

ABSTRACT
Tumor necrosis factor receptor (TNFR)-associated factor 6 (TRAF6) is an important adaptor molecule that mediates the TNFR family and interleukin-1 (IL-1)/Toll-like receptor (TLR) signaling cascades. These pathways are important for the host to control viral infections. In this report, we demonstrated that hepatitis C virus (HCV) depleted TRAF6 from its host cells through a posttranslational mechanism. This depletion was independent of proteasomes, as it was not affected by the proteasome inhibitor MG132, but it was suppressed by bafilomycin A1, which led to the association of TRAF6 with autophagosomes. As bafilomycin A1 is a vacuolar ATPase inhibitor that inhibits autophagic protein degradation, these results suggested that HCV depleted TRAF6 via autophagy. The degradation of TRAF6 was apparently mediated by the p62 sequestosome protein, which is a factor important for selective autophagy, as it could bind to TRAF6 and its silencing stabilized TRAF6. The depletion of TRAF6 suppressed activation of NF-κB and induction of proinflammatory cytokines and enhanced HCV replication. In contrast, the overexpression of TRAF6 suppressed HCV replication. These results revealed a novel mechanism that was used by HCV to disrupt the host innate immune responses for viral replication and persistence.
IMPORTANCE HCV can cause severe liver diseases and is one of the most important human pathogens. It establishes chronic infections in the great majority of patients that it infects, indicating that it has evolved sophisticated mechanisms to evade host immunity. TRAF6 is an important signaling molecule that mediates activation of NF-κB and expression of proinflammatory cytokines and interferons. In this study, we found that HCV infection suppressed the host innate immune response through the induction of autophagic degradation of TRAF6. This finding provided important information for further understanding how HCV evades host immunity to establish persistence.
INTRODUCTION
Hepatitis C virus (HCV) chronically infects 170 million people worldwide. It is a hepatotropic virus, and its chronic infection can cause progressive inflammation-associated liver diseases to result in liver failure and hepatocellular carcinoma (1, 2). HCV infection of hepatocytes triggers various host cellular responses and activates innate immunity signaling pathways, which are known to assist the host to eradicate viral infections (3). In spite of these host responses, HCV is able to evade them to establish chronic infection in the great majority of patients.
Pattern recognition receptors (PRRs) that include retinoic acid-inducible gene I product (RIG-I)-like receptors (RLRs) and Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns (PAMPs) associated with microbial pathogens. Upon activation by PAMPs, PRRs will trigger the downstream signaling events, leading to the production of antiviral cytokines, including interferons. PRRs can also recognize HCV RNAs or proteins (4–9). HCV infection can activate RIG-I, which recognizes the poly(U) motif in the 3′ untranslated region of the HCV genome (10), melanoma differentiation-associated protein 5 (MDA5) (11), which is another member of the RLR family, the double-stranded RNA-dependent kinase PKR (protein kinase R) (12), Toll-like receptor 3 (TLR3) (13), TLR7, and TLR8 (9). However, HCV has also developed mechanisms to suppress the host innate immune responses. For example, the HCV NS3/4A protease can cleave mitochondrial antiviral signaling protein (MAVS), the downstream adaptor molecule of RIG-I, to suppress the induction of interferons (14). It can also cleave TRIF (Toll–interleukin-1 [IL-1] receptor domain containing adaptor-inducing interferon beta), another signaling adaptor protein, to disrupt the signaling of TLR3 (15), which senses double-stranded RNA.
Autophagy is another type of cellular innate immune responses that can remove intracellular microbial pathogens. It begins with the appearance of membrane crescents known as phagophores or isolation membranes, which will grow to sequester part of the cytoplasm to form enclosed double-membrane vesicles known as autophagosomes. Autophagosomes mature by fusing with lysosomes to form autolysosomes, in which the cargoes of autophagosomes are digested (16). HCV infection can induce the autophagic response and temporally regulate the maturation of autophagosomes in its host cells (17). Rather than suppressing HCV replication, autophagy induced by HCV enhances HCV replication (for reviews, see references 18 and 19).
In this report, we studied the interplay between HCV and the innate immune response of its host cells. We found that HCV could deplete tumor necrosis factor receptor-associated factor 6 (TRAF6) via autophagy to suppress the induction of cytokines to enhance its replication. TRAF6 belongs to the tumor necrosis factor receptor-associated factor (TRAF) family and participates in interleukin-1 receptor and TLR signaling. It is an important factor that mediates the activation of NF-κB after TLR is activated. Our studies thus revealed a novel mechanism used by HCV to disrupt host innate immune responses for the enhancement of its replication.
MATERIALS AND METHODS
Cells and viruses.
Huh7 cells, a human hepatoma cell line, was maintained in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (10,000 IU/ml), amphotericin (25 μg/ml), streptomycin (10,000 μg/ml), and nonessential amino acids (0.1 mM). Huh7N1b replicon cells harboring an HCV subgenomic RNA replicon were maintained in the same medium containing 0.4 mg/ml G418 (20). Huh7 cells that stably expressed the green fluorescent protein (GFP)–microtubule-associated protein light chain 3 (LC3) fusion protein had been previously described (21).
Plasmid and siRNAs.
The Flag-tagged TRAF6 expression plasmid was constructed by inserting the Flag-tagged TRAF6 coding sequence (NCBI accession number NM_004620) into the pEF-Myc/His version C vector (Life Technologies). The small interfering RNA (siRNA) targeting TRAF6 was purchased from Santa Cruz Biotech (catalog no. SC-36717), and siRNA targeting p62 was purchased from Sigma (catalog no. EHU027651). Lipofectamine RNAiMax from Invitrogen was used to transfect the DNA plasmids and the siRNAs. For DNA transfections, we could routinely obtain transfection efficiencies of >50%.
Confocal immunofluorescence microscopy.
Huh7-GFP-LC3 cells were plated on glass slides for 24 h and then infected with HCV. The cells were fixed with 3.7% formaldehyde in phosphate-buffered saline (PBS) for 10 min, permeabilized with 0.02% Triton X-100 in PBS for 10 min, and blocked with PBS containing 5% bovine serum albumin (BSA) for 1 h. Immunofluorescence assays were performed using the rabbit anti-TRAF6 (catalog no. SC-7221; Santa Cruz Biotech) and mouse anti-NS5A (gift from C. M. Rice) primary antibodies in PBS containing 3% BSA for 1 h, followed by incubation with goat anti-mouse Alexa Fluor 405-conjugated and Immunopure goat anti-rabbit rhodamine-conjugated secondary antibodies from ThermoFisher Scientific (catalog no. A-31553 and R-6394). The slides were mounted with 70% glycerol in PBS. All images were taken with a Carl Zeiss LSM 510 confocal microscope system at the Cell and Tissue Imaging Core of the University of Southern California Research Center for Liver Diseases. All images were analyzed with Zen Black Edition lite, version 2009.
Immunoblot analysis and antibodies.
All cell samples were lysed in modified radioimmunoprecipitation assay buffer (150 mM NaCl, 0.5% sodium deoxycholate, 10 mM Tris [pH 7.5], 1% Triton X-100) supplemented with the complete protease inhibitor cocktail (Roche). After the cells were lysed, protein samples were subjected to electrophoresis in an SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride (PVDF) membrane, and blocked with PBS containing 5% nonfat milk for 1 h. The membranes were incubated overnight at 4°C or 1 h at room temperature with the antibodies. The antibodies used in this study included anti-TRAF6 from Santa Cruz Biotech (catalog no. SC-7221), anti-TRAF2 and anti-SQSTM1 (sequestosome-1)/p62 from Cell Signaling (catalog no. 4712 and 5114), antiactin from Abcam (ab8227), anti-LC3 from MBL (PM036), anti-Flag from Sigma-Aldrich (F3165), and anti-HCV core (22).
Gene expression analysis, reporter assay, and ELISA.
RNA was extracted using Qiagen RNeasy kit. Real-time quantitative PCR was conducted using the SYBR green-based one-step reverse transcription-PCR (RT-PCR) method (catalog no. 4392653; Applied Biosciences). The forward and reverse primers used for the different factors or proteins were as follows: for TRAF6, 5′-TTTGCTCTTATGGATTGTCCCC-3′ and 5′-CATTGATGCAGCACAGTTGTC-3′, respectively; for tumor necrosis factor alpha (TNF-α), 5′-ATCAATCGGCCCGACTATCTC-3′ and 5′-GCAATGATCCCAAAGTAGACCTG-3′, respectively; for IL-6, 5′-ACTCACCTCTTCAGAACGAATTG-3′ and 5′-CCATCTTTGGAAGGTTCAGGTTG-3′, respectively; and for glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-ACAACTTTGGTATCGTGGAAGG-3′ and 5′-GCCATCACGCCACAGTTTC-3′, respectively. Relative RNA levels were determined after normalization against the GAPDH RNA. The analysis of the NF-κB promoter was conducted using the firefly luciferase reporter in the plasmid constructed from pGL3-basic vector purchased from Promega (catalog no. E1751) with a NF-κB binding motif, GGGAAGTTC. The plasmid containing the renilla luciferase linked to the cytomegalovirus (CMV) promoter was used for cotransfection to monitor the transfection efficiency. The luciferase activities were measured using the Dual-Luciferase reporter assay system (catalog no. E1910; Promega) according to the manufacturer's protocol. The firefly luciferase activities were normalized against the renilla luciferase activities. The concentration of TNF-α was measured using Human TNF-α ELISA (enzyme-linked immunosorbent assay) Ready-Set-Go kit (catalog no. 88734622; eBioscience) and FLUOstar Omega microplate reader.
RESULTS
Suppression of TRAF6 expression by HCV.
To study how HCV might interfere with the innate immune response of its host cells, we examined the possible effect of HCV on TRAF6, an important mediator of the TLR signaling pathway. We infected Huh7 cells, a human hepatoma cell line, with a cell culture-adapted HCV JFH1 variant (23) and analyzed the protein level of TRAF6 at different time points after infection. As shown in Fig. 1A, the TRAF6 protein level was not significantly affected by HCV at 24 h postinfection, but it was significantly reduced at 48 h and not detected at 72 h. In contrast, TRAF2, another member of the TRAF family, was not affected by HCV at any time point. When the TRAF6 mRNA was analyzed by real-time RT-PCR, no significant difference of TRAF6 mRNA levels was detected at different time points (Fig. 1B). For a positive control, we also measured the HCV RNA levels, which increased and reached the peak at 36 h postinfection. These results indicated that HCV likely suppressed the expression of TRAF6 via a posttranslational mechanism.
FIG 1.

Suppression of TRAF6 expression by HCV. Huh7 cells were infected with HCV at a multiplicity of infection (MOI) of 1. At 3 h postinfection (hpi), the inoculum was removed, and cells were incubated in fresh medium. Cells were harvested at the time points indicated for immunoblot analysis (A) or for real-time RT-PCR analysis for TRAF6 mRNA (B) or HCV RNA (C). In panel B, the TRAF6 mRNA level in mock-infected cells was arbitrarily set at 1. The values shown in panels B and C are the means plus standard errors of the means (SEM) (error bars) from at least three independent experiments.
Degradation of TRAF6 by HCV-induced autophagy.
To determine the mechanism that might be responsible for the depletion of TRAF6 by HCV, we treated HCV-infected cells with MG132, which inhibits the proteolytic activity of the 26S proteasome. As shown in Fig. 2A, the treatment of cells with 5 or 10 μM MG132 slightly increased the TRAF6 protein level in mock-infected cells, but it did not restore the protein level of TRAF6 in HCV-infected cells. These results indicated that it was unlikely that HCV induced the degradation of TRAF6 via proteasomes. As the peak time points of TRAF6 loss (i.e., 48 and 72 h postinfection) coincided with the peak activity of autophagic protein degradation induced by HCV (17), we tested the possible role of autophagy in the reduction of TRAF6 by treating cells with bafilomycin A1, a vacuolar ATPase inhibitor that inhibits the autophagic protein degradation (24). As shown in Fig. 2B, bafilomycin A1 increased the levels of LC3-II (the lipidated form of LC3, the microtubule-associated protein light chain 3) and p62 in both mock-infected and HCV-infected cells. Its nonlipidated form (i.e., LC3-I) is localized to the cytosol, and its lipidated form (i.e., LC3-II) is localized to autophagosomes. LC3-II is delipidated and released back into the cytosol for recycling after the maturation of autophagosomes or digested in autolysosomes if it resides in the inner membrane of autophagosomes (25). The p62 sequestosome-1 (p62/SQSTM1) protein is degraded by autophagy. The increases in the protein levels of LC3-II and p62 were consistent with the inhibitory effect of bafilomycin A1 on the maturation of autophagosomes. Interestingly, as also shown in Fig. 2B, although bafilomycin A1 had no effect on the TRAF6 protein level in mock-infected cells, it partially restored the TRAF6 protein level in HCV-infected cells at 48 h postinfection, supporting a role of autophagy in the degradation of TRAF6. To determine whether this effect of autophagy on TRAF6 was HCV specific, we treated Huh7 cells with rapamycin, which induced autophagy (26). As shown in Fig. 2C, rapamycin increased the level of LC3-II at different time points after the treatment, in agreement with its ability to induce autophagy. However, this treatment had no effect on the TRAF6 levels, indicating that the effect of autophagy on TRAF6 was HCV specific. We also analyzed the level of TRAF6 in stable Huh7 cells that contained an HCV subgenomic RNA replicon. As shown in Fig. 2D, the level of TRAF6 in HCV replicon cells was not different from that in the control Huh7 cells. This result was not surprising, as the HCV subgenomic RNA replicon inhibited the maturation of autophagosomes and hence the autophagic protein degradation (17, 19). To further test the possibility that HCV induced autophagy to degrade TRAF6, we conducted confocal microscopy to examine the possible colocalization of TRAF6 with autophagosomes. Huh7 cells stably expressing the GFP-LC3 fusion protein were mock infected or infected with HCV. As shown in Fig. 3A, TRAF6 in mock-infected cells displayed a diffused staining pattern in the whole cell, in agreement with the previous reports (27, 28). Few autophagic vacuoles (i.e., GFP-LC3 puncta) could be detected in mock-infected cells. The TRAF6 signal was largely not detected in HCV-infected cells at 48 h postinfection, also in agreement with its depletion by HCV at this time point as shown in Fig. 1A. In these HCV-infected cells, GFP-LC3 puncta became apparent, confirming the previous reports that HCV could induce autophagic vacuoles (17). Bafilomycin A1 did not significantly affect the subcellular localization of TRAF6 in mock-infected cells, although it did cause the accumulation of GFP-LC3 autophagic puncta, which was expected due to its inhibitory effect on the maturation of autophagosomes. Approximately 10% of TRAF6 was found to colocalize with GFP-LC3 puncta in these cells (Fig. 3B). In HCV-infected cells that were treated with bafilomycin A1, the TRAF6 signals became bright and punctate, and approximately 50% of the signals colocalized with GFP-LC3 puncta. The association of TRAF6 with autophagic puncta when the autophagic protein degradation was suppressed by bafilomycin A1 lends further support to the degradation of TRAF6 by HCV-induced autophagy.
FIG 2.
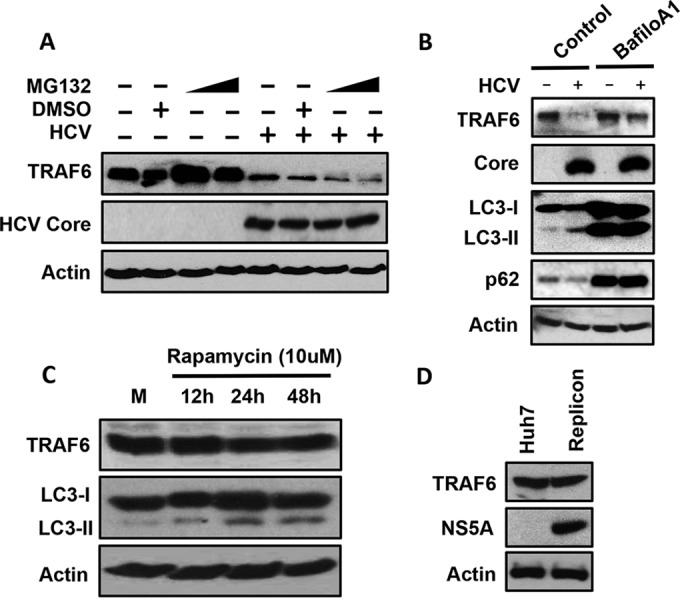
Stabilization of TRAF6 in HCV-infected cells by bafilomycin A1 but not by MG132. Huh7 cells were infected with HCV for 24 h and then treated with 5 or 10 μM MG132 dissolved in dimethyl sulfoxide (DMSO) for 16 h (A) or with 100 nM bafilomycin A1 (BafiloA1) for 24 h (B). Cells were then lysed for immunoblot analysis. (C) Huh7 cells were treated with rapamycin (10 μM) for the indicated lengths of time and lysed for immunoblot analysis. M, mock-treated cells. (D) Huh7 cells and HCV subgenomic RNA replicon cells were lysed for immunoblot analysis.
FIG 3.

Colocalization analysis of TRAF6 and autophagic vacuoles in mock- and HCV-infected cells. (A) Huh7-GFP-LC3 cells were mock infected or infected with HCV for 24 h and then treated with either DMSO or bafilomycin A1 (BafoA1) for 24 h. TRAF6 (red) and the HCV NS5A protein (blue) were then immunostained for confocal microscopy. Bars, 20 μm. (B) Percentage of TRAF6 colocalized with GFP-LC3 puncta. The results represent the means plus standard errors of the means (SEM) (error bars) from >30 cells analyzed.
Autophagic degradation of TRAF6 mediated by p62/SQSTM1.
To further investigate how TRAF6 was targeted to autophagic vacuoles for degradation, we performed the siRNA knockdown experiment to analyze the role of p62, which could bind to TRAF6 to mediate the activation of NF-κB and also interact with LC3 to target ubiquitinated proteins to autophagosomes for degradation (29–31). As shown in Fig. 4A, treating Huh7 cells with the control siRNA had no effect on the depletion of TRAF6 by HCV at 48 h after infection. However, the treatment of Huh7 cells with the p62 siRNA, which significantly inhibited expression of p62, prevented HCV from depleting TRAF6 at 48 h postinfection. These results indicated an important role of p62 in mediating the depletion of TRAF6 induced by HCV. To determine whether p62 could also bind to TRAF6 in HCV-infected cells, we conducted coimmunoprecipitation experiments. Huh7 cells were infected with HCV and immunoprecipitated with either the control antibody or the anti-p62 antibody, followed by immunoblot analysis using the anti-TRAF6 antibody. As shown in Fig. 4B, TRAF6 could be coimmunoprecipitated with p62 in mock-infected cells, indicating that these two proteins could bind to each other. This interaction was enhanced by HCV at 24 h postinfection and was also detected at 48 h postinfection, even though the levels of TRAF6 and p62 were significantly reduced at this time point, suggesting an enhanced interaction between these two proteins at this time point. Note that the p62 protein level immunoprecipitated by the anti-p62 antibody was not reduced at 48 h postinfection. This might be due to the increased binding affinity of the antibody to p62 after its binding to TRAF6. The results shown in Fig. 4 provided strong support to the argument that TRAF6 was recruited by p62 to autophagic vacuoles for their simultaneous degradation in HCV-infected cells.
FIG 4.
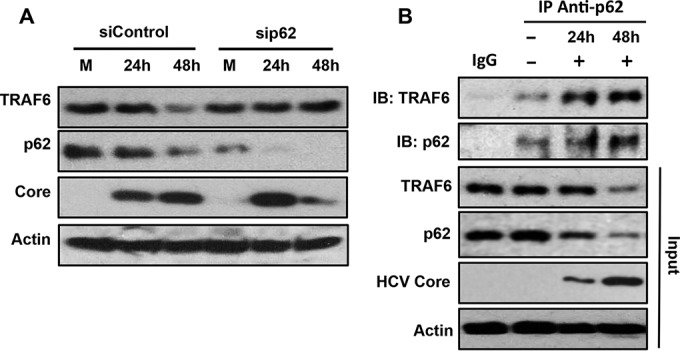
Analysis of the interaction between p62 and TRAF6 in HCV-infected cells. (A) Huh7 cells were transfected with the control siRNA (siControl) or the p62 siRNA (sip62) twice on consecutive days and then infected with HCV for 24 or 48 h using an MOI of 1. The cells were then lysed for immunoblot analysis. (B) Huh7 cells were infected with HCV for 24 or 48 h. The cells were then lysed and immunoprecipitated (IP) with either the control IgG or the anti-p62 antibody followed by immunoblot analysis (IB) using the anti-TRAF6 and anti-p62 antibodies (top two blots). The total cell lysates were also used for immunoblotting to serve as the input control.
Negative regulation of HCV replication by TRAF6.
The depletion of TRAF6 by HCV prompted us to investigate the possible effect of TRAF6 on HCV replication. We transfected a plasmid that expressed the Flag-tagged TRAF6 into Huh7 cells and then infected the cells with HCV for either 24 or 48 h. As for endogenous TRAF6, the protein level of Flag-tagged TRAF6 was not affected by HCV at 24 h postinfection, but it was significantly reduced by HCV at 48 h postinfection (Fig. 5A). As the expression of Flag-tagged TRAF6 was driven by the EF1α promoter, this result was consistent with the observation that the depletion of TRAF6 by HCV was mediated by autophagy and not by transcriptional repression. This expression of Flag-tagged TRAF6 reduced the HCV core protein level at both 24 and 48 h postinfection (Fig. 5A) and significantly reduced the HCV RNA levels in cells (Fig. 5B), indicating that TRAF6 suppressed the HCV replication. To confirm this finding, we also performed the siRNA knockdown experiment. Huh7 cells were transfected with either the control siRNA or the TRAF6 siRNA and then infected with HCV for 24 or 48 h. This suppression of TRAF6 expression increased HCV core protein level (Fig. 5C) and RNA level (Fig. 5D) and the amount of progeny virus released into the incubation medium (Fig. 5E), confirming that TRAF6 is a negative regulator of HCV replication.
FIG 5.

Effects of TRAF6 on HCV replication. (A) Huh7 cells were transfected with the control vector or the plasmid that expressed the Flag-tagged TRAF6 (FLAG-TRAF6) for 24 h and then mock infected or infected with HCV for 24 h or 48 h. Cell lysates were then collected for immunoblot analysis. (B) Huh7 cells infected by HCV as mentioned above for panel A were lysed for quantification of HCV RNA using real-time RT-PCR. The HCV RNA levels were normalized against GAPDH RNA. (C to E) Huh7 cells were transfected twice with the control siRNA or the TRAF6 siRNA and then infected with HCV for 24 or 48 h with an MOI of 1. The cells were then lysed for immunoblot analysis (C) or real-time RT-PCR analysis for quantification of HCV RNA (D and E). The incubation medium was also harvested at 24 and 48 h for titration of infectious HCV particles using the focus formation assay as previously described (46). Values that are significantly different are indicated by asterisks as follows: *, P < 0.05; **, P < 0.005.
TRAF6 supports cytokine response in HCV-infected cells.
HCV infection can activate NF-κB and induce the expression of cytokines (9, 32, 33). TRAF6 is an important mediator for the activation of NF-κB and expression of proinflammatory cytokines (34–37). To determine whether TRAF6 is also important for the activation of NF-κB and expression of proinflammatory cytokines in HCV-infected cells, we first analyzed the effect of TRAF6 on the NF-κB promoter by transfecting Huh7 cells with either a control siRNA or the TRAF6 siRNA and a firefly luciferase reporter construct linked to the NF-κB promoter. The cells were then infected with HCV for 24 or 48 h. As shown in Fig. 6A, similar to our previous report (9), HCV infection could activate the NF-κB promoter, but this activation was abolished when expression of TRAF6 was suppressed. We then analyzed the effect of TRAF6 knockdown on the expression of IL-6 and TNF-α, two proinflammatory cytokines, by analyzing their RNA levels using real-time RT-PCR. As shown in Fig. 6B, HCV could induce expression of IL-6 and TNF-α, but this induction was abolished and reduced below the basal level when expression of TRAF6 was suppressed. Similar results were obtained when IL-6 and TNF-α secreted into the incubation medium were analyzed by ELISA (Fig. 6C). As our previous studies indicated that TNF-α could suppress HCV replication (9), these results provided an explanation to how TRAF6 suppressed HCV replication.
FIG 6.

Effects of TRAF6 on the NF-κB promoter and the expression of cytokines in HCV-infected cells. (A) Huh7 cells were transfected with the NF-κB-luciferase reporter and either the control siRNA or the TRAF6 siRNA for 24 h. The CMV-renilla luciferase reporter was also used for cotransfection to monitor transfection efficiencies. The cells were then infected with HCV for 24 h or 48 h and then lysed for measuring the luciferase activities using the Dual-Luciferase assay kit (Promega). The results represent the means plus SEM from three independent experiments. Huh7 cells transfected with the control siRNA or the TRAF6 siRNA were infected with HCV for 24 or 48 h. (B) Cells were then lysed for quantification of IL-6 or TNF-α (TNFa) mRNAs using real-time RT-PCR. The levels of IL-6 and TNF-α mRNAs of mock-infected cells transfected with the control siRNA were arbitrarily set at 1. (C) The incubation medium was collected and filtered at 24 and 48 h postinfection for analysis of IL-6 and TNF-α by ELISA.
DISCUSSION
HCV infection can induce inflammatory responses (38). In spite of this, HCV establishes persistent infections in the great majority of patients that it infects. The ability of HCV to suppress the host innate immune responses likely plays an important role for HCV to establish its persistence. In this report, we demonstrated that TRAF6, an important molecule that mediates the signaling pathways of TLRs and the activation of NF-κB for the expression of antiviral cytokines, is targeted by HCV for degradation. We found that TRAF6 was depleted by HCV via a posttranslational mechanism in a time-dependent manner (Fig. 1). This mechanism was independent of proteasomes, as the depletion of TRAF6 by HCV could not be abolished by the proteasome inhibitor MG132 (Fig. 2). However, it was dependent on autophagy, as it could be inhibited by bafilomycin A1, which inhibits the maturation of autophagosomes and the autophagic protein degradation (Fig. 2). The argument that TRAF6 was degraded by autophagy was also supported by the observation that it colocalized with autophagic vacuoles when its degradation was suppressed by bafilomycin A1 (Fig. 3). Our further analysis indicated that the depletion of TRAF6 by HCV was dependent on p62 (Fig. 4A), which could bind to TRAF6 (Fig. 4B). p62 contains an LC3-interacting region (LIR). It also contains an ubiquitin-associated domain and can bind to ubiquitinated proteins. Due to these dual activities, p62 can target ubiquitinated protein aggregates and organelles to autophagic vacuoles for removal and plays a very important role in mediating selective autophagy. It is conceivable that HCV used a similar pathway and the ability of TRAF6 to bind to p62 to deplete TRAF6, which led to suppression of the NF-κB promoter and the expression of proinflammatory cytokines in the later stage of infection and the enhancement of HCV replication (Fig. 5 and 6).
Note that it had previously been reported that p62 could bind to TRAF6 after the stimulation of cells with IL-1, and this binding was important for the activation of NF-κB (29). More recently, it was also demonstrated that p62 could recruit TRAF6 to the mammalian target of rapamycin complex 1 (mTORC1) for its activation after the stimulation of nutrients to promote cell growth and suppress autophagy (39). Our results shown in Fig. 4 also indicated that HCV could stimulate the interaction between p62 and TRAF6, although in this case, the outcome of this interaction was the autophagic degradation of TRAF6. Thus, it is apparent that different stimuli can promote the interactions between p62 and TRAF6 to lead to drastically different outcomes.
The depletion of TRAF6 by HCV was delayed until the later time points of HCV infection. This is likely due to temporal regulation of the autophagic flux by HCV (17). Autophagy is ongoing at the basal level in cells, and it can be stimulated by the infection of microbial pathogens and used by the host cells to remove intracellular pathogens, including viruses (40). HCV could also induce autophagy, although similar to a number of RNA viruses (41), this induction of autophagy enhanced rather than suppressed HCV replication (19). Our recent studies indicated that HCV induced the autophagic response of its host cells but suppressed the maturation of autophagosomes at the early time points of infection. This suppression of autophagosomal maturation allowed autophagosomes to accumulate in cells, which is beneficial to HCV replication, as HCV could use autophagosomal membranes for its RNA replication (42). HCV allowed the autophagosomes to mature and fuse with lysosomes in the later stage of infection, likely because the importance of autophagosomes for its RNA replication diminished in the later stage of infection either due to the shift of the stage of the life cycle from RNA replication to the assembly of progeny viral particles or the extensive reorganization of cellular membranes, such as the appearance of smaller double-membrane vesicles that were found to also support HCV RNA replication (19, 43). The results of our studies presented in this report indicated that the maturation of autophagosomes in the later stage of infection was also important for HCV, as it allowed HCV to deplete TRAF6 to suppress the host innate immune response to favor HCV persistence. This finding, plus the previous observations that autophagy induced by HCV could suppress the induction of type I interferons and interferon-stimulated genes via the suppression of RIG-I (44, 45), clearly demonstrated that HCV had developed sophisticated mechanisms to control the host autophagic response to maximize its replication.
ACKNOWLEDGMENTS
We thank Michelle MacVeigh-Aloni and the Cell and Tissue Imaging Core of the USC Research Center for Liver Diseases for help with the confocal microscopy studies.
REFERENCES
- 1.Negro F, Alberti A. 2011. The global health burden of hepatitis C virus infection. Liver Int 31(Suppl 2):1–3. doi: 10.1111/j.1478-3231.2011.02537.x. [DOI] [PubMed] [Google Scholar]
- 2.Lavanchy D. 2009. The global burden of hepatitis C. Liver Int 29(Suppl 1):74–81. doi: 10.1111/j.1478-3231.2008.01934.x. [DOI] [PubMed] [Google Scholar]
- 3.Fredericksen B, Akkaraju GR, Foy E, Wang C, Pflugheber J, Chen ZJ, Gale M Jr. 2002. Activation of the interferon-beta promoter during hepatitis C virus RNA replication. Viral Immunol 15:29–40. doi: 10.1089/088282402317340215. [DOI] [PubMed] [Google Scholar]
- 4.Horner SM, Gale M Jr. 2013. Regulation of hepatic innate immunity by hepatitis C virus. Nat Med 19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lemon SM. 2010. Induction and evasion of innate antiviral responses by hepatitis C virus. J Biol Chem 285:22741–22747. doi: 10.1074/jbc.R109.099556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi O, Akira S. 2009. Innate immunity to virus infection. Immunol Rev 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang S, Dolganiuc A, Szabo G. 2007. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J Leukoc Biol 82:479–487. doi: 10.1189/jlb.0207128. [DOI] [PubMed] [Google Scholar]
- 8.Dolganiuc A, Oak S, Kodys K, Golenbock DT, Finberg RW, Kurt-Jones E, Szabo G. 2004. Hepatitis C core and nonstructural 3 proteins trigger Toll-like receptor 2-mediated pathways and inflammatory activation. Gastroenterology 127:1513–1524. doi: 10.1053/j.gastro.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Tian Y, Chan ST, Kim JY, Cho C, Ou JH. 2015. TNF-alpha induced by hepatitis C virus via TLR7 and TLR8 in hepatocytes supports interferon signaling via an autocrine mechanism. PLoS Pathog 11:e1004937. doi: 10.1371/journal.ppat.1004937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saito T, Owen DM, Jiang F, Marcotrigiano J, Gale M Jr. 2008. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao XZ, Ding Q, Lu J, Tao WY, Huang B, Zhao YN, Niu JQ, Liu YJ, Zhong J. 2015. MDA5 plays a critical role in interferon response during hepatitis C virus infection. J Hepatol 62:771–778. doi: 10.1016/j.jhep.2014.11.007. [DOI] [PubMed] [Google Scholar]
- 12.Kang JI, Kwon SN, Park SH, Kim YK, Choi SY, Kim JP, Ahn BY. 2009. PKR protein kinase is activated by hepatitis C virus and inhibits viral replication through translational control. Virus Res 142:51–56. doi: 10.1016/j.virusres.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 13.Wang N, Liang Y, Devaraj S, Wang J, Lemon SM, Li K. 2009. Toll-like receptor 3 mediates establishment of an antiviral state against hepatitis C virus in hepatoma cells. J Virol 83:9824–9834. doi: 10.1128/JVI.01125-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li XD, Sun L, Seth RB, Pineda G, Chen ZJ. 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc Natl Acad Sci U S A 102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferreon JC, Ferreon AC, Li K, Lemon SM. 2005. Molecular determinants of TRIF proteolysis mediated by the hepatitis C virus NS3/4A protease. J Biol Chem 280:20483–20492. doi: 10.1074/jbc.M500422200. [DOI] [PubMed] [Google Scholar]
- 16.Levine B, Kroemer G. 2008. Autophagy in the pathogenesis of disease. Cell 132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L, Tian Y, Ou JH. 2015. HCV induces the expression of Rubicon and UVRAG to temporally regulate the maturation of autophagosomes and viral replication. PLoS Pathog 11:e1004764. doi: 10.1371/journal.ppat.1004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ploen D, Hildt E. 2015. Hepatitis C virus comes for dinner: how the hepatitis C virus interferes with autophagy. World J Gastroenterol 21:8492–8507. doi: 10.3748/wjg.v21.i28.8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L, Ou JHJ. 2015. Hepatitis C virus and autophagy. Biol Chem 396:1215–1222. doi: 10.1515/hsz-2015-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi J, Lee KJ, Zheng Y, Yamaga AK, Lai MM, Ou JH. 2004. Reactive oxygen species suppress hepatitis C virus RNA replication in human hepatoma cells. Hepatology 39:81–89. doi: 10.1002/hep.20001. [DOI] [PubMed] [Google Scholar]
- 21.Sir D, Chen WL, Choi J, Wakita T, Yen TS, Ou JH. 2008. Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48:1054–1061. doi: 10.1002/hep.22464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lo SY, Masiarz F, Hwang SB, Lai MM, Ou JH. 1995. Differential subcellular localization of hepatitis C virus core gene products. Virology 213:455–461. doi: 10.1006/viro.1995.0018. [DOI] [PubMed] [Google Scholar]
- 23.Jiang J, Luo G. 2012. Cell culture-adaptive mutations promote viral protein-protein interactions and morphogenesis of infectious hepatitis C virus. J Virol 86:8987–8997. doi: 10.1128/JVI.00004-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23:33–42. doi: 10.1247/csf.23.33. [DOI] [PubMed] [Google Scholar]
- 25.Klionsky DJ. 2016. Stepping back from the guidelines: where do we stand? Autophagy 12:223–224. doi: 10.1080/15548627.2016.1139264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. 2009. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ 16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- 27.Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H, Hong L, Xie C, Li X, Zhao H, Liu Q, Jiang M, Chen Q, Zhang J, Li Y, Song S, Wang HR, Zhou R, Johnson RL, Chien KY, Lin SC, Han J, Avruch J, Chen L, Zhou D. 2015. Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat Immunol 16:1142–1152. doi: 10.1038/ni.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vilotti S, Codrich M, Dal Ferro M, Pinto M, Ferrer I, Collavin L, Gustincich S, Zucchelli S. 2012. Parkinson's disease DJ-1 L166P alters rRNA biogenesis by exclusion of TTRAP from the nucleolus and sequestration into cytoplasmic aggregates via TRAF6. PLoS One 7:e35051. doi: 10.1371/journal.pone.0035051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. 2000. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J 19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. 2005. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katsuragi Y, Ichimura Y, Komatsu M. 2015. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J 282:4672–4678. doi: 10.1111/febs.13540. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida H, Kato N, Shiratori Y, Otsuka M, Maeda S, Kato J, Omata M. 2001. Hepatitis C virus core protein activates nuclear factor kappa B-dependent signaling through tumor necrosis factor receptor-associated factor. J Biol Chem 276:16399–16405. doi: 10.1074/jbc.M006671200. [DOI] [PubMed] [Google Scholar]
- 33.Israelow B, Narbus CM, Sourisseau M, Evans MJ. 2014. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 60:1170–1179. doi: 10.1002/hep.27227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu H, Arron JR. 2003. TRAF6, a molecular bridge spanning adaptive immunity, innate immunity and osteoimmunology. Bioessays 25:1096–1105. doi: 10.1002/bies.10352. [DOI] [PubMed] [Google Scholar]
- 35.Sun LJ, Deng L, Ea CK, Xia ZP, Chen ZJJ. 2004. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 14:289–301. doi: 10.1016/S1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 36.Deng L, Wang C, Spencer E, Yang LY, Braun A, You JX, Slaughter C, Pickart C, Chen ZJ. 2000. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103:351–361. doi: 10.1016/S0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 37.Darnay BG, Ni J, Moore PA, Aggarwal BB. 1999. Activation of NF-kappaB by RANK requires tumor necrosis factor receptor-associated factor (TRAF) 6 and NF-kappaB-inducing kinase: identification of a novel TRAF6 interaction motif. J Biol Chem 274:7724–7731. doi: 10.1074/jbc.274.12.7724. [DOI] [PubMed] [Google Scholar]
- 38.Zampino R, Marrone A, Restivo L, Guerrera B, Sellitto A, Rinaldi L, Romano C, Adinolfi LE. 2013. Chronic HCV infection and inflammation: clinical impact on hepatic and extra-hepatic manifestations. World J Hepatol 5:528–540. doi: 10.4254/wjh.v5.i10.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Linares JF, Duran A, Yajima T, Pasparakis M, Moscat J, Diaz-Meco MT. 2013. K63 polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol Cell 51:283–296. doi: 10.1016/j.molcel.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Knodler LA, Celli J. 2011. Eating the strangers within: host control of intracellular bacteria via xenophagy. Cell Microbiol 13:1319–1327. doi: 10.1111/j.1462-5822.2011.01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sir D, Ou JHJ. 2010. Autophagy in viral replication and pathogenesis. Mol Cells 29:1–7. doi: 10.1007/s10059-010-0014-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sir D, Kuo CF, Tian Y, Liu HM, Huang EJ, Jung JU, Machida K, Ou JH. 2012. Replication of hepatitis C virus RNA on autophagosomal membranes. J Biol Chem 287:18036–18043. doi: 10.1074/jbc.M111.320085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul D, Hoppe S, Saher G, Krijnse-Locker J, Bartenschlager R. 2013. Morphological and biochemical characterization of the membranous hepatitis C virus replication compartment. J Virol 87:10612–10627. doi: 10.1128/JVI.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. 2011. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 53:406–414. doi: 10.1002/hep.24073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ke PY, Chen SS. 2011. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest 121:37–56. doi: 10.1172/JCI41474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yi M. 2010. Hepatitis C virus: propagation, quantification, and storage. Curr Protoc Microbiol Chapter 15:Unit 15D.1. doi: 10.1002/9780471729259.mc15d01s19. [DOI] [PMC free article] [PubMed] [Google Scholar]