μM for 24 h. The error bars represent SEM. *, P < 0.05; **, P < 0.01.
Importantly, exogenous XPB expression partially restored Tat-dependent HIV-1 transactivation in SP-treated Jurkat T cells (Fig. 6D), confirming that inhibition by SP relies at least partially on XPB degradation.
To study viral gene expression from an integrated promoter, we took advantage of a cellular system in which the HIV-1 LTR is stably integrated into the host genome of Jurkat cells and drives the expression of GFP (JLTRG-R5 cells). Basal and Tat-dependent GFP expression levels were compared by adding increasing concentrations of Tat to cells treated or not with SP. Basal transcription levels from the integrated LTR promoter were identical in the presence or absence of SP (Fig. 6E, 0 ng of Tat). However, Tat-dependent transactivation was very low in the presence of SP compared to control cells (Fig. 6E). In contrast, SP had no effect on tumor necrosis factor alpha (TNF-α)-mediated LTR activation (Fig. 6F). These results strongly suggest that the inhibition of HIV-1 infection observed in SP-treated cells is mainly due to inhibition of Tat-dependent HIV-1 LTR transactivation.
Together, these results support the conclusion that SP specifically inhibits Tat-dependent HIV-1 LTR transactivation through XPB degradation in lymphocyte Jurkat T cells.
DISCUSSION
In this study, we describe the ability of SP to inhibit HIV-1 and HIV-2 infection of T cell lines and primary CD4+ T cells. We provide evidence that this inhibition relies on XPB degradation and occurs at the level of Tat-dependent transactivation. XPB degradation is specific for SP and independent of its aldosterone antagonist effect, since the derived EPL molecule does not have this property. These results suggest that XPB acts as a cofactor of viral replication rather than a host defense factor, as previously described (21, 22), and highlight a potential role for SP as an antiviral compound.
In Jurkat and primary CD4+ T cells, we observed that SP specifically induces the degradation of XPB without cell toxicity, since cellular mRNA levels, cell viability, and cell proliferation were not affected. SP treatment does not affect the expression of proteins of the transcription machinery, such as CDK7, Pol II, and CDK9. These results are surprising, considering that SP targets XPB, a central component of the TFIIH transcription complex. One possibility is that a cellular protein takes the place of XPB to carry out its activity in the presence of SP. Another possibility is that a very low level of XPB is sufficient to play a role within TFIIH. As the degradation of XPB induced by SP is reversible (25), it could be that XPB is constantly kept at a minimal protein level.
Several lines of evidence support the hypothesis that XPB is a cofactor of viral replication: (i) inhibition of HIV-1 and HIV-2 transduction and infection by SP correlates with XPB degradation in Jurkat, HeLa, and primary CD4+ T cells; (ii) exogenous XPB expression partially restores HIV-1 LTR transcription from SP inhibition in both HeLa and Jurkat T cells; (iii) SP inhibits Tat-dependent transcription of the integrated HIV LTR promoter, which is likely due to the role of TFIIH in transcription.
We show here that SP slightly increases HIV-1 transduction of the single-cycle virus HIV-1-CMV-GFP, in agreement with previous studies with the virus (21, 22). These results probably reflect the effect of XPB on gene expression from the CMV promoter. However, with HIV LTR-encoding viruses, as in this study and in previous reports (23, 24), the results reflect the role of XPB in LTR-dependent viral gene expression and highlight a positive effect of XPB on the virus. In conclusion, the present study provides evidence for revisiting the role of XPB as a cofactor of HIV-1 and HIV-2 infection in their natural target cells.
Nonetheless, we cannot completely rule out the possibility that SP may have additional targets other than XPB. Global methods, such as proteomic stable-isotope labeling with amino acids in cell culture (SILAC), may lead to the identification of other cellular proteins that could be targeted to the degradation pathway under SP treatment.
Our study shows that basal transcription from the integrated or transfected HIV-1 LTR promoter is unchanged by SP treatment. However, Tat-dependent transcription from the same HIV-1 LTR is very low in SP-treated cells compared to control cells. We conclude that the inhibition of HIV-1 infection observed in SP-treated cells is due to inhibition of Tat-dependent transcription. However, the precise molecular mechanism of action of SP in Tat-dependent HIV transcription remains to be elucidated. Does SP inhibit initiation and elongation of transcription through XPB degradation? SP could also inhibit other TFIIH actions, such as posttranslational modifications of histones, DNA demethylation, or chromatin remodeling (27).
Could it be that in the absence of XPB and in the presence of Tat, P-TEFb contributes to the function of TFIIH? In line with this possibility, it has been reported that Tat modifies the substrate specificity of P-TEFb, allowing it to phosphorylate the Pol II CTD at positions 2 and 5 (28). Other studies have reported that Tat may bind an additional CTD kinase, the Tat-associated T-cell-derived kinase (CDK2) that regulates the target-specific phosphorylation of the Pol II CTD and stimulates Tat-dependent transcription of the HIV-1 LTR (29). These findings open several possibilities for a role of XPB, together with Tat, in both initiation and elongation of transcription.
Recently, it was reported that SP has anti-Epstein-Barr virus (EBV) activity by inhibiting the function of the EBV protein known as SM protein, required for expression of several late lytic EBV genes and for infectious-EBV production (30). The mechanism of action of SP in this model remains incompletely characterized. Though SP was shown to inhibit RNA accumulation of SM targets, XPB levels were not analyzed. It would be interesting to know whether in EBV-infected cells SP induces inhibition of SM-activated RNA transcription and if this inhibition is associated with XPB degradation.
A long history of more than 50 years of daily use of SP as a diuretic and antihypertensive, and also as an inhibitory androgen drug, confirms that it is not toxic for human cells. SP action does not affect normal cells in long-term treatments (31). These results highlight the potential advantage of using SP in the treatment of viral infections, in particular of EBV and HIV.
Triptolide has also been reported to target XPB (32). It is a natural product isolated from a traditional Chinese medicinal plant with anti-inflammatory, immunosuppressive, contraceptive, and antitumor activities. Triptolide binds to XPB and inhibits RNA Pol II transcription. However, triptolide has been reported to promote proteasomal degradation of RNA Pol II (33) and Tat (34). Triptolide probably interferes with important cellular functions, and its clinical potential in its current form is limited due to potential safety concerns. HIV-1 infection can be suppressed by antiretroviral therapy, but the persistence of latent virus reservoirs limits viral eradication. Future experiments should aim at examining the possibility that SP provides an alternative to inhibit transcription from a latent HIV promoter, as recently reported for the Tat inhibitor didehydrocortistatin (dCA), to prevent viral reactivation from latent reservoirs (35, 36). Targeting HIV transcription could overcome the emergence of drug resistance and improve antiretroviral therapy with the aim of a functional cure (37).
ACKNOWLEDGMENTS
We thank all the members of our Retrovirus, Quiescence, and Proliferation group for fruitful discussions. We thank A. Cimarelli for HIV-1-CMV-GFP and N. Manel and A. Adachi for HIV-2. We also thank D. A. Donahue, A.-L. Haenni, and O. Schwartz for helpful discussions and critical reading of the manuscript. We thank the NIH AIDS Research and Reference Reagent Program for kindly providing reagents. We acknowledge the Immunobiology and the Genome and Sequencing facilities of the Cochin Institute and of the Paris Descartes University.
Funding Statement
This work was supported by a grant from the “Fondation pour la Recherche Medicale” (FRM; grant number DEQ20140329528) attributed to F.M.G. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.Drapkin R, Reardon JT, Ansari A, Huang J-C, Zawel L, Ahn K, Sancar A, Reinberg D. 1994. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature 368:769–772. doi: 10.1038/368769a0. [DOI] [PubMed] [Google Scholar]
- 2.Compe E, Egly JM. 2016. Nucleotide excision repair and transcriptional regulation: TFIIH and beyond. Annu Rev Biochem 85:265–290. doi: 10.1146/annurev-biochem-060815-014857. [DOI] [PubMed] [Google Scholar]
- 3.Dvir A, Conaway RC, Conaway JW. 1997. A role for TFIIH in controlling the activity of early RNA polymerase II elongation complexes. Proc Natl Acad Sci U S A 94:9006–9010. doi: 10.1073/pnas.94.17.9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giglia-Mari G, Coin F, Ranish JA, Hoogstraten D, Theil A, Wijgers N, Jaspers NG, Raams A, Argentini M, van der Spek PJ, Botta E, Stefanini M, Egly JM, Aebersold R, Hoeijmakers JH, Vermeulen W. 2004. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet 36:714–719. doi: 10.1038/ng1387. [DOI] [PubMed] [Google Scholar]
- 5.Oksenych V, Coin F. 2010. The long unwinding road: XPB and XPD helicases in damaged DNA opening. Cell Cycle 9:90–96. doi: 10.4161/cc.9.1.10267. [DOI] [PubMed] [Google Scholar]
- 6.Moreland RJ, Tirode F, Yan Q, Conaway JW, Egly JM, Conaway RC. 1999. A role for the TFIIH XPB DNA helicase in promoter escape by RNA polymerase II. J Biol Chem 274:22127–22130. doi: 10.1074/jbc.274.32.22127. [DOI] [PubMed] [Google Scholar]
- 7.Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. 2006. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell 22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 8.Lu H, Zawel L, Fisher L, Egly JM, Reinberg D. 1992. Human general transcription factor IIH phosphorylates the C-terminal domain of RNA polymerase II. Nature 358:641–645. doi: 10.1038/358641a0. [DOI] [PubMed] [Google Scholar]
- 9.Zhou M, Nekhai S, Bharucha DC, Kumar A, Ge H, Price DH, Egly J-M, Brady JN. 2001. TFIIH Inhibits CDK9 phosphorylation during human immunodeficiency virus type 1 transcription. J Biol Chem 276:44633–44640. doi: 10.1074/jbc.M107466200. [DOI] [PubMed] [Google Scholar]
- 10.Arya S, Guo C, Josephs S, Wong-Staal F. 1985. Trans-activator gene of human T-lymphotropic virus type III (HTLV-III). Science 229:69–73. doi: 10.1126/science.2990040. [DOI] [PubMed] [Google Scholar]
- 11.Sodroski J, Rosen C, Wong-Staal F, Salahuddin S, Popovic M, Arya S, Gallo R, Haseltine W. 1985. Trans-acting transcriptional regulation of human T-cell leukemia virus type III long terminal repeat. Science 227:171–173. doi: 10.1126/science.2981427. [DOI] [PubMed] [Google Scholar]
- 12.Berkhout B, Silverman R, Jeang K. 1989. Tat trans-activates the human immunodeficiency virus through a nascent RNA target. Cell 59:273–282. doi: 10.1016/0092-8674(89)90289-4. [DOI] [PubMed] [Google Scholar]
- 13.Demarchi F, d'Adda di Fagagna F, Falaschi A, Giacca M. 1996. Activation of transcription factor NF-kappaB by the Tat protein of human immunodeficiency virus type 1. J Virol 70:4427–4437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brady J, Kashanchi F. 2005. Tat gets the “green” light on transcription initiation. Retrovirology 2:1–8. doi: 10.1186/1742-4690-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cujec TP, Okamoto H, Fujinaga K, Meyer J, Chamberlin H, Morgan DO, Peterlin BM. 1997. The HIV transactivator TAT binds to the CDK-activating kinase and activates the phosphorylation of the carboxy-terminal domain of RNA polymerase II. Genes Dev 11:2645–2657. doi: 10.1101/gad.11.20.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.García-Martínez LF, Mavankal G, Neveu JM, Lane WS, Ivanov D, Gaynor RB. 1997. Purification of a Tat-associated kinase reveals a TFIIH complex that modulates HIV-1 transcription. EMBO J 16:2836–2850. doi: 10.1093/emboj/16.10.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herrmann CH, Rice AP. 1993. Specific interaction of the human immunodeficiency virus Tat proteins with a cellular protein kinase. Virology 197:601–608. doi: 10.1006/viro.1993.1634. [DOI] [PubMed] [Google Scholar]
- 18.Kao SY, Calman AF, Luciw PA, Peterlin BM. 1987. Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330:489–493. doi: 10.1038/330489a0. [DOI] [PubMed] [Google Scholar]
- 19.Marciniak RA, Sharp PA. 1991. HIV-1 Tat protein promotes formation of more-processive elongation complexes. EMBO J 10:4189–4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall NF, Price DH. 1995. Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 270:12335–12338. doi: 10.1074/jbc.270.21.12335. [DOI] [PubMed] [Google Scholar]
- 21.Yoder KE, Roddick W, Hoellerbauer P, Fishel R. 2011. XPB mediated retroviral cDNA degradation coincides with entry to the nucleus. Virology 410:291–298. doi: 10.1016/j.virol.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoder K, Sarasin A, Kraemer K, McIlhatton M, Bushman F, Fishel R. 2006. The DNA repair genes XPB and XPD defend cells from retroviral infection. Proc Natl Acad Sci U S A 103:4622–4627. doi: 10.1073/pnas.0509828103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Espeseth A, Fishel R, Hazuda D, Huang Q, Xu M, Yoder K, Zhou H. 2011. siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS One 6:e17612. doi: 10.1371/journal.pone.0017612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brass A, Dykxhoorn D, Benita Y, Yan N, Engelman A, Xavier R, Lieberman J, Elledge S. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 25.Alekseev S, Ayadi M, Brino L, Egly J-M, Larsen Annette K, Coin F. 2014. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol 21:398–407. doi: 10.1016/j.chembiol.2013.12.014. [DOI] [PubMed] [Google Scholar]
- 26.He J, Choe S, Walker R, Di Marzio P, Morgan DO, Landau NR. 1995. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J Virol 69:6705–6711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singh A, Compe E, Le May N, Egly J-M. 2015. TFIIH subunit alterations causing xeroderma pigmentosum and trichothiodystrophy specifically disturb several steps during transcription. Am J Hum Genet 96:194–207. doi: 10.1016/j.ajhg.2014.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou M, Halanski MA, Radonovich MF, Kashanchi F, Peng J, Price DH, Brady JN. 2000. Tat modifies the activity of CDK9 to phosphorylate serine 5 of the RNA polymerase II carboxyl-terminal domain during human immunodeficiency virus type 1 transcription. Mol Cell Biol 20:5077–5086. doi: 10.1128/MCB.20.14.5077-5086.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nekhai S, Shukla RR, Fernandez A, Kumar A, Lamb NJ. 2000. Cell cycle-dependent stimulation of the HIV-1 promoter by Tat-associated CAK activator. Virology 266:246–256. doi: 10.1006/viro.1999.0035. [DOI] [PubMed] [Google Scholar]
- 30.Verma D, Thompson J, Swaminathan S. 2016. Spironolactone blocks Epstein-Barr virus production by inhibiting EBV SM protein function. Proc Natl Acad Sci U S A 113:3609–3614. doi: 10.1073/pnas.1523686113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mackenzie IS, Macdonald TM, Thompson A, Morant S, Wei L. 2012. Spironolactone and risk of incident breast cancer in women older than 55 years: retrospective, matched cohort study. BMJ 345:e4447. doi: 10.1136/bmj.e4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Titov DV, Gilman B, He Q-L, Bhat S, Low W-K, Dang Y, Smeaton M, Demain AL, Miller PS, Kugel JF, Goodrich JA, Liu JO. 2011. XPB, a subunit of TFIIH, is a target of the natural product triptolide. Nat Chem Biol 7:182–188. doi: 10.1038/nchembio.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y, Lu J-J, He L, Yu Q. 2011. Triptolide (TPL) inhibits global transcription by inducing proteasome-dependent degradation of RNA polymerase II (Pol II). PLoS One 6:e23993. doi: 10.1371/journal.pone.0023993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan Z, Chen X. 2014. Triptolide inhibits human immunodeficiency virus type 1 replication by promoting proteasomal degradation of Tat protein. Retrovirology 11:88. doi: 10.1186/s12977-014-0088-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mousseau G, Kessing CF, Fromentin R, Trautmann L, Chomont N, Valente ST. 2015. The Tat inhibitor didehydro-cortistatin A prevents HIV-1 reactivation from latency. mBio 6:e00465. doi: 10.1128/mBio.00465-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mousseau G, Valente S. 2012. Strategies to block HIV transcription: focus on small molecule Tat inhibitors. Biology 1:668. doi: 10.3390/biology1030668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Le Douce V, Ait-Amar A, Forouzan Far F, Fahmi F, Quiel J, El Mekdad H, Daouad F, Marban C, Rohr O, Schwartz C. 18 June 2016. Improving combination antiretroviral therapy by targeting HIV-1 gene transcription. Expert Opin Ther Targets doi: 10.1080/14728222.2016.1198777. [DOI] [PubMed] [Google Scholar]